
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali halides as nucleophilic reagent sources for N-directed palladium-catalysed ortho-C–H halogenation of s-tetrazines and other heteroaromatics†
Ahmad Daher,
Oumaima Abidi,
Jean-Cyrille Hierso * and
Julien Roger*
* and
Julien Roger*
Institut de Chimie Moléculaire de l′Université de Bourgogne (UMR-CNRS 6302), Université Bourgogne Franche-Comté (UBFC), 9 Avenue Alain Savary 21078 Dijon, France. E-mail: julien.roger@u-bourgogne.fr; jean-cyrille.hierso@u-bourgogne.fr
First published on 26th October 2022
Abstract
A general palladium-catalysed selective C–H halogenation reaction is reported, which was successfully achieved for a large variety of functionalized aromatic rings incorporating diverse N-directing groups. By using simple alkali halides of MX type as the nucleophilic reagent source (M = Li, Na, K, Cs and X = I, Br and Cl), and phenyliodanediacetate oxidant, clean C–H-iodination, bromination and chlorination reactions were performed. This general protocol of selective ortho-monohalogenation, which complements but contrasts with the classical methods using electrophilic reagents, is achievable in a short time (30 min) with microwave irradiation assistance. The reaction was extended to substrates bearing N-directing pyridine, pyrimidine, pyrazole, oxazoline, naphtho[1,2-d]thiazole, and azobenzene groups. Notably, the topical and selectivity-challenging s-tetrazine, as a nitrogen-rich heteroaromatic, was successfully halogenated by this protocol.
Introduction
Organic halides are essential synthetic precursors to reach highly functionalized pharmaceuticals, agrochemicals, and operative materials.1 The synthetic and industrial routes to form these important building blocks, like heteroaromatic halides, generally involve multiple step reactions, often under harsh conditions.2 The use of expensive and toxic reagents and solvents, under energy-consuming conditions, is an issue that is tackled by the search for more sustainable chemistry processes.3 An alternative pathway to overcome the lack of selectivity encountered in the traditional organic halogenation protocols has been to exploit useful transmetallation processes such as boron-based,4 albeit these are generally synthesized from an halide precursor, and in fine invariably generates undesirable stoichiometric metallic waste.Ligand-directed C–H bond functionalization that is achieved by using the versatility of transition metal chemistry allowed much progress for the mild and selective introduction of halogens in synthetic organics (Scheme 1, top).5 The number of synthetic steps is reduced, and so are the side-reactions and purification procedures.
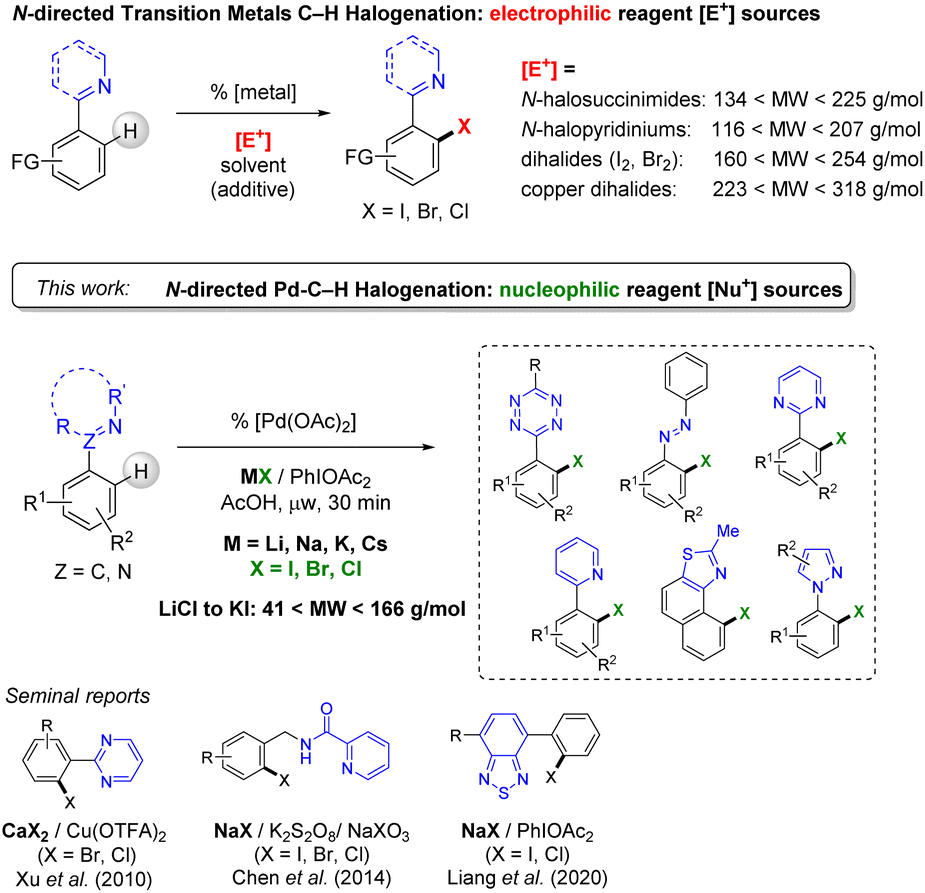 | ||
| Scheme 1 Electrophilic N-directed halogenation (top) and N-directed palladium-catalysed mono-halogenation using alkali halide nucleophilic sources (this work). | ||
The amount of waste is also minored by improving the regioselectivity of the functionalization, and the global tolerance to varied substituents. Following this strategy, our group and others illustrated the efficiency of palladium catalysts for the ortho-halogenation on different N-containing heteroarenes, including challenging substrates of high nitrogen-content.5,6
The current ligand-directed C–H halogenation approach has been mainly focused on the use of highly reactive preformed electrophilic reagents such as N-halosuccinimides, N-halopyridiniums, dihalides or transition metal halides (CuX2 for instance).7 These reagents, despite their undisputable synthetic usefulness, suffer from several limitations that include their cost and relatively low atom-economic aspect (see their range of molecular weight, MW, Scheme 1, top).
In addition to the amount of waste generated, a critical limitation of this electrophilic reagent synthetic approach is that concurrent non-catalysed electrophilic aromatic substitution occurs in many instance.8,9 This is favored by the presence of activating substituent on the heteroaromatic substrates. Thus, such a competition with the targeted regioselective metal catalysed C–H activation is detrimental to atom-economy.
With the view to decrease undesired side-reactions, and the total cost of the transformation, the employment of widely available, intrinsically less reactive, or more selective, halogen sources appears to be highly desirable. These may include alkali halide nucleophilic sources, if suitable general synthetic routes are established (Scheme 1, middle). Several groups pioneered an approach of “nucleophilic” C–H halogenation promoted by palladium catalysts with selected N-directing groups. In 2010, Xu et al. reported calcium halide CaX2 reagents for the o-functionalization of pyrimidine derivatives with cupric trifluoroacetate as the oxidant (Scheme 1, bottom).10 A selective monohalogenation was achieved using 6.0 to 8.0 equiv. of calcium bromide. In 2014, Chen et al. reported the o-halogenation of an aromatic in the presence of sodium halides NaX (X = I, Br, Cl) using benzylamine derivatives.11 A mixture of NaX, NaXO3 and K2S2O8 was used for the introduction of the halide on benzylamine derivatives, albeit with a difficult control of regioselectivity when several halogenation sites were available. Most recently, Liang and co-workers reported the employment of phenyliodanediacetate (PIDA) as the oxidant in the presence of NaCl and NaI for the N-directed Pd-catalysed halogenation of benzothiadiazoles.12 This useful and promising catalytic system was surprisingly found ineffective for C–H direct bromination, and ultimately N-bromosuccinimide (NBS) has to be used. In 2020, Jiao et al. reported the bromination of pyridine and pyrimidine aryls in the presence of HBr as the nucleophilic halide with DMSO, which played the role of solvent and oxidant.13 Additionally, few works on the nucleophilic C–H halogenation that are promoted by Cu, Ni or Rh were also reported.14 Therefore, the generalization of these approaches to a larger range of useful directing-groups, and achieved from cheap and widely available nucleophiles is highly desirable.
We now report the conditions for a wide scope palladium-catalysed N-directed o-halogenation of heteroaromatics based on simple alkali metal halides as “nucleophilic” sources, using PIDA as the oxidant (Scheme 1, bottom). In this generalized protocol, the heteroaromatic substrates suitable for direct C–H halogenation are functionalized pyridines, pyrimidines, pyrazoles, oxazolines, naphtho[1,2-d]thiazoles, azobenzenes, and notably the selectivity-challenging and clickable/bioconjugable nitrogen-rich s-aryltetrazine. These substrates including N-directing groups were successfully employed for highly selective monohalogenation (X = I, Br and Cl) using the full range of alkali metals Li, Na, K and Cs. A good tolerance to substituents on the C–H-halogenated aryl was probed and the use of microwave conditions efficiently reduced long-time synthesis to less than one hour.
Results and discussion
N-directed palladium catalysed nucleophilic iodination of heteroaromatics
S-aryltetrazine were arguably the most challenging substrates for selective monohalogenation. The substrates can undergo up to four concurrent C–H functionalization reactions, and we previously disclosed its practical and very efficient o-C–H-fluorination, as well as multistep analogous halogenation using electrophilic reagents.15–17 We envisioned, however, some room for improvement in terms of reagents and selectivity.For the direct C–H iodination of 3,6-bis(2- fluorophenyl)-1,2,4,5-tetrazine 1 (Scheme 2) we explored the performances of various palladium-based catalysts, the full set of MI alkali halides as source (where M = Li, Na, K and Cs), and the oxidizing agents PIDA, PIFA, and K2S2O8 within various solvents (see also Table S1 in ESI†). N-directed C–H bond activation was best achieved in the presence of [Pd(OAc)2] and PIDA, in acetic acid at 110 °C for 30 min under microwave conditions (μw, 200 W). All the alkali metal sources LiI, NaI, KI, CsI, and even N(t-Bu)4I, were found compatible with the s-aryltetrazine substrate for its C–H iodination in excellent conversion up to 72% (Scheme 2), with the formation of ca 15–25% of the dihalogenated product. Overall, as fairly good selectivity is achieved for the monoiodinated product 1a (67 to 72%), especially concerning the concurrent reaction of C–H acetoxylation that is found deleterious when electrophilic sources are used for this reaction.15
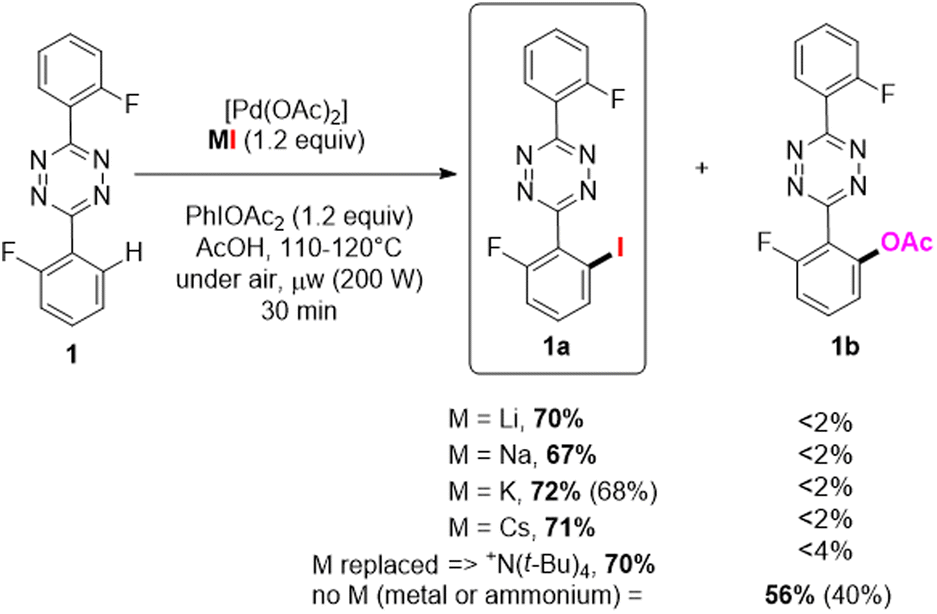 | ||
| Scheme 2 o-C–H iodination palladium-catalysed with alkali halides or ammonium salt as nucleophilic reagents: LiI, NaI, KI, CsI, [N(t-Bu)4]I. | ||
The cheapest potassium iodide was selected as most suitable reagent. Notably, KI was neither used in the pioneering works above-mentioned as “nucleophilic” source of halide. This new monoiodination methodology was found superior to our previous results according its conversion, selectivity, and importantly for the purification procedure.
For instance, electrophilic reaction using NIS furnished 57% of 1a due to a slightly better selectivity in favor of monofunctionalization observed with the “nucleophilic” C–H halogenation approach (typically, the use of 2 equiv. of NaI yielded 1a and 1a′ in 67/33 ratio vs. 57/43 using NIS16). Without alkaline metal halide present the acetoxylated product 1b was preferentially formed, and could be isolated in 40% yield (Scheme 2).
With these optimized conditions in hands, we further investigated the scope of heteroaromatic compounds as coupling partners in the C–H iodination reaction (Scheme 3).15,16 We mainly achieved o-C–H monoiodination of s-aryltetrazines and azobenzene 1–5 (optimization details in Table S1 in ESI†). These substrates are arguably the most challenging substrates for selective monohalogenation since up to four concurrent C–H functionalization reactions may potentially occur. Pleasingly, alkali salts, which had neither been used before for such substrates (s-aryltetrazines), are clearly suitable halogen sources. Overall, the monoiodination was achieved in moderate to good isolated yields of 61% and 51% for 2a and 3a respectively. C3-substituted s-aryltetrazines are also suitable for C–H halogenation, the functionalization occurred in the para-position from the C3 group. Only traces of the C2-iodinated product was detected and the targeted C6-halogenated tetrazine 4a was obtained in 35% isolated yield with satisfying purity (+99%). The presence of functional groups at the meta-position (C3) of the arene induces a dominant selectivity for the functionalization at C6, presumably the concurrent C2-position is sterically disfavoured since this effect is observed both for electron-donating or electron-withdrawing groups at C3, as previously reported by our group and others.6a,8,18 Using a wider scope of heteroaromatic substrates, only minor reactivity changes were observed. [Pd(OAc)2] at 10 mol% was found to be necessary for the conversion of azobenzene 5, giving 5a in 39% isolated yield. The aryl-pyrimidine 6, with a six-membered ring directing group, achieved the aryl C–H o-iodination to give the pure 6a in 65% yield.
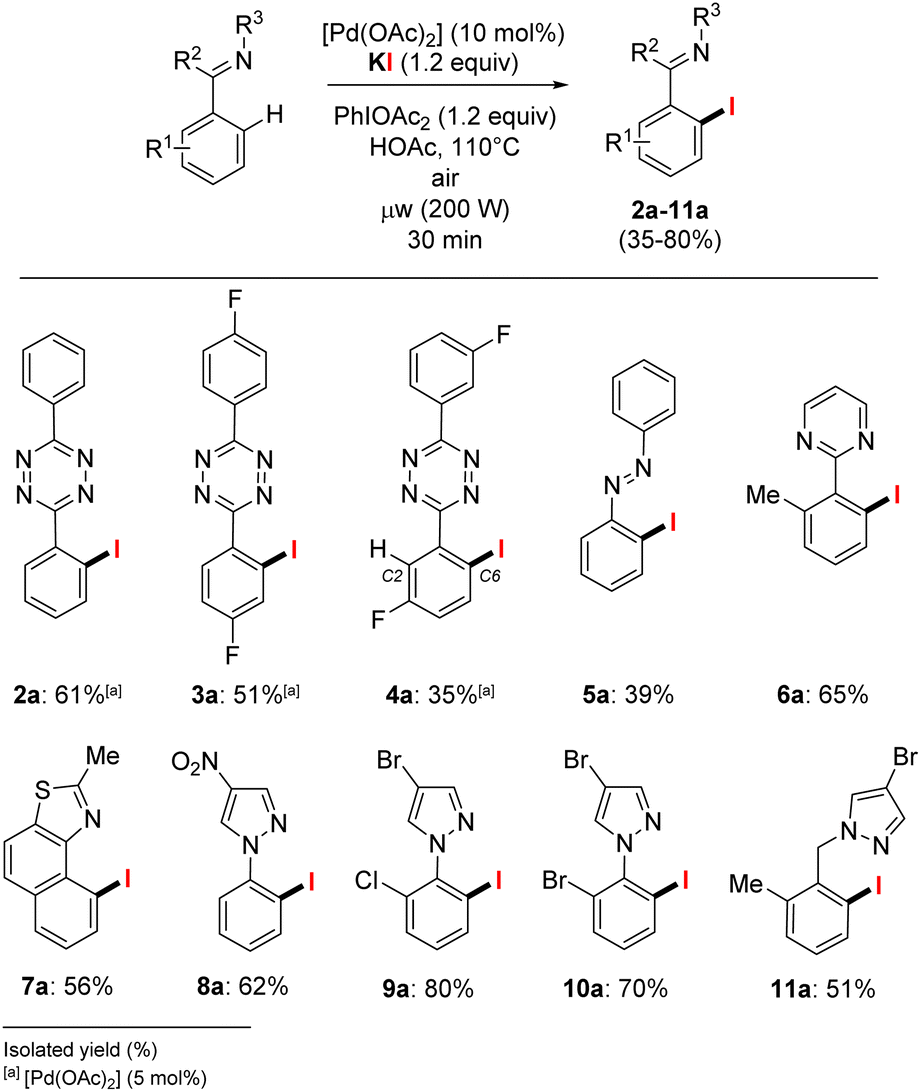 | ||
| Scheme 3 o-C–H iodination of heteroaromatics from KI halogen source. | ||
The other substrates with five-membered N-directing groups also achieved C–H o-iodination in satisfactory to good yields. The naphtho[1,2-d]thiazole 7 was successfully o-iodinated for 7a in 56% isolated yield after side-products removal, including compounds from sp3C–H iodination of the methyl group. The o-iodinated arylpyrazoles 8a to 10a were obtained in isolated yields above 60% and tolerated bromo and nitro functions, and 11a with a remote directing bromopyrazole was isolated in 51% yield. Overall, the conversion of these various heteroaromatics is mostly achieved between 70–90% yields, with the formation of less than 15% of dihalogenated product (see Table S1 in ESI†). Accordingly, the need for separation with a high final purity led to isolated yields of 2a–11a between 35 and 80%.
N-directed palladium catalysed bromination of heteroaromatics
Liang and co-workers reported a lack of reactivity of NaBr in the N-directed palladium catalysed o-bromination of benzothiadiazole derivatives.12 We investigated the use of KBr as the halide source on various heteroaromatic substrates (Scheme 4). Under our conditions, the challenging bromination of s-aryltetrazines 1–4 was achieved, with good to moderate yields obtained for 1c–4c in 60% to 35% isolated yields. This method also proceeded properly for the o-halogenation of 3,6-bis(benzylic) s-tetrazine 12, giving pure 12c in 54% yield. The azobenzene 5 gave 60% of brominated 5c, a much higher yield than obtained for its iodinated counterpart 5a (39%, Scheme 3). The pyridine and pyrimidine heteroaromatic substrates, which are in general broadly employed in C–H halogenation, were also successfully brominated in 51% and 61% isolated yield for 13c and 6c, respectively. This is valuable, since conversely for the catalysed C–H functionalization of 2,4-difluorophenyl pyridine in the presence of KI, the homocoupling reaction was achieved instead of the expected halogenation.19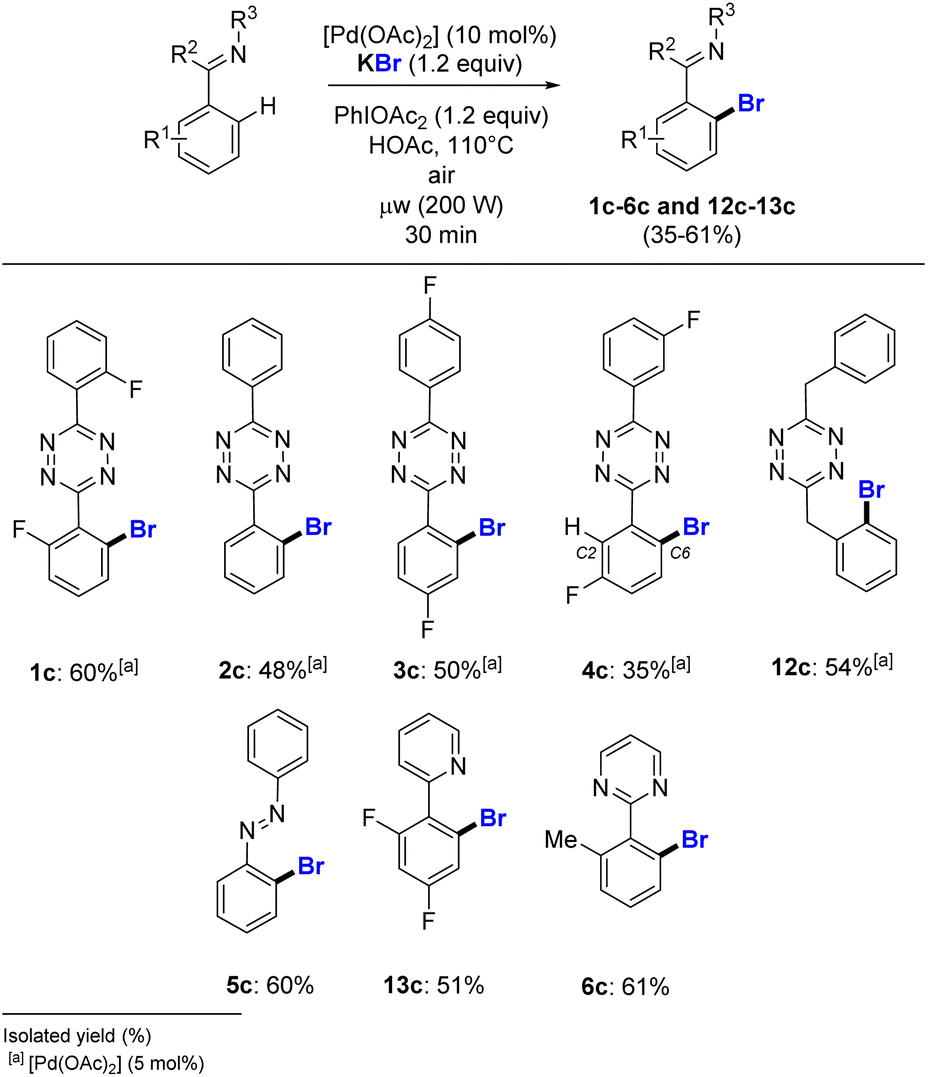 | ||
| Scheme 4 o-C–H bromination of heteroaromatics from KBr halogen source. | ||
N-directed palladium catalysed chlorination of heteroaromatics
The palladium catalysed ortho-halogenation with nucleophilic alkali halides was further extended to chlorination. The selective o-C–H-monochlorination was achieved on a set of heteroaromatic substrates from the use of 1.2 equivalent of the low-mass KCl (MW = 74.6 g mol−1). By using a reduced amount of palladium catalyst, [Pd(OAc)2] 5 mol%, the s-tetrazines 1–4 were chlorinated to give isolated yields of pure 1d–4d, in 55%, 35%, 45% and 28% yield, respectively (Scheme 5). The 2,4-difluorophenyl pyridine and naphtho[1,2-d]thiazole substrates also successfully achieved a selective o-aryl monochlorination, giving 70% and 53% isolated yields for 13d and 7d, respectively.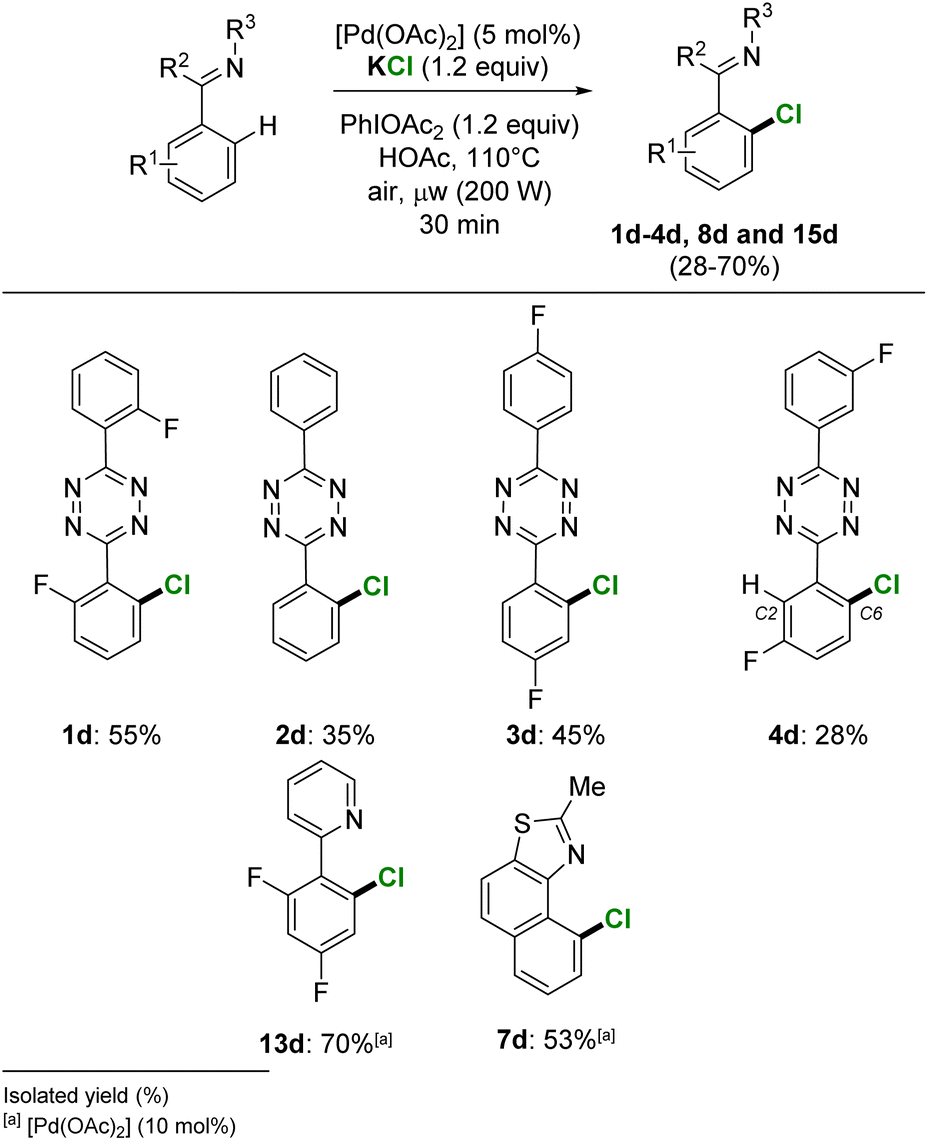 | ||
| Scheme 5 o-C–H chlorination of heteroaromatics from KCl halogen source. | ||
Remarkably, the full range of alkali metals Li, Na, K and Cs with the various halides displayed very similar reactivity. While in-depth mechanistic investigation is out of the scope of the present study, we hypothesize that classical Pd(II)/Pd(IV) process may occur, with the possible formation of PhI(X)2 or PhI(X)(OAc) as “genuine” halogenation reagents by ligand exchange with OAc (X = I, Br and Cl). This would be consistent with the apparent unicity of reactivity. Experimental and computational reports are available concerning ligand exchanges between salts and hypervalent iodine, and the related radical (non-electrophilic) reactivity.20
Finally, we used our new alkali halide-based C–H functionalization protocol to access the unsymmetrical trihalogenated pyrazole 16, starting from the phenyl 4-chloropyrazole 14 and by sequential C–H bond activation (Scheme 6). The first halogenation takes place in the presence of 10 mol% of [Pd(OAc)2], KBr (1.2 equiv.), PIDA (1.2 equiv.) in HOAc at 110 °C for 30 min under microwave irradiation. The mono o-brominated pyrazole 15 was obtained in 46% isolated yield, which allowed the second functionalization under similar conditions using KI instead of KBr and a high yield for 16 was obtained without any other byproduct formed.
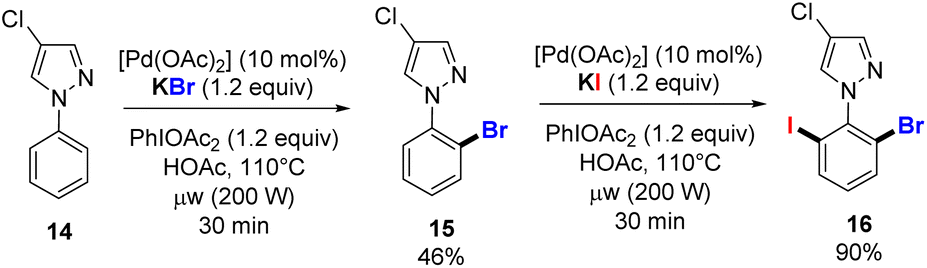 | ||
| Scheme 6 Sequential palladium-catalysed o-C–H halogenation. | ||
Conclusion
We reported a novel convenient protocol for a palladium-catalysed N-directed o-halogenation of heteroaromatic based on the use of simple alkali metal halides as nucleophilic sources and PIDA as oxidant. Monofunctionalization is achieved in majority, in general together with the formation of ca 15–25% of the dihalogenated product.The reaction was found suitable for a large range of structurally different N-heteroaromatic substrates, which includes pyridines, pyrimidines, pyrazoles, oxazolines, naphtho[1,2-d]thiazoles, azobenzenes, and specifically the topical (in click chemistry and bioconjugation applications15,16) and challenging nitrogen-rich heteroaromatic s-tetrazine. Highly selective o-aryl monohalogenation was achieved (halogen = I, Br and Cl), which compared favorably to the classical protocols using electrophilic reagents in palladium-catalysed N-directed ortho-C–H halogenation. These results paved the way for the use of simple alkali halides in both inexpensive (cheaper salts) and more atom-economic (mass economy) conditions for selective palladium-catalysed C–H halogenation.
Authors and distributions
A. D. and O. A. performed the experiments, and J. R. and J.-C. H. conceived the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS, the Université de Bourgogne, the Conseil Régional de Bourgogne and the Fonds Européen de Développement Regional (FEDER). Financial supports from UBFC (I-SITE UB180013. MUB. IS_SmartTZ, PhD AD) and by the ANR (JCJC ANR-18-CE07-0015 – FIT-Fun, PhD OA) are acknowledged. Thanks is due to the PACSMUB platform for analyses (SATT SAYENS) especially M.-J. Penouilh, Q. Bonnin and T. Régnier.Notes and references
- (a) T. Kosjek, E. Heath, J. Iskra, ed. Springer, New York, Halogenated Heterocycles, 2012 Search PubMed; (b) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS; (c) M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- (a) H. H. Hodgson, Chem. Rev., 1947, 40, 251–277 CrossRef CAS PubMed; (b) J. Clayden, N. Greeves, S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn, 2012, pp. 471–497 Search PubMed.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC; (b) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC; (c) K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed; (d) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC.
- C. Zhu and J. R. Falck, Adv. Synth. Catal., 2014, 356, 2395–2410 CrossRef CAS PubMed.
- (a) C. Testa, J. Roger, P. Fleurat-Lessard and J.-C. Hierso, Eur. J. Org. Chem., 2019, 2019, 233–253 CrossRef CAS; (b) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524–1541 CrossRef CAS; (c) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed; (d) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS.
- (a) C. Testa, J. Roger, S. Schieb, P. Fleurat-Lessard and J.-C. Hierso, Adv. Synth. Catal., 2015, 357, 2913–2923 CrossRef CAS; (b) J. Guilbaud, M. Labonde, H. Cattey, S. Contal, C. Montalbetti, N. Pirio, J. Roger and J.-C. Hierso, Adv. Synth. Catal., 2017, 21, 3792–3804 CrossRef; (c) J. Guilbaud, A. Selmi, M. Kammoun, S. Contal, C. Montalbetti, N. Pirio, J. Roger and J.-C. Hierso, ACS Omega, 2019, 4, 20459–20469 CrossRef CAS PubMed.
- X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416–7417 CrossRef CAS PubMed.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 CrossRef CAS.
- (a) L. Ping, D. S. Chung, J. Bouffard and S.-G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC; (b) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524–1541 CrossRef CAS.
- B. Song, X. Zheng, J. Mo and B. Xu, Adv. Synth. Catal., 2010, 352, 329–335 CrossRef CAS.
- C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Tetrahedron, 2014, 70, 4197–4203 CrossRef CAS.
- H. He, J. Guo, W. Sun, B. Yang, F. Zhang and G. Liang, J. Org. Chem., 2020, 85, 3788–3798 CrossRef CAS PubMed.
- Y. Yuan, Y. Liang, S. Shi, Y.-F. Liang and N. Jiao, Chin. J. Chem., 2020, 38, 1245–1251 CrossRef CAS.
- (a) Y. Lu, R. Wang, X. Qiao and Z. Shen, Synlett, 2011, 7, 1038–1042 Search PubMed; (b) S. Mo, Y. Zhu and Z. Shen, Org. Biomol. Chem., 2013, 7, 2756–2760 RSC; (c) B.-B. Zhan, Y.-H. Liu, F. Hu and B.-F. Shi, Chem. Commun., 2016, 52, 4934–4937 RSC; (d) S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100–104 RSC.
- C. Testa, E. Gigot, S. Genc, R. Decreau, J. Roger and J.-C. Hierso, Angew. Chem., Int. Ed., 2016, 55, 5555–5559 CrossRef CAS PubMed.
- C. D. Mboyi, C. Testa, S. Reeb, S. Genc, H. Cattey, P. Fleurat-Lessard, J. Roger and J.-C. Hierso, ACS Catal., 2017, 7, 8493–8501 CrossRef CAS.
- For examples on Pd-catalysed electrophilic iodination, see: (a) L. Hu, H. Xu, Q. Yang, Z. Deng, C.-Y. Yu and Y. Peng, J. Organomet. Chem., 2018, 9, 100–104 Search PubMed; (b) S. Gupta, J. A. Melanson, L. Vaillancourt, W. A. Nugent, G. J. Tanoury, G. Schatte and V. Snieckus, Org. Lett., 2018, 20, 3745–3748 CrossRef CAS PubMed; (c) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326–10329 CrossRef CAS PubMed.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 CrossRef CAS PubMed.
- W. Li, Y. Zhu and C.-J. Li, Synthesis, 2016, 48, 1616–1621 CrossRef.
- See: (a) K. Kang, S. Lee and H. Kim, Asian J. Org. Chem., 2015, 4, 137–140 CrossRef CAS; (b) Z.-W. Qu, H. Zhu and S. Grimme, ChemCatChem, 2020, 12, 6186–6190 CrossRef CAS; (c) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Tetrahedron, 2009, 65, 10797–10815 CrossRef CAS , and references therein..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06169d |
| This journal is © The Royal Society of Chemistry 2022 |