
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully wood based novel translucent and thermoplastic materials by solvent-free esterification
Prabu Satria Sejati ab,
Firmin Obounou Akong*a,
Camile Torlotingc,
Frédéric Fradetc and
Philippe Gérardin*a
ab,
Firmin Obounou Akong*a,
Camile Torlotingc,
Frédéric Fradetc and
Philippe Gérardin*a
aLERMAB, INRAE, Université de Lorraine, 54000 Nancy, France. E-mail: philippe.gerardin@univ-lorraine.fr; firmin.obounou-akong@univ-lorraine.fr
bResearch Center for Biomass and Bioproducts, National Research and Innovation Agency (BRIN), 16911 Bogor, Indonesia
cPLASTINNOV, IUT de Moselle-Est, Université de Lorraine, 57500 Saint-Avold, France
First published on 9th December 2022
Abstract
Wood has been investigated for bioplastic production because of its abundance and biorenewability to reduce dependence on petro-based plastics. A series of experiments have been carried out to graft myristic acid, chosen as the fatty acid model, onto spruce sawdust using trifluoroacetic anhydride (TFAA) as the impelling agent without any solvent. The reaction was performed rapidly, leading to high ester content. Most of the hydroxyl groups in wood structure reacted with myristic acid, as demonstrated by FTIR and CPMAS 13C NMR. XRD measurements indicated a decrease in wood crystallinity. Myristic acid-esterified wood showed higher thermal stability by TGA and DSC and delivered several softening temperatures, as observed by TMA. Thermoplastic and translucent films were obtained after pressing at a high temperature. Scanning electron micrographs revealed that pressed esterified wood at the high temperature showed complete disappearance of fibrous structure to a smooth and homogenous surface, indicating that thermal fluidity was achieved during pressing. Esterified sawdust film also showed surface hydrophobicity by contact angle measurements.
Introduction
The dependence of human life on plastic puts pressure on nature, as evidenced by more than 8 million tons of plastic waste entering the oceans every year.1 Recycling of plastic waste alone is not enough to solve this pollution. Furthermore, the replacement of petro-based polymer plastics with bio-based polymers is a promising alternative to provide plastics with similar characteristic, which competes with petro-based plastics.2 Bio-based polymers for plastic production come from various natural resources ranging from animals, crustaceans, and plants in the form of polyester (poly lactic acid, PLA and poly-3-hydroxybutyrate P3HB), polysaccharides (starch, lignocellulosic polymer, and chitosan), proteins, and natural fiber.3 Lignocellulosic materials play an important role as the source for bioplastic production because of their abundance and biorenewability. Using lignocellulosic materials, especially from the forestry sector, contributes to the reduction of recourse to petrochemical resources.4–6 Wood is an abundant lignocellulosic material consisting of three main constituents, namely cellulose, hemicellulose, and lignin, arranged together in a sophisticated way, which give high strength and a rigid cell wall.7 Therefore, contrary to plastic materials in native condition, wood does not easily solubilize or melt through conventional extrusion or molding processes.Without modification, compared with other materials, wood has some drawbacks because of its affinity to water such as dimensional instability and susceptibility to bio and photodegradation. Hydroxyl groups that are present in all major component of wood such as cellulose, hemicellulose, and lignin are responsible not only for these drawbacks but also for it chemical reactivity.8 The reactivity of these hydroxyl groups makes wood a potential sustainable material for chemical modification to obtain thermoplastic properties.
Chemical modification to improve wood thermoplasticity was firstly conducted by the esterification of wood with acid chloride and fatty acid using N2O4-dimethylformamide (DMF) as a pretreatment to decrystallize cellulose in pyridine solvent.9,10 Esterified wood with appropriate number of carbon atoms showed high ester content and lower softening temperature compared to non-modified wood. Matsuda11 continued these work using esterified dicarboxylic acid and anhydride in the same medium. Wood esterification with octanoyl and palmitoyl chlorides in various solvents (DMF, CHCl3, and methyl tert-butyl ether/MTBE) was reported by Zhang et al.12 and showed that greater amount of acid chloride gives lower mole fraction of esterification; moreover, the grafted ester group increased with the polarity of the solvent.
The other promising medium to make wood thermoplastic is using trifluoroacetic anhydride (TFAA). Arni et al.13 introduced the utilization of TFAA as an impelling agent to esterify dicarboxylic acids to wood using benzene as the solvent to improve wood hydrophobicity. Wood esterification with unsaturated carboxylic acids using TFAA and the same solvent also led to decreased moisture content and the crystallinity of modified wood.14,15 The thermofluidity of esterified wood was achieved by Shiraishi et al.16 using various acyl chlorides in DMF, resulting in lower melting temperature of esterified wood. Several years later, Shiraishi and Yoshioka17 reported the utilization of trifluoroacetic acid (TFA) for the acetylation of wood to obtain thermoplastic properties. The thermoplasticity mechanism of TFAA esterified wood in benzene was explained by Nakano18 using the free volume approach. These early works on wood esterification were performed using non-ecofriendly organic solvent (dimethylformamide, pyridine, or benzene). To encounter this issue, Thiebaud and Borredon,19 Thiebaud et al.,20 and Wu et al.21 introduced fatty acid chlorides to esterify wood without any solvent. The reaction took place in a complex reactor with nitrogen bubbling system at high temperature and high content of fatty acid chloride using sodium hydroxide solution to trap the hydrogen chloride byproduct. Most previous studies grafted fatty acid derivatives onto wood in the presence of toxic solvent to obtain thermoplastic materials. This present research is therefore conducted to develop new methodologies to confer spruce sawdust thermoplastic and translucent properties using myristic acid and TFAA as the impelling agent without any solvent.
Experimental
Materials and sample preparation
Wood sawdust (18 mesh) of spruce wood (Picea abies) was extracted from a soluble fraction with mixed solvent of toluene/ethanol (1/2, v/v) using Soxhlet extraction for 4 h, followed by ethanol solvent extraction for 4 h, and was dried at 103 °C for 24 h. Saturated carboxylic acids used was myristic acid (C14) was purchased from Alfa Aesar and trifluoroacetic anhydride (TFAA) was purchased from Sigma-Aldrich.Wood esterification
A solution of equimolar proportions of myristic acid and TFAA was prepared for 30 min to form the mixed anhydride. A varied proportion of oven dried wood mass (m0) than the solution (Table 1) was then added to the mixed anhydride at room temperature in a closed tube for a duration varying between 30 min and 24 h. The esterified sample was successively rinsed with ethanol and water, followed by Soxhlet extraction with ethanol for 24 h and then with water for 48 h, changing water every 24 h. The treatment of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mass ratio of wood and TFAA and 4 h of reaction time were selected for further characterization. For comparison, a series of reaction was also carried out in CH2Cl2 as the solvent for 24 h at 50 °C. The sample was then dried at 103 °C for 24 h to obtain the final mass (m1). The weight percent gain (WPG) was calculated as follows.
:4 mass ratio of wood and TFAA and 4 h of reaction time were selected for further characterization. For comparison, a series of reaction was also carried out in CH2Cl2 as the solvent for 24 h at 50 °C. The sample was then dried at 103 °C for 24 h to obtain the final mass (m1). The weight percent gain (WPG) was calculated as follows.| WPG (%) = 100 × (m1 − m0)/m0 |
| Wood | TFAA | Myristic acid |
|---|---|---|
| 1 | 0.5 | 0.5 |
| 1 | 1 | 1 |
| 1 | 2 | 2 |
| 1 | 4 | 4 |
| 1 | 6 | 6 |
| 1 | 10 | 10 |
Based on the WPG result, the ester content were calculated from the molecular mass of fatty acid grafted on the wood after esterification minus the molecular mass of OH substituted (m2) as follows.
| Ester content (mmol ester per g of dry wood) = WPG/m2 × 1000 |
Sheet formation of esterified wood
Esterified spruce sawdust was placed in a 2 × 3 × 0.1 cm3 mold between two parchment papers, then hot pressed using laboratory press LabManual 300 from Fontijne at 120–200 °C under 10 MPa pressure for 10 min to obtain a transparent sheet or film.Fourier transform infrared (FTIR) spectroscopy analysis
The FTIR spectra were recorded on an ATR PerkinElmer Spectrum 2000 (PerkinElmer Ltd, Beaconsfield, UK) equipped with a diamond cell. The spectra of the non-modified and esterified sawdust were measured in the range of 4000–650 cm−1 at a resolution of 4 cm−1 and were then baseline-corrected and normalized by means of the dedicated Spectrum 10 software (PerkinElmer Ltd, Beaconsfield, UK).Cross-polarization/magic angle spinning (CP/MAS) 13C NMR analysis
Solid-state CP/MAS 13C NMR spectra were recorded on a Bruker MSL300 spectrometer at a frequency of 75.47 MHz. The acquisition time was 0.026 s with a number of transients of about 1200. All the spectra were run at a relaxation delay of 5 s, CP time of 1 ms, and spectral width of 20000 Hz. The spinning rates were 5 kHz. The interferograms were processed with Top Spin 3.6.2 using an apodization value of 21 Hz. The chemical shifts were expressed in parts per million (ppm).
X-ray diffraction (XRD) analysis
The diffraction diagrams were made directly on modified and non-modified wood powders. The assembly used comprises a SEIFERT XRD-3000 type generator, working at 40 kV and 30 mA, and according to a Bragg–Brentano (q/q) geometry. CuKα1 radiation is selected from a copper source 5 using a graphite monochromator at a wavelength h = 0.1540598 nm. The diffraction patterns were recorded for angles q between 1 and 30°. The crystallinity index was measured by calculating the ratio between the diffraction of the 002 peak at about 22.15° and at the minimum found between the 002 peak and the 101 lattice peak for an amorphous background at about 18.67°.22Thermal properties (TGA, DSC, TMA)
Thermogravimetric analysis (TGA) measurements were performed using a TGA/DSC 1 LF/1100 instrument from Mettler Toledo equipped with the STARe V.14 system program. Approximately 5 mg sample powder were placed in a 70 μL aluminum oxide sample pan and measured in a programmed temperature ranged from 30 to 600 °C in a nitrogen atmosphere (50 mL min−1) and a heating rate of 10 °C min−1.Differential scanning calorimetry (DSC) measurements were carried out with a DSC 1/700 instrument from Mettler Toledo equipped with the STARe V.14 system program. Using the same amount of the sample, the sample was placed in a 40 μL aluminum sample pan with a cover, and then performed in a nitrogen atmosphere (50 mL min−1) at a heating rate of 5 °C min−1 from 25–500 °C.
Thermomechanical analysis (TMA) of esterified wood was performed using a Mettler Toledo TMA SDTA 840 instrument. A pressed esterified wood sample was compressed under a constant load of 0.1 N in a heated chamber in the range of 30–260 °C at 10 °C min−1 heating rate. As the temperature increased, the load moved and recorded the deformation. The maximum value of deformation was then considered as the softening temperature. The results were analyzed using the STARe software.
Contact angle measurements
Contact angle measurements were performed to study the hydrophobicity of the pressed esterified wood surface using a goniometer (Kruss FM40 EasyDrop, Hamburg, Germany) equipped with the Easy Drop software. A drop of water was placed on the surface of the film, then the contact angle between the water drop surface and the baseline was measured every 3 s from 0 to 60 s. The left and right contact angles data were averaged based on each capturing time. This measurement was repeated three times.Scanning electron microscopy (SEM)
The pressed esterified wood sample and a pressed non-modified wood samples were observed with a Hitachi S4800 scanning electron microscope (SEM). The whole examination was carried out at room temperature and the micrographs of surfaces were produced at various magnifications.Results and discussion
The chemical modification of wood and the characterizations of the resulting materials were studied. As presented in Fig. 1 and 2, myristic acid was mixed with TFAA to form unsymmetrical anhydride, which was then reacted with wood sawdust. A swelled wood with ester function and long fatty acid chain will be formed as the main product, and the side products were trifluoracetylated wood and a volatile compound. Thermoplastic and translucent material was obtained after hot pressing. | ||
| Fig. 1 Chemical reaction of mixed anhydride formation and esterification of wood. | ||
 | ||
| Fig. 2 Schematic overview of the study, esterification of wood by introduction ester function and fatty acid chain to the hydroxyl group to obtain thermoplastic and translucent materials. | ||
Mass gain
The amount of acyl groups grafted to the wood can be calculated by the difference mass of esterified wood compared to the initial mass before esterification that was expressed as WPG and ester content. Fig. 3 shows the WPG and ester content of esterified wood in different quantities of reactants, either in CH2Cl2 solvent or without solvent. Generally, treatment without solvent resulted in a higher ester content than the treatment in CH2Cl2 in all quantities of reactants used. A higher quantity of reactive leads to higher ester content, which tends to stabilize for a ratio of 1:4 wood and TFAA/FA, resulting in 214.7% of WPG when no solvent was used in the reaction. The same WPG was obtained using 1:10 ratio of wood and TFAA/FA in CH2Cl2. These indicated that the maximum hydroxyl group variable for esterification reacted, leading to 10.17 mmol ester per g wood. This maximum ester content was reached in a lower quantity of TFAA/FA, when no solvent was used in the reaction than the reaction in the CH2Cl2 solvent.
 | ||
| Fig. 3 WPG and ester content of myristicated wood in different quantities of reactants used. | ||
The reaction rate of esterification of wood without the solvent, as present in Fig. 4, shows that after 1 h of reaction, 7.05 mmol ester per g of wood was grafted to the wood, stabilized after 4 h of reaction, and reached maximum ester content after 24 h by 11.34 mmol ester per g of wood. This result is within the molarity substituent reported by Shiraishi et al.10,23 of wood esterified with lauroyl chloride (C12) and palmitoyl chloride (C16) 11.1 and 8.5, respectively, in N2O2-DMF-Pyridine medium. Nakano et al.18 reported a lower ester content of about 5 mmol per g of wood for the esterification of wood using lauric acid and palmitic acid with the TFAA method in benzene. A lower weight increase of about 50% also reported by Thiebaud and Borredon19 for the esterification of wood using TFAA method with myristoyl chloride.
 | ||
| Fig. 4 WPG and ester content of myristicated wood at different reaction durations. | ||
Chemical structure transformation of esterified wood
Fourier-transform infrared spectroscopy (FTIR), cross-polarization/magic angle spinning nuclear magnetic resonance (CP/MAS 13C NMR), and X-ray diffraction (XRD) analysis were performed to confirm the chemical changes of wood before and after esterification. As presented in Fig. 5, before modification, the absorption of the O–H stretching vibration band (3500–3300 cm−1) was obvious and almost disappeared after modification. In esterified wood, there are three new strong bands recorded, first at 1744 cm−1, which corresponds to the adsorption of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl ester groups, second at 2921 cm−1 and 2851 cm−1, which is associated with symmetric and asymmetric aliphatic chain (–CH2–), respectively, from myristic acid ester, and third at 720 cm−1, which is characteristic of at least four linearly connected –CH2– groups, which shows that myristic acid has been directly grafted onto wood by an ester junction.21,24–27 These observations revealed that by the method used successfully, hydroxyl groups of wood were substituted with acyl groups of myristic acid.
O carbonyl ester groups, second at 2921 cm−1 and 2851 cm−1, which is associated with symmetric and asymmetric aliphatic chain (–CH2–), respectively, from myristic acid ester, and third at 720 cm−1, which is characteristic of at least four linearly connected –CH2– groups, which shows that myristic acid has been directly grafted onto wood by an ester junction.21,24–27 These observations revealed that by the method used successfully, hydroxyl groups of wood were substituted with acyl groups of myristic acid.
 | ||
| Fig. 5 FTIR spectra of wood sawdust before and after esterification. | ||
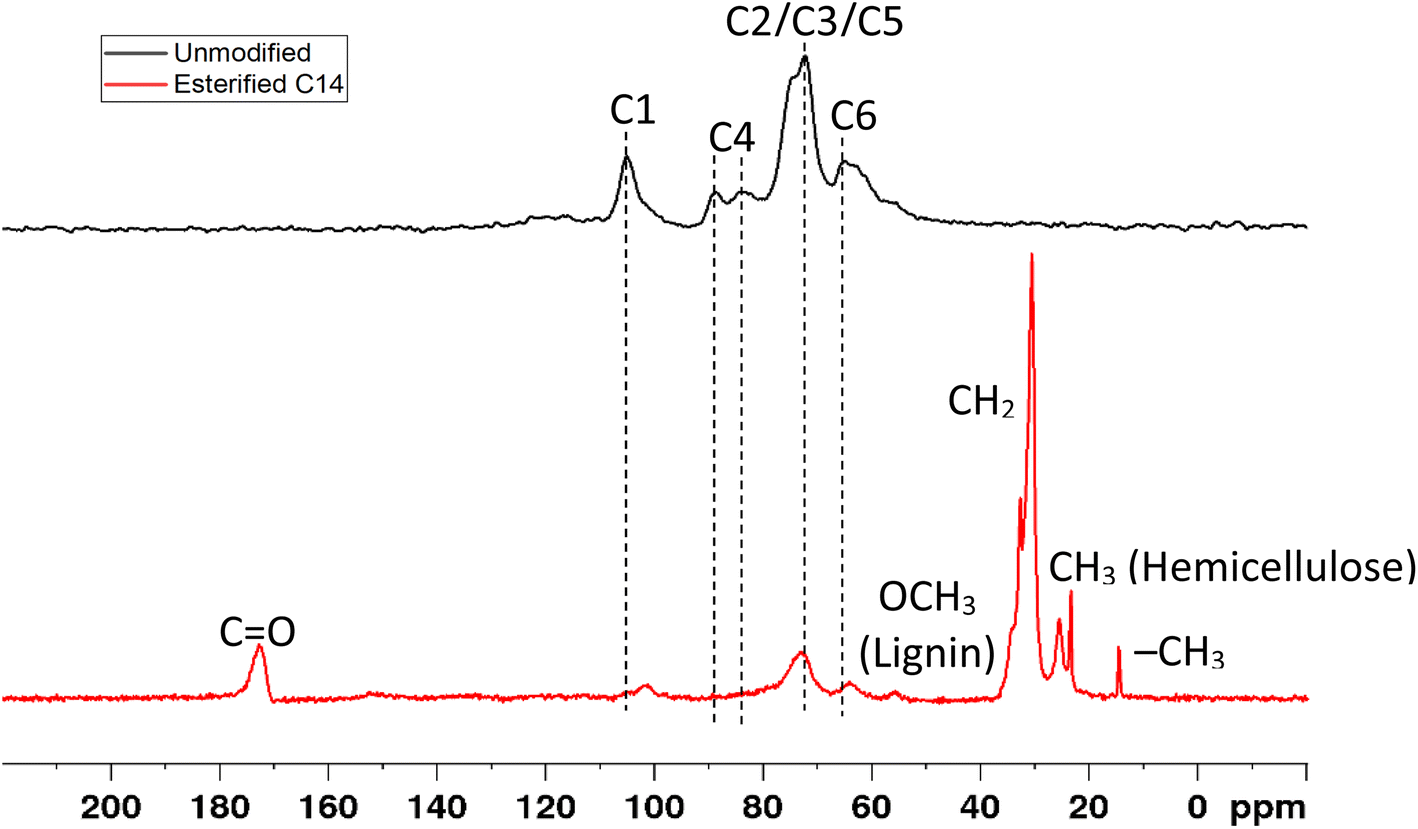
The change in the chemical structure was also confirmed by solid state CP/MAS 13C NMR spectroscopy, as shown in Fig. 6. Dominant signals of carbohydrate pattern appear before modification, which correspond to cellulose C1 (105.2 ppm),28–31 C4 crystalline cellulose (88.7 ppm),28–32 C4 amorphous cellulose (83.6 ppm),28–32 C2, C3, C5 cellulose (72.4 ppm),28–32 hemicellulose,30 and lignin30–33 and C6 crystalline cellulose (65.0 ppm).28,29,31,32 After esterification with myristic acid, the peak C1 of cellulose was shifted to 102.0 ppm C1 of hemicellulose,28–31 the peak C4 of cellulose crystalline at 88.7 ppm disappeared, decreased, and the peak for C6 crystalline cellulose shifted to that of C6 amorphous cellulose at 64.2 ppm28–32 and hemicellulose,28,30 and decreased the intensities of peaks C2, C3, and C5 of cellulose. The decrease and shifting of these signal intensities confirm the success of esterification.21,34
 | ||
| Fig. 6 CP/MAS 13C NMR spectra of wood sawdust before and after esterification. | ||
The appearance of new peaks was observed in esterified wood at 14.6 ppm due to the methyl group (–CH3)35 at 23.6 ppm, which corresponded to the CH3 carbon of the acetyl group in hemicellulose,36 at 30.8 ppm from the methylene group (–CH2–),34 and methylene group adjacent to carbonyl group (–CH2–CO),35 at 55.9 ppm that corresponds to the methoxy group (OCH3) of aromatic moieties in lignin,28–33,36 and at 172.7 ppm from the carbonyl group in the myristicated substitution group in the modified wood21,25,34,35 either in cellulose, hemicellulose,28,31 or lignin.28,30 This result indicates using the proposed esterification method of wood by myristic acid changes the structure of hemicellulose and lignin, decrystallization of the crystalline cellulose, and also introduces new acyl and carbonyl groups from myristic acid onto modified wood (Table 2).
| Chemical shift (ppm) | Assignment | |
|---|---|---|
| Observed | Reference | |
| 172.7 | 171.6–180 | Carbonyl groups (CO) in the modified wood,21,25,34,35 carboxyl groups of hemicellulose,28,31 carbonyl groups of lignin28,30 |
| 105.2 | 104.9–106 | C1 cellulose28–31 |
| 102.0 | 101–103 | C1 hemicellulose28–31 |
| 88.7 | 88–89 | C4 of crystalline cellulose28–32 |
| 83.6 | 83.2–86 | C4 of amorphous cellulose28–32 |
| 72.4 | 70–80 | C2, C3, C5 of cellulose,28–32 C2, C3, C5 of lignin,30–33 C2, C3, C5 of hemicellulose30 |
| 65.0 | 64.7–66 | C6 of crystalline cellulose28,29,31,32 |
| 64.2 | 61.8–66 | C6 of amorphous cellulose,28–32 C6 of hemicellulose28,30 |
| 55.9 | 55–57 | Methoxyl group (OCH3) in lignin28–33,36 |
| 30.8 | 30–30.6 | Methylene group (–CH2–) in modified wood34,37 methylene adjacent to carbonyl group (–CH2–CO)35 |
| 23.6 | 23.4 | Methyl group (–CH3) in hemicellulose36 |
| 14.6 | 14.8 | Methyl group (–CH3) in modified wood35 |
The study of the X-ray diffraction (XRD) of the wood samples before and after esterification was carried out to highlight the decrystallization of the crystalline cellulose fraction during the chemical modification of the wood, a phenomenon which would be at the origin of the thermoplasticity of the latter.
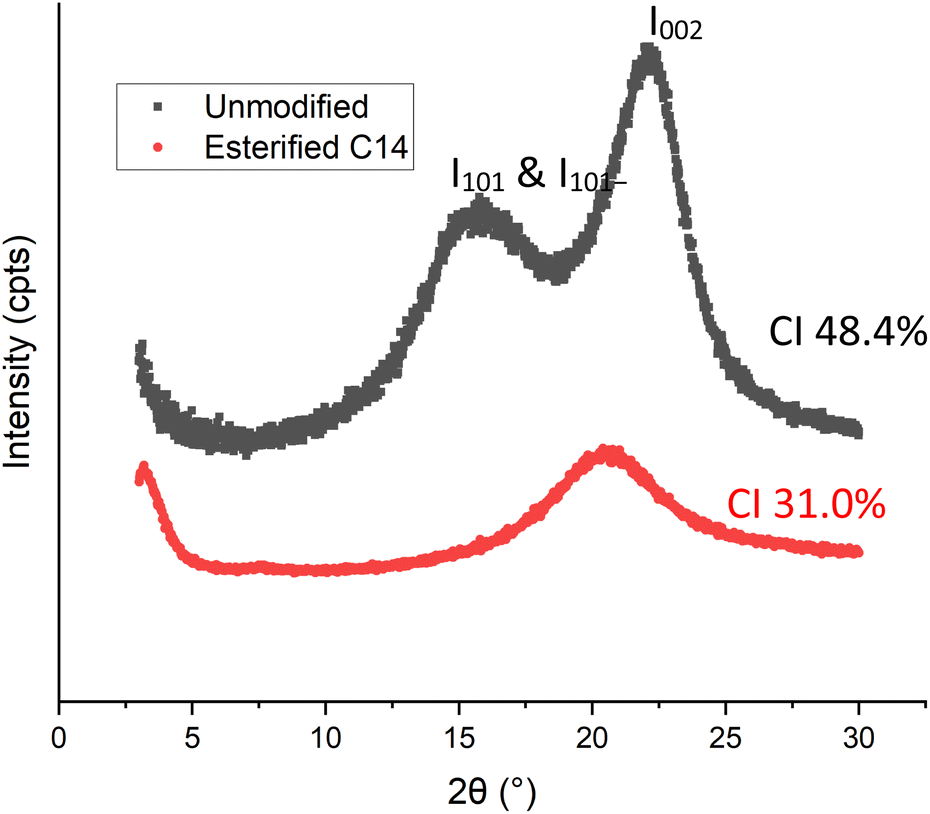
Fig. 7 showed that before esterification, native spruce sawdust exhibited an X-ray scattering pattern that had two diffraction planes for 2θ = 22.15° and 15.77°. After esterification with myristic acid, the first crystalline ray at 22.15° due to the 002 plane decreased, shifted to amorphous at 20.9°, and was enlarged. This result shows that the shape and position of the peak were close to those of the completely decrystallized cellulose,38 and the esterification of lignocellulose with acyl chloride,20,21 ionic liquid,39 and symmetrical anhydride.40
 | ||
| Fig. 7 X-ray diffraction diagram of wood sawdust before and after esterification. | ||
The second peak of spruce sawdust before modification at 15.77°, corresponding to the reflection of the planes 101 and 10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) of the native cellulosic network, disappeared with the appearance of a new 2θ peak at low angles of about 3.19°. The position of this peak was found by Heritage et al.38 as a function of the size of the substituent introduced at the hydroxyl groups of cellulose. This peak was attributed to the lateral spaces present between the long molecular chains of cellulose created by the functionalization of the glucopyranosic rings by fatty acids of different chain lengths.
of the native cellulosic network, disappeared with the appearance of a new 2θ peak at low angles of about 3.19°. The position of this peak was found by Heritage et al.38 as a function of the size of the substituent introduced at the hydroxyl groups of cellulose. This peak was attributed to the lateral spaces present between the long molecular chains of cellulose created by the functionalization of the glucopyranosic rings by fatty acids of different chain lengths.
The crystallinity index (CI) based on the empirical approach of untreated spruce sawdust obtained in this study was 48.4%. This value is in the range of 39.6–50.2% of native spruce CI reported by Andersson et al.41 After esterification with myristic acid, the CI of esterified wood decreased to 31.0%, below the ranged as mentioned before.
These modifications in the X-ray scattering spectrum of the wood after esterification with myristic acid are indicative of the decrystallization of cellulose. This is essentially controlled by the extent of esterification. Indeed, cellulose basically exists in crystalline and non-crystalline form. The chemical reactions take place first in the amorphous regions and at the end of the chains or on the surface of the crystallites because the reactants do not diffuse easily in the crystalline region. This results in the opening of some of the hydrogen-bonded cellulosic chains, thus producing the formation of amorphous cellulose. Myristic acid then diffuse into these new amorphous regions to react with the newly accessible hydroxyl groups and consequently generate more amorphous cellulose.10
Thermal behavior of esterified wood
The thermal properties of esterified wood were observed by TGA, DSC, and TMA. The thermal degradation of spruce sawdust before and after esterification is presented in Fig. 8. Before modification, a weight loss of 2.67% was recorded as water evaporation of absorbed water in the cell walls between 30 and 120 °C, as observed in the DSC result (Fig. 9) as an endothermic phenomenon; then, the mass starts to decrease sharply to 70.64% from 200 to 400 °C with two slopes observed in this range of temperature. A weight loss of 24.41% was obtained between 200 and 330 °C, which is attributed to the decomposition of thermally fragile hemicellulose and weight loss of 46.23% between 330 and 400 °C corresponding to cellulose degradation. Wood degradation of 7.94% with a smaller slope continued to occur between 400 and 600 °C corresponding to the degradation of lignin.20 At the end of the measurement, there was 18.25% residue. | ||
| Fig. 8 TGA and DTG of wood sawdust before and after esterification. | ||
 | ||
| Fig. 9 DSC of wood sawdust before and after esterification. | ||
After being esterified, water evaporation was not recorded at the same range of temperature observed in the TGA and DSC results. The absence of water evaporation was an indication that the hydrophobicity of the material was obtained after esterification with myristic acid. This behavior was already reported by Thiebaud et al.20 and Wu et al.21 The thermal degradation temperature range of esterified wood was between 260 and 440 °C with weight loss of 97.35% and the midpoint increase was 28 °C from 350 to 378 °C compared to non-modified wood. The higher degradation temperature of esterified wood could be explained by the greater thermal stability of esterified hemicellulose20 and crystallization of the long aliphatic side chains of esterified wood.42
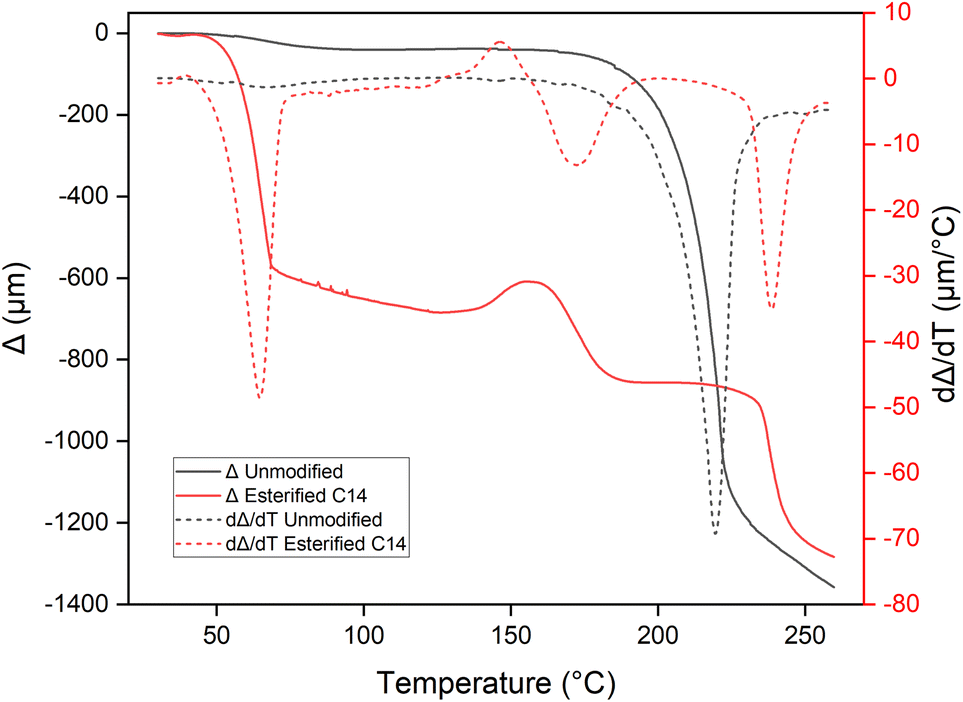
Fig. 10 showed the deformation of the pressed wood sample before and after esterification at various temperatures under constant loading, which represent the softening temperature. Unmodified pressed wood only showed single deformation after 200 °C. This deformation could be caused by the beginning of decomposition of wood component, as explained in the TGA results. After esterification, the first deformation appeared very early between 36 and 76 °C, indicating that the film losses its rigidity because the myristic acid grafted to the wood. The second deformation arose between 148 and 198 °C. These two softening temperatures were also recorded from the DSC result as an endothermic phenomena at 32.3–103.4 °C and 100–160 °C. Hou et al.26 reported the glass transition temperature at the same range of temperature corresponding to the reorganization of fatty acid chain and cellulose backbones in oleic acid-esterified cellulose. The third deformation was found between 227 and 265 °C. Three softening points with the same temperature ranges were also reported by Funakoshi9 for esterified wood, which explained the first deformation or T1 corresponds to the melting point of lauroyl cellulose within the wood structure, second deformation or T2 corresponds to the thermal softening of lignin, and T3 is considered to the melting point of the chemically-modified wood meal. Using the same atom number carbon, Shiraishi et al.16 reported the apparent melting of myristoyl wood at 200 °C, while Thiebaud and Borredon19 reported a higher apparent melting temperature at 310 °C.
 | ||
| Fig. 10 TMA of wood film before and after esterification. | ||
Moreover, the possibility of measuring an apparent melting temperature for our samples as soon as the degree of substitution is sufficient, which confirms that the grafting of a myristyl chain ester group brings thermoplastic properties to the wood.
Visual aspect and surface properties of esterified film
To confirm the thermoplastic and translucent properties of wood delivered by myristic acid esterification, visual inspection was performed. Non-modified pressed spruce sawdust showed brownish opaque (Fig. 11A) and fibrous surface, which was clearly observed from scanning electronic micrographs (Fig. 12A). After esterification and pressing at 120 °C, the color of the film drastically turns to yellowish and fairly translucent (Fig. 11B). Fig. 12B also shows the disappearance of fibrous structure of wood, which indicated thermal melting with some part of aggregation of wood fiber. When the temperature of the press was increased to 140 °C, a clear yellow translucent film aspect was obtained. Surface homogeneous and smoothness improvement were observed (Fig. 12C) comparatively to the sample pressed at 120 °C, indicating that sufficient thermal fluidity was achieved when myristic acid-esterified spruce sawdust was pressed at 140 °C, which corresponds to the second deformation temperature observed in TMA. Shiraishi et al.16 reported the same temperature of pressing for lauroylated wood. The melting temperature of high ester content lauroylated wood sawdust before pressing was reported at 275 °C (ref. 9) (around the third deformation temperature of TMA); meanwhile, complete translucent film in our research was obtained far below at 140 °C. The color of the film will get darker when the press temperature increases to 200 °C. | ||
| Fig. 11 Visual appearance of pressed spruce sawdust before esterification (A) and after esterification at different press temperatures (B). | ||
 | ||
| Fig. 12 Scanning electron micrograph of pressed unmodified wood at 200 °C (A), myristic acid-esterified wood after pressing at 120 °C (B), after pressing at 140 °C (C), and after pressing at 160 °C (D). | ||
Contact angle measurement by the water droplet of pressed spruce film before and after esterification was conducted to understand the surface properties. Fig. 13 shows that before esterification, the water droplet formed a low contact angle right after the measurement began. The myristic acid-esterified sample exhibited a drastic improvement of the contact angle from 96° at the beginning of measurement and 93° after 60 s of measurement, as also reported by Thiebaud et al.20 for C14 fatty acid chloride-esterified wood. The contact angle obtained proved that myristic acid esterification of wood sawdust promotes the hydrophobicity of the surface and confirms the disappearance of the hydroxyl group from the FTIR result and water evaporation from the TGA and DSC results.
 | ||
| Fig. 13 Contact angle of pressed wood film before and after esterification. | ||
Conclusion
The esterification of wood sawdust has been successfully achieved by myristic acid at room temperature without a solvent using TFAA as the impelling agent. The evaluation of chemical properties of modified sawdust revealed the disappearance of hydroxyl groups of the wood substituted by the alkyl group of myristic acid and ester groups, change in the carbohydrate structure, and decrystallization of cellulose. These chemical changes improved the thermal stability of esterified wood, which exhibited a softening temperature that allowed esterified wood to be pressed and form a hydrophobic translucent film. The first softening temperature corresponds to the melting of myristic acid grafted to wood, which decreased the film rigidity. The second softening temperature corresponds to the softening of wood component together with myristic acid, which allowed esterified wood to be pressed and form a translucent film. The third softening temperature corresponds to the complete softening of esterified wood, which can be observed only in the presence of pressure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Région GrandEst and Lorraine University for the doctoral fellowship granted to the first author. UR 4370 LERMAB is supported by a grant overseen by the French National Research Agency (ANR) as part of the “Investissements d’Avenir” program (ANR-11-LABX-0002-01, Lab of Excellence ARBRE) in the frame of the project “Woodstic”. The authors also thank ICEEL for the financial in the frame of the project “BoisPlast” (CARN 0013 01). The authors warmly thank Pierrick Durand for technical support during X-Ray Diffraction analysis.References
- D. E. MacArthur, Science, 2017, 358, 843 CrossRef CAS PubMed.
- S. O. Cinar, Z. K. Chong, M. A. Kucuker, N. Wieczorek, U. Cengiz and K. Kuchta, Int. J. Environ. Res. Public Health, 2020, 17, 1–21 Search PubMed.
- M. Bocqué, C. Voirin, V. Lapinte, S. Caillol and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 11–33 CrossRef.
- J. Yang, Y. C. Ching and C. H. Chuah, Polymers, 2019, 11, 1–26 Search PubMed.
- R. Reshmy, D. Thomas, E. Philip, S. A. Paul, A. Madhavan, R. Sindhu, R. Sirohi, S. Varjani, A. Pugazhendhi, A. Pandey and P. Binod, Rev. Environ. Sci. Biotechnol., 2021, 20, 167–187 CrossRef CAS.
- J. A. Okolie, S. Nanda, A. K. Dalai and J. A. Kozinski, Waste Biomass Valorization, 2021, 12, 2145–2169 CrossRef CAS.
- L. Koehler and F. W. Telewski, Am. J. Bot., 2006, 93, 1433–1438 CrossRef CAS PubMed.
- P. Gerardin, in Lignocellulosic Fibers and Wood Handbook: Renewable Materials for Today's Environment, John Wiley & Sons, 2016, pp. 313–322 Search PubMed.
- H. Funakoshi, N. Shiraishi, M. Norimoto, T. Aoki and H. Hayashi, Holzforschung, 1979, 33, 159–166 CrossRef CAS.
- N. Shiraishi, T. Matsunaga, T. Yokota and Y. Hayashi, J. Appl. Polym. Sci., 1979, 24, 2347–2359 CrossRef CAS.
- H. Matsuda, Wood Sci. Technol., 1987, 21, 75–88 CrossRef CAS.
- Y. Zhang, Y. Xue, H. Toghiani, J. Zhang and C. U. Pittman, Compos. Interfaces, 2009, 16, 671–686 CrossRef CAS.
- P. C. Arni, J. D. Gray and R. K. Scougall, J. Appl. Chem., 1961, 11, 157–163 CrossRef CAS.
- T. Nakagami, H. Amimoto and T. Yokota, Bull. Kyoto Univ. For., 1974, 46, 217–224 Search PubMed.
- T. Nakagami and T. Yokota, Bull. Kyoto Univ. For., 1975, 47, 178–183 Search PubMed.
- N. Shiraishi, A. Tutomu, M. Norimoto and M. Okumura, in Graft Copolymerization of Lignocellulosic Fibers, 1982, pp. 321–348 Search PubMed.
- N. Shiraishi and M. Yoshioka, Sen'i Gakkaishi, 1986, 42, 346–355 CrossRef.
- T. Nakano, Holzforschung, 1994, 48, 318–324 CrossRef CAS.
- S. Thiebaud and M. E. Borredon, Bioresour. Technol., 1995, 52, 169–173 CrossRef CAS.
- S. Thiebaud, M. E. Borredon, G. Baziard and F. Senocq, Bioresour. Technol., 1997, 59, 103–107 CrossRef CAS.
- J. H. Wu, T. Y. Hsieh, H. Y. Lin, I. L. Shiau and S. T. Chang, Wood Sci. Technol., 2004, 37, 363–372 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- N. Shiraishi, T. Matsunaga and T. Yokota, J. Appl. Polym. Sci., 1979, 24, 2361–2368 CrossRef CAS.
- H. Hanabusa, E. I. Izgorodina, S. Suzuki, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, Green Chem., 2018, 20, 1412–1422 RSC.
- M. Pei, X. Peng, Y. Shen, Y. Yang, Y. Guo, Q. Zheng, H. Xie and H. Sun, Green Chem., 2020, 22, 707–717 RSC.
- D. F. Hou, M. L. Li, C. Yan, L. Zhou, Z. Y. Liu, W. Yang and M. B. Yang, Green Chem., 2021, 23, 2069–2078 RSC.
- L. Duchatel-Crépy, N. Joly, P. Martin, A. Marin, J. F. Tahon, J. M. Lefebvre and V. Gaucher, Carbohydr. Polym., 2020, 234, 115912 CrossRef PubMed.
- G. Gilardi, L. Abis and A. E. G. Cass, Enzyme Microb. Technol., 1995, 17, 268–275 CrossRef CAS.
- H. Wikberg and S. L. Maunu, Carbohydr. Polym., 2004, 58, 461–466 CrossRef CAS.
- S. G. Kostryukov, P. S. Petrov, V. S. Tezikova, Y. Y. Masterova, T. J. Idris and N. S. Kostryukov, Cellul. Chem. Technol., 2021, 55, 461–468 CAS.
- C. M. Popescu, P. T. Larsson and C. Vasile, Carbohydr. Polym., 2011, 83, 808–812 CrossRef CAS.
- C. M. Popescu, D. E. Demco and M. Möller, Polym. Degrad. Stab., 2013, 98, 2730–2734 CrossRef CAS.
- M. Bardet, M. F. Foray and Q. K. Trân, Anal. Chem., 2002, 74, 4386–4390 CrossRef CAS PubMed.
- L. Wei, A. G. McDonald, C. Freitag and J. J. Morrell, Polym. Degrad. Stab., 2013, 98, 1348–1361 CrossRef CAS.
- G. K. Prakash and K. M. Mahadevan, Appl. Surf. Sci., 2008, 254, 1751–1756 CrossRef CAS.
- J. Chen, C. Tang, Y. Yue, W. Qiao, J. Hong, T. Kitaoka and Z. Yang, Ind. Crops Prod., 2017, 108, 286–294 CrossRef CAS.
- S. Link, S. Arvelakis, H. Spliethoff, P. De Waard and A. Samoson, Energy Fuels, 2008, 22, 3523–3530 CrossRef CAS.
- K. J. Heritage, J. Mann and L. Roldan-Gonzalez, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 671–685 CrossRef CAS.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2010, 58, 11302–11310 CrossRef CAS.
- S. Borysiak, J. Appl. Polym. Sci., 2013, 127, 1309–1322 CrossRef CAS.
- S. Andersson, R. Serimaa, T. Paakkari, P. Saranpää and E. Pesonen, J. Wood Sci., 2003, 49, 531–537 CrossRef.
- U. Ratanakamnuan, D. Atong and D. Aht-Ong, Carbohydr. Polym., 2012, 87, 84–94 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |