DOI:
10.1039/D2RA06572J
(Paper)
RSC Adv., 2022,
12, 35794-35808
Experimental and theoretical study on the regioselective bis- or polyalkylation of 6-amino-2-mercapto-3H-pyrimidin-4-one using zeolite nano-gold catalyst and a quantum hybrid computational method†
Received
18th October 2022
, Accepted 3rd December 2022
First published on 15th December 2022
Abstract
The synthetic utility of 6-amino-2-mercapto-3H-pyrimidin-4-one 3 as building blocks for new poly (pyrimidine) by alkylation using the bis(halo) compounds and zeolite nano-gold as a catalyst was investigated. Furthermore, the experimental findings by the theoretical Density functional theory (DFT) computations at the DFT/B3LYP level of theory, utilizing the 6-311++G (d,p) basis set in the gas phase, were used to investigate the distinct phases for Regio isomer 11a & 12a and 11b & 12b compounds was fair and of good quality. The stability of the 12a and 12b phases is higher than the other Regio isomer 11a and 11b phases, according to DFT modelling. By computing HOMO and LUMO pictures, the electronic parameters: dipole moment of these compounds in the ground state were theoretically investigated. Non-linear optical (NLO) characteristics and quantum chemical parameters were examined using frontier molecular orbital (FMO) analysis. Natural bond orbital analysis was used to characterize the charge transfer of the electron density in the investigated compounds (NBO). The molecular electrostatic potential surfaces (MEPS) plots have been generated, and absorption spectral analysis in different solvents has been theoretically and experimentally examined to better understand the reactivity spots. At the B3LYP/6-311G (d,p) level of theory, thermodynamic properties were also calculated. Finally, DFT calculations were used to connect the structure–activity relationship (SAR) with real antibacterial results for compounds 12a and 12b.
1 Introduction
Pyrimidines are one of the most important heterocycles with impressive pharmacological properties.1 According to a review of the literature, compounds with pyrimidine nuclei have a wide range of pharmacological activities, including antifungal,1,2 anticancer,3,4 antioxidant,5 antidiabetic,6 antiviral,7 antihypertensive,8,9 and anti-inflammatory properties.10 In the last two decades, several pyrimidine derivatives have been reported as antibacterial drugs.11–18 As a result, the continued development of diverse pyrimidine derivative compounds remains in high demand. Some previously published protocols for the formation of analogues of the target moiety using acid,19–21 basic,22 metal catalysts,23 or catalyst-free protocols under microwave24 or thermal conditions. Unfortunately, all previous methods produce very low yields; thus, it was our goal to develop new conditions based on the use of a nanocatalyst in the hope of improving yields. The use of a nanocatalytic system would enable rapid and selective chemical transformations, as well as easy catalyst separation and recovery.25 The contact between the reactants and the catalyst is greatly increased by using a nano-size catalyst (high surface area) (this phenomenon is close to homogeneous catalysis).26,27 Because the catalyst is insoluble in the reaction solvent, the process is heterogeneous, and thus the catalyst can be easily separated from the reaction mixture (this phenomenon is close to heterogeneous catalysis).28–31 Metal NPs' catalytic properties are known to be size and shape dependent.32–36
It has been reported that gold nanoparticles are stable and well dispersed on a variety of supports (metal oxides,37–42 carbon materials,43–51 metal-organic frameworks,52–54 zeolites,55 modified aluminium,43,55–58 ionic liquids,59 and so on) and can successfully catalyze several reactions. Several studies60–64 have shown that zeolites have a wide range of applications as catalysts and adsorbents. These microporous materials are three-dimensional and crystalline hydrated alumino silicates,65 and they are highly rigid under dehydration as well as various aggressive environmental actions.66 Zeolite's important structural, physical, and chemical properties, with tailored channels and cavities on the molecular scale, make them versatile and valuable as adsorbents and catalysts in industrial, agricultural, and environmental applications.67 Furthermore, zeolite nano gold has high thermal stability and serves a dual purpose in stabilizing nanoparticles against sintering, and their distinct pore structure can facilitate shape-selective catalysis. For cyclohexane oxidation, zeolite nano-shell encapsulating gold nanoparticles have been successfully used, and these catalysts show better conversion with increased reusability.55
All these properties promoted us to investigate how we might employ zeolite with nano-gold (Fig. 2) in process of making thiazolo[3,2-a] pyrimidine 12a and pyrimido[2,1-a][1,3]thiazine 12b through the simple reaction of 6-amino-2-mercapto-3H-pyrimidin-4-one (3) with the appropriate diabromoalkane 4a, 4b (Scheme 2). Transmission electron microscopy (TEM) and X-ray diffraction (TEM) confirmed the catalyst structure and morphology. Because DFT and computational methods for predicting the electronic structure of molecular systems have excellent accuracy, experimental values of molecular geometry, vibrational frequencies, atomic charges, dipole moment, thermodynamical properties, and so on may be reproduced.68–73
The experimental and computational vibrational spectroscopy on compounds 12a and 12b has not been published in the literature, thus the study examines the thermodynamic, and electrical properties of compounds 12a and 12b. In continuation of our prior work,74–76 which aimed to investigate the characteristics of chemicals 12a and 12b and predict their uses. We use bond natural orbital (NBO) charge delocalization and chemical shift (NMR), FT-IR, and vibrational investigations to provide a broad description of the chemical reactivity of compounds 12a and 12b. The HOMO-LUMO energy gap, chemical hardness, and chemical potential were estimated as electronic characteristics. Furthermore, utilizing CAM-B3LYP/6-311++G (d,p), the potential molecular electrostatic (MEP) and UV-visible analysis of compounds 12a and 12b were investigated using theoretical and experimental calculations to yield spectra and electronic structure of compounds 12a and 12b. The electronic dipole moment (μ), first order hyperpolarizability (β), hyper-Rayleigh scattering (βHRS), and the depolarization ratio (DR) were derived on the same level of theory as the NLO non-linear optical characteristics. The DFT method was used to perform all computations in this study at the B3LYP/6-311++G (d,p) theoretical level. The antibacterial application was also used to investigate the structure–activity relationship (SAR) for the present compounds 12a and 12b.
2 Experimental
2.1. General
Melting points were measured on Gallenkamp electrothermal melting point apparatus and are uncorrected. IR spectra were recorded as KBr discs using a Shimadzu FTIR-prestige 21 spectrophotometers. 1H NMR spectra were recorded in DMSO-d6 as solvents at 300 MHz on a Varian Mercury NMR spectrometer using TMS as internal standard. Chemical shifts δ are reported in parts per million units (ppm), and J values are given in hertz. The mass spectra were recorded on a GCeMS-QP1000 EX mass spectrometer at 70 eV. Elemental analyses were carried out at the Micro-analytical Centre of Cairo University, Giza, Egypt.
22. Synthesis of thiazolo[3,2-a]pyrimidine 12a and pyrimido[2,1-a][1,3]thiazine 12b
The dibromoalkanes 4a, 4b (2 mmol) was added to a solution of 6-amno-2-thioxo-2 3-dihydro-1H-pyrimidin-4-one 3 (2 mmol), KOH (20 mmol), or zeolite nano-gold (20 mmol) in ethanol (10 mL). After a period of one hour of refluxing. The mixture was allowed to cool to room temperature. After that, it was poured over crushed ice and the precipitate obtained was filtered, dried, and purified by recrystallization.
2.2.1 7-Amino-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidin-5-one (12a). Yellow needles crystals (77%; 81% yield); (DMF), mp 284 °C; IR (KBr) ν 3450, 3250, 1695 cm−1; 1H NMR (DMSO-d6) δ 3.57 (t, 2H, CH2 J = 7.6 Hz), 4.24 (t, 2H, CH2 J = 7.3 Hz), 5.98 (s, 1H), 7.22 (br., s, 2H, NH2); 13C NMR (DMSO-d6) δ 26.9, 49.6, 82.6, 157.6, 163.5, 168.3; MS m/z (%) 171,170, 169, 142, 84, 55, 54, 28. Anal. calcd for C6H7N3OS (169.2) C, 42.59; H, 4.17; N, 24.83; S, 18.95. Found: C, 42.48; H, 4.09; N, 24.77; S, 18.81.
2.2.2 8-Amino-3,4-dihydro-2H,6H-pyrimido[2,1-a][1,3]thiazin-6-one (12b). Yellow needles crystals (79%; 83% yield); (DMF), mp 294 °C; IR (KBr) ν 3465, 3272, 1701 cm−1; 1H NMR (DMSO-d6) δ 2.22–2.29 (m, 2H, CH2), 3.27 (t, 2H, CH2 J = 5.7 Hz), 4.12 (t, 2H, CH2 J = 5.4 Hz), 6.22 (s, 1H), 7.37 (br., s, 2H, NH2); 13C NMR (DMSO-d6) δ 23.1, 27.3, 46.9, 85.1, 161.6, 163.3, 168.7; MS m/z (%) 184, 183, 182, 143, 109, 68, 54, 43, 28. Anal. calcd for C7H9N3OS (183.2) C, 45.89; H, 4.95; N, 22.93; S, 17.50. Found: C, 48.78; H, 4.79; N, 22.77; S, 17.61.
2.3. Synthesis of nano Au zeolite
In a typical synthesis procedure, an exact amount of NaOH solution was added to Pyramid sodium silicate (Crossfield chemicals) under stirring at room temperature, then a mixture solution of NaAlO2 and TPAOH were added individually with vigorous stirring. The reaction kept under stirring for at least 2 hours and then transferred into a 250 mL Teflon-lined steel autoclave. Reaction kept at 170 °C under pressure for 24 h. The product was filtered, rinsed with deionized water, dried, and calcined at 540 °C for 8 hours to remove the template. A certain amount of gold was added, and stirred for 30 min, 2 g of calcined zeolite was flooded in the above solution after heat treatment at 150 °C under vacuum over-night. The total mixture was left at 80 °C under vigorous stirring for 6 hours under reflux. Mixture was then transferred to rotary evaporator and waster was completely evaporated. The obtained composite dried at 70 °C for 8 hours in air.77
2.4. Characterization of nano Au zeolite
The particle size and the morphology of the prepared catalysts were characterized by High Transmission Electron Microscope (TEM) (Philips). The measuring mode of the sample in TEM instrument depends on its suspension in water followed by ultrasonication for 600 s in ultra-8050-H Clifton. It was then applied in the TEM instrument on 100 mesh copper grades coated with carbon. Powder X-ray diffraction (XRD) patterns were recorded with an Analytical: X'Pert PRO diffractometer using Cu Kα radiation source for the investigation of crystalline structure and phase.
2.5. Physicochemical characterization of nanocomposite
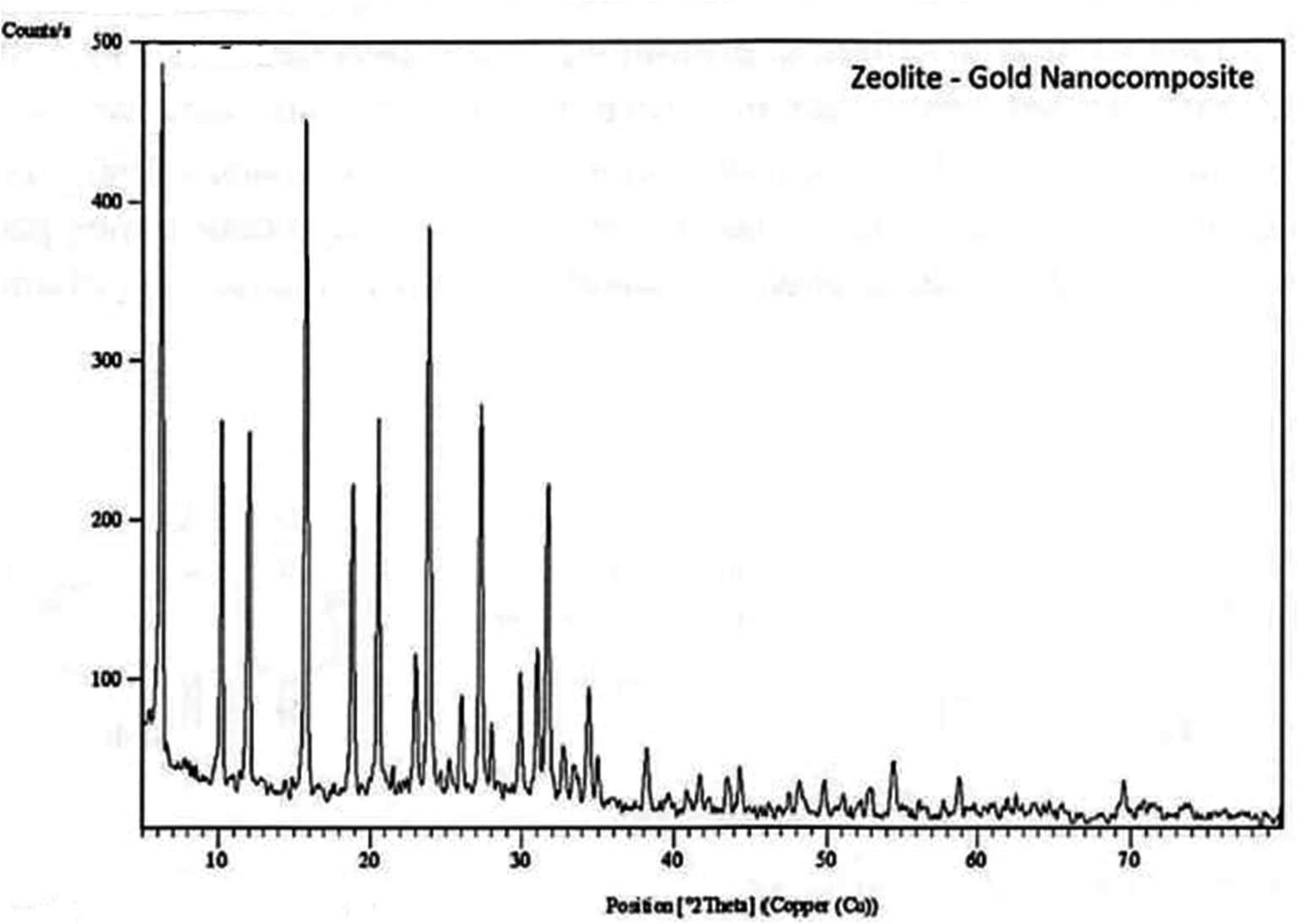
Low-angle XRD patterns corresponding to the prepared zeolite Au nanocomposite (Fig. 1). The sum of reflection intensities at 2θ of 15.5°, 24.0°, 28.0°, 32.0° and 63.0° corresponding to (4 5 0), (3 09), (2 0 7), and (202) planes of cubic crystal system as shown in (Fig. 1) for prepared nano-gold indicate the formation of cubic crystal of zeolite nano Au, the intensities of the peaks sample are relatively high as indication of high crystallinity. The formed zeolite is a mixture of sodium aluminum silicate and sodium aluminum oxide silicates, which is confirmed by standard data for zeolite (ref's: 00-045-0437 & 01-076-0591).
 |
| | Fig. 1 XRD patterns of zeolite-gold nanocomposite. | |
 |
| | Fig. 2 TEM micrographs nanocomposite formed by zeolite doped nanogold. | |
The preparation method followed in this work produced a uniformly dispersion of small particles around 4–6 nm of gold nanoparticles on zeolite as shown in TEM images in (Fig. 2). The formation of small nanoparticles may help the incorporation of gold nanoparticles within zeolite framework as also indicated by XRD measurements.
2.6. Solvents
Methanol, DMSO, and 1,2-dichloromethane polar solvents and 1,4-dioxane non-polar solvents were employed without purification in Merck, AR-grade.
2.7. Apparatus
A PerkinElmer lambda 4B spectrophotometer with 1.0 cm fused quartz cells was used to detect the absorption electronic spectra in the region 200–900 nm.
2.8. Antimicrobial study
Antibacterial and antifungal capabilities of produced compounds 12a and 12b were investigated against several bacteria, including Gram-positive S. aureus and B. subtilis, and Gram-negative S. typhimurium and E. coli, as well as yeast, C. albicans. A. fumigatus is a species of A. fumigatus.
2.9. Computational details
Using “A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP),” at the 6-311++G (d,p) bases set, the maximum excitation wavelength (λmax) and relative intensities (oscillator strengths, f) of the electronic transition properties were calculated using the time dependent density functional theory (TD-DFT)78–82 is used to optimize the geometry and other parameters of quantum chemistry. As a result, in this paper, the B3LYP approach was employed to execute quantum calculations using the Gaussian 09 program83 and the Gauss View 5.084 or chem craft 1.685 software packages molecular visualization program. From the literature,86–88 the total static dipole moment (μ), 〈Δα〉, 〈β〉, βHRS, and DR values were determined.
2.10. Global descriptors
Electron Affinity (AE) = −ELUMO, Ionization Potential (PI) = −EHOMO calculate the energies of the border orbital in the context of Koopmans' theorem's validity.89 Chemical hardness (η) = (PI − AE)/2, electronegativity (X) = (PI + AE)/2, chemical potential (V) = −(PI + AE)/2, electrophilicity (ω) = μ2/2η and global softness (S) = 1/2η values were calculated in Table 2. The ability of a molecule to take more electrons in an unoccupied molecular orbital with the lowest LUMO value. The HOMO and LUMO energies, as well as gap energy, can be used to forecast the evolution of reactivity and chemical stability of molecules 12a and 12b. The energy gap (ΔE) lower value indicates a greater reactivity of the molecule and conversely low chemical stability.
2.11. NBO (natural bond orbital)
NBO intra and intermolecular interactions between donors and acceptors are given in NBO analysis between (full Lewis or binding NBO and empty Lewis or anti-binder) and their energy is estimated using second order perturbation theory.90,91 According to the equation below, the stabilization energy E(2)91 associated with electron delocalization between the donor NBO (i) of electrons and the acceptor NBO (j) of electrons is calculated according to the equation below.| | |
E(2) = ΔEij = qi(F(ij)2/εj − εi),
| (1) |
F(ij) is an element of the off-diagonal NBO Fock matrix, qi represents the occupation of the donor orbital, εi and εj are the energies of the diagonal elements of NBO orbitals of the acceptor and the donor, respectively.
3 Result and discussion
3.1. Chemistry
In the present study the reaction of 6-amino-2-mercapto-3H-pyrimidin-4-one 3 with the suitable dibromoalkanes 4a, 4b in catalytic medium is investigated. Compound 3 was produced using the well-known method of reacting thiourea and ethyl cyanoacetate in the presence of sodium ethylate (Scheme 1).92
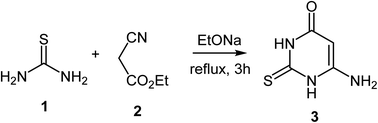 |
| | Scheme 1 Synthesis of 6-amino-2-mercapto-3H-pyrimidin-4-one 3. | |
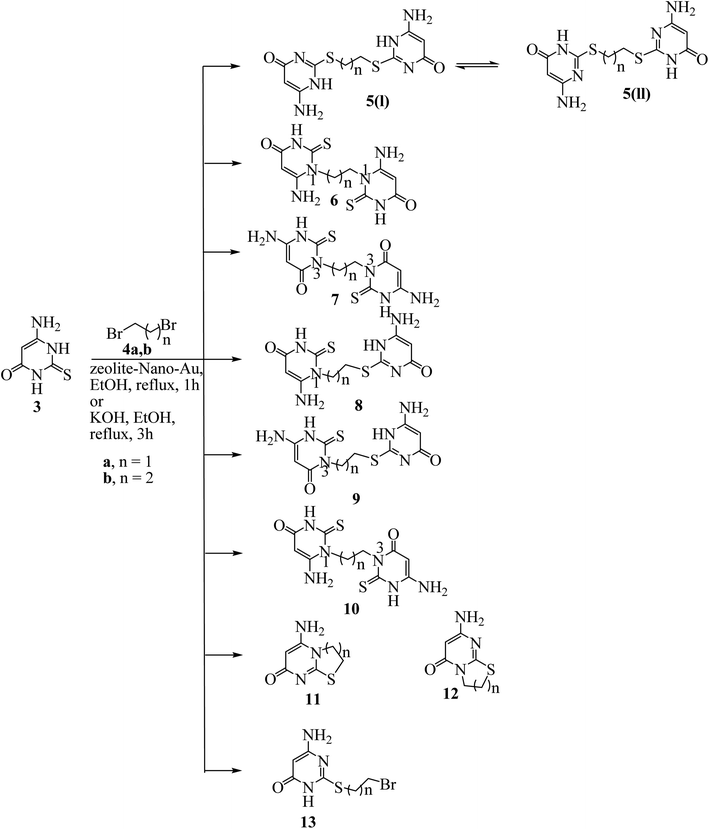 |
| | Scheme 2 Possible reaction products from the reaction of 3 with the appropriate dibromo alkane. | |
Our research started with a test of zeolite-nano-gold catalyst in a model reaction of 6-amino-2-mercapto-3H-pyrimidin-4-one 3 with 1,2-dibromoethane (4a) (Scheme 2). The reaction was carried out in ethanol, methanol, isopropyl alcohol, and DMF, as different solvents (Table 1). Among the solvents tested, ethanol proved to be the most effective, yielding the highest product yield (Table 1, entry 2). After complete consumption of the starting material, TLC analysis of the reaction mixtures revealed the existence of only one component, which was detected after conventional workup, which was proposed to be the symmetrical bis-derivatives (S-, S-), (1N-,1N-), or (3N-, 3N-), or the asymmetrical derivatives (S-, 1N-), (S-, 3N-) or (1N-, 3N-). Reaction at sulfur accompanied by cyclization at nitrogen (1-N-) or nitrogen (3-N-) or not was one of the potential reaction products (Scheme 2).
Table 1 Catalyst and solvent effects on the reaction of 6-amino-2-mercapto-3H-pyrimidin-4-one 3 with 1,2-dibromoethane 4aa
| Run |
Catalyst |
Solvent |
Yield% |
| Reaction conditions: 6-amino-2-mercapto-3H-pyrimidin-4-one/1,2-dibromomethane/catalyst/solvent (10 mL): 2/2/1 mmol, at reflux temp. (1 h). |
| 1 |
Zeolite-nano-Au |
MeOH |
66 |
| 2 |
Zeolite-nano-Au |
EtOH |
81 |
| 3 |
Zeolite-nano-Au |
Isopropyl alcohol |
71 |
| 4 |
Zeolite-nano-Au |
DMF |
78 |
| 5 |
KOH |
MeOH |
55 |
| 6 |
KOH |
EtOH |
77 |
| 7 |
KOH |
Isopropyl alcohol |
67 |
| 8 |
KOH |
DMF |
74 |
| 9 |
NaOEt |
EtOH |
71 |
| 10 |
K2CO3 |
EtOH |
73 |
| 11 |
Pyridine |
Pyridine |
42 |
Table 2 The optimized calculations of different Regio isomer 11a & 12a and 11b & 12b compounds at the B3LYP/6-311++G(d,p)
| Parameters |
11a |
12a |
11b |
12b |
| Total energy, (ET) (a.u) |
−870.680 |
−870.709 |
−909.999 |
−910.030 |
| Energy of highest occupied molecular orbital (EHOMO) (eV) |
−6.38466 |
−6.16760 |
−6.33216 |
−6.03867 |
| Energy of lowest unoccupied molecular orbital (ELUMO) (eV) |
−1.05536 |
−1.07766 |
−1.03170 |
−1.03061 |
| Energy gap, (Eg) (eV) |
5.32930 |
5.08994 |
5.30046 |
5.00806 |
| Dipole moment, (μ) (Debye) |
10.2769 |
2.4774 |
10.7446 |
2.7461 |
| I (eV) |
6.38466 |
6.16760 |
6.33216 |
6.03867 |
| A (eV) |
1.05536 |
1.07766 |
1.03170 |
1.03061 |
| X (eV) |
3.72001 |
3.62263 |
3.68193 |
3.53464 |
| V (eV−1) |
−3.72001 |
−3.62263 |
−3.68193 |
−3.53464 |
| η (eV) |
2.66465 |
2.54497 |
2.65023 |
2.50403 |
| S (eV−1) |
0.18764 |
0.19646 |
0.18866 |
0.19968 |
| ω (eV) |
2.59668 |
2.57831 |
2.55763 |
2.49471 |
The regioselectivity was determined by NMR spectroscopy and supported by theoretical calculation at DFT level.
The 1H-NMR spectrum of the reaction product shows two triplet signals at d 3.57 and d 4.24, both integrating for two protons characteristic of the two methylene groups of the ethylene spacer, as well as a singlet signal at 5.38 integrating for the methine proton of the pyrimidine ring. Moreover, the 13C NMR spectrum displayed two methylene carbons at δ 26.9 and at δ 49.6, respectively, characteristic for the methylene carbons attached to sulfur and nitrogen atoms.
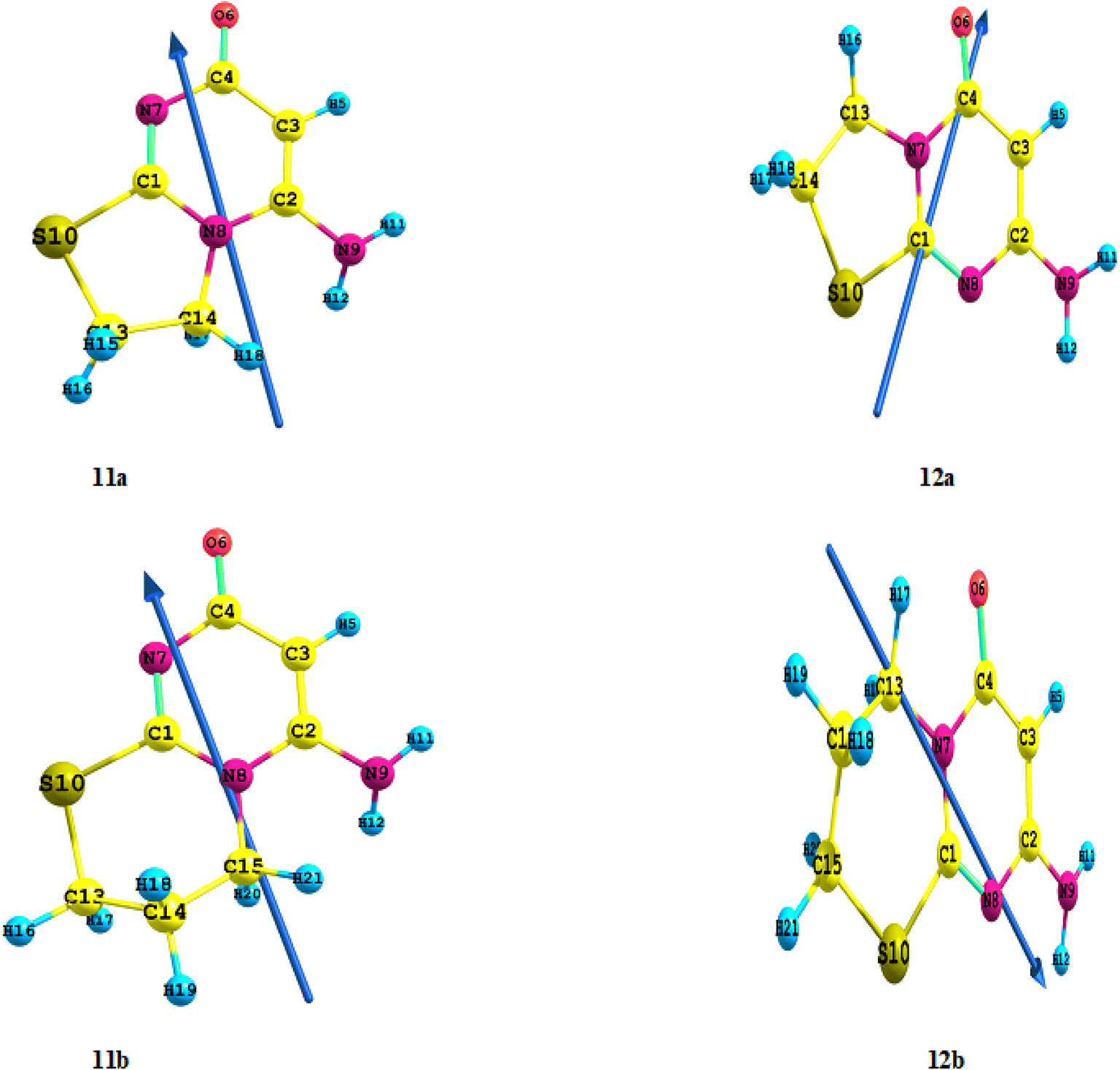
These findings support the presence of the fused compounds 11a or 12a thus ruling out all other bis-derivatives. Mass spectrometry revealed a molecular ion peak at 169 (34.8, M) corresponding to the mass of compounds 11a or 12a, providing further confirmation of the proposed structure (Fig. 3) (c.f. Section 2.2). The formation of the Regio isomer 12a is strongly supported by DFT calculations (c.f. Section 3.2).
 |
| | Fig. 3 Possible cyclic products from the reaction of 3 with 1,2-dibromoethane. | |
Next, we looked at several basic conditions, such as K2CO3 in ethanol, sodium ethoxide in ethanol, KOH in (ethanol, methanol, isopropyl alcohol, and DMF) and pyridine, which acted as both a catalyst and a solvent (Table 1, entries 5–11). The reactions produced regioselectivity, the same alkylated product in all cases, though the reaction was most efficiently carried out with KOH in ethanol under reflux for 3 hours and zeolite-nano-gold catalyst in ethanol under reflux condition for 1 h.
When 3 was refluxed with 1,3-dibromopropane (4b, n = 2) under the same reaction conditions, similar results were obtained. The structure of reaction product has been suggested as 11b or 12b (Fig. 4). (c.f. Section 2.2). DFT calculations confirm the development of Regio isomer 12b once more (c.f. Section 3.2).
 |
| | Fig. 4 Possible cyclic products from the reaction of 3 with 1,3-dibromopropane. | |
3.2. Molecular orbital calculations
To better understand the observed high activity and reactivity for different Regio isomer 11a or 12a and 11b or 12b compounds (c.f. Fig. 3 and 4) toward Regio isomer formation, we achieved a DFT study at the DFT/B3LYP level of theory using the 6-311++G (d,p) as a basis set into the interaction between compound 3 and 1,2-dibromoethane by the various catalyst compounds, the total energies ET of the four Regio isomers were determined in this reaction, and their values were presented in Table 2. The stability of the compounds increased as the estimated ET (a.u) decreased. The order of increasing activity in Table 2 was 12a > 11a and 12b > 11b, indicating that the 12a and 12b compounds had a strong chance of being more active solids with high stability and separated formation in the reaction media. “Furthermore, the computed energy gap differences between 12a and 11a, is 23.1 kJ mol−1 (5.52 kcal mol−1) while 12b and 11b is 28.2 kJ mol−1 (6.74 kcal mol−1)”. So that compounds 12a and 12b have higher reactivity than compounds 11a and 11b. In summary, the global descriptors revealed that among the 11a and 11b compounds investigated, while the compounds 12a and 12b are the highly reactive, the more stable, the softest, and have the greatest electronic exchange capacity. Furthermore, the estimated dipole moment (μ) (Debye) for compounds 12a and 12b is lower than for compounds 11a and 11b, and the order of dipole moment increases; 11b > 11a > 12b > 12a; the direction is also the same (c.f. Fig. 5). Furthermore, in contrast to compounds 11a and 11b, where the charge density of the HOMO is localized on all the moiety of the compound, all the charge density of the HOMO is focused on the pyrimidine moiety in 12a and pyrimido moiety in 12b (c.f. Fig. S1†). Furthermore, the high stability of the synthesized compounds 12a and 12b as determined by computed total energy agrees well with data obtained from mass spectrometry of this compound, which revealed the molecular ion peak as a base peak, indicating the stability of the synthesized compounds 12a and 12b. At the DFT/B3LYP level of theory, using the 6-311++G (d,p) as a basis set by Gaussian 09 program package, these theoretical studies were also used for the first time to calculate the geometry, global, ground state properties, natural bond orbital (NBO), thermodynamics, and non-linear optical (NLO) properties of different Regio isomer compounds 12a and 12b that were related to the stability.
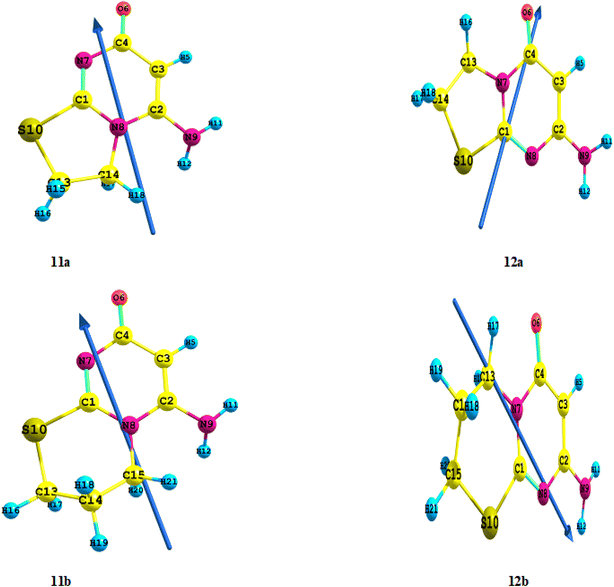 |
| | Fig. 5 Optimized geometry, numbering system, and vector of dipole moment for different Regio isomer 11a & 12a and 11b & 12b compounds at the B3LYP/6-311++G(d,p). | |
3.3 Electronic structures
3.3.1. Geometry structure. The geometrically optimized and ground state energies of the compounds 12a and 12b determined using the B3LYB/6-311++G (d,p) level are shown in Fig. 5 and Table S1.† The optimized bond length of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the phenyl ring falls in the range of 1.360 to 1.479 Å, which is in good agreement with the experimental data 1.481 Å, while the optimized bond length of CO is slightly lower than the experimental data 1.229 Å, Exp.93–95. The existence of a CO group in C4 has a significant impact on the computed bond angles in two compounds 12a and 12b, particularly <C4C3O6 in compound 12a is 128.98° and compound 12b is 128.90°; <C4N7O6 in compound 12a is 118.56° and compound 12b is 118.53°, respectively (c.f. Table S1†). The planar structure is the most stable shape of the examined compounds 12a and 12b.
C in the phenyl ring falls in the range of 1.360 to 1.479 Å, which is in good agreement with the experimental data 1.481 Å, while the optimized bond length of CO is slightly lower than the experimental data 1.229 Å, Exp.93–95. The existence of a CO group in C4 has a significant impact on the computed bond angles in two compounds 12a and 12b, particularly <C4C3O6 in compound 12a is 128.98° and compound 12b is 128.90°; <C4N7O6 in compound 12a is 118.56° and compound 12b is 118.53°, respectively (c.f. Table S1†). The planar structure is the most stable shape of the examined compounds 12a and 12b.
3.3.2. Global reactivity descriptors and ground state properties. The calculation of global indices derived from electronic characteristics is used to study the global chemical reactivity of compounds. The ionization energy (IE) which measures the donating property (oxidation power), the electron affinity (EA) which measures the accepting property (reduction power), and the polarity or charge separation measurement theoretically by computed dipole moment (μ) are given in Fig. 5 and S1† and Table 2. Compound 12b, which has the lowest energy difference values (ΔE = 5.00806 eV), is the most reactive and less stable molecule, according to Table 2. As a result, in order of decreasing reactivity, the following sequence can be established: 12b > 12a (ΔE). The chemical hardness (η) is another characteristic examined, with compound 12b having the lowest value (2.50 eV) compared to the other 12a compound, indicating that it is the softest of the 12a compound. Furthermore, with the highest electronegativity (X) or chemical potential (V) (3.53 eV), this molecule 12b has the maximum electronic exchange capacity. In conclusion, the global descriptors revealed that compound 12b is the most reactive, least stable, softest, and has the highest electronic exchange capacity of the other 12a compound tested.
3.3.3. NBO analysis. Table S2† shows the donor and acceptor orbital and second-order interaction energies. The stronger the interaction between the donor and the acceptor, the greater the interaction energy value. Furthermore, as the interaction energy E(2) increases, the molecule structure is more stabilized by this contact. Strong intermolecular hyper conjugative interactions are created via an orbital overlap, according to the second-order perturbation theory of the Fock matrix. The results of NBO analysis of compounds 12a and 12b tabulated in Table S2† indicate that there is a strong hyper conjugative interactions LP (1) N9 → π*C2–C3, π*C1–N8 → π*C2–C3, and LP (1) N7 →.π*C1–N8, for 12a is 48.68, 57.47, and 67.41 kcal mol−1, respectively, and LP (1) N9 → π*C2–C3, π*C1–N8 → π*C2–C3, and LP (1) N7 → π*C1–N8, for 12b is 43.73, 58.61, and 66.54 kcal mol−1, respectively. The pyrimidine ring interacts equally well with the C–N π orbital and the amino group. In fact, it has a stronger contact with the pyrimidine ring. Furthermore, the nitrogen atom's lone pair orbital is hyperconjugation with the C4–O6 and C2–C3 π* orbitals. The oxygen lone pair orbital, on the other hand, interacts mostly with the pyrimidine ring's C1–N8 and C2–N9 π orbitals. The population of the NBO C1–N7, C3–C4, C4–O6, C4–N7, and C2–C3 decreases unexpectedly, indicating a charge transfer away from the pyrimidine ring. Finally, the linear conjugation that is responsible for the observed spectrum is enjoyed by 12a and 12b.
3.3.4. Natural charge. The distribution of electrons in distinct subshells of their atomic orbital as specified by natural population analysis96 applied to the electronic structures of compounds 12a and 12b. Tables S3 and S4† show the distinct atom charges. The most negative centers in our examined compounds 12a and 12b are O6, N7, N8, and N9-atoms, respectively. These negative atoms tend to contribute an electron from the molecule's electrostatic point of view. The most electropositive atoms, such as the S10-atom, are more likely to accept an electron.
3.3.5. Nonlinear optical (NLO) analysis. There has been no experimental or theoretical work on NLO for the compounds studied in the literature, therefore this study is of particular interest. The ability of any chemical to transform light of a longer wavelength into light of a shorter wavelength is known as non-linear optical characteristics. Semiconductors, infrared detectors, solid-state lasers, photosensitive materials, and crystalline thin films for microelectronics are among the most common applications of single crystals of any nonlinear materials.97 The electronic structure relationship and NLO characteristics of the examined compounds 12a and 12b were analyzed theoretically using DFT/B3LYP/6-311++G (d,p). Table S5† lists the total static dipole moment (μ), mean polarizability (α), polarizability anisotropy (Δα), and mean first-order hyperpolarizability (β) of the examined compounds 12a and 12b. There were no experimental values for the examined compounds' NLO characteristics, so P-nitro aniline (PNA) was used as a reference. The values of, α, β, in Table S5† shows that the order of rising α with respect to PNA is as follows: compounds 12a and 12b are ∼1.5 and 2 times higher than (PNA), respectively. Compounds 12a and 12b are 2 and 2.5 times greater than (PNA), for β parameter respectively, according to the parameter analysis.98–100 As a result, the chemicals investigated have promising optical characteristics.
3.3.6. Molecular electrostatic potential (MEP). Electrostatic molecular potential (MEP), which simultaneously displays molecular form, size, and electrostatic potential in terms of color gradation, can be used to identify a molecule's chemical reactivity. The charge distribution generates an electrostatic potential in the space around a molecule, which can be used to figure out if a molecule is electrophilic or nucleophilic.101 The examined compounds 12a and 12b were calculated 3D MEP and ESP from the optimized molecular structure using the DFT/B3LYP/6-311++G (d,p) technique Fig. S2, S3 and S4†. Potential increases in the order following: red < orange < yellow < green < blue.102,103 The negative region (red) is centered on the N, and O atomic sites, owing to the contribution of lone-pair electrons from nitrogen and oxygen atoms, while the positive (blue) potential sites are centered on hydrogen, sulfur, and carbon atoms. A part of the molecule with a negative electrostatic potential is vulnerable to electrophilic assault, the more negative the electrostatic potential, the greater the likelihood of electrophilic attack.
3.4. Thermodynamic properties
Table S6† shows the calculated thermodynamic parameters (such as zero-point vibrational energy (ZPVE), thermal energy, specific heat capacity, rotational constants, and entropy SVib (T)), determined using B3LYP/6-311++G (d,p) of compounds 12a and 12b at ambient temperature 298 K and 1.00 atm pressure. From 12a to 12b, the rotational constants decrease in value. In comparison to another chemical 12a and 12b has a greater entropy value. The thermal energies are likewise following the global minimum energy trend. ZPVEs (zero-point vibrational energy) appear to vary significantly. 12b has a higher ZPVE value of 109.447 kcal mol−1 than 12a, which has a value of 91.126 kcal mol−1. The basic statistical thermodynamic functions: heat capacity (C), entropy (S), and enthalpy changes (H) for the compounds 12a and 12b were determined from the theoretical harmonic frequencies using vibrational analysis at the B3LYP/6-311++G(d,p) level and are listed in Table S7.† Because the molecule vibrational intensities rise with temperature,97 these thermodynamic functions increase with temperature ranging from 200 to 600 K, as shown in Table S7.† Quadratic formulas were used to fit the correlation equations between heat capacities, entropies, enthalpy changes, and temperatures; the corresponding fitting factors (R2) for these thermodynamic properties are 0.99988, 0.99999, and 0.99988, respectively. The related fitting equations and correlation graphs of those given in Fig. 6 are as follows.| | |
C0p,m, 12a = 7.80349 + 0.12608T − 2.79319 × 10−4T2; (R2 = 0.99998),
| (2) |
| | |
C0p,m, 12b = 7.94349 + 0.21608T − 2.85319 × 10−4T2; (R2 = 0.99998),
| (3) |
| | |
S0m, 12a = 64.89967 + 0.15765T + 1.82708 × 10−4T2; (R2 = 0.99999),
| (4) |
| | |
S0m, 12b = 65.75467 + 0.18665T + 1.98708 × 10−4T2; (R2 = 0.99999),
| (5) |
| | |
H0m, 12a = 85.61953 − 0.04695T + 2.29806 ×10−4T2; (R2 = 0.99988),
| (6) |
| | |
H0m, 12b = 86.72153 − 0.05695T + 2.31806 ×10−4T2; (R2 = 0.99988),
| (7) |
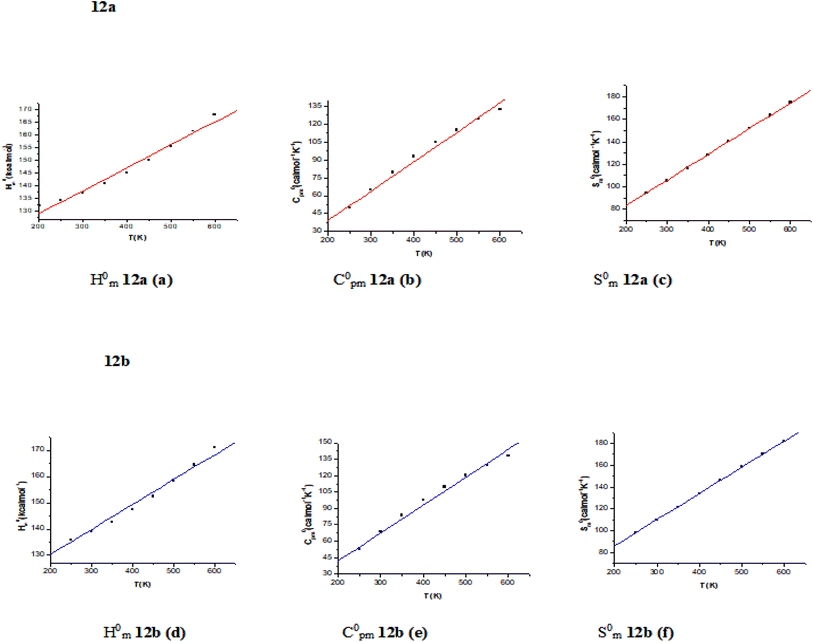 |
| | Fig. 6 Correlation graphics of thermodynamic properties and temperatures (a–f) for the studied compounds 12a and 12b at the B3LYP/6-311++G (d,p). | |
All the thermodynamic data is useful for further research into the compounds 12a and 12b. They can be used to calculate other thermodynamic energies based on thermodynamic function relationships and estimate chemical reaction directions based on the second law of thermodynamics in the thermochemical field. All thermodynamic calculations were performed in the gas phase and could not be applied to a solution.
3.5. NMR analysis
The 1H and 13C chemical shifts of the title molecules with the experimental NMR in DMSO solvent were computed using DFT theory in the gas and DMSO solvent are gathered in Fig. S5–S8.† Using the hybrid B3LYP technique and GIAO, the whole geometry optimization of the molecules was first done at the gradient corrected DFT.104–106 The 1H and 13C chemical shifts were calculated using the same approach, with a 6–311++ G (d,p) basis set in gas and DMSO solvent. H5, H11, H12, H17, and H18 attached to the carbon atoms of the methylene groups of the ethylene spacer have smaller values than the other proton for the pyrimidine ring (H19, H20, & H21) are due to the electronic charge density around the ring. The value of δ (chemical shift) of carbon atoms is absorbed at 26.91 and 49.46 ppm in the experimental 13C NMR spectrum (DMSO). The molecule has two methylene carbons, yet these carbons are consistent with the structure and molecular symmetry of methylene carbons attached to sulfur and nitrogen atoms, respectively.
3.6. Electronic UV-spectra of studied compounds 12a and 12b
Fig. 7 depicts the effect of the solvent on the electronic spectra of compounds 12a and 12b for both computed and experimental data. The charge density maps of the occupied and vacant MO,s is shown in Fig. S9 and S10.† The spectra of 12a and 12b is made up of four bands with wavelengths ranging from 200 to 400 nm. The spectra contain four non-polar solvent (dioxane) bands, which were seen at 220 nm, 211 nm, 180 nm, and 150 nm for compound 12a, and at 220 nm, 215 nm, 182 nm, and 151 nm for compound 12b. A blue shift occurs as the polarity of the solvent is increased from dioxane to methanol. In PCM simulations, the electronic absorption spectra of compounds 12a and 12b show little solvent dependence and a substantial blue shift difference when compared to the experiment. Vertical excitation at the TD-B3LYP/6-311G(d,p) level is carried out in both solvents to account for the experimentally observed blue shift as a result of changing solvent polarity for compounds 12a and 12b. When the polarity of the solvent is increased to (DMSO, dichloromethane, and methanol), the range of spectrum bands for the excited and ground states has the same values, and the intensity of the bands likewise increases with polar solvents, so all the band's shifts to (π–π*) and (n–π*). The electron stimulation of the fifteen MO molecular orbital's φ57−1φ75, for compounds 12a and 12b, creates a convergence between theoretical and experimental data. The first (n–π*) 1 band in non-polar solvents (dioxane) is detected at (220 nm) and theoretically at (256 nm) through a configuration of φ57–−1φ75 for compounds 12a and 12b, as shown in Tables S8–S9 and Fig. S9–S10.† Other sides, in polar solvents (DMSO, dichloromethane, and methanol), at (212, 217, 211 nm) band, where computed bands at (252, 253, 251 nm) for compounds 12a and 12b, respectively. The electron density's characteristics are derived from the molecular orbital's nature, according to the electronic transformation. Fig. S9 and S10† apparent the delocalization of the electron density and the charge transfer characteristic. The absorption bands in the visible region are typical transitions of n–π* and π–π*.
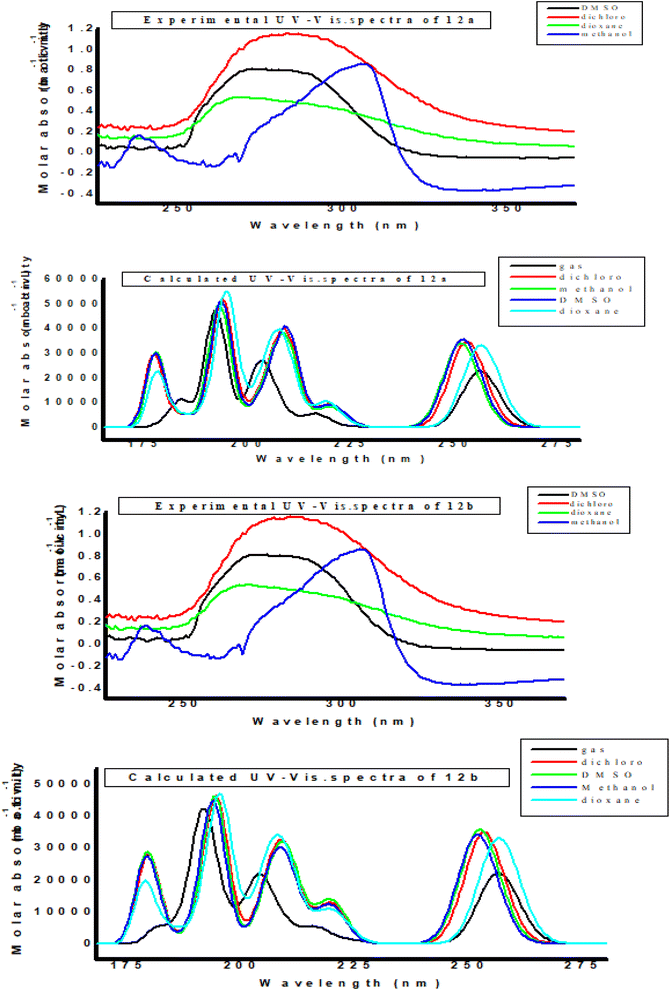 |
| | Fig. 7 Experimental UV-Vis. spectra and calculated electronic absorption spectra of the studied compounds 12a and 12b in different solvents. | |
3.7. Antimicrobial activity
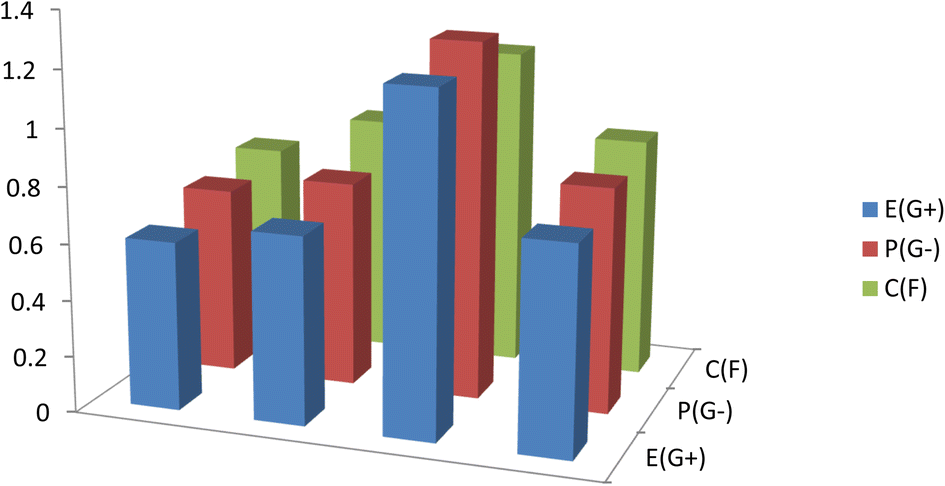
Antibacterial and antifungal capabilities of produced compounds 12a and 12b were investigated against several bacteria, including Gram-positive S. aureus and B. subtilis, and Gram-negative S. typhimurium and E. coli, as well as yeast, C. albicans. A. fumigatus is a species of A. fumigatus. Measuring the growth inhibition (zone of inhibition) around the disc of material that has been recorded the results in Table 3 and Fig. 8. Antibiotics were tested for antibacterial activity, and the results showed that the small size of compound 12b boosts its absorption ability on the surface of the cell wall of microorganisms and the cell respiration process, which explains why it has such significant antibacterial action against all bacteria and fungi tested in both compounds 12a and 12b. As a result, compound 12b is required for the growth-inhibitor effect to occur.
Table 3 In vitro antimicrobial activities of the synthesized compounds at 500 and 1000 μg mL−1 and the MIC values for some selected compounds 12a and 12b
| Compd |
Conc. (μg ml−1) |
Zone of inhibition in mma and (MIC values in μg mL−1) |
| Bacteria Gram (+) ve |
Bacteria Gram (–) ve |
Yeast |
Fungi |
| S. aureus |
B. subtilis |
S. typhimurium |
E. coli |
C. albicans |
A. fumigatus |
| Low active: 6–12 mm; moderately active: 13–19 mm; highly active: 20–30 mm; —: no inhibition or inhibition less than 5 mm. S**: standard drugs. |
| 12a |
500 |
— |
— |
— |
— |
14 (250) |
— |
| 1000 |
— |
— |
— |
— |
20 |
— |
| 12b |
500 |
— |
— |
— |
— |
12 |
— |
| 1000 |
— |
— |
— |
— |
16 |
— |
| S**b |
500 |
26 |
25 |
28 |
27 |
28 |
26 |
| 1000 |
35 |
35 |
36 |
38 |
35 |
37 |
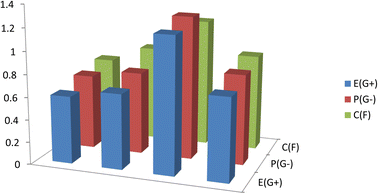 |
| | Fig. 8 Antimicrobial activity for the studied compounds 12a and 12b against Gram-positive bacteria (G+), Gram-negative bacteria (G−), and Fungi (F). | |
3.7.1 Structure activity relationship (SAR). The predicted ground state energetic and global properties can be linked to the biological activity of the produced molecules 12a and 12b. The biological activity of the examined compound 12a and 12b produced experimentally follows the order 12b > 12a, against G+, G−, and fungi, according to (Tables 2 and 3). The energy gap, Eg, of the researched compound computed at B3LYP/6-311++G (d,p) follows the same sequence determined experimentally, suggesting that Eg is one factor contributing to the reactivity of the studied compounds, EHOMO, which measures the donating power, the order of EHOMO 12b > 12a, and dipole moment which measure the charge separation, the order of the dipole moment 12a > 12b (c.f. Table 2). The examined compounds theoretically estimated global softness (S), global electrophilicity index (ω), electronegativity (χ), and chemical potential (V) follow the same order as their practical biological activity, which is 12b > 12a. The chemical hardness (η) follows the experimental biological activity 12a > 12b in reverse order. Natural charge from NBO and mean first-order hyperpolarizability (β) have orders of 12b > 12a and 12b > 12a, respectively, which are incompatible with the experimental biological activity order. Finally, the biological activity of compounds 12a and 12b is increased by the substituent.
4 Summary and conclusion
We have developed a simple method for the preparation of poly(pyrimidines) via alkylation of the appropriate 6-amino-2-mercapto-3H-pyrimidin-4-one with the corresponding bis(halo) compounds using a zeolite-nanogold catalyst. Although several Regio isomers are possible from the alkylation reaction, spectroscopic analyses as well as theoretical calculations confirmed the formation of one Regio isomer formed by preferential S-alkylation in good yield. Full characterization of these compounds is reported. A range of quantum chemical simulations were used to determine the structure and symmetry properties of the named molecules 12a and 12b. UV-Vis spectral investigations were examined using theoretical computations. To understand the electronic transitions of the current compounds 12a and 12b, TD-DFT calculations were performed on electronic absorption spectra in gas phase and solvent (DMSO, dichloromethane, methanol and dioxane). In comparison to the PNA molecule, the created molecule offers good benefits in technology-related applications, according to the NLO study. According to the ESP and MEP maps, negative potential sites are found around oxygen and nitrogen atoms, while positive potential sites are found around hydrogen atoms. The charges predicted by MEP were validated by the chemical shift analysis from the NMR study. NBO analysis is used to find the most likely transitions in the compounds. The thermodynamic characteristics of the chemical were also calculated. Relationships between statistical thermodynamics and temperature were also discovered. Heat capacity, entropy, and enthalpy all increase as the intensities of molecular vibrations increase with rising temperature. Compounds 12a and 12b explain why a compound has so significant antibacterial activity against all tested bacteria and fungi.
Conflicts of interest
There are no conflicts to declare.
References
- N. Agarwal, S. K. Raghuwanshi, D. N. Upadhyay, P. K. Shukla and V. Ram, Significance and Biological Importance of Pyrimidine in the Microbial World, J. Bioorg. Med. Chem. Lett., 2014, 10, 703–706 CrossRef PubMed.
- H. S. Basavaraj, G. M. Sreenivasa, E. Jayachandran, L. V. G. Nargund and D. S. Rao, Indian Room Temperature ICl-Induced Dehydration/Iodination of 1-Acyl-5-hydroxy-4,5-dihydro-1H pyrazoles. A Selective Route to Substituted 1-Acyl-4-iodo-1H-pyrazoles, J. Heterocycl. Chem., 2008, 15, 69–70 Search PubMed.
- F. Xie, H. Zhao, L. Zhao, L. Lou and Y. Hu, Synthesis, and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity, Bioorg. Med. Chem. Lett., 2009, 19, 275–278 CrossRef CAS PubMed.
- R. Kaur, P. Kaur and S. Sharma, et al., Anti-cancer pyrimidines in diverse scaffolds: a review of patent literature, Recent Pat. Anti-Cancer Drug Discovery, 2014, 10, 23–71 CrossRef PubMed.
- A. A. Abu-Hashem, M. F. El-Shehry and F. A.-E. Badria, Synthesis, Antioxidant, Antituomer Activities of Some New Thiazolopyrimidines, Pyrrolothiazolopyrimidines and Triazolopyrrolothiazolopyrimidines Derivatives, Acta Pharm., 2011, 60, 311–323 Search PubMed.
- V. Ukrainets, S. G. Taran, L. V. Sidorenko, O. V. Gorokhova, A. A. Ogirenko, A. V. Turov and N. I. Filimonova, Khim. Geterotsikl. Soedin., 1996, 8, 1113 (Chem. Abstr., 1997, 126, 59840) Search PubMed.
- J. Balzarini and C. McGuigan, Bicyclic pyrimidine nucleoside analogues (BCNAs) as highly selective and potent inhibitors of varicella-zoster virus replication, J. Antimicrob. Chemother., 2002, 50, 5–9 CrossRef CAS PubMed.
- D. R. Hannah and M. F. G. Stevens, Structural Studies on Bioactive Compounds. Part 38.1 Reactions of 5-Aminoimidazole-4-Carboxamide: Synthesis of Imidazo[1,5-a] Quinazoline-3Carboxamides, J. Chem. Res., 2003, 1, 398–401 CrossRef.
- K. Rana, B. Kaur and B. Kumar, Synthesis and antihepatotoxic activity of dihydropyrimidinone derivatives linked with 1,4-benzodioxane, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2019, 43, 1553–1557 Search PubMed.
- S. M. Sondhi, S. Jain, A. D. Dwivedi, R. Shukla and R. Raghubir, Structural and theoretical studies of 4-chloro-2-methyl-6-oxo-3,6-dideuteropyrimidin-1-ium chloride (d 6), Indian J. Chem. Sect. B Org. Incl. Med., 2021, 47, 136–143 Search PubMed.
- I. K. von Wick, Sesquiterpenoids from Costus Root Oil (Saussurea lappa CLARKE) Helv A, Chim. Acta., 1977, 60, 3025–3034 CrossRef.
- S. Hawser, S. Lociuro and K. Islam, Dihydrofolate reductase inhibitors as antibacterial agents, Biochem. Pharmacol., 2006, 71, 941–948 CrossRef CAS PubMed.
- P. Schneider, S. Hawser and K. Islam, 3-cyanoindole-based inhibitors of inosine monophosphate dehydrogenase: synthesis and initial structure-activity relationships, Bioorg. Med. Chem. Lett., 2003, 13, 4217–4221 CrossRef CAS PubMed.
- B. Roth and C. C. Cheng, Recent progress in the medicinal chemistry of 2,4-diaminopyrimidines, Prog. Med. Chem., 1982, 19, 269–331 CrossRef PubMed.
- P. Sharma, N. Rane and V. K. Gurram, Synthesis and QSAR studies of pyrimido[4,5-d] pyrimidine-2,5-dione derivatives as potential antimicrobial agents, Bioorg. Med. Chem. Lett., 2004, 14, 4185–4190 CrossRef CAS PubMed.
- O. Prakash, V. Bhardwaj, R. Kumar, P. Tyagi and K. R. Aneja, Simple oxidation of pyrimidinylhydrazones to triazolopyrimidines and their inhibition of Shiga toxin trafficking, Eur. J. Med. Chem., 2009, 39, 1073–1077 CrossRef PubMed.
- N. Agarwal, P. Srivastava and S. K. Raghuwanshi, et al., 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods, Bioorg. Med. Chem. Lett., 2002, 10, 869–874 CrossRef CAS PubMed.
- M. Doaa Khaled, E. Mohamed Elshakre, A. Mahmoud Noamaan, H. Butt, M. Marwa Abdel Fattah and A. Dalia Gaber, A Computational QSAR, Molecular Docking and In Vitro Cytotoxicity Study of Novel Thiouracil-Based Drugs with Anticancer Activity against Human-DNA Topoisomerase II, Int. J. Mol. Sci., 2022, 23, 11799 CrossRef PubMed.
- Z. Khosravi, H. Mohammadi and H. R. Shaterian, Preparation of pyrimido[4,5-b] [1,6] naphthyridin-4(1H)-one derivatives using a zeolite-nanogold catalyst and their in vitro evaluation as anticancer agent, J. Chin. Chem. Soc., 2019, 1–8 Search PubMed.
- M. S. Moustafa, S. M. Al-Mousawi and N. M. Hilmy, et al., Unexpected Behavior of Enaminones: Interesting New Routes to 1,6-Naphthyridines, 2-Oxopyrrolidines and Pyrano[4,3,2-de] [1,6] naphthyridines, Molecules, 2013, 18, 276–286 CrossRef CAS PubMed.
- N. A. Abdel Hafez, T. A. Farghaly and M. A. Al-Omar, et al., Synthesis of bioactive polyheterocyclic ring systems as 5α-reductase inhibitors, Eur. J. Med. Chem., 2010, 45, 4838–4844 CrossRef CAS PubMed.
- Z. G. Han, G. Zhang and B. Jiang, et al., Diversity Synthesis of Pyrimido[4,5-b] [1,6] Naphthyridine and Its Derivatives under Microwave Irradiation, J. Comb. Chem., 2009, 11, 809–812 CrossRef CAS PubMed.
- D. M. Chen, J. Y. Liu and M. Wei, et al., Synthesis of indazolo [5,4-b][1, 6] naphthyridine and indazolo [6, 7-b][1, 6] naphthyridine derivatives, Heterocycl. Commun., 2019, 25, 15–21 CrossRef CAS.
- B. Insuasty, D. Becerra and J. Quiroga, et al., Microwave-assisted synthesis of pyrimido [4,5-b][1, 6] naphthyridin-4 (3H)-ones with potential antitumor activity, Eur. J. Med. Chem., 2013, 60, 1–9 CrossRef CAS.
- M. B. Gawande, A. Goswami and F. X. Felpin, et al., Cu and Cu-based nanoparticles: synthesis and applications in catalysis, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- M. A. Zolfigol and R. Ayazi-Nasrabadi, Synthesis of the first magnetic nanoparticles with a thiourea dioxide-based sulfonic acid tag: application in the one-pot synthesis of 1,1,3-tri(1H-indol-3-yl) alkanes under mild and green conditions, RSC, 2016, 6, 69595 CAS.
- S. Gandhi, N. Nagalakshmi and I. Baskaran, et al., Synthesis and characterization of nano-sized NiO and its surface catalytic effect on poly (vinyl alcohol), J. Appl. Polym. Sci., 2010, 118, 1666–1674 CAS.
- S. G. Babu and R. Karvembu, Copper based nanoparticles-catalyzed organic transformations, Catal. Surv. Asia, 2013, 17, 156–176 CrossRef CAS.
- P. K. Tandon and S. B. Singh, Catalytic applications of copper species in organic transformations: a review, J. Catal. Catal., 2014, 1, 1–14 Search PubMed.
- S. B. Singh and P. K. Tandon, Catalysis: a brief review on nano-catalyst, J. Energy Chem. Eng., 2014, 2, 106–115 Search PubMed.
- K. Yan, G. Wu and C. Jarvis, et al., Facile synthesis of porous microspheres composed of TiO2 nanorods with high photocatalytic activity for hydrogen production, Appl. Catal. B Environ., 2014, 148, 281–287 CrossRef.
- P. Priecel, H. A. Salami and R. H. Padilla, et al., Anisotropic gold nanoparticles: preparation and applications in catalysis, Chin. J. Catal., 2016, 37, 1619–1650 CrossRef CAS.
- Y. Sun and Y. Xia, Shape-controlled synthesis of gold and silver nanoparticles, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- J. Yan, X. Zhang and Y. Liu, et al., Shape-controlled synthesis of liquid metal nanodroplets for photothermal therapy, Nano Res., 2019, 12, 1313–1320 CrossRef CAS.
- C. Lin, K. Tao and D. Hua, et al., Size effect of gold nanoparticles in catalytic reduction of p-nitrophenol with NaBH4, Molecules, 2013, 18, 12609–12620 CrossRef CAS PubMed.
- A. R. M. N. Afrooz, S. T. Sivalapalan and C. J. Murphy, et al., Spheres vs. rods: The shape of gold nanoparticles influences aggregation and deposition behavior, Chemosphere, 2013, 91, 93–98 CrossRef CAS PubMed.
- J. T. Carneiro, T. J. Savenije and J. A. Moulijn, et al., The effect of Au on TiO2 catalyzed selective photocatalytic oxidation of cyclohexane, J. Photochem. Photobiol. A Chem., 2011, 217, 326–332 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, an Attempt to Selectively Oxidize Methane over Supported Gold Catalysts, Catal. Lett., 2011, 141, 1429–1434 CrossRef CAS.
- A. Alshammari, A. Koeckritz and V. N. Kalevaru, et al., Potential of supported gold bimetallic catalysts for green synthesis of adipic acid from cyclohexane, Cat. Chem., 2012, 4, 1330–1336 CAS.
- M. Conte, X. Liu and D. M. Murphy, et al., Density functional theory study of selective aerobic oxidation of cyclohexane: the roles of acetic acid and cobalt ion, Phys. Chem., 2012, 14, 16279–16285 CAS.
- X. Liu, M. Conte and M. Sankar, et al., Liquid phase oxidation of cyclohexane using bimetallic Au–Pd/MgO catalysts, Appl. Catal. A Gen., 2015, 504, 373–380 CrossRef CAS.
- L. M. D. R. de Sousa Martins, S. A. C. Carabineiro and J. Wang, et al., Preparation of pyrimido[4,5-b][1,6]naphthyridin-4(1H)-one derivatives using a zeolite-nanogold catalyst and their in vitro evaluation as anticancer agent, Cat. Chem., 2017, 9, 1211–1221 Search PubMed.
- S. Chen, H. Fu and L. Zhang, et al., Nanospherical mesoporous carbon-supported gold as an efficient heterogeneous catalyst in the elimination of mass transport limitations, Appl. Catal. B Environ., 2019, 248, 22–30 CrossRef CAS.
- A. C. Ribeiro, L. S. de Sousa Martins and S. A. C. Carabineiro, et al., Nanodiamonds/poly (vinylidene fluoride) composites for tissue engineering applications, Molecules, 2017, 22, 37–44 CrossRef PubMed.
- Y. Dong, J. Luo and S. Li, et al., Honeycomb Carbon Nanofibers: A Superhydrophilic O2-Entrapping Electrocatalyst Enables Ultrahigh Mass Activity for the Two-Electron Oxygen Reduction Reaction, Catal. Commun., 2020, 133 Search PubMed.
- A. P. C. Ribeiro, L. M. D. R. de Sousa Martins and S. A. C. Carabineiro, et al., Gold nanoparticles deposited on surface modified carbon materials as reusable catalysts for hydrocarboxylation of cyclohexane, Appl. Catal. A Gen., 2017, 547, 124–131 CrossRef CAS.
- X. Wan, C. Zhou and J. Chen, et al., Base-free aerobic oxidation of 5-hydroxymethyl-furfural to 2, 5-furandicarboxylic acid in water catalyzed by functionalized carbon nanotube-supported Au–Pd alloy, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- S. A. C. Carabineiro, L. M. D. R. de Sousa Martins and M. Avalos-Borja, et al., Gold nanoparticles supported on carbon materials for cyclohexane oxidation with hydrogen peroxide, Appl. Catal. A Gen., 2013, 467, 279–290 CrossRef CAS.
- M. P. De Almeida, L. M. D. R. de Sousa Martins and S. A. C. Carabineiro, et al., Homogeneous and heterogenised new gold C-scorpionate complexes as catalysts for cyclohexane oxidation, Catal. Sci. Technol., 2013, 3, 3056–3069 RSC.
- E. G. Rodrigues, S. A. C. Carabineiro and J. J. Delgado, et al., Gold supported on carbon nanotubes for the selective oxidation of glycerol, J. Catal., 2012, 285, 83–91 CrossRef CAS.
- S. Wang, Q. Zhao and H. Wei, et al., Aggregation-free gold nanoparticles in ordered mesoporous carbons: toward highly active and stable heterogeneous catalysts, J. Am. Chem. Soc., 2013, 135, 11849–11860 CrossRef CAS PubMed.
- H. García and S. Navalón, in Applications in separations and catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2018 Search PubMed.
- Z. Sun, G. Li and L. Liu, et al., Au nanoparticles supported on Cr-based metal-organic framework as bimetallic catalyst for selective oxidation of cyclohexane to cyclohexanone and cyclohexanol, Catal. Commun., 2012, 27, 200–205 CrossRef CAS.
- R. Wu, X. Qian and K. Zhou, et al., Highly dispersed Au nanoparticles immobilized on Zr-based metal–organic frameworks as heterostructured catalyst for CO oxidation, Chem A, 2013, 1, 14294–14299 CAS.
- S. Saxena, R. Singh and R. G. S. Pala, et al., Sinter-resistant gold nanoparticles encapsulated by zeolite nanoshell for oxidation of cyclohexane, RSC Adv., 2016, 6, 8015–8020 RSC.
- L. X. Xu, C. H. He and M. Q. Zhu, et al., A highly active Au/Al2O3 catalyst for cyclohexane oxidation using molecular oxygen, Catal. Lett., 2007, 114, 202–205 CrossRef CAS.
- L. X. Xu, C. H. He and M. Q. Zhu, et al., Surface stabilization of gold by sol–gel post-modification of alumina support with silica for cyclohexane oxidation, Catal. Commun., 2008, 9, 816–820 CrossRef CAS.
- J. Huang, Y. Wang and J. Zheng, et al., Influence of support surface basicity and gold particle size on catalytic activity of Au/γ-AlOOH and Au/γ-Al2O3 catalyst in aerobic oxidation of α,ω-diols to lactones, Appl. Catal. B Environ., 2011, 103, 343–350 CrossRef CAS.
- H. Hosseini-Monfared, H. Meyer and C. Janiak, Dioxygen oxidation of 1-phenylethanol with gold nanoparticles and N-hydroxyphthalimide in ionic liquid, J. Mol. Catal. A Chem., 2013, 372, 72–78 CrossRef CAS.
- V. Van Speybroeck, K. Hemelsoet and L. Joos, et al., Advances in theory and their application within the field of zeolite chemistry, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- P. A. Jacobs, M. Dusselier and B. F. Sels, Will Zeolite-Based Catalysis be as Relevant in Future Biorefineries as in Crude Oil Refineries?, Angew. Chem., Int. Ed., 2014, 53, 8621–8626 CrossRef CAS PubMed.
- M. Pera-Titus, Porous Inorganic Membranes for CO2 Capture: Present and Prospects, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- V. Javanbakht, S. M. Ghoreishi and N. Habibi, et al., Synthesis of zeolite/magnetite nanocomposite and a fast experimental determination of its specific surface area, Prot. Met. Phys. Chem. Surfaces, 2017, 53, 693–702 CrossRef CAS.
- A. M. Zihlif, Z. Elimat and G. Ragosta, Thermal and mechanical characterization of polymer composites filled with dispersed zeolite and oil shale, J. Compos. Mater., 2011, 45, 1209–1216 CrossRef CAS.
- H. Faghihian, M. Moayed and A. Firooz, et al., Evaluation of a new magnetic zeolite composite for removal of Cs+ and Sr2+ from aqueous solutions: kinetic, equilibrium and thermodynamic studies, Compt. Rendus Chem., 2014, 17, 108–117 CrossRef CAS.
- P. Kowalczyk, M. Sprynskyy and A. P. Terzyk, et al., Porous structure of natural and modified clinoptilolites, J. Colloid Interface Sci., 2006, 297, 77–85 CrossRef CAS PubMed.
- M. Pera-Titus, Thermodynamic Analysis of Type VI Adsorption Isotherms in MFI Zeolites, J. Phys. Chem. C, 2011, 115, 3346–3357 CrossRef CAS.
- M. Karabacak, E. Sahin, M. Cinar, I. Erol and M. Kurt, X-ray, FT-Raman, FT-IR spectra and ab initio HF, DFT calculations of 2-[(5-methylisoxazol-3-yl) amino]-2-oxo-ethyl methacrylate, J. Mol. Struct., 2008, 886, 148–157 CrossRef CAS.
- M. Karabacak, M. Cinar, Z. Unal and M. Kurt, FTIR UV spectroscopic and DFT quantum chemical study on the molecular conformation, vibrational and electronic transitions of 2-aminoterephthalic acid, J. Mol. Struct., 2010, 982, 22–27 CrossRef CAS.
- M. Kurt, T. R. Sertbakan, M. Ozduran and M. Karabacak, Infrared and Raman spectrum, molecular structure, and theoretical calculation of 3,4-dichlorophenylboronic acid, J. Mol. Struct., 2009, 921, 178–187 CrossRef CAS.
- M. Karabacak, M. Kurt and A. Atac, Experimental and theoretical FT-IR and FT-Raman spectroscopic analysis of N1-methyl-2-chloroaniline, J. Phys. Org. Chem., 2009, 22, 321–330 CrossRef CAS.
- M. Govindarajan, K. Ganasan, S. Periandy and M. Karabacak, Experimental (FT-IR and FT-Raman), electronic structure and DFT studies on 1-methoxynaphthalene, Spectrochim. Acta A, 2011, 79, 646–653 CrossRef CAS PubMed.
- M. Karabacak, M. Cinar and M. Kurt, DFT based computational study on the molecular conformation, NMR chemical shifts and vibrational transitions for N-(2-methylphenyl) methanesulfonamide and N-(3-methylphenyl) methanesulfonamide, J. Mol. Struct., 2010, 968, 108–114 CrossRef CAS.
- S. Abdel Halim and A. Kh. Khalil, TD-DFT calculations, NBO analysis and electronic absorption spectra of some thiazolo[3,2-a] pyridine derivatives, J. Mol. Struct., 2017, 1147, 651–667 CrossRef.
- S. Abdel Halim, M. A. Ibrahim and Synthesis, Density Functional Theory Band Structure Calculations, Optical, and Photoelectrical Characterizations of the Novel (9-Bromo-3-cyano-5-oxo-1,5-dihydro-2H-chromeno[4,3-b] pyridin-2-ylidene) propanedinitrile, J. Heterocyclic. Chem., 2019, 56, 2542–2554 CrossRef CAS.
- S. Abdel Halim, L. I. Ali and S. G. Sanad, Theoretical calculations of solvation 12-Crown-4 (12CN4) in aqueous solution and its experimental interaction with nano CuSO4, Int. J. Nano Dimens, 2017, 8, 142–158 Search PubMed.
- T. A. Salah El-Din, A. A. Elzatahry and D. M. Aldhayan, et al., Synthesis and Characterization of Magnetite Zeolite Nano Composite, Int J Electrochem Sci, 2011, 6, 6177–6183 CAS.
-
(a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B Condens. Matter., 1988, 37, 785 CrossRef CAS PubMed;
(b) B. Miehlich, A. Savin, H. Stolt and H. Preuss, Chem.Phys. Lett., 1989, 157, 200 CrossRef CAS.
-
(a) A. D. Becke, J. Chem. Phys, 1993, 98, 5648 CrossRef CAS;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- B. Stefanov, B. G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 2003.
- M. Karabacak, A. Coruh and M. Kurt, FT-IR, FT-Raman, NMR spectra, and molecular structure investigation of 2,3-dibromo-N-methylmaleimide: A combined experimental and theoretical study, J. Mol. Struct., 2008, 892, 125–131 CrossRef CAS.
- N. Sundaraganesan, S. Illakiamani, H. Saleem, P. M. Wojciechowski and D. Michalska, FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine, Spectrochim. Acta, Part A, 2005, 61, 2995–3001 CrossRef CAS PubMed.
- M. Frisch, J. G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., Gaussian, Inc., Wallingford CT, 2009.
- , R. Dennington, T. Keith and J. Millam, in GaussView, Version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- http://www.chemcraftprog.com.
- D. Avci, Second and third-order nonlinear optical properties and molecular parameters of azo chromophores: semiempirical analysis, Spectrochim. Acta, Part A, 2011, 82, 37–43 CrossRef CAS PubMed.
- D. Avci, A. Başoğlu and Y. Atalay, Ab initio HF and DFT calculations on an organic non-linear optical material, Struct. Chem., 2010, 21, 213–219 CrossRef CAS.
- D. Avci, H. Cömert and Y. Atalay, Ab initio Hartree-Fock calculations on linear and second-order nonlinear optical properties of new acridine-benzothiazoly- lamine chromophores, J. Mol. Mod., 2008, 14, 161–169 CrossRef CAS PubMed.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries, Adv. Mater., 2018, 30, 1803075–1803082 CrossRef PubMed.
- J. Chocholoušová, V. Špirko and P. Hobza, First local minimum of the formic acid dimer exhibits simultaneously red-shifted O–H…O and improper, blue-shifted C–H…O hydrogen bonds, Phys. Chem., 2004, 6, 37–41 Search PubMed.
- M. Szafran, A. Komasa and E. Bartoszak-Adamska, Crystal and molecular structure of 4-carboxypiperidinium chloride (4-piperidinecarboxylic acid hydrochloride), J. Mol. Struct., 2007, 827, 101–107 CrossRef CAS.
- D. Sajan, L. Joseph, N. Vijayan and M. Karabacak, Natural bond orbital analysis, electronic structure, non-linear properties, and vibrational spectral analysis of l-histidinium bromide monohydrate: a density functional theory, Spectrochim. Acta A, 2011, 81, 85–98 CrossRef CAS PubMed.
- H. M. E. Hassaneen, A novel one-pot three-components reaction: synthesis of indeno[2',1':5,6] pyrido[2,3:4'',5'']pyrimido[2'',1''-c]triazole-5,7-dione, A new ring system Arkivoc, I, 2007, 154–163 CAS.
- R. Ditchfield, Molecular orbital theory of magnetic shielding and magnetic susceptibility, J. Chem. Phys., 1972, 56, 5688–5691 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, Efficient implementation of the gauge independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
-
(a) S. Natorajan, G. Shanmugam and S. A. MartinCryst, Res. Technal., 2008, 43, 561 CrossRef;
(b) D. S. Chemiaand J. Zysss, TD-DFT Calculations, NBO, NLO Analysis and Electronic Absorption Spectra of Some Novel Thiazolo[3,2-a]Pyridine Derivatives
Bearing Anthracenyl Moiety, Orlando, FL, 1987 Search PubMed;
(c) D. S. Bradshow and D. L. Andrews, J. Nonlinear Opt. Phys. Matter, 2009, 18, 285–295 CrossRef.
- L. T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, Experimental investigations of organic molecular nonlinear optical polarizabilities. 1. Methods and results on benzene and stilbene derivatives, J. Phys. Chem., 1991, 95, 10631–10643 CrossRef CAS.
- P. Kaatz, E. A. Donley and D. P. Shelton, A comparison of molecular hyperpolarizabilities from gas and liquid phase measurements, J. Chem. Phys., 1998, 108, 849–856 CrossRef CAS.
- T. Gnanasambandan, S. Gunasekaran and S. Seshadri, Spectrochimica. Acta Part A: Molecular and Biomolecular Spectroscopy, 2014, 117, 557–567 CrossRef CAS PubMed.
-
(a) J. S. Murray and K. Sen, in Molecular Electrostatic Potentials, Conseptsana Applications, Elsevier, Amsterdam, 1996, p. 7 Search PubMed;
(b) E. Sscrocco and J. Tomasi, An Euristic Interpretation, Adv. Quant. Chem., 1978, 11, 115–123 CrossRef.
- P. Politzer and J. S. Murray, the fundamental nature and role of the electrostatic potential in atoms and molecules, Theor. Chem. Acc., 2002, 108, 134–142 Search PubMed.
- T. Yanai, D. Tew and N. A. Handy, New hybrid exchange–correlation functional was using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. Ditchfield, Molecular orbital theory of magnetic shielding and magnetic susceptibility, J. Chem. Phys., 1972, 56, 5688–5691 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, Efficient implementation of the gauge independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- H. O. Kalinowski, S. Berger and S. Braun, in Carbon-13 NMR Spectroscopy, John Wiley &Sons, Chicheser, 1988 Search PubMed.
- V. R. T. Verbiest and K. Clays, Synthesis, FT-IR, structural, thermochemical, electronic absorption spectral, and NLO analysis of the novel 10-methoxy-10H-furo[3,2-g]chromeno[2,3-b][1,3]thiazolo[5,4-e]pyridine-2,10(3H)-dione (MFCTP): a DFT/TD-DFT study, CRC Press, New York, 2009 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Huwaida M. E. Hassaneenb
*a and
Huwaida M. E. Hassaneenb