
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutocatalytic methylthiomethylation of carboxylic acid/phenol involving the formation of DMSO enolate: convenient synthesis of methylthiomethyl ester/ether†
Hongshi Liuab,
Enhua Wangc,
Juan Yangab,
Mei Pengab,
Ming Gaoab,
Yangming Jiangab,
Enming Huab,
Guangyan Liangab,
Lishou Yang*ab and
Xiaosheng Yang *ab
*ab
aState Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University, Guiyang 550014, P. R. China. E-mail: gzcnp@sina.cn; 1039160204@qq.com
bThe Key Laboratory of Chemistry for Natural Products of Guizhou Province and Chinese Academy of Sciences, Guiyang 550014, P. R. China
cDepartment of Food and Medicine, Guizhou Vocational College of Agriculture, Qingzhen 551400, P. R. China
First published on 18th November 2022
Abstract
This work reported a simple and practical protocol for the preparation of methylthiomethyl (MTM) esters/ethers directly from carboxylic acid/phenol and dimethylsulfoxide (DMSO) as solvent and methylthiomethyl source. With different types of carboxylic acids/phenols the reactions underwent smooth transformation to afford the corresponding MTM esters/ethers in moderate to excellent yields. This method features catalyst-free, easy to operate, broad substrate scope, good functional group tolerance and involvement of the formation of DMSO enolate.
Introduction
Methylthiomethyl (MTM) groups exist in some functional molecules and have been used as important protecting groups for carboxylic acids.1 The MTM group could serve as activating group for the amidation of acids.2 MTM esters also used as flavor additives in some dairy and oil products.3 In addition, the compounds that contain MTM esters groups displayed varied bioactivities such as anti-viral,4 anti-inflammatory,5 and dopaminergic6 properties (Fig. 1). Consequently, the preparation of MTM esters have gained widespread attention.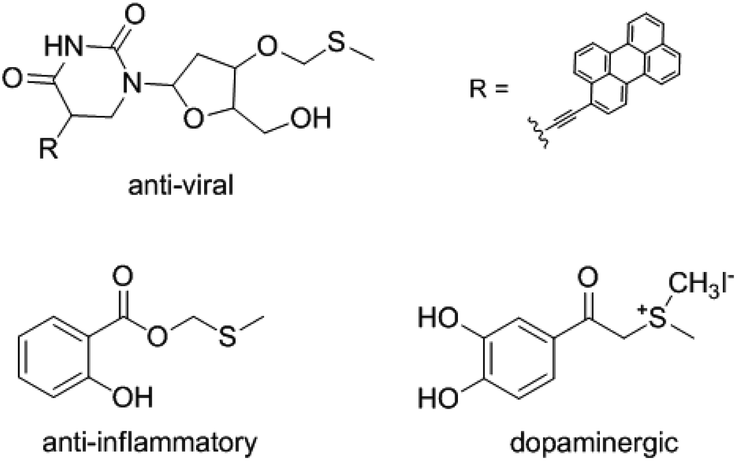 | ||
| Fig. 1 Selected bioactive MTM ester/ether. | ||
Traditionally, the MTM esters were synthesized from methylthiomethyl chloride (MTM-Cl) and carboxylic acid catalyzed by base and 18-crown-6.7 However, the application of this method was limited by the use of toxic reagents, MTM-Cl and 18-crown-6. Another typical strategy for the synthesis of MTM esters from activated DMSO and carboxylic acid via a Pummerer rearrangement have been explored. However, this methodology need using activating reagents, such as tert-butyl bromide,5 dicyclohexylcarbodiimide,8 sulfuryl chloride,9 and N-chlorosuccinimide.10
Recently, several procedures for the preparation of MTM esters using unactivated DMSO and carboxylic acids/acyl chlorides as the starting materials have been reported.11 Despite significant advances, these methods suffer some disadvantages such as need of microwave-assisted11a and the use of catalysts.11b–f Therefore, it is still desirable to develop new convenient, efficient and environment-friendly methods.
We found that MTM esters could be efficiently prepared from carboxylic acid and DMSO under traditional heating conditions. This methylthiomethylation involving the formation of DMSO enolate. Phenols were also compatible with this transformation to give corresponding MTM phenyl ethers which were commonly synthesized in the presence of base catalyst from phenols and DMSO/chloromethyl methyl sulfide.12 The method has several additional advantages such as easy to operate, broad substrate scope and good functional group tolerance.
Herein, we wish to report an autocatalytic methylthiomethylation of carboxylic acids/phenols involving the formation of DMSO enolate for the synthesis of MTM esters/ethers.
Results and discussion
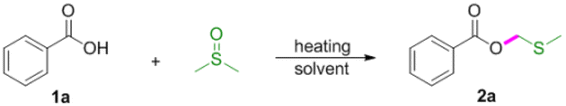
Initially, our studies were carried out with readily available benzoic acid 1a as the model substrate to optimize the reaction conditions. A mixture of 1a (1 equiv.) in DMSO refluxed for 2 h under an air atmosphere to give the desired product 2a in 23% yield (Table 1, entry 1). To further improve the reaction yield, we then investigated the impact of the reaction time. To our delight, the yield could be extremely improved when the reaction time was decreased and found that 15 min was the best choice (Table 1, entries 2–5). However, further decreasing the reaction time (10 min) had a reduction in yield (Table 1, entry 6). Thus it can be seen that this reaction depends mainly upon the reaction time. Decreasing the reaction temperature led to decreased isolated yields (Table 1, entries 7–8). Subsequently, various solvents such as DMF, 1,4-dioxane, THF and EtOH were screened, and no improvement in transformation was observed (Table 1, entries 9–12). Therefore, the optimized reaction conditions were determined as 1a (0.3 mmol) in DMSO (1 mL) refluxed for 15 min under an air atmosphere (Table 1, entry 5).
|
||||
|---|---|---|---|---|
| Entry | T (°C) | Time (min) | Solvent | 2ab (%) |
| a Reaction conditions: 1a (0.3 mmol), DMSO (1 mL), solvent (1 mL).b Isolated yield.c Reaction carried out in a pressure-resistant reaction bottle. | ||||
| 1 | Reflux | 120 | — | 23 |
| 2 | Reflux | 60 | — | 37 |
| 3 | Reflux | 30 | — | 70 |
| 4 | Reflux | 20 | — | 79 |
| 5 | Reflux | 15 | — | 85 |
| 6 | Reflux | 10 | — | 68 |
| 7c | 180 | 15 | — | 11 |
| 8 | 190 | 15 | — | 66 |
| 9 | Reflux | 15 | DMF | 12 |
| 10 | Reflux | 15 | 1,4-Dioxane | — |
| 11 | Reflux | 15 | THF | — |
| 12 | Reflux | 15 | EtOH | — |
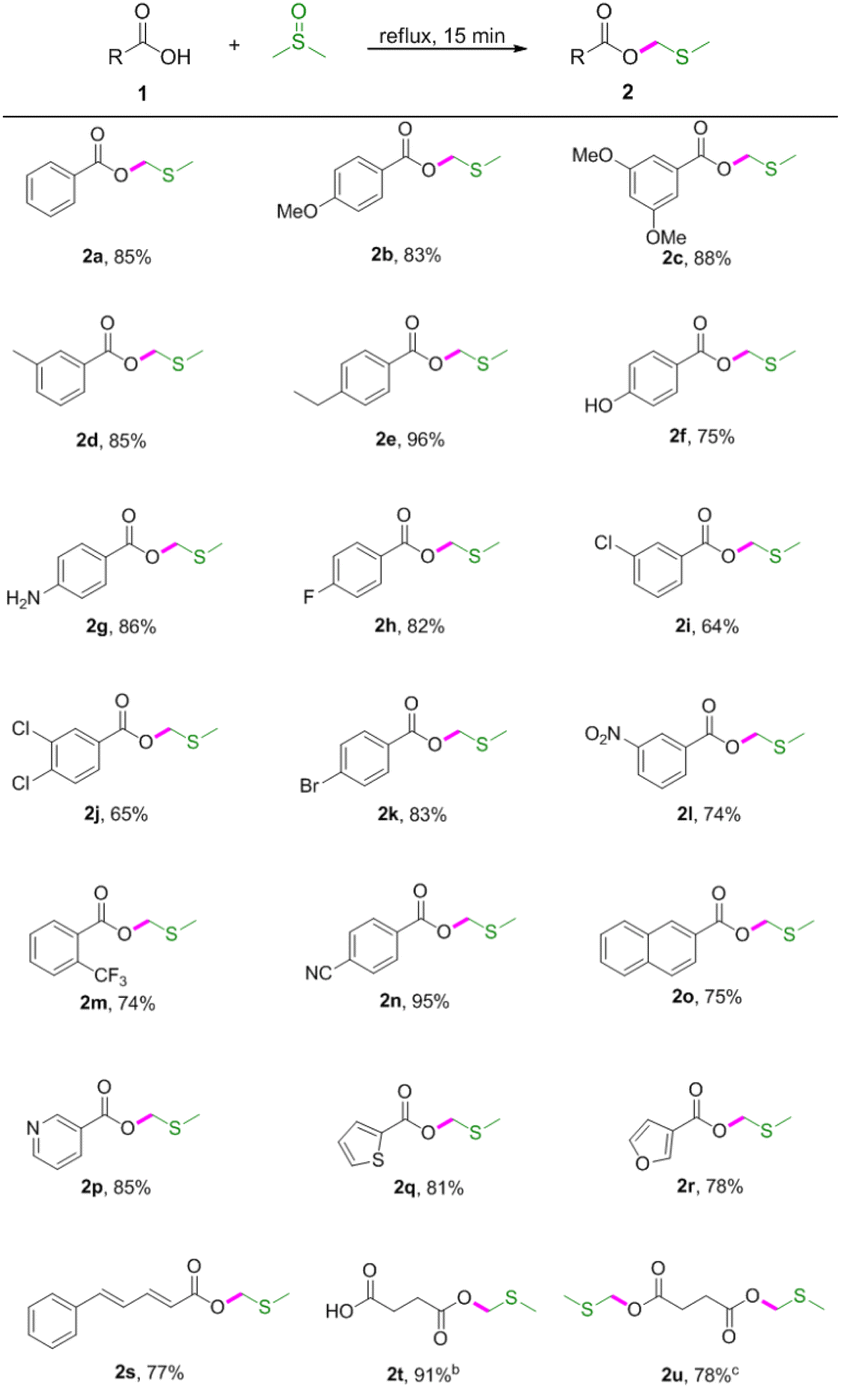
With the optimised conditions in hand, the scope of the substrates was explored by varying carboxylic acids 1 (Table 2). As shown in Table 2, this protocol was suitable for various carboxylic acids, including aromatic, aliphatic, unsaturated and heteroaromatic carboxylic acids. Specifically, electron-donating groups (OMe, Me, ethyl and NH2) on the aromatic acids provided excellent yields (Table 2, 2b–2e and 2g). However, the hydroxyl group had limited influence on the yield (Table 2, 2f). It was observed that the stronger electron-donating effect provided better yields (Table 2, 2b vs. 2c and 2d vs. 2e). Electron-withdrawing groups, such as F, Cl, Br, NO2 and CF3, led to lower yields (Table 2, 2h–2m). Cyano-substituted benzoic acid gave the desired product in 95% yield (Table 2, 2n). It was also found that this procedure was efficient for converting 2-naphthalenecarboxylic acid, niacin, 2-thiophenecarboxylic acid, 3-furoic acid, 5-phenyl-2,4-pentadienoic acid and succinic acid to the corresponding MTM esters in good yields (Table 2, 2o–2u), and that hydroxyl and amino groups could be tolerated (Table 2, 2f and 2g). Interestingly, the dimethylthiomethyl ester 2u was detected as the main product when prolonging the reaction time to 60 min.
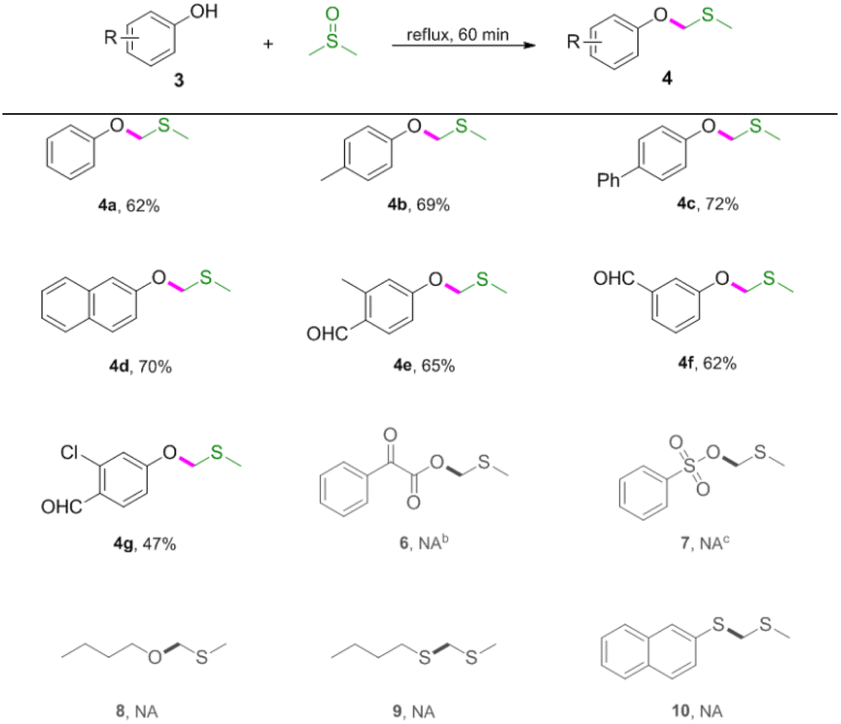
Next, reactions between phenols 3 and DMSO were carried out (Table 3). Phenols were also amenable to this autocatalytic methylthiomethylation to give MTM phenyl ethers in moderate yields (4a–4g, reaction condition optimization see Table S1†). We envisioned that the enol form of DMSO was generated in this transformation. In order to gain a clear insight into this reaction, the preliminary mechanistic studies were carried out (Scheme 1). The DMSO enolate 5 was detected from DMSO-d6 (Scheme 1, eqn (1)), and the desired product 2a was not detected when methyl benzoate 1ab was used (Scheme 1, eqn (2)). These observations collectively suggested that the enolization of DMSO was occurred in the presence of benzoic acid. Then, the radical scavenger TEMPO and BHT were employed for this reaction. The reactions were not inhibited which demonstrated that this procedure might rule out the radical pathway (Scheme 1, eqn (3)). The scope and limitation of the reaction was further explored regarding phenylglyoxylic acid, benzenesulfonic acid, n-butanol, n-butyl mercaptan and 2-naphthalenethiol. Unfortunately, corresponding MTM products were not detected (Table 3, 6–10).
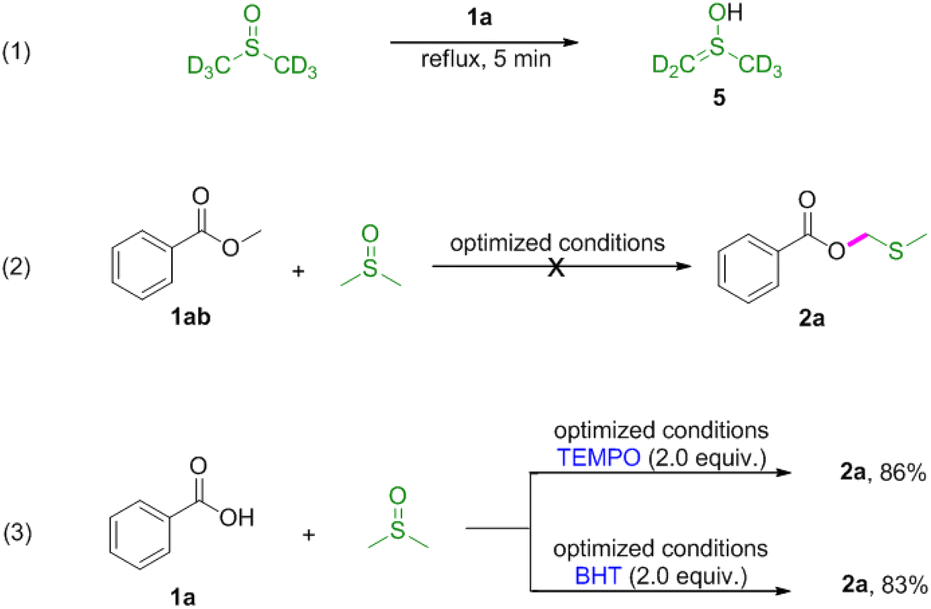 | ||
| Scheme 1 Control experiments. | ||
The important synthetic value of this method was further examined by its application in several biologically relevant molecules to prepare corresponding MTM esters (Table 4). Oleanolic acid and 1α,2β-dihydroxyursolic acid were compatible with the reaction, giving rise to the target MTM products 2v, 2w in good yields. Coumalic acid is a lactone compound that could be converted to MTM ester in good yield (Table 4, 2x). Ferulic acid also proved to give modulate yield of the desired product (Table 4, 2y). Furthermore, biotin and 6-carboxyoxindole were also applicable to the reaction system, and the desired products 2z and 2aa were assembled in 62% and 97% yields, respectively.
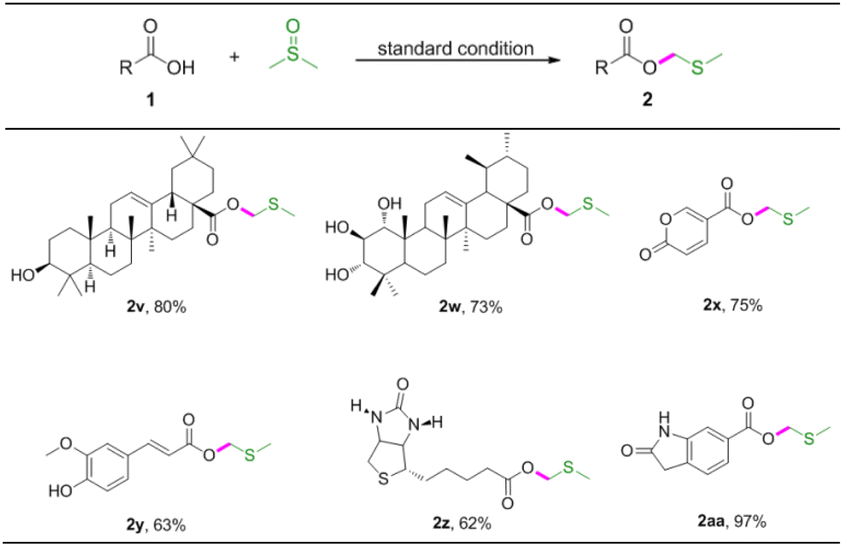 |
To demonstrated the synthetic potential of this methodology, reactions were conducted on gram scale (Scheme 2). The gram-scale reactions also proceeded well to afford corresponding products in moderate to good yields.
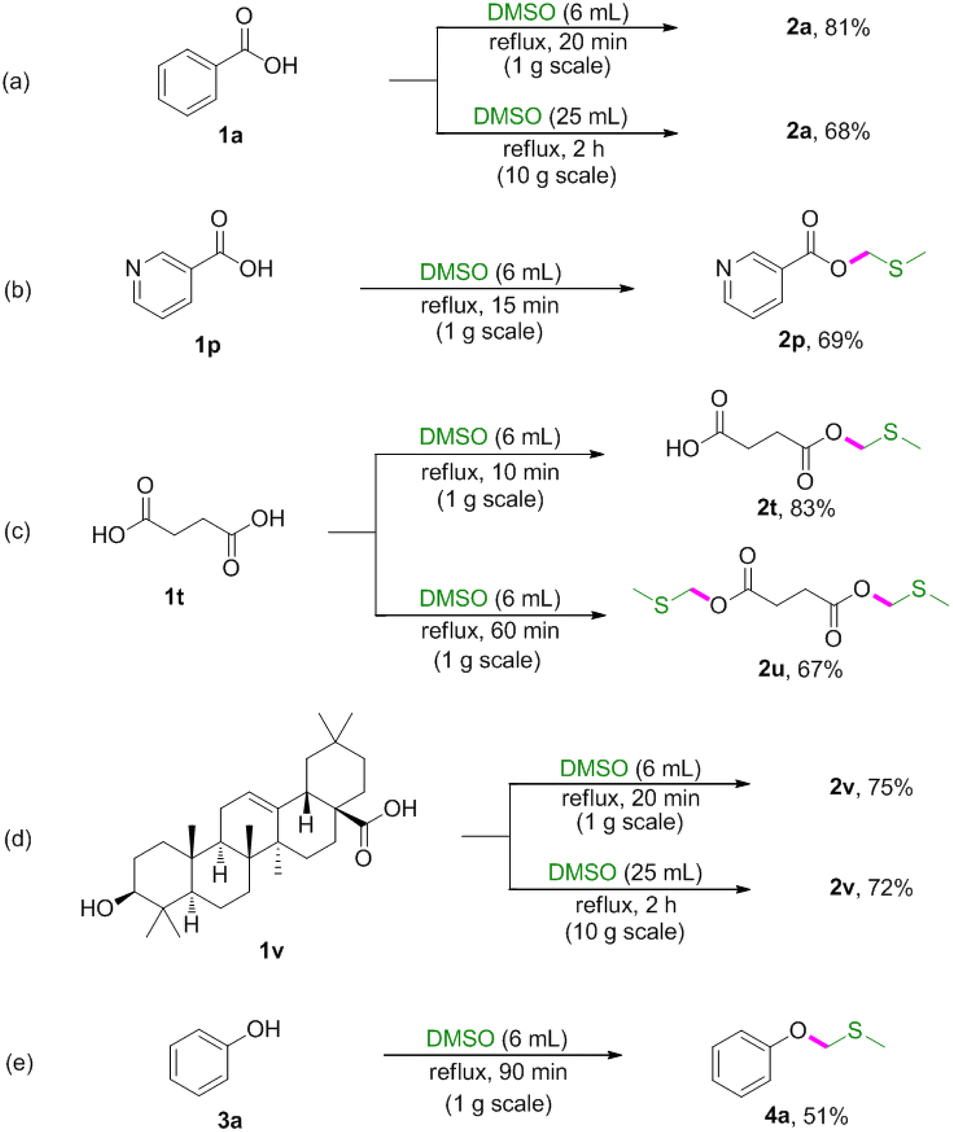 | ||
| Scheme 2 Gram-scale reactions. | ||
A plausible mechanism was proposed as described in Scheme 3. Intermediate 11 is formed from the enolization of DMSO, which after subsequent dehydration gives the thionium species 12. Then, intermediate 12 undergoes addition with carboxylate or phenoxy anion to produce 2a or 4a.
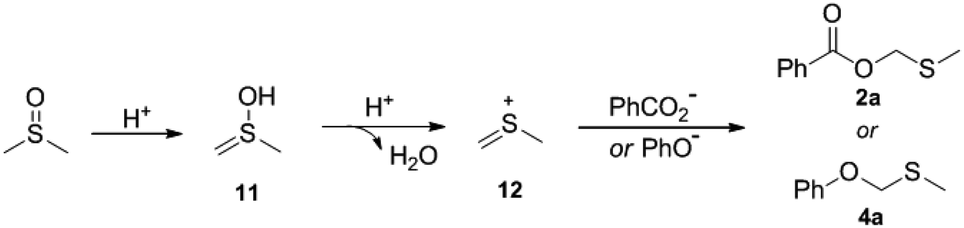 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In summary, herein we developed a convenient and practical procedure for the preparation of MTM esters/ethers directly from carboxylic acids/phenols and DMSO under traditional heating conditions. This autocatalytic methylthiomethylation showed a broad substrate scope and good tolerance for functional groups such as hydroxyl, amino and amide group. This protocol also features easy to operate, catalyst-free and involvement of the formation of DMSO enolate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the staff at the Key Laboratory of Chemistry for Natural Products of Guizhou Province and the Chinese Academy of Sciences. The work here was supported by the National Natural Science Foundation of China (No. 81860609 and 32160104), the Project of Guizhou Science and Technology Platform and Talent Team (No. QKHZC[2021] General 424), the Project of Key Laboratory for Characteristics of Colleges and Universities of Guizhou Provincial Department of Education (No. QJHKYZ[2020]018) and the Plan Project of Science and Technology in Guizhou Province [No. QKHFQ[2020]4013].Notes and references
- (a) T. L. Ho and C. M. Wong, J. Chem. Soc., Chem. Commun., 1973, 224 RSC; (b) S. Kim, Y. H. Park and I. S. Kee, Tetrahedron Lett., 1991, 32, 3099 CrossRef CAS.
- (a) U. Ghosh, R. Bhattacharyya and A. Keche, Tetrahedron, 2010, 66, 2148 CrossRef CAS; (b) J. Liu and Y. Tao, Tetrahedron, 1992, 48, 6739 CrossRef.
- (a) S. Bandenbooshu, E. B. Rando and S. Yan, JPS627820B2-02-19, 1987; (b) S. Vandenbosch, E. Van't Land and J. Stoffelsma, Chem. Abstr., 1980, 93, 113981 Search PubMed; (c) S. Vandenbosch, E. Van't Land and J. Stoffelsma, Chem. Abstr., 1984, 100, 84460 Search PubMed; (d) M. Moir, I. M. Gallacher, J. Hobkirk, J. C. Seaton and A. Suggett, Tetrahedron Lett., 1980, 21, 1085 CrossRef CAS.
- G. V. Proskurin, A. A. Orlov, V. A. Brylev, L. I. Kozlovskaya, A. A. Chistov, G. G. Karganova and A. V. Aralov, Eur. J. Med. Chem., 2018, 155, 77 CrossRef CAS.
- T. Loftsson, J. J. Kaminski and N. Bodor, J. Pharm. Sci., 1981, 70, 743 CrossRef CAS.
- K. Anderson, A. Kuruvilla, N. Uretsky and D. D. Miller, J. Med. Chem., 1981, 24, 683 CrossRef CAS.
- L. G. Wade, J. H. Gerdes and R. P. Wirth, Tetrahedron Lett., 1978, 8, 731 CrossRef.
- K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1965, 87, 5661 CrossRef CAS.
- T. L. Ho, Synth. Commun., 1979, 9, 267 CrossRef CAS.
- A. Dossena, R. Marchelli and G. Casnati, J. Chem. Soc., Chem. Commun., 1979, 370 RSC.
- (a) A. McCarthy, R. Spatney, M. Manpadi, B. J. Myers and J. R. Zimmerman, Tetrahedron Lett., 2012, 53, 4782 CrossRef CAS; (b) H. Xing, L. Chen, Y. Jia, Z. Jiang and Z. Yang, Tetrahedron Lett., 2017, 58, 2199 CrossRef CAS; (c) S. Yu, K. C. Nam and S. Lee, Bull. Korean Chem. Soc., 2018, 39, 906 CrossRef CAS; (d) S. Wang, Z. Fu, Y. Jiang, Y. Liang and H. Cai, Synth. Commun., 2019, 49, 950 CrossRef CAS; (e) B. B. P. de Puga Carvalho, A. A. P. Amaral, P. P. de Castro, F. C. M. Ferreira, B. A. C. Horta and G. W. Amarante, Org. Biomol. Chem., 2020, 18, 5420 RSC; (f) S. B. Jadhav and U. Ghosh, Tetrahedron Lett., 2007, 48, 2485 CrossRef CAS.
- (a) R. A. Holton and R. G. Davis, Tetrahedron Lett., 1977, 6, 533 CrossRef; (b) W. Du, W. Liu, X. Ma, H. Cheng and Y. Jiang, Synth. Commun., 2019, 49, 2572 CrossRef CAS; (c) L. A. Oparina, O. V. Vysotskaya, N. A. Kolyvanov, N. K. Gusarova and B. A. Trofimov, Russ. J. Org. Chem., 2014, 50, 1216 CrossRef; (d) A. Dossena, R. Marchelli and G. Casnati, J. Chem. Soc., Perkin Trans. 1, 1983, 1141 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06618a |
| This journal is © The Royal Society of Chemistry 2022 |