
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La2O3 oxides for ammonia synthesis†
Hubert Ronduda a,
Magdalena Zyberta,
Wojciech Patkowskia,
Kamil Sobczakb,
Dariusz Moszyńskic,
Aleksander Albrechtc,
Adam Sarneckic and
Wioletta Raróg-Pilecka*a
a,
Magdalena Zyberta,
Wojciech Patkowskia,
Kamil Sobczakb,
Dariusz Moszyńskic,
Aleksander Albrechtc,
Adam Sarneckic and
Wioletta Raróg-Pilecka*a
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664, Warsaw, Poland. E-mail: wiola@ch.pw.edu.pl; Tel: +48 22 234 57 66
bUniversity of Warsaw Biological and Chemical Research Centre, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cWest Pomeranian University of Technology in Szczecin, Faculty of Chemical Technology and Engineering, 42 Piastów Ave, 71-065 Szczecin, Poland
First published on 25th November 2022
Abstract
Synthesis of ammonia from nitrogen and hydrogen is one of the largest manmade chemical processes, with annual production reaching 170 million tons. The Haber–Bosch process is the main industrial method for producing ammonia, which proceeds at high temperatures (400–600 °C) and pressures (20–40 MPa) using an iron-based catalyst. It is thus highly desirable to develop new catalysts with sufficient activity and stability under mild conditions. In this work, we report cobalt catalysts supported on magnesium–lanthanum mixed oxide with different Co loading amounts synthesised via a simple wet impregnation method. We have found a clear relationship between the ammonia synthesis rate and the Co loading amount. Specifically, the NH3 synthesis rate increased on increasing cobalt loading and reached a maximum at 40 wt% Co deposition. A further increase in Co loading did not change the activity significantly. Interestingly, the surface-specific activity (TOF) remained almost unchanged regardless of the Co loading amount in the catalysts. It revealed that the resultant ammonia synthesis rate over the studied catalysts did not depend on the size and structure of Co nanoparticles but strongly on the Co loading amount. Finally, it is believed that the use of this type of catalyst will be a starting point toward energy-efficient ammonia production.
1. Introduction
With the rapid population growth and continuous environmental deterioration, human beings are facing increasing demand for efficient solutions for energy conversion and storage applications. As a clean and sustainable energy carrier, hydrogen is considered the most promising method to address the energy crisis in the upcoming decades.1–13 However, the problem of hydrogen storage and transportation is the main obstacle to the widespread application of hydrogen as a feeding fuel since hydrogen has a very low volumetric density (0.0899 kg m−3) under ambient conditions.14 Thus, the development of safe and effective hydrogen storage materials is a step towards a hydrogen-based energy economy.15–24 Recently, ammonia has been considered a promising energy vector that can store the high energy density of hydrogen. This is because ammonia is liquid at room temperature and has high gravimetric (17.6 wt%) and volumetric (121 kg m−3) hydrogen densities.25,26 In industrial-scale production, ammonia is formed in the Haber–Bosch process at high pressures (20–40 MPa) and high temperatures (400–600 °C) from nitrogen extracted from the air and hydrogen obtained from steam reforming of methane. Feeding the process with fossil fuel consumes a lot of energy globally produced, i.e., ∼2% of the world's energy supply. It also releases ∼2.5% of worldwide fossil fuel-based CO2 emissions (670 million tonnes of CO2 emissions per year with a production scale of ∼174 million tonnes per year).27,28 In light of these facts, replacing the conventional fossil-fuelled Haber–Bosch process with renewable source-powered ammonia production is of increasing interest, especially in view of more restrictive environmental requirements regarding emissions reduction. Nevertheless, the ever-growing population (predicted 9.7 billion people in 2050) and the ever-increasing demand for fertilisers make it impossible to turn away from the traditional industrial ammonia synthesis process. The development of a catalytic system enabling the efficient synthesis of ammonia is thus of great importance and has attracted much interest.27,29–34The ammonia synthesis reaction is generally mediated by transition metals (such as Fe, Ru, Os, Mo, Re, Co, Rh) owing to their advantageous combination of occupied and unoccupied d-orbitals that can backdonate/accept electrons to/from the N2 molecule.29 Currently, the industrial process is carried out over the iron-based catalyst and, to some extent, the ruthenium catalyst. Some of their limitations, such as the low activity in the high conversion region for the former and high price and high requirements for the purity of synthesis gas for the latter, cause that research is still ongoing to develop alternative catalysts. Recently, cobalt catalysts that are used in many catalytic reactions35–42 are getting more attention for their use in ammonia synthesis owing to their relatively high activity and stability.43–49 Cobalt-based catalysts are commonly used with the support materials. It gives a possibility of using a lower amount of active metal, which is advantageous from an economic point of view. However, a necessary condition is to ensure optimal dispersion and stabilisation of the active phase during catalyst long-term operation. In the current literature, one can find reports mostly on using activated carbons,50,51 cerium oxide,52–54 magnesium oxide55 or mixed magnesium–lanthanide oxides.48,56–59 There are also many papers concerning electrides (e.g. C12A7:e−),60,61 hydride support materials (e.g. LiH, BaH2)43,62 and rare-earth metal nitrides (e.g. CeN) as very effective supports promoting ammonia synthesis over a Co-based catalyst.63 However, as reported in the literature,60 they suffer from disadvantages associated with the tendency to aggregation of active metal species, causing a rapid decay of activity and thus inferior durability. Moreover, their activity in conditions of low temperature and atmospheric pressure limits the possibilities of their wider application. Recently, much effort has been devoted to optimising cobalt-based systems deposited on various supports due to their tunability in terms of basicity, which is a key property from the point of view of the mechanism of ammonia synthesis reaction. In this regard, the support systems based on mixed magnesium–lanthanide oxides are particularly interesting.48,57–59
It is well-known that the effectiveness of supported catalysts depends not only on the support but even more on the metal loading. This factor determines the relative concentration of the active species formed on the support surface and may change the nature of active sites. For example, Fan et al.64 studied γ-Al2O3-supported FeOOH catalysts with different Fe loadings (5–25 wt%). The inverse correlation between the activity in the ammonia synthesis reaction and the Fe-loading was observed. The highest activity and the lowest activation energy exhibited FeOOH/Al2O3 (5% Fe). They indicated that increasing activity with decreasing metal loading is not only due to the effect of dispersion but also the changes in the active sites' nature. For the carbon-supported ruthenium catalyst, Rossetti et al.65 studied the influence of metal loading in the range of 1.5–9 wt%. The studies revealed that a significant increase in ammonia productivity is noticed when metal loading increases to about 4 wt% Ru, while for higher loading, activity seems to attain a plateau. Similar conclusions were also reported for potassium-promoted Ru/C catalysts by Liang et al.66 The authors explained this effect with the appropriate metal dispersion, ensuring the optimal size of metal particles exposing a sufficient number of active B5-type sites. Increasing Ru loading beyond a certain value is useless because it does not lead to an increase in these sites.65
In this work, we present the continuation of our comprehensive studies concerning cobalt catalysts supported on mixed MgO–La2O3 oxides.48,56–59 This study addresses the influence of the active phase loading (metallic cobalt) on the physicochemical properties and catalytic activities of the systems in the ammonia synthesis reaction. This work aims to find the threshold value of cobalt loading required for achieving high catalyst activity. The appropriate loading amount of cobalt is crucial for maximising the availability of active cobalt surface sites participating in the ammonia synthesis reaction. The optimisation of the catalyst composition in terms of the active phase content is crucial from a technological point of view – finding an appropriate activity-to-price ratio, especially when the active phase of the catalyst is expensive metals, is crucial for the development of efficient and cost-effective catalysts. The activity of the supported Co catalysts with different Co loading amounts was tested in ammonia synthesis at 400–470 °C under pressures of 6.3 and 9.0 MPa. The detailed characterisation studies using N2 physisorption, X-ray powder diffraction (XRPD), scanning transmission electron microscopy (STEM) coupled with energy dispersive X-ray (EDX), X-ray photoelectron spectroscopy (XPS), and temperature-programmed desorption (TPD) were conducted to understand the interrelationship between the active phase properties and its catalytic performance.
2. Experimental section
2.1. Catalyst preparation
MgO–La2O3 mixed oxide synthesised in our previous report48 was used as the support for the preparation of the cobalt catalysts. The detailed synthesis and characterisations of this support have been reported in ref. 48. Co(x)/MgO–La2O3 catalysts with different Co loadings were synthesised by the wet impregnation method (x wt% stands for the nominal weight loading of Co in the reduced catalyst; x = 10, 20, 30, 40, and 50). Typically, a certain amount of Co(NO3)2·6H2O (Acros Organics, 98+%) was dissolved in distilled water under stirring, and the oxide support was added to this solution. The mixture was shaken to obtain a homogenous mixture and then kept at room temperature for 18 h. Next, water was removed using a rotary evaporator under reduced pressure at 60 °C. The solid was then dried in an oven at 120 °C for 18 h and calcined in a muffle furnace at 500 °C for 18 h. The samples were activated at 600 °C in pure hydrogen before the catalytic reaction and characterisations (see section below for details on characterisation). The reduction temperature was selected based on the TPR profiles (Fig. S1†).Unless otherwise specified (e.g. the catalyst precursors), the supported Co catalysts mentioned in this paper refer to the freshly reduced catalysts. The full list of the prepared samples with their chemical composition is presented in Table S1.† The determined Co loading amounts are in good agreement with the nominal expected values (Table S1†).
2.2. Catalyst characterisation
Temperature-programmed reduction was carried out on a NETZSCH STA449C thermal analyser equipped with a quadrupole mass spectrometer (NETZSCH QMS Aëolos 403C) detector. The sample (50 mg) was heated up to 600 °C with a ramp of 3 °C min−1 under H2 + Ar (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar = 1:1) flow of 100 cm3 min−1 and kept in the isothermal conditions for 90 minutes. The signals at m/z = 16 (CH4) and 18 (H2O) were registered.
:Ar = 1:1) flow of 100 cm3 min−1 and kept in the isothermal conditions for 90 minutes. The signals at m/z = 16 (CH4) and 18 (H2O) were registered.
N2 physisorption was performed at −196 °C using a Micromeritics ASAP2020 instrument. Before the experiment, each catalyst precursor sample (500 mg) was reduced at 600 °C for 10 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under He flow (40 cm3 min−1). Next, the catalyst was degassed at 200 °C under a high vacuum for 2 h. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method in the relative partial pressure range (P/P0) from 0.05 to 0.3.
In situ X-ray powder diffraction (XRPD) experiments were carried out using Anton Paar XRK 900 reaction chamber mounted to Philips X'pert PRO diffractometer working in Bragg–Brentano geometry and equipped with CoKα radiation source (λKα1 = 1.789010 Å, λKα2 = 1.792900 Å). XRPD measurements were performed to identify the phase composition of the catalyst precursors and the reduced catalysts. To obtain the metallic Co catalysts, each catalyst precursor sample was reduced at 600 °C for 18 h under H2 flow of 100 cm3 min−1. Diffraction data were collected at ambient temperature and reduction temperature (600 °C), respectively, in the scattering 2θ range of 20–140°. PANalytical High Score Plus software with ICDD PDF-4+ 2021 database was used for data analysis.
TEM investigations were conducted using a Talos F200X (FEI) microscope operated at 200 kV. The measurements were performed in TEM and scanning TEM (STEM) modes using the high-angle annular dark-field (HAADF) detector and energy-dispersive X-ray spectroscopy on a Brucker BD4 spectrometer. Each catalyst precursor sample was reduced at 600 °C for 18 h under H2 flow (40 cm3 min−1) and next crushed and powdered. The powdered catalyst was dispersed in ethanol under ambient conditions, and several drops of the resulting dispersion were dropped on a carbon-coated copper-mesh TEM grid and dried out overnight at room temperature. STEM-EDX images were used to determine the Co nanoparticle size distributions. The particle size histograms were obtained by assuming the shape of Co nanoparticles as spherical in the calculation. The obtained histograms were fitted to log-normal distributions.
X-ray photoelectron spectroscopy (XPS) analyses were carried out for the catalyst precursors and the reduced catalysts. The measurements were conducted using Al Kα (hν = 1486.6 eV) radiation in a Prevac system equipped with Scienta SES 2002 electron energy analyser operating at constant transmission energy (Ep = 50 eV). The pressure in the analysis chamber was kept under 10−9 mbar. The reduction of precursors was conducted in a High-Pressure Cell (HPC) of an ultra-high vacuum (UHV) system. A small tablet of a sample, about 10 mm in diameter, was placed on a sample holder and introduced into HPC. During the reduction, hydrogen (99.999 vol%) was passed through the volume with the catalyst precursor sample (constant flow 20 cm3 min−1). The inlet gas was preheated to 300 °C. The sample was in this experiment only heated to 550 °C because of the instrument limitation. The process was carried out for 5 h. After reduction, the HPC was evacuated, and the sample was transferred under UHV to the analysis chamber of the electron spectrometer.
H2 temperature-programmed desorption was conducted on a Micromeritics AutoChem II 2920 instrument. Before the experiment, each catalyst precursor sample (500 mg) was reduced at 600 °C for 18 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under Ar flow (40 cm3 min−1). After cooling to 150 °C, H2 was introduced at a flow rate of 40 cm3 min−1 for 15 min, then cooled to 0 °C and kept at that temperature for 15 min. Next, the sample was purged with Ar at 0 °C until the baseline was stable. H2-TPD was carried out in a flow of Ar (40 cm3 min−1) at a heating rate of 5 °C min−1 up to 800 °C, and the TCD signal was recorded continuously. The error in the amount of H2 desorbed is ±3%.
CO2 temperature-programmed desorption was carried out on a Micromeritics AutoChem II 2920 instrument. Before the experiment, each catalyst precursor sample (500 mg) was reduced at 600 °C for 18 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under He flow (40 cm3 min−1). After cooling to 40 °C, CO2 was introduced at a flow of 40 cm3 min−1 and kept for 2 h. Next, the sample was purged with He flow at 40 °C until the baseline was stable. CO2-TPD was carried out in a flow of He (40 cm3 min−1) at a heating rate of 5 °C min−1 up to 800 °C, and the TCD signal was recorded continuously. The error in the amount of CO2 desorbed is ±3%.
2.3. Catalytic activity evaluation
Catalytic synthesis of ammonia was conducted in a tubular flow reactor.19 Before the catalytic test, each catalyst precursor sample (500 mg) was reduced at the following temperatures for the time indicated in the brackets: 470 °C (72 h); 520 °C (24 h); 550 °C (48 h); 600 °C (72 h) in a mixture of 75% H2 and 25% N2 (70 dm3 h−1) at atmospheric pressure. After the reduction, the system was pressurised to 6.3 or 9.0 MPa in the reaction mixture and heated to different target temperatures (400, 430, and 470 °C). The resulting NH3 gas was analysed interferometrically at accuracy and precision levels of ±0.01 mol%.67 The ammonia synthesis rate was calculated from the mass balance for a plug-flow differential reactor, as defined by the equation described in ref. 56. The error of the determined reaction rate is below 1%.56 A thermal stability test was performed by repeating the catalytic test at 470 °C under the pressure of 6.3 MPa after overheating at 600 °C for 75 h in the reaction mixture (30 dm3 h−1) at atmospheric pressure.3. Results and discussion
3.1. Structural and textural properties
The MgO–La2O3 support used for the synthesis of the Co catalysts showed the surface area and pore volume values of 93.8 m2 g−1 and 0.50 cm3 g−1, respectively. The type IV adsorption–desorption isotherm with type H3 hysteresis loop (Fig. S2†) indicates that the support was mesoporous with slit-shaped pores. The corresponding pore size distribution curve (Fig. S2†) indicates that most of pores centred at 4 and 20 nm. According to BET (Brunauer–Emmett–Teller) analysis, the measured catalyst surface areas were between 25 to 33 m2 g−1, which were lower than the surface area of the mixed MgO–La2O3 oxide support. The observed decrease in the surface area could be due to the pores being blocked by Co species and, to some extent, to support sintering. However, one can see that the surface area of the catalysts increased as the Co loading amount increased (Table 1). This observation can be explained by the fact that after the support pores are filled with Co species, the remaining Co species with a certain porosity are built up on the support surface. After reducing the Co catalyst precursor, the Co nanoparticles with a well-developed surface are formed. This is an interesting observation since, as reported by previous studies,68,69 an increase in metal loading resulted in a decrease in the surface area. The results indicate that Co catalysts with a high metal loading (>20 wt%) proved to be quite resistant to sintering. This was also observable in the average Co particle size (Table 1). Considering that the Co loading amount increased fivefold (Table 1), the observed increase in the Co particle size was not so significant.| Catalyst | Surface area (m2 g−1) | Particle size (nm) | Crystallite sizec (nm) | Number of basic sitesd (μmol g−1) | |
|---|---|---|---|---|---|
| H2-TPDa | STEMb | ||||
| a Average Co particle size calculated from the total number of hydrogen adsorption sites (Table 4) assuming spherical metal particles and the hydrogen adsorption ratio Co/H = 1.70,71b Average Co particle size calculated based on the STEM-EDX particle size distribution (Fig. S3, see ESI). The values were rounded to the nearest 1 nm.c Average Co crystallite size calculated based on the FWHM value obtained from full pattern refinement utilising the Rietveld method (size-only in HighScore Plus software).d Calculated from the total number of carbon dioxide adsorption sites. | |||||
| Co(10)/Mg–La | 25 | 13 | 15 | 15 | 200 |
| Co(20)/Mg–La | 25 | 15 | 17 | 16 | 207 |
| Co(30)/Mg–La | 30 | 18 | 17 | 14 | 223 |
| Co(40)/Mg–La | 33 | 19 | 20 | 13 | 255 |
| Co(50)/Mg–La | 33 | 21 | 22 | 13 | 236 |
Fig. 1 shows the XRPD patterns obtained for samples treated with flowing hydrogen at 600 °C. Only three crystallographic phases were identified in all materials: cubic magnesium oxide (01-074-1225), hexagonal lanthanum oxide (04-007-0681) and cubic metallic cobalt (01-077-7452). Reflections from all crystallographic phases did not shift their position, indicating that there were no sizeable changes to the lattice parameters attributed to regular and repetitive substitution or vacancies. No significant changes in half-widths of these reflections were observed either, which suggests that there was no substantial change in the average particle sizes of these phases. Based on the reflection peaks originating from metallic cobalt, the average size of their crystallites was calculated, and the obtained values are shown in Table 1. The average Co crystallite sizes were in the range of 13–16 nm and showed no systematic variation. It was characteristic that the intensity of diffraction reflections varied with increasing cobalt loading amount in the sample. Reflections originating from phases of the support decreased their intensity while those originating from cobalt metal increased. On this basis, the concentrations of each phase in the samples were calculated, and the results are presented in Table 2. The amount of Co loading determined by diffraction data in all cases was lower than that calculated from chemical composition studies (Table S1†). This discrepancy may be explained by the occurrence of some cobalt atoms in the form of quasi-amorphous structures, which are not recorded by the diffraction method.
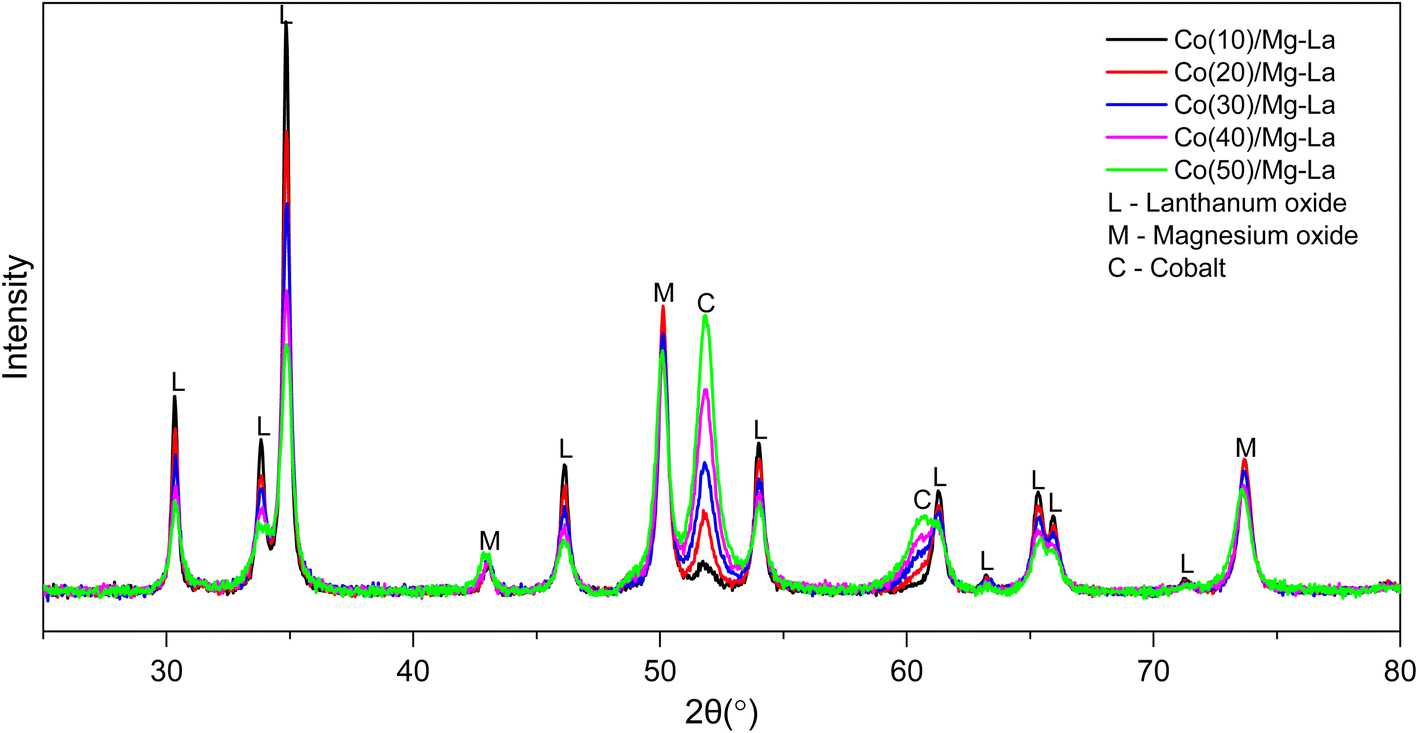 | ||
| Fig. 1 XRPD patterns of the Co catalysts with different Co loadings after the treatment in hydrogen flow at 600 °C. | ||
STEM-EDX investigations were performed to analyse the distribution of the atomic elements within the catalysts (Fig. 2 and Fig. S4–S8†). Generally, a homogeneous distribution of cobalt, magnesium and lanthanum elements was observed. This homogeneity was visible regardless of the amount of Co deposited (Fig. S4–S8†). The analysis of the oxygen distribution on the catalyst surface presented in Fig. 2e indicated that the Co nanoparticles were partially oxidised on the surface, and the core of the particle was metallic Co (Fig. 2b). Considering that the catalysts were exposed to air before imaging (for details, see section Catalyst characterisation), a partial reoxidation of the Co nanoparticles was expected. The TEM images suggest that the Co nanoparticles had a polycrystalline structure (Fig. 3 and S9†).
 | ||
| Fig. 2 (a) HAADF-STEM image of representative Co(30)/Mg–La catalyst and corresponding EDX distribution maps of (b) Co–K, (c) Mg–K, (d) La–L, (e) O–K, and (f) reconstructed overlay image of Co, Mg, and La. | ||
 | ||
| Fig. 3 (a) TEM image of representative Co(30)/Mg–La catalyst with (b) HR-TEM image of Co nanoparticles. In the inset, the edge of the Co nanoparticle with a polycrystalline structure is presented. | ||
XPS studies showed that the surface of the analysed samples contained oxygen, cobalt, magnesium, lanthanum and potassium. Magnesium, lanthanum, and oxygen elements formed the support of the studied catalysts. Cobalt was the active phase. The presence of potassium atoms on the surface of catalysts was due to the use of potassium carbonate as a precipitating agent in the support preparation process.48 The surface composition of these materials is shown in Table 3. It should be noted that the concentration values in Table 3 are based on the assumption of a homogeneous distribution of all elements in the area analysed by the XPS method.
| Catalyst | O (wt%) | Co (wt%) | La (wt%) | K (wt%) | Mg (wt%) |
|---|---|---|---|---|---|
| Co(10)/Mg–La | 27 | 5 | 20 | 8 | 40 |
| Co(20)/Mg–La | 25 | 12 | 23 | 8 | 32 |
| Co(30)/Mg–La | 24 | 16 | 19 | 8 | 33 |
| Co(40)/Mg–La | 23 | 21 | 18 | 8 | 30 |
| Co(50)/Mg–La | 20 | 25 | 20 | 10 | 25 |
Fig. 4 shows the binding energy range of the photoelectron lines coming from the Co 3p and Mg 2p shells. The spectra contained therein were collected after treating materials containing from 10 to 50 wt% cobalt with hydrogen at 550 °C. For comparison, a photoelectron spectrum obtained for a sample containing 50 wt% cobalt before hydrogen treatment is also provided. The presented plots were normalised to the maximum intensity of the Mg 2p lines.
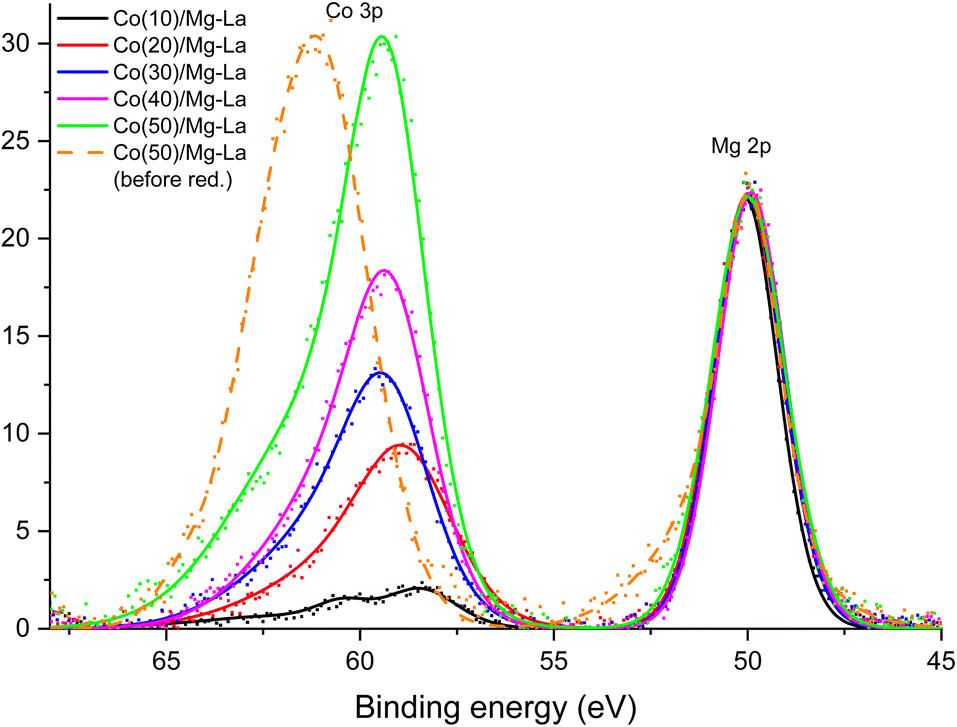 | ||
| Fig. 4 The binding energy range containing the photoelectron lines characteristic for Co 3p and Mg 2p electrons acquired by in situ XPS analysis of the Co catalysts with different Co loadings treated in hydrogen flow at 550 °C. | ||
Mg 2p lines had an identical envelope, indicating an identical chemical state of the magnesium atoms in all samples. The position of the maximum of the Mg 2p line corresponded to the presence of Mg2+ ions.72 The only element capable of forming anions identified on the studied surfaces is oxygen, so it can be assumed that magnesium atoms were present in the form of MgO oxide. The crystallographic results presented earlier support this conclusion (Fig. 1). According to diffraction studies, the second component of the support of catalysts was lanthanum oxide. Fig. S10,† which shows XPS spectra from La 3d electrons, can be found in the ESI.† After normalising the intensities, they were virtually identical, proving that there was no change in the chemical environment associated with a change in the cobalt loading. The location of the main maximum of the La 3d line at 834 eV corresponded to La3+ ions in lanthanum oxide.73
Much more relevant changes are observed for the XPS Co 3p line. Fig. 4 shows the Co 3p line for a material containing 50 wt% cobalt before hydrogen treatment at 550 °C. The maximum of this line was located at a binding energy of about 61 eV, which corresponded to the presence of cobalt in the oxides.74 Both the preparation method and the results of diffraction analysis of the catalysts before reduction, not presented in this paper, confirmed the presence of cobalt oxides in the samples. The treatment with hydrogen at elevated temperatures should lead to the reduction of cobalt oxides. This was confirmed by XPS spectra obtained after reduction at 550 °C under a hydrogen atmosphere. A distinct shift of XPS Co 3p line maximum to the binding energy of about 59 eV was observed. It corresponded to the reduction of cobalt ions to metallic cobalt.75 In the spectrum of the catalyst with 10% Co deposition, the local inflexion of the Co 3p line can be distinguished at the binding energy of about 61 eV. This may indicate the incomplete reduction of cobalt oxides. This conclusion can be confirmed by the broadening of the Co 2p line on the high binding energy side (Fig. S11†) observed for this sample. In the case of the three catalysts with the highest loadings of cobalt (30, 40, and 50 wt%), the envelope of the XPS Co 3p lines was identical after normalising their intensities but slightly (about 0.5 eV) shifted towards the higher binding energy. Such a shift can be explained by the presence of a certain insignificant number of cobalt atoms whose chemical environment was modified by the presence of oxygen atoms. That modification can occur, for example, at the boundary between cobalt crystallites and metal oxide support grains.
Based on the presented results of the surface composition of the catalysts and crystallographic investigations, it can be concluded that the support consisting of magnesium oxide and lanthanum oxide was blended with crystallites of metallic cobalt. Thus, all the catalysts were macroscopically treated as a homogeneous system. As for the XPS, on the close-to-atomic level, the samples were not made of homogenously distributed atoms of each element, so the approach was intentionally used as a generalisation.
3.2. Adsorption properties
In order to measure the number of active sites for hydrogen adsorption, the H2 temperature-programmed desorption (H2-TPD) measurements were carried out. The resultant desorption profiles could be divided into a low-temperature region with a maximum at ∼100 °C and a well-separated high-temperature region with a maximum at 700 °C (Fig. 5). Each region describes different hydrogen adsorption sites present on the Co surface.44 However, modest hydrogen adsorption on the support surface was observed48 and was taken into account when determining the number of hydrogen adsorption sites on the Co surface (Table 4). The low-temperature peak at ∼100 °C with a discernible shoulder at ∼230 °C was ascribed to the desorption of H2 weakly and intermediately bound to the Co nanoparticles (Hα), while the high-temperature peak accounted for the strong chemisorption of H2 on the Co nanoparticles (Hβ). It is observed that the number of Co sites capable of hydrogen adsorption increased with the increasing amount of Co loading (Table 4), probably because of the higher Co surface concentration (Table 3). Another observation is that the intensity of the low-temperature peak increased as the Co loading amount increased, indicating an increase in the weak and intermediate hydrogen adsorption sites on the Co nanoparticle surfaces (Fig. 5). This was also confirmed by the ratio of the number of weak and intermediate to strong hydrogen adsorption sites on the Co nanoparticles (Hα/Hβ), as shown in Table 4. Such differences can be explained by an increase in the size of Co particles (Table 1). It is in line with the literature reports48 indicating that the Co particles of larger size provide more weak and intermediate hydrogen adsorption sites on their surfaces. This provides a variety of surface cobalt active sites for H2 adsorption, which is advantageous in view of the catalyst activity in ammonia synthesis.76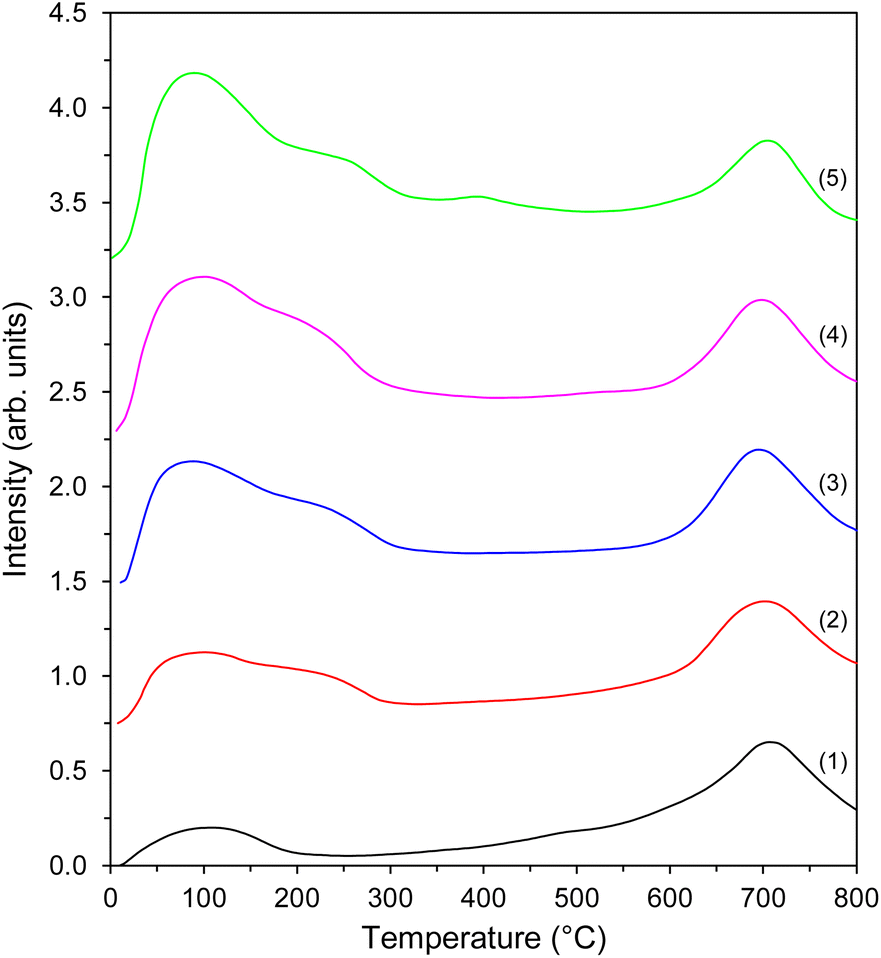 | ||
| Fig. 5 H2-TPD profiles of the Co catalysts with different Co loadings: (1) Co(10)/Mg–La, (2) Co(20)/Mg–La, (3) Co(30)/Mg–La, (4) Co(40)/Mg–La, and (5) Co(50)/Mg–La. | ||
| Catalyst | Hαa (μmol g−1) | Hβb (μmol g−1) | Htotalc (μmol g−1) | Hα/Hβd |
|---|---|---|---|---|
| a Number of the weak and intermediate hydrogen adsorption sites.b Number of the strong hydrogen adsorption sites.c Total number of the hydrogen adsorption sites.d Ratio of the number of the weak and intermediate to strong hydrogen adsorption sites. | ||||
| Co(10)/Mg–La | 14 | 74 | 88 | 0.2 |
| Co(20)/Mg–La | 59 | 89 | 148 | 0.7 |
| Co(30)/Mg–La | 103 | 77 | 180 | 1.3 |
| Co(40)/Mg–La | 143 | 81 | 224 | 1.8 |
| Co(50)/Mg–La | 187 | 65 | 252 | 2.9 |
To evaluate the surface basicity of the supported Co catalysts, the CO2 temperature-programmed desorption (CO2-TPD) measurements were conducted. One can see that the resultant CO2-TPD profiles had a similar shape, with only slight differences in the peak intensities (Fig. 6). However, this could be expected since, as reported previously,48 the deposition of Co nanoparticles on the mixed magnesium–lanthanum oxide support did not change the nature of the basic sites significantly. The profiles could be divided into three temperature regions: <150 °C (low-temperature region), 150–400 °C (middle-temperature region), and >400 °C (high-temperature region). Each region describes different carbon dioxide adsorption sites present on the catalyst surface. The desorption peak at around 100 °C was assigned to the interactions between CO2 and surface hydroxyl groups on MgO (Brønsted basicity). Two desorption peaks, i.e., the distinct peak at 250 °C and the broad peak at 400–600 °C, were attributed to the CO2 chemisorption on the different surface oxygen species on both MgO and La2O3 (Lewis basicity).77–79 The origin of the last broad peak at 750 °C was considered to be the result of the decomposition of lanthanum carbonate species formed by the CO2 chemisorption on La2O3.79 It is, however, worth mentioning that only Lewis-type basic sites can donate electrons to the d-orbitals of transition metals, thus accelerating the rate-determining step of ammonia synthesis, that is, the cleavage of N2 molecules.80 For each catalyst, the total amount of CO2 desorbed from its surface was used as a metric of basicity. The results are given in Table 1. As the Co loading amount increased, a gradual increase in the basicity was observed up to 40 wt% Co. A further increase in the amount of Co deposited from 40 to 50 wt% caused a slight decrease in the number of basic sites. Considering the similarity in the nature of the basic sites in these catalysts (Fig. 6), the observed relatively high number of the basic sites in the catalysts with 40 and 50 wt% Co were probably due to improvement in their surface areas (Table 1).48
 | ||
| Fig. 6 CO2-TPD profiles of the Co catalysts with different Co loadings: (1) Co(10)/Mg–La, (2) Co(20)/Mg–La, (3) Co(30)/Mg–La, (4) Co(40)/Mg–La, and (5) Co(50)/Mg–La. | ||
3.3. Catalytic activity
The temperature dependence of the activity of the Co catalysts for ammonia synthesis is presented in Fig. 7a. The NH3 synthesis rate increased with the Co loading and reached a maximum at 40 wt% Co deposition across the examined temperature range. A further increase in the Co loading from 40 to 50 wt% caused only slight (∼4%) decrease in the activity. When the total pressure increased from 6.3 to 9.0 MPa, the activity increased by about 20% (Fig. 7b). The obtained catalysts were also found to be highly stable and resistant to heat treatment. They continuously produced ammonia with a negligible decrease in activity, even after overheating at 600 °C for 75 h (Table 5). Such a durable performance of these catalysts was also observed for the literature-reported Co catalysts.44,55,81 | ||
| Fig. 7 Evaluation of ammonia synthesis activities of the Co catalysts with different Co loadings. (a) Temperature dependence of ammonia synthesis rate at 6.3 MPa. (b) Pressure dependence of ammonia synthesis rate at 470 °C. NH3 synthesis conditions: catalyst, 0.5 g; reaction mixture, H2/N2 = 3 with a flow rate of 70 dm3 h−1. The error range was ±1%. | ||
| Catalyst | NH3 synthesis ratea (mmolNH3 gcat−1 h−1) | TOFb (s−1) | Eappc (kJ mol−1) |
|---|---|---|---|
| a NH3 synthesis rate at 400 °C and 6.3 MPa. The numbers in the brackets represent the ammonia formation rate after overheating at 600 °C for 75 h.b TOF was calculated from the total number of hydrogen adsorption sites (Table 4) and the NH3 synthesis rate shown in this table. The error range was ±4%.c Apparent activation energy was derived from the Arrhenius plots in Fig. S12. The error range was ±3%. | |||
| Co(10)/Mg–La | 16.0 (15.7) | 0.025 | 62 |
| Co(20)/Mg–La | 24.2 (23.7) | 0.023 | 58 |
| Co(30)/Mg–La | 31.8 (30.9) | 0.025 | 57 |
| Co(40)/Mg–La | 40.3 (39.5) | 0.025 | 59 |
| Co(50)/Mg–La | 38.2 (37.5) | 0.021 | 60 |
The superior performance of the Co catalysts for NH3 synthesis is further revealed by a comparison to the literature results with similar reaction conditions (Table S2†). In particular, the ammonia formation rate over Co(40)/Mg–La reached 40 mmolNH3 gcat−1 h−1, which is two times that of commercial iron catalyst (ZA-5).
Table 5 presents the activity, expressed as the turnover frequencies (TOF) and apparent activation energies (Eapp). A key observation is that there was no correlation between TOF and Co loading amount. The calculated TOF values were similar, indicating that for all the catalysts, the surface cobalt active sites were of almost the same strength. As a consequence, similar values of apparent activation energies determined from the slopes of the Arrhenius plots (Fig. S12†) were observed for all the catalysts (Table 5). This is an interesting finding since previous studies have evidenced that ammonia synthesis is a structure-sensitive reaction.82 However, this can be explained by the fact that, regardless of the Co loading amount, the Co crystallites were almost of the same size (13–16 nm) with the same crystal structure of face-centred cubic (fcc) (Fig. 1). And as revealed by Rambeau et al., the ammonia synthesis reaction takes place on metallic Co at different rates depending on the crystal structure.82 It was found that hexagonal-close packed (hcp) cobalt exhibits higher activity (8 × 10−4 s−1) on ammonia formation than face-centred cubic (fcc) cobalt (4 × 10−4 s−1). The reason why hcp Co was not found in the catalysts can be explained by two factors. First, as reported by Kitakami et al.,83 there is a close relationship between the size and crystallographic structure of the cobalt. That is, the Co particles of <20 nm size are of cubic structure, 20–30 nm are a mixture of cubic and hexagonal structures, and >40 nm are of hexagonal structure. In fact, for all the catalysts studied, the calculated Co particle sizes were below 21 nm, as revealed by H2 chemisorption studies (Table 1) and STEM particle size measurements (Table 1 and Fig. S3†). Second, there was a possibility that the support influenced the formation of a particular crystal structure of cobalt, here cubic structure. For instance, Jacobsen et al. reported that the support type plays a decisive role in controlling the Ru structure and the resulting changes in the abundance of B5-type sites. This is the main reason for the significant changes in the activity levels of Ru catalysts on different supports.84
From the above discussion, it is clear that the ammonia synthesis rate over supported Co catalysts was dependent on the amount of Co deposited. The Co loading of 40 wt% Co was the threshold amount of Co loading, allowing to obtain the highest reaction rate (Fig. 7), and a further increase of Co loading caused only slight decrease in the activity. It is not so clear what caused the slight decrease in the ammonia formation rate for the catalyst with 50 wt% Co deposition. The results suggest that despite the highest number of Co sites capable of hydrogen adsorption (Table 4), the decreased ammonia formation rate for this catalyst was associated with a decrease in the number of the basic sites (Table 1). This, in turn, reduced the electron-donating ability of the Co surface and resulted in the lower intrinsic activity of the active sites (average TOF of 0.021 s−1).
Another interesting outcome of this work is the possibility of obtaining high-loaded supported Co catalysts with well-dispersed Co nanoparticles. Despite the increase in Co loading amount from 10 to 50 wt%, the Co particle and crystallite sizes remained almost unchanged (Table 1). This suggests that the obtained catalysts were resistant to sintering, showing a stable performance during the continuous ammonia synthesis process (Table 5). This is of major importance for the industrial process since the harsh conditions applied in the Haber–Bosch process can result in excessive sintering and loss of the active metal centres.85 Taken together, the obtained results suggest that the developed catalyst with 40 wt% Co deposition can be regarded as a promising candidate for efficient ammonia synthesis.
4. Conclusions
In summary, a series of Co catalysts supported on mixed MgO–La2O3 oxides with different Co loading amounts (in the range from 10 to 50 wt%) was designed, systematically characterised, and tested in the ammonia synthesis reaction. A clear relationship between the ammonia synthesis rate and the amount of deposited Co was observed. Specifically, the ammonia synthesis rate increased with increasing Co loading and reached a maximum at 40 wt% Co deposition. A further increase in Co loading caused only slight decrease in the activity. However, interestingly, there were almost no differences in TOF values calculated for these catalysts. We attributed these discrepancies to the fact that the size and structure of Co nanoparticles in the catalysts remained nearly unchanged despite the fivefold increase in the Co loading amount. These results suggest that the ammonia synthesis rate on the studied catalysts was not dependent on the average size of Co nanoparticles between 13 to 21 nm. Thus, the increase in the ammonia synthesis rate for the catalyst with 10–40 wt% Co deposition was mainly the result of the increased amount of deposited Co. However, the observed decrease in the ammonia formation rate for the catalyst with 50 wt% Co deposition was most likely caused by a decrease in the number of the surface basic sites as compared to the catalyst with 40 wt% Co deposition.Author contributions
H. R. conceived of the idea for this study and designed the experiments. H. R. and M. Z. prepared the samples. H. R., and M. Z. performed the gas sorption experiments. H. R., M. Z., W. P., and W. R.-P. performed the catalytic measurements. D. M. and A. A. carried out the XRPD measurements. D. M. and A. S. performed the XPS measurements. K. S. conducted the STEM characterisation. M. Z., K. S., D. M., and W. R.-P. provided suggestions and feedback on the study. H. R. prepared the manuscript with input from M. Z., K. S. and D. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was funded by POB Technologie Materiałowe of Warsaw University of Technology within the Excellence Initiative: Research University (IDUB) programme.References
- F. Dawood, M. Anda and G. M. Shafiullah, Hydrogen production for energy: An overview, Int. J. Hydrogen Energy, 2020, 45, 3847–3869, DOI:10.1016/j.ijhydene.2019.12.059
.
- J. S. Wallace and C. A. Ward, Hydrogen as a fuel, Int. J. Hydrogen Energy, 1983, 8, 255–268, DOI:10.1016/0360-3199(83)90136-2
- J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Hydrogen energy, economy and storage: Review and recommendation, Int. J. Hydrogen Energy, 2019, 44, 15072–15086, DOI:10.1016/j.ijhydene.2019.04.068
- M. Momirlan and T. N. Veziroglu, The properties of hydrogen as fuel tomorrow in sustainable energy system for a cleaner planet, Int. J. Hydrogen Energy, 2005, 30, 795–802, DOI:10.1016/j.ijhydene.2004.10.011
- M. V. Rodionova, A. M. Bozieva, S. K. Zharmukhamedov, Y. K. Leong, J. Chi-Wei Lan, A. Veziroglu, T. N. Veziroglu, T. Tomo, J.-S. Chang and S. I. Allakhverdiev, A comprehensive review on lignocellulosic biomass biorefinery for sustainable biofuel production, Int. J. Hydrogen Energy, 2022, 47, 1481–1498, DOI:10.1016/j.ijhydene.2021.10.122
- K. Bolatkhan, B. D. Kossalbayev, B. K. Zayadan, T. Tomo, T. N. Veziroglu and S. I. Allakhverdiev, Hydrogen production from phototrophic microorganisms: Reality and perspectives, Int. J. Hydrogen Energy, 2019, 44, 5799–5811, DOI:10.1016/j.ijhydene.2019.01.092
- S. Kalantarifard, S. I. Allakhverdiev and M. M. Najafpour, Water oxidation by a nickel complex: New challenges and an alternative mechanism, Int. J. Hydrogen Energy, 2020, 45, 33563–33573, DOI:10.1016/j.ijhydene.2020.09.111
- B. D. Kossalbayev, T. Tomo, B. K. Zayadan, A. K. Sadvakasova, K. Bolatkhan, S. Alwasel and S. I. Allakhverdiev, Determination of the potential of cyanobacterial strains for hydrogen production, Int. J. Hydrogen Energy, 2020, 45, 2627–2639, DOI:10.1016/j.ijhydene.2019.11.164
- H. Feizi, R. Bagheri, Z. Song, J.-R. Shen, S. I. Allakhverdiev and M. M. Najafpour, Cobalt/Cobalt Oxide Surface for Water Oxidation, ACS Sustainable Chem. Eng., 2019, 7, 6093–6105, DOI:10.1021/acssuschemeng.8b06269
- M. M. Najafpour, S. Mehrabani, R. Bagheri, Z. Song, J.-R. Shen and S. I. Allakhverdiev, An aluminum/cobalt/iron/nickel alloy as a precatalyst for water oxidation, Int. J. Hydrogen Energy, 2018, 43, 2083–2090, DOI:10.1016/j.ijhydene.2017.12.025
- M. Singh, D. Zappa and E. Comini, Solid oxide fuel cell: Decade of progress, future perspectives and challenges, Int. J. Hydrogen Energy, 2021, 46, 27643–27674, DOI:10.1016/j.ijhydene.2021.06.020
- F. S. da Silva and T. M. de Souza, Novel materials for solid oxide fuel cell technologies: A literature review, Int. J. Hydrogen Energy, 2017, 42, 26020–26036, DOI:10.1016/j.ijhydene.2017.08.105
- S. Mekhilef, R. Saidur and A. Safari, Comparative study of different fuel cell technologies, Renewable Sustainable Energy Rev., 2012, 16, 981–989, DOI:10.1016/j.rser.2011.09.020
- R. Balderas-Xicohténcatl, P. Schmieder, D. Denysenko, D. Volkmer and M. Hirscher, High Volumetric Hydrogen Storage Capacity using Interpenetrated Metal–Organic Frameworks, Energy Technol., 2018, 6, 510–512, DOI:10.1002/ente.201700608
- D. V. Schur, A. Veziroglu, S. Yu. Zaginaychenko, Z. A. Matysina, T. N. Veziroglu, M. T. Gabdullin, T. S. Ramazanov, An. D. Zolonarenko and Al. D. Zolonarenko, Theoretical studies of lithium–aluminum amid and ammonium as perspective hydrogen storage, Int. J. Hydrogen Energy, 2019, 44, 24810–24820, DOI:10.1016/j.ijhydene.2019.07.205
- Z. A. Matysina, S. Yu. Zaginaychenko, D. V. Schur, T. N. Veziroglu, A. Veziroglu, M. T. Gabdullin, Al. D. Zolotarenko and An. D. Zolotarenko, The mixed lithium–magnesium imide Li2Mg(NH)2 a promising and reliable hydrogen storage material, Int. J. Hydrogen Energy, 2018, 43, 16092–16106, DOI:10.1016/j.ijhydene.2018.06.168
- H. M. A. Hunter, J. W. Makepeace, T. J. Wood, O. S. Mylius, M. G. Kibble, J. B. Nutter, M. O. Jones and W. I. F. David, Demonstrating hydrogen production from ammonia using lithium imide – Powering a small proton exchange membrane fuel cell, J. Power Sources, 2016, 329, 138–147, DOI:10.1016/j.jpowsour.2016.08.004
- D. D. Papadias and R. K. Ahluwalia, Bulk storage of hydrogen, Int. J. Hydrogen Energy, 2021, 46, 34527–34541, DOI:10.1016/j.ijhydene.2021.08.028
- N. A. Ali, N. A. Sazelee and M. Ismail, An overview of reactive hydride composite (RHC) for solid-state hydrogen storage materials, Int. J. Hydrogen Energy, 2021, 46, 31674–31698, DOI:10.1016/j.ijhydene.2021.07.058
- W. Zhang, Y. Cheng, D. Han and S. Han, The hydrogen storage properties of MgH2–Fe3S4 composites, Energy, 2015, 93, 625–630, DOI:10.1016/j.energy.2015.09.080
- R. Pedicini, B. Schiavo, P. Rispoli, A. Saccà, A. Carbone, I. Gatto and E. Passalacqua, Progress in polymeric material for hydrogen storage application in middle conditions, Energy, 2014, 64, 607–614, DOI:10.1016/j.energy.2013.11.073
- J. R. Morse, D. A. Zugell, E. Patterson, J. W. Baldwin and H. D. Willauer, Hydrogenated graphene: Important material properties regarding its application for hydrogen storage, J. Power Sources, 2021, 494, 229743, DOI:10.1016/j.jpowsour.2021.229734
- C. Pohlmann, L. Röntzsch, F. Heubner, T. Weißgärber and B. Kieback, Solid-state hydrogen storage in Hydralloy–graphite composites, J. Power Sources, 2013, 231, 97–105, DOI:10.1016/j.jpowsour.2012.12.044
- L. Schlapbach and A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 2001, 414, 353–358, DOI:10.1038/35104634
- J. Feng, X. Zhang, J. Wang, X. Ju, L. Liu and P. Chen, Applications of rare earth oxides in catalytic ammonia synthesis and decomposition, Catal. Sci. Technol., 2021, 11, 6330–6343, 10.1039/d1cy01156a
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, A technological roadmap to the ammonia energy economy: Current state and missing technologies, Chem. Eng. J., 2021, 408, 127310, DOI:10.1016/j.cej.2020.127310
- A. Daisley, J. S. J. Hargreaves, R. Hermann, Y. Poya and Y. Wang, A comparison of the activities of various supported catalysts for ammonia synthesis, Catal. Today, 2020, 357, 534–540, DOI:10.1016/j.cattod.2019.06.009
- P. H. Pfromm, Towards sustainable agriculture: Fossil-free ammonia, J. Renewable Sustainable Energy, 2017, 9, 034702, DOI:10.1063/1.4985090
- Q. Wang, J. Guo and P. Chen, The impact of alkali and alkaline earth metals on green ammonia synthesis, Chem, 2021, 7, 3203–3220, DOI:10.1016/j.chempr.2021.08.021
- J. Ni, S. Shi, C. Zhang, B. Fang, X. Wang, J. Lin, S. Liang, B. Lin and L. Jiang, Enhanced catalytic performance of the carbon-supported Ru ammonia synthesis catalyst by an introduction of oxygen functional groups via gas-phase oxidation, J. Catal., 2022, 409, 78–86, DOI:10.1016/j.jcat.2022.03.026
- H. Fang, D. Liu, Y. Luo, Y. Zhou, S. Liang, X. Wang, B. Lin and L. Jiang, Challenges and Opportunities of Ru-Based Catalysts toward the Synthesis and Utilisation of Ammonia, ACS Catal., 2022, 12, 3938–3954, DOI:10.1021/acscatal.2c00090
- Y. Luo, S. Liang, X. Wang, B. Lin, C. Chen and L. Jiang, Facile Synthesis and High-Value Utilisation of Ammonia, Chin. J. Chem., 2022, 40, 953–964, DOI:10.1002/cjoc.202100826
- W. Liao, L. Qi, Y. Wang, J. Qin, G. Liu, S. Liang, H. He and L. Jiang, Interfacial Engineering Promoting Electrosynthesis of Ammonia over Mo/Phosphotungstic Acid with High Performance, Adv. Funct. Mater., 2021, 31, 2009151, DOI:10.1002/adfm.202009151
- W. Liao, K. Xie, L. Liu, X. Wang, Y. Luo, S. Liang, F. Liu and L. Jiang, Triggering in-plane defect cluster on MoS2 for accelerated dinitrogen electroreduction to ammonia, J. Energy Chem., 2021, 62, 359–366, DOI:10.1016/j.jechem.2021.03.043
- G. Melaet, W. T. Ralston, C.-S. Li, S. Alayoglu, K. An, N. Musselwhite, B. Kalkan and G. A. Somorjai, Evidence of Highly Active Cobalt Oxide Catalyst for the Fischer–Tropsch Synthesis and CO2 Hydrogenation, J. Am. Chem. Soc., 2014, 136, 2260–2263, DOI:10.1021/ja412447q
- F. Hebrard and P. Kalck, Cobalt-Catalyzed Hydroformylation of Alkenes: Generation and Recycling of the Carbonyl Species, and Catalytic Cycle, Chem. Rev., 2009, 109, 4272–4283, DOI:10.1021/cr8002533
- F. Chen, C. Topf, J. Radnik, C. Kreyenschulte, H. Lund, M. Schneider, A.-E. Surkus, L. He, K. Junge and M. Beller, Table and Inert Cobalt Catalysts for Highly Selective and Practical Hydrogenation of C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and C
N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O Bonds, J. Am. Chem. Soc., 2016, 138, 8781–8788, DOI:10.1021/jacs.6b03439
O Bonds, J. Am. Chem. Soc., 2016, 138, 8781–8788, DOI:10.1021/jacs.6b03439 - A. Rokicińska, M. Drozdek, E. Bogdan, A. Węgrzynowicz, P. Michorczyk and P. Kuśtrowski, Combustion of toluene over cobalt-modified MFI zeolite dispersed on monolith produced using 3D printing technique, Catal. Today, 2021, 375, 369–376, DOI:10.1016/j.cattod.2020.04.005
- A. Rokicińska, T. Berniak, M. Drozdek and P. Kuśtrowski, Search of Factors Determining Activity of Co3O4 Nanoparticles Dispersed in Partially Exfoliated Montmorillonite Structure, Molecules, 2021, 26, 3288, DOI:10.3390/molecules26113288
- A. Rokicińska, M. Żurowska, P. Łątka, M. Drozdek, M. Michalik and P. Kuśtrowski, Design of Co3O4@SiO2 Nanorattles for Catalytic Toluene Combustion Based on Bottom-Up Strategy Involving Spherical Poly(styrene-co-acrylic Acid) Template, Catalysts, 2021, 11, 1097, DOI:10.3390/catal11091097
- A. Rokicińska, P. Natkański, B. Dudek, M. Drozdek, L. Lityńska-Dobrzyńska and P. Kuśtrowski, Co3O4-pillared montmorillonite catalysts synthesised by hydrogel-assisted route for total oxidation of toluene, Appl. Catal., B, 2016, 195, 59–68, DOI:10.1016/j.apcatb.2016.05.008
- A. Rokicińska, M. Drozdek, B. Dudek, B. Gil, P. Michorczyk, D. Brouri, S. Dzwigaj and P. Kuśtrowski, Cobalt-containing BEA zeolite for catalytic combustion of toluene, Appl. Catal., B, 2017, 212, 59–67, DOI:10.1016/j.apcatb.2017.04.067
- W. Gao, P. Wang, J. Guo, F. Chang, T. He, Q. Wang, G. Wu and P. Chen, Barium Hydride-Mediated Nitrogen Transfer and Hydrogenation for Ammonia Synthesis: A Case Study of Cobalt, ACS Catal., 2017, 7, 3654–3661, DOI:10.1021/acscatal.7b00284
- X. Wang, L. Li, T. Zhang, B. Lin, J. Ni, C.-T. Au and L. Jiang, Strong metal–support interactions of Co-based catalysts facilitated by dopamine
for highly efficient ammonia synthesis: in situ XPS and XAFS spectroscopy coupled with TPD studies, Chem. Commun., 2019, 55, 474–477, 10.1039/C8CC07130F
- X. Wang, X. Peng, W. Chen, G. Liu, A. Zheng, L. Zheng, J. Ni, C.-T. Au and L. Jiang, Insight into dynamic and steady-state active sites for nitrogen activation to ammonia by cobalt-based catalyst, Nat. Commun., 2020, 11, 653, DOI:10.1038/s41467-020-14287-z
- M. Karolewska, E. Truszkiewicz, M. Wściseł, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Ammonia synthesis over a Ba and Ce-promoted carbon-supported cobalt catalyst. Effect of the cerium addition and preparation procedure, J. Catal., 2013, 303, 130–134, DOI:10.1016/j.jcat.2013.03.005
- A. Tarka, M. Zybert, E. Truszkiewicz, B. Mierzwa, L. Kępiński, D. Moszyński and W. Raróg-Pilecka, Effect of a Barium Promoter on the Stability and Activity of Carbon-Supported Cobalt Catalysts for Ammonia Synthesis, ChemCatChem, 2015, 7, 2836–2839, DOI:10.1002/cctc.201500309
- H. Ronduda, M. Zybert, W. Patkowski, A. Tarka, P. Jodłowski, L. Kępiński, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Tuning the catalytic performance of Co/Mg–La system for ammonia synthesis via the active phase precursor introduction method, Appl. Catal., A, 2020, 598, 117553, DOI:10.1016/j.apcata.2020.117553
- J. Humphreys, R. Lan and S. Tao, Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process, Adv. Energy Sustainability Res., 2021, 2, 2000043, DOI:10.1002/aesr.202000043
- W. Raróg-Pilecka, E. Miśkiewicz, L. Kępiński, Z. Kaszkur, K. Kielar and Z. Kowalczyk, Ammonia synthesis over barium-promoted cobalt catalysts supported on graphitised carbon, J. Catal., 2007, 249, 24–33, DOI:10.1016/j.jcat.2007.03.023
- M. Karolewska, E. Truszkiewicz, M. Wściseł, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Ammonia synthesis over a Ba and Ce-promoted carbon-supported cobalt catalyst. Effect of the cerium addition and preparation procedure, J. Catal., 2013, 303, 130–134, DOI:10.1016/j.jcat.2013.03.005
- B. Lin, Y. Qi, K. Wei and J. Lin, Effect of pretreatment on ceria-supported cobalt catalyst for ammonia synthesis, RSC Adv., 2014, 4, 38093–38102, 10.1039/C4RA06175F
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, Effect of ceria morphology on the catalytic activity of Co/CeO2 catalyst for ammonia synthesis, Catal. Commun., 2017, 101, 15–19, DOI:10.1016/j.catcom.2017.07.015
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, Effect of barium and potassium promoter on Co/CeO2 catalysts in ammonia synthesis, J. Rare Earths, 2018, 36, 703–707, DOI:10.1016/j.jre.2018.01.017
- K. Sato, S.-I. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, Barium Oxide Encapsulating Cobalt Nanoparticles Supported on Magnesium Oxide: Active Non-Noble Metal Catalysts for Ammonia Synthesis under Mild Reaction Conditions, ACS Catal., 2021, 11, 13050–13061, DOI:10.1021/acscatal.1c02887
- H. Ronduda, M. Zybert, W. Patkowski, A. Tarka, A. Ostrowski and W. Raróg-Pilecka, Kinetic studies of ammonia synthesis over a barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide, J. Taiwan Inst. Chem. Eng., 2020, 114, 241–248, DOI:10.1016/j.jtice.2020.09.020
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański and W. Raróg-Pilecka, Co supported on Mg–La mixed oxides as an efficient catalyst for ammonia synthesis, Int. J. Hydrogen Energy, 2022, 47, 35689–35700, DOI:10.1016/j.ijhydene.2022.08.144
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, A high performance barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide for ammonia synthesis, RSC Adv., 2021, 11, 14218–14228, 10.1039/D1RA01584B
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, Development of cobalt catalyst supported on MgO–Ln2O3 (Ln = La, Nd, Eu) mixed oxide systems for ammonia synthesis, Int. J. Hydrogen Energy, 2022, 47, 6666–6678, DOI:10.1016/j.ijhydene.2021.12.022
- J. Wu, J. Li, Y. Gong, M. Kitano, T. Inoshita and H. Hosono, Intermetallic Electride Catalyst as a Platform for Ammonia Synthesis, Angew. Chem., Int. Ed., 2019, 58, 825–829, DOI:10.1002/anie.201812131
- Y. Inoue, M. Kitano, M. Tokunari, T. Taniguchi, K. Ooya, H. Abe, Y. Niwa, M. Sasase, M. Hara and H. Hosono, Direct Activation of Cobalt Catalyst by 12CaO·7Al2O3 Electride for Ammonia Synthesis, ACS Catal., 2019, 9, 1670–1679, DOI:10.1021/acscatal.8b03650
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation, Nat. Chem., 2017, 9, 64–70, DOI:10.1038/nchem.2595
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, J. Wu, M. Sasase, M. Kitano and H. Hosono, Dissociative and Associative Concerted Mechanism for Ammonia Synthesis over Co-Based Catalyst, J. Am. Chem. Soc., 2021, 143, 12857–12866, DOI:10.1021/jacs.1c06657
- H. Fan, X. Huang, K. Kähler, J. Folke, F. Girgsdies, D. Teschner, Y. Ding, K. Hermann, R. Schlögl and E. Frei, In-Situ Formation of Fe Nanoparticles from FeOOH Nanosheets on γ-Al2O3 as Efficient Catalysts for Ammonia Synthesis, ACS Sustainable Chem. Eng., 2017, 5, 10900–10909, DOI:10.1021/acssuschemeng.7b02812
- I. Rossetti and L. Forni, Effect of Ru loading and of Ru precursor in Ru/C catalysts for ammonia synthesis, Appl. Catal., A, 2005, 282, 315–320, DOI:10.1016/j.apcata.2004.12.024
- C. Liang, Z. Wei, Q. Xin and C. Li, Ammonia synthesis over Ru/C catalysts with different carbon supports promoted by barium and potassium compounds, Appl. Catal., A, 2001, 208, 193–201, DOI:10.1016/S0926-860X(00)00713-4
- Z. Kowalczyk, Effect of potassium on the high pressure kinetics of ammonia synthesis over fused iron catalyst, Catal. Lett., 1996, 37, 173–179, DOI:10.1007/BF00807750
- Y. Zhao, Y. Zhang, J. Chen, J. Li, K. Liew and M. R. B. Nordin, SBA-16-Supported Cobalt Catalyst with High Activity and Stability for Fischer–Tropsch Synthesis, ChemCatChem, 2012, 4, 265–272, DOI:10.1002/cctc.201100223
- S. Hinokuma, H. Shimanoe, Y. Kawabata, S. Matsuki, S. Kiritoshi and M. Machida, Effects of support materials and silver loading on catalytic ammonia combustion properties, Catal. Today, 2018, 303, 2–7, DOI:10.1016/j.cattod.2017.08.010
- R. C. Reuel and C. H. Bartholomew, The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation, J. Catal., 1984, 85, 63–77, DOI:10.1016/0021-9517(84)90110-6
- A. Borodziński and M. Bonarowska, Relation between Crystallite Size and Dispersion on Supported Metal Catalysts, Langmuir, 1997, 21, 5613–5620, DOI:10.1021/la962103u
- S. Ardizzone, C. L. Bianchi, M. Fadoni and B. Vercelli, Magnesium salts and oxide: an XPS overview, Appl. Surf. Sci., 1997, 119, 253–259, DOI:10.1016/S0169-4332(97)00180-3
- Y. A. Teterin and A. Y. Teterin, Structure of X-ray photoelectron spectra of lanthanide compounds, Russ. Chem. Rev., 2002, 71, 347–381, DOI:10.1070/RC2002v071n05ABEH000717
- A. Sarnecki, P. Adamski, A. Albrecht, A. Komorowska, M. Nadziejko and D. Moszyński, XPS study of cobalt-ceria catalysts for ammonia synthesis – The reduction process, Vacuum, 2018, 155, 434–438, DOI:10.1016/j.vacuum.2018.06.034
- M. Zybert, M. Wyszyńska, A. Tarka, W. Patkowski, H. Ronduda, B. Mierzwa, L. Kępiński, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Surface enrichment phenomenon in the Ba-doped cobalt catalyst for ammonia synthesis, Vacuum, 2019, 168, 108831, DOI:10.1016/j.vacuum.2019.108831
- A. Tarka, W. Patkowski, M. Zybert, H. Ronduda, P. Wieciński, P. Adamski, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Synergistic Interaction of Cerium and Barium-New Insight into the Promotion Effect in Cobalt Systems for Ammonia Synthesis, Catalysts, 2020, 10, 658, DOI:10.3390/catal10060658
- C. Chizallet, M. L. Bailly, G. Costentin, H. Lauron-Pernot, J. M. Krafft, P. Bazin, J. Saussey and M. Che, Thermodynamic brønsted basicity of clean MgO surfaces determined by their deprotonation ability: Role of Mg2+–O2− pairs, Catal. Today, 2006, 116, 196–205, DOI:10.1016/j.cattod.2006.01.030
- C. Chu, Y. Zhao, S. Li and Y. Sun, Correlation between the acid–base properties of the La2O3 catalyst and its methane reactivity, Phys. Chem. Chem. Phys., 2016, 18, 16509–16517, 10.1039/C6CP02459A
- J. Ni, L. Chen, J. Lin, M. K. Schreyer, Z. Wang and S. Kawi, High performance of Mg–La mixed oxides supported Ni catalysts for dry reforming of methane: The effect of crystal structure, Int. J. Hydrogen Energy, 2013, 38, 13631–13642, DOI:10.1016/j.ijhydene.2013.08.041
- K. Sato, K. Imamura, Y. Kawano, S.-I. Miyahara, T. Yamamoto, S. Matsumura and K. Nagaoka, A low-crystalline ruthenium nano-layer supported on praseodymium oxide as an active catalyst for ammonia synthesis, Chem. Sci., 2017, 8, 674–679, 10.1039/C6SC02382G
- P. Adamski, D. Moszyński, M. Nadziejko, A. Komorowska, A. Sarnecki and A. Albrecht, Thermal stability of catalyst for ammonia synthesis based on cobalt molybdenum nitrides, Chem. Pap., 2019, 73, 851–859, DOI:10.1007/s11696-018-0642-0
- G. Rambeau, A. Jorti and H. Amariglio, Catalytic activity of a cobalt powder in NH3 synthesis in relation with the allotropic transformation of the metal, J. Catal., 1985, 94, 155–165, DOI:10.1016/0021-9517(85)90091-0
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Size effect on the crystal phase of cobalt fine particles, Phys. Rev. B, 1997, 56, 13849–13854, DOI:10.1103/PhysRevB.56.13849
- C. J. H. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, H. Topsøe, D. V. Prip, P. B. Møenshaug and I. Chorkendorff, Structure sensitivity of supported ruthenium catalysts for ammonia synthesis, J. Mol. Catal. A: Chem., 2000, 163, 19–26, DOI:10.1016/S1381-1169(00)00396-4
- H. Fan, J. M. Folke, Z. Liu, F. Girgsdies, R. Imlau, H. Ruland, S. Heumann, J. Granwehr, R.-A. Eichel, R. Schlögl, E. Frei and X. Huang, Ultrathin 2D Fe-Nanosheets Stabilized by 2D Mesoporous Silica: Synthesis and Application in Ammonia Synthesis, ACS Appl. Mater. Interfaces, 2021, 13, 30187–30197, DOI:10.1021/acsami.1c06771
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06053a |
| This journal is © The Royal Society of Chemistry 2022 |