
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual activity inhibition of threonine aspartase 1 by a single bisphosphate ligand†
Alexander Höing a,
Robin Struthb,
Christine Beuckc,
Neda Rafieiolhosseinid,
Daniel Hoffmannd,
Roland H. Staubere,
Peter Bayerc,
Jochen Niemeyer*b and
Shirley K. Knauer*a
a,
Robin Struthb,
Christine Beuckc,
Neda Rafieiolhosseinid,
Daniel Hoffmannd,
Roland H. Staubere,
Peter Bayerc,
Jochen Niemeyer*b and
Shirley K. Knauer*a
aMolecular Biology II, Center of Medical Biotechnology (ZMB)/Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstrasse 5, 45141 Essen, Germany. E-mail: shirley.knauer@uni-due.de
bOrganic Chemistry, Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstrasse 7, 45141 Essen, Germany. E-mail: jochen.niemeyer@uni-due.de
cStructural and Medicinal Biochemistry, Center for Medical Biotechnology (ZMB), University of Duisburg-Essen, Universitätsstrasse 5, 45141 Essen, Germany
dBioinformatics and Computational Biophysics, Center for Medical Biotechnology (ZMB), University of Duisburg-Essen, Universitätsstrasse 5, 45141 Essen, Germany
eMolecular and Cellular Oncology/ENT, University Medical Center Mainz (UMM), Langenbeckstrasse 1, 55101 Mainz, Germany
First published on 30th November 2022
Abstract
Therapy resistance remains a challenge for the clinics. Here, dual-active chemicals that simultaneously inhibit independent functions in disease-relevant proteins are desired though highly challenging. As a model, we here addressed the unique protease threonine aspartase 1, involved in various cancers. We hypothesized that targeting basic residues in its bipartite nuclear localization signal (NLS) by precise bisphosphate ligands inhibits additional steps required for protease activity. We report the bisphosphate anionic bivalent inhibitor 11d, selectively binding to the basic NLS cluster (220KKRR223) with high affinity (KD = 300 nM), thereby disrupting its interaction and function with Importin α (IC50 = 6 μM). Cell-free assays revealed that 11d additionally affected the protease's catalytic substrate trans-cleavage activity. Importantly, functional assays comprehensively demonstrated that 11d inhibited threonine aspartase 1 also in living tumor cells. We demonstrate for the first time that intracellular interference with independent key functions in a disease-relevant protein by an inhibitor binding to a single site is possible.
Introduction
‘Hit once better hit twice!’ this saying from the clinical bedside is accepted when treating viral (e.g., COVID) or bacterial infections as well as live threatening diseases, such as cancer.1–3 Hence, combination therapies are practiced to improve treatment success and minimize the complications of therapy resistances.1–3 Thus, modern chemistry in basic and applied research seeks to expand our treatment repertoire by investigating chemical structures that target multiple independent functions in disease relevant proteins, ideally by a single inhibitor.2,3 So far, compounds affecting multiple (unrelated) proteins by ‘off target’ effects or inhibiting protein function by binding to different sites as well as hybrid drugs combining pharmacophores simultaneously targeting various protein functions have been explored.2,4–7 The concept of ‘two for the price of one’ is clearly under intense investigation.8 For example, it has been shown that inhibiting two different enzymes in inflammatory pathways with a single peptide reduced inflammation.9 However, proven examples of molecules effectively targeting independent functions in proteins by binding to a single site not only in vitro but also in vivo are to the best of our knowledge missing to date.To thus expand the repertoire of such dual-active compounds, a trans-approach combining synthetic and analytical chemistry with a profound knowledge of disease-relevant molecular structures and functions is obligatory. Moreover, as shown by others and us, rational chemical design strategies now allow to precisely target and selectively inhibit small, functionally pivotal protein domains.10–12 Based on our previous work,13–20 we here choose the cancer-relevant protease threonine aspartase 1 (Taspase 1) as a model for the development of dual active inhibitors for following reasons: For one, proteases are central for life and protease deregulation is associated with a variety of diseases.21,22 As shown by others and us, Taspase 1 is not only critically involved in the regulation of cellular development but also in hematological malignancies and solid tumors.15,23,24
Although it belongs to the group of threonine proteases, Taspase 1 is a unique enzyme. In contrast to the exclusively cis-active type 2 asparaginases, only Taspase 1 is also able to cleave other substrates in trans by hydrolyzing its target proteins at conserved Q3[FILV]2D1↓G1X′2′D3′D4′ motifs.18,25–27 Thus, the discovery of Taspase 1 founded a new class of endopeptidases that utilize the N-terminal threonine (Thr234, Fig. 1) of its mature β-subunit as the active site (Fig. 1). Besides the proto-oncogene Mixed Lineage Leukemia (MLL),25 other essential proteins such as the precursor of the general transcription factor IIA (TFIIA)18,28–30 or the Upstream Stimulatory Factor 2 (USF2)18 could be identified as bona fide Taspase 1 targets in the protease's degradome.18 Since Taspase 1 is normally expressed mainly during embryonic development, interference with its activity would not affect healthy adult tissue.23,27,31 In contrast, liquid as well as solid tumor cells have been shown to re-express Taspase 1,23,27,31 rendering this protease as a promising therapeutic target.32–34 Importantly, this increased dependency on the protease's activity correlates with increased tumor cell proliferation and reduced apoptosis, as shown by RNAi depletion studies.24,31 Therefore, Taspase 1 is classified as a ‘non-oncogene addiction’ protease.23,24 However, the full repertoire of physiological or pathological pathways regulated in humans by Taspase 1 still remains to be understood.
 | ||
| Fig. 1 In Taspase 1, the basic clusters (blue) constituting the bipartite NLS are arranged on two neighbouring α-helices on a surface-exposed loop, close to the catalytically active threonine (Thr234, red).36 Proposed tertiary structure (A) and amino acid sequence (B) including residues 183–233 constituting the loop in the unprocessed proenzyme are shown. After cleavage, Thr243 acts as N-terminal nucleophile of the β-subunit. NLS-targeting ligands may not only interfere with Importin α-binding but also with the loops' flexibility and the protease's catalytic activity. N-/C-termini are indicated. | ||
Second, although Taspase 1 belongs to the group of threonine proteases, such as the proteasome, it is not affected by general protease inhibitors.23,24 To date, some more or less specific inhibitors have been suggested, including ubiquitous Cl− anions.13,16,24,25,31 However, their mode of action is often not understood, they need to be used in high concentrations, and completely fail to inhibit Taspase 1 in living systems.13,16,24,25,31 As no effective in vivo Taspase 1 inhibitors are available, this caveat not only hampers the dissection of Taspase 1's disease mechanisms, but also precludes the assessment/translation of its clinical relevance.9,14–17 Hence, Taspase 1 not only represents a highly relevant disease target but remains a challenging biochemical model for the design of novel inhibitors.16,35
Third, we discovered that Taspase 1 has to undergo a distinct, multistep activation process to execute its pathobiological cleavage activity (see ESI, Fig. S1†).19,23 The protease is expressed as an inactive α/β-proenzyme (45 kDa) in the cytoplasm and depends on active nuclear transport for its autoproteolytic activation, prerequisite for trans-cleavage of its degradome.19,23 Active nuclear import is mediated by Taspase 1's bipartite nuclear localization signal (NLS) consisting of two distinct basic amino acid clusters containing lysines and arginines.19,23 These are located on neighbouring α-helices arranged on a surface-exposed loop in the α-subunit (Fig. 1), mediating binding to its cognate nuclear transport receptor Importin α.19 Of note, this interacting loop comprising residues 183–233 behaves flexible in solution as shown by NMR analyses36 although some but not all crystal structures indicate more rigid helical structures.26,37 Inside the nucleus, autoproteolysis results in the formation of the two subunits α (25 kDa) and β (20 kDa), which reassemble into the active heterodimer capable of cleaving substrates.19,25 Thus, the Taspase 1/Importin α-axis is prerequisite for intracellular protease activation, and besides the catalytically active Thr234, represents an alternative target for chemical intervention strategies inside cells (see ESI, Fig. S1,† and 1).
Indeed, we just recently developed different supramolecular interference strategies (see ESI, Fig. S2 and Table S1†) utilizing the guanidiniocarbonylpyrrole (GCP)38 motif introduced by Carsten Schmuck to address surface-accessible glutamate/aspartate residues for different target proteins including Taspase 1,10,14,35,39–43 and thus significantly advanced its applicability previously focused on gene delivery.44–53 In particular for Taspase 1, we could rely on the presence of a substantial number of acidic amino acids dispersed on its loop structure in direct vicinity of the NLS (Fig. 1). More specifically, several GCP units were multivalently arranged either by direct covalent linkage54 or presented on sequence-controlled macromolecules35 to interfere with this pivotal protein–protein interaction (PPI). Both rational design strategies (see SI, Fig. S2 and Table S1†) turned out to be rather effective in vitro at low micromolar concentrations: the macromolecule 3GLP (ref. 35) composed of three Lys-GCP side chains and 3 kDa poly(ethylene glycol) (PEG) stealth polymer as well as the bivalent ligand 2GC.54 The latter was even active in different tumor cell lines (EC50 ∼ 40–70 μM), although Taspase 1's proteolytic activity was only affected in unphysiologically high concentrations in a semi-in vitro Taspase 1 substrate cleavage assay (∼500 μM). However, both studies utilizing GCP moieties as such did not target the interacting amino acids itself but rather the region adjacent to the PPI interface. Notably, they revealed to be only fruitful when ligands allowed an adequate coverage and thus masking of a sufficient portion of the protein surface. Here, this could be achieved by the power of multivalent binding that emerged prerequisite for efficient PPI disturbance.
As already suggested, a more straightforward approach would certainly aim at directly addressing the basic residues constituting the NLS. In particular its bipartite nature renders Taspase 1's import signal prone to multivalent targeting with eligible supramolecular moieties. Insofar, we first utilized bisphosphate ligands providing a hydrophobic cavity that can strongly bind to Lys- and Arg-residues, the so-called molecular tweezers.55 Here, newly established, laborious synthesis routes enabled us to covalently fuse several of such tweezer molecules into multivalent ligand systems including bi-up to pentavalent constructs (see ESI, Fig. S2 and Table S1†).56 Indeed, a stepwise increase in valency was robustly reflected by the ligands' gradually enhanced potency to disrupt the Taspase 1/Importin α interaction and correlated with both higher binding affinity and inhibition of proteolytic activity. As such, highest binding affinities (KD = 60 μM) and most effective interference with Importin α interaction as well as proteolytic activity was revealed for the pentavalent ligand system. Notably, molecular weights of the largest multivalent tweezer systems are already in the low kDa range and approach the size of polypeptides (20–40 mers).
This study however for the first time characterized multivalent phosphates as a potent class of ligands suited to target Taspase 1. Hence, based on this knowledge and the available structural information,26,36 we hypothesized that also other compounds with multiple anionic binding sites might in general be suited to rationally address the NLS-loop in Taspase 1. Desirably, such ligands should feature a smaller molecular weight, making them most likely not only synthetically more readily available, but supposedly also cell permeable. In addition, an increased rigidity of such small molecules compared to the multivalent tweezer system should still enable the restriction of the enzyme's molecular motion and thus to inhibit its catalytic activity (Fig. 1). Likewise, such chemical structures should be indeed prone to function as ‘dual inhibitors', enabling to address two independent enzyme functions by binding to a single site.
Results and discussion
Synthesis and characterization of NLS-targeting ligands
For applications in chemosensing and organocataylsis, we have recently established a library of structurally related, low molecular weight bis- and trisphosphates.57–60 These organic, multivalent ligands are based on rigid aromatic backbones, which allows a control of the relative positioning of the anionic phosphate groups in space, possibly offering a way to influence the strength and selectivity of binding to the NLS domain comprising clusters of lysine and arginine residues (Fig. 1B) by systematic structural variations. Initially, three chiral bis- and triphosphates allowing the selective recognition of cations were synthesized based on the covalent linkage of 1,1′-binaphthylphosphoric acids (1d/e/f, see ESI, Fig. S3,† numbering of all ligands based on earlier publications).60 Here, the bisphosphoric acid ligands 1e and in particular 1d allowed for a highly stereoselective binding of D-lysine.60 Subsequently, our library of linked aromatic phosphoric acid diesters was complemented by a series of related monoesters (11d/e/f, Fig. 2, and see ESI, Fig. S3†).60 In contrast to the diesters, the latter class of ligands could be successfully utilized as chemosensors for different amino sugars.57–59 They partially even revealed selective binding to certain sugar species, but have not yet been tested for their interaction with cationic protein-residues. Notably, all of these hosts ligands are synthetically readily available also on gram-scale which makes them exceptionally attractive for a broad range of potential biomedical applications.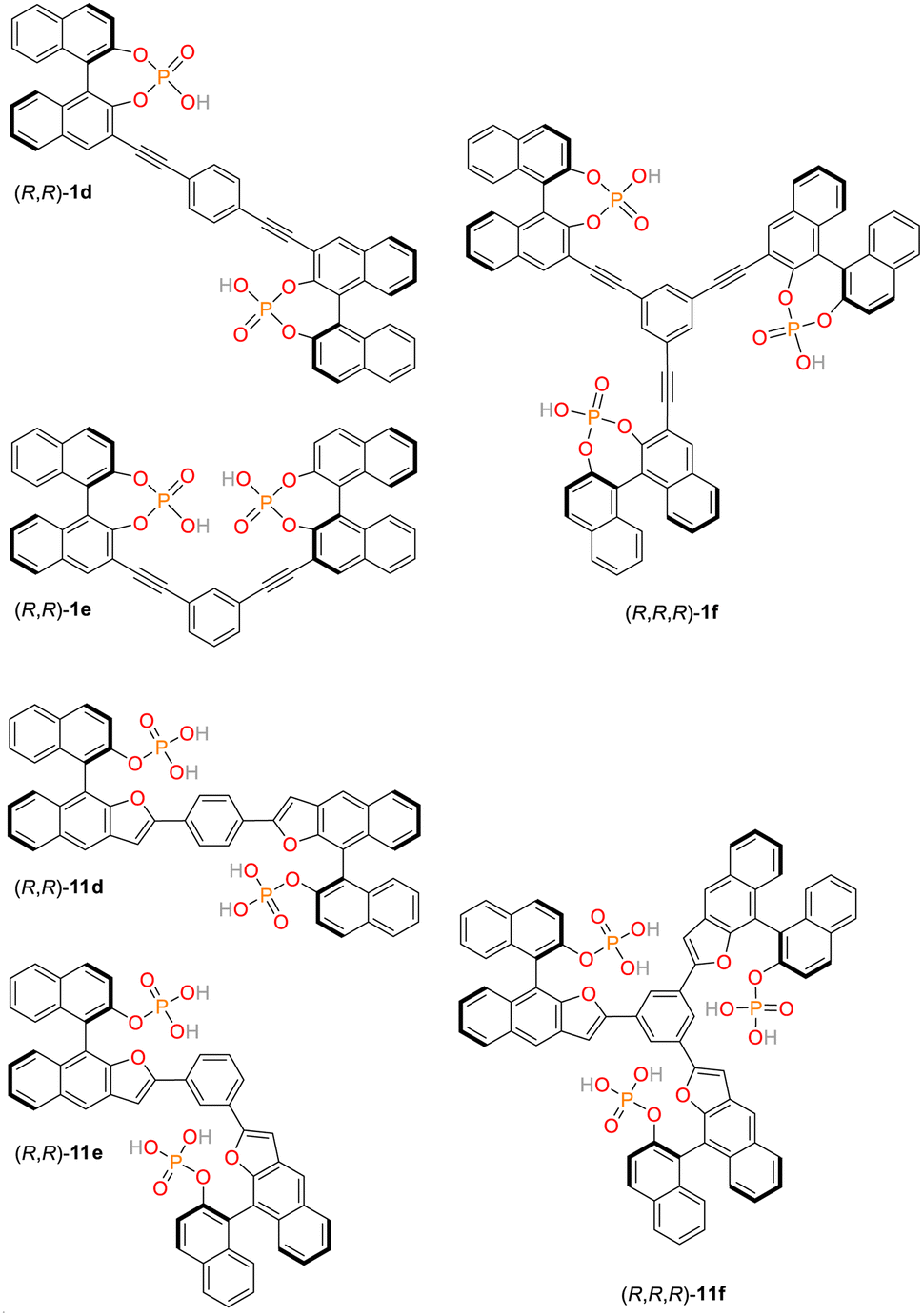 | ||
| Fig. 2 Chemical structures of phosphate-based ligands (all-R)-1d/e/f and (all-R)-11d/e/f. The ligands were exclusively used as the (R,R)- and (R,R,R)-isomers respectively (no further stereodescriptors are given in the text). | ||
Importantly, the bis/trisphosphate-diesters 1d/e/f can feature two or three negative charges on their phosphate groups, while the bis/trisphosphate-monoesters 11d/e/f can even possess four or six negative charges, respectively. This potentially enables strong and selective binding to the positively charged lysine and arginine residues constituting the NLS domain.
In terms of photophysical properties, ligands 1d/e/f and 11d/e/f show blue fluorescence with emission wavelengths in the range of λem = 450–470 nm (λexc = 380/405 nm, see Fig. S5†). Although quantum yields of these compounds are similar (Φ = 0.21–0.70 at λexc = 305 nm, see Table S2†), the strongest relative fluorescence intensities are found for 1d and 11d due to their stronger UV/vis-absorption at 380/405 nm (in comparison to 1e/f and 11e/f) (see Fig. S5†). This enables their exploitation as fluorescence-based chemosensors in vitro as well as in vivo.61,62
Screening of the library for promising candidates
Consequently, we first applied a customized biochemical pull-down assay employing pure, recombinantly expressed proteins (see ESI, Fig. S9†) to analyze the effects of our small focused library of phosphate-based ligands on the binding of Taspase 1 to Importin α (see ESI for details, Fig. S10 and S11†).35,54 As this interaction crucially relies on the basic NLS clusters present in the Taspase 1 loop, the assay serves as a reliable and direct readout to support our ligand binding hypothesis. Briefly, Importin α is expressed with an N-terminal glutathione S-transferase (GST) tag and immobilized on a sepharose column. Subsequently, binding of Taspase 1-His in the presence or absence of ligands allows to quantify the compounds inhibitory potential (see ESI, Fig. S11†). Of note, since wildtype Taspase 1 partially undergoes autoproteolysis during protein purification, we here used a proteolytically inactive Taspase 1 mutant (D233A/T234A) ensuring a stable concentration of the proenzyme for robust and reliable detection.With this assay, we could identify 11d as the most promising candidate to perturb the Taspase 1/Importin α interaction at a concentration of 100 μM when compared to the other ligands. Notably, the chemical properties of 1f resulted in a strong interference with the chemiluminescent protein detection (data not shown). Thus, this ligand was be omitted to allow a robust evaluation of the ligands' performance. Based on our results, and to ensure an optimal comparability, we decided to continue our studies with the sub-library 11d/e/f. As a perfect set of structurally related ligands, the paramount performance of 11d would thus be nicely accompanied by a moderately active compound (11e) and a presumably unactive control (11f).
Molecular modelling of NLS-targeting ligands
To now manifest our assumption that in particular biphosphate ligand 11d might allow the molecular recognition of Taspase 1's NLS domain we next employed in silico molecular modelling on ligands 11d/e/f (Fig. 2, and see ESI†). Using a recently developed tool for scanning the surface of proteins with fragments of large ligands called Epitopsy followed by a Simulated Annealing Monte Carlo Simulation (SAMC), we identified the energetically most favorable binding sites for these ligands on the surface of Taspase 1. Therefore, we made use of a bead-spring model where each respective group is represented by one bead. Accordingly, ligands 11d and 11e are substituted by 5, and ligands 11f by 7 beads. Spring constants and equilibrium lengths between the beads are the same for all ligands, and so were all other simulation parameters. Notably, the only difference between ligands 11d and 11e is the angles that are not considered in the bead-spring model. As such, our model cannot distinguish between these structures, and simulation results are the same for both bisphosphate ligands. However, SAMC simulations indeed located the energetically most favorable binding site of 11d/e at the loop optimally contacting the basic NLS residues (Fig. 3; see ESI, Fig. S6 and S7†). Interestingly, also the minimum energy conformation of 11f was found near the loop, although seemingly more displaced towards the proximal turn region with less contact to the helix (Fig. S8†). From the modelling data, bisphosphate ligands might allow a tighter and more stable association with the loop and thus reveal a higher inhibitory potential.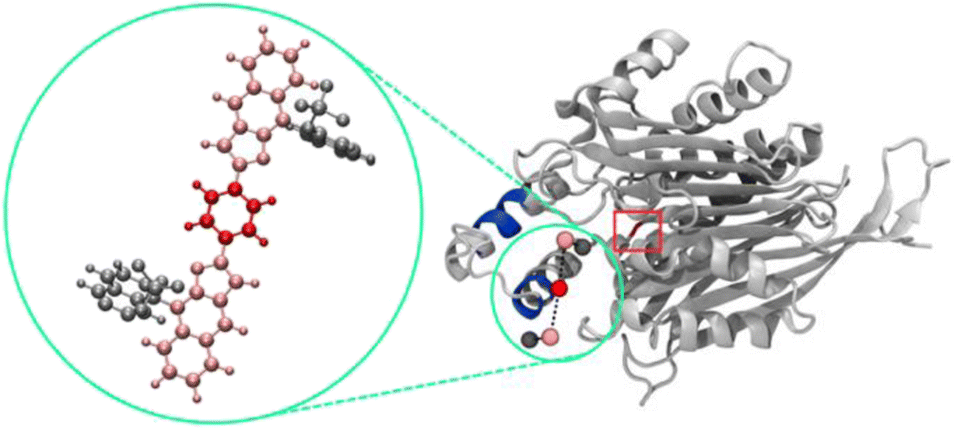 | ||
| Fig. 3 SAMC simulations predict the energetically most favourable binding site of 11d on Taspase 1, with the ligand represented as a bead-spring model. The magnified image (green circle) is the atomistic view of 11d with each colour indicating the respective group represented by one bead. Blue, bipartite NLS; red square: active site. | ||
11d is a potent inhibitor of the Taspase 1/Importin α interaction
To subsequently substantiate our findings from the molecular simulations, we again made use of our customized biochemical pull-down assay (see ESI, Fig. S10 and S11†) employing pure recombinant proteins (see ESI, Fig. S9†) to analyze the ligands' effects on the Taspase 1/Importin α interaction (Fig. 4, and see ESI for details†).35,54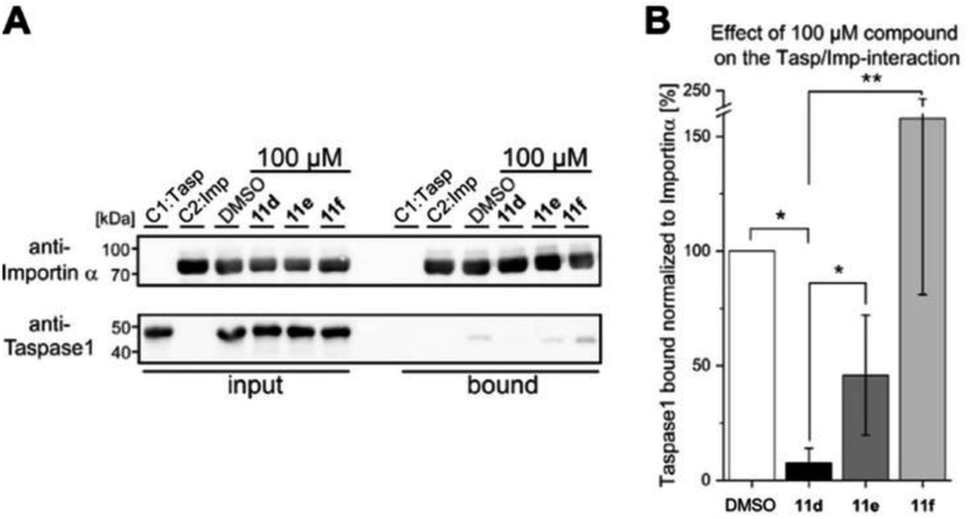 | ||
| Fig. 4 The interaction between Taspase 1 and Importin α is effectively disrupted by 11d. (A) Pull-down assay with 100 μM of indicated ligands. Controls: Taspase 1-His (C1) or GST-Importin α (C2) alone were added to the column, and a DMSO-treated sample served as reference. (B) Densitometric quantification of pull-down assays, comprising the mean of three replicates ± standard deviation (*p < 0.05, **p < 0,01) compared to the DMSO control. | ||
As shown in Fig. 4, 11d could indeed be verified as the most potent inhibitor of the Taspase 1/Importin α interaction, reducing the amount of bound Taspase 1 by more than 93% at a concentration of 100 μM (Fig. 4). In contrast, compound 11e was less effective (reduction by 50%), and the trivalent derivate 11f did not reduce binding, thereby again confirming the results of our previous library screen (see ESI Fig. S12†). Therefore, we now finally decided to merely focus on 11d as the most promising candidate for further characterization. To increase our resolution, we also performed pull-down assays with low micromolar concentrations of 11d (2–8 μM; Fig. 5A). Notably, 11d significantly interfered with the Taspase 1/Importin α interaction already at 6 μM, and densitometric quantification of the respective immunoblots resulted in an IC50 of 6.10 ± 0.27 μM (Fig. 5, see ESI, Fig. S14†). As an important control, the binding of GST-Importin α to the matrix was not affected by the ligand itself (see ESI, Fig. S13†).
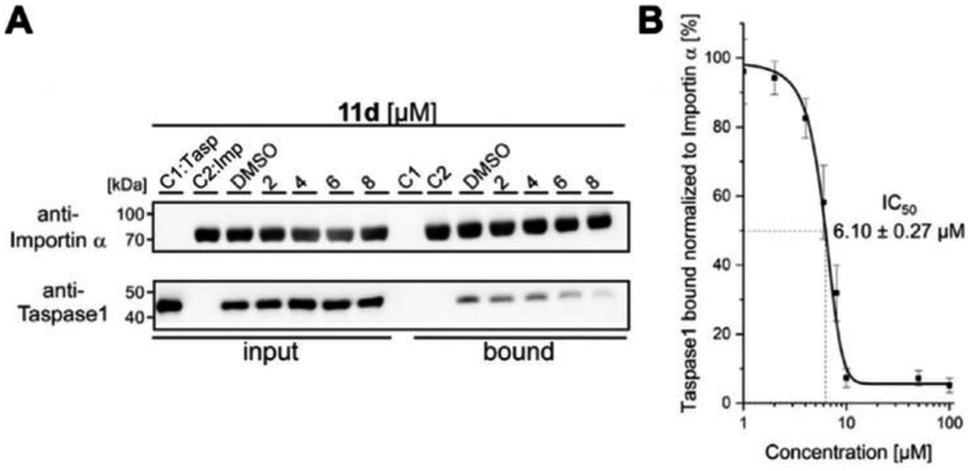 | ||
| Fig. 5 11d efficiently prevents Taspase 1/Importin α interaction. (A) Pull-down assay with increasing concentrations of 11d (2–8 μM). Controls: Taspase 1-His (C1) or GST-Importin α (C2) alone were added to the column; a DMSO-treated sample served as reference. (B) Densitometric quantification of pull-down assays reveals a half maximal inhibitory concentration of 11d in the low micromolar range (IC50 = 6.10μM ± 0.27 μM). Quantification comprises the mean of three replicates ± standard deviation (see ESI†). | ||
11d is a Taspase 1 loop binder
Our assumption that 11d might specifically recognize the NLS domain was comprehensively supported by independent evidence: first, to prove the direct binding of 11d to the protease experimentally, we used a quantitative fluorescence anisotropy assay. Here, recombinant full-length Taspase 1 was titrated to a constant concentration of the fluorescent ligand 11d, which revealed a strong binding KD in the nanomolar range (KD = 300 ± 50 nM) (see ESI, Fig. S15†). Second, to further narrow down 11d's binding site, we coupled an N-terminal FAM-label to the NLS peptide loop (aa S181-D233) and performed fluorescence titration experiments. A decrease in fluorescence due to compound binding in close proximity to the label was observed (KD = 3 μM), in contrast to the negative control (FAM-label without peptide; see ESI, Fig. S16†). Third, NMR titration experiments with the isolated Taspase 1 NLS loop and 11d revealed a change in relative signal intensities and thus additionally verified binding of 11d to the NLS loop (see ESI, Fig. S17†).Dual activity: 11d also inhibits Taspase 1's proteolytic function
We convincingly demonstrated that 11d efficiently disrupts the Taspase 1/Importin α interaction (activity #1). Next, we tested our hypothesis that binding of 11d to the loop region also affects the protease's proteolytic activity. To verify this second, independent effect (activity #2), we first employed a biochemical colorimetric cleavage assay using a recombinant fusion protein harbouring the MLL CS2 substrate cleavage site (Fig. 6, and see SI†). As shown in Fig. 6A, addition of recombinant, proteolytically active Taspase 1 resulted in substrate cleavage that was efficiently inhibited by 11d in a concentration-dependent manner (Fig. 6A). 11d turned out to act as a highly potent inhibitor, already preventing cleavage at low micromolar concentrations as revealed by densitometric quantification (IC50 ≈ 2 μM) (Fig. 6B).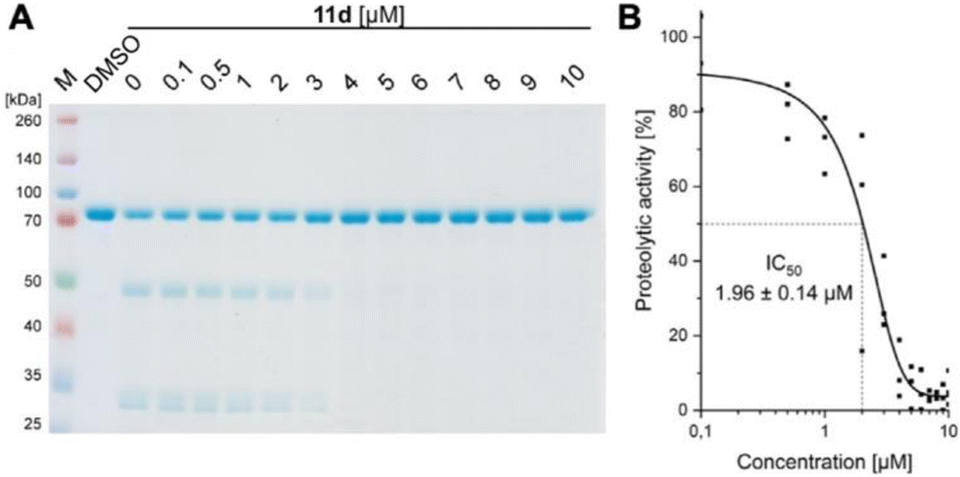 | ||
| Fig. 6 11d interferes with Taspase 1's proteolytic activity. (A) Cleavage of a recombinant substrate by Taspase 1 was assessed via SDS-PAGE and Coomassie staining after 90 min reaction time in absence and presence of 11d (0.1–10 μM). Inhibition of proteolysis by 11d was already evident at a concentration between 3 and 4 μM. A DMSO-treated sample served as reference. (B) Densitometric quantification of the uncleaved substrate (asterisk) and its cleaved products (arrows) verifies an IC50 in the lower micromolar range (1.96 ± 0.14 μM). | ||
To independently substantiate the dual inhibition activity, we additionally established a FRET-based assay. Here, a MLL CS2 cleavage site peptide was coupled to a quencher and a fluorophore.
Addition of proteolytically active, wild-type Taspase 1 resulted in cleavage and an increase in fluorescence (see ESI, Fig. S18†). In contrast, 11d efficiently inhibited Taspase 1's proteolytic activity, indicated by a lack of signal increment. To next mimic a cellular environment in which other proteins may act as (potent) competitors, we added different amounts of cancer cell lysates to our assay. Notably, 11d was still able to reduce Taspase 1's proteolytic activity to 50% even in the presence of a 30-fold excess of cellular proteins, underlining 11d's specificity and setting the stage for subsequent assays in living tumour cells.
Dual inhibition in vivo: 11d suppresses Taspase 1 function also in living cancer cells
An absolute requirement for an inhibitor's in vivo activity and potential therapeutic/clinical relevance in drug development is its access to the relevant target protein inside disease cells. Notably, most suggested Taspase 1 inhibitors reported to date failed to convincingly take this key hurdle.16,23,63 Hence, we first controlled the uptake of 11d in live cells utilizing its autofluorescence. Fluorescence microscopy confirmed that 11d efficiently entered Taspase 1 expressing carcinoma cells already 1 h following incubation. Notably, to substantiate the internalization of the compound, we included a fluorescent outer plasma membrane staining following fixation (Fig. 7), and in addition generated maximum projection images from stacks (see ESI, Fig. S19†). Hereby, we could confirm that the compound was completely surrounded by the outer plasma membrane following successful intracellular uptake. Although no ample proof, the mainly homogeneous cytoplasmic distribution of 11d indicates for its intracellular bioavailability.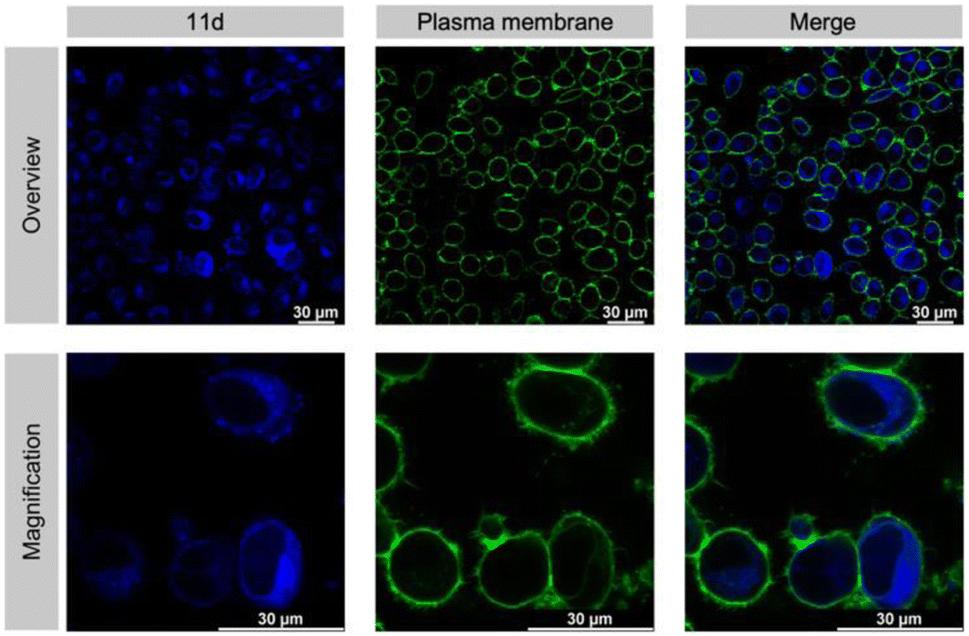 | ||
| Fig. 7 11d enters living tumour cells. Cells were incubated with 50 μM 11d for 1 h, and the plasma membrane was stained with CellBrite® (green). Confocal microscopy detects the blue-fluorescent compound 11d inside HeLa Kyoto cells. Scale bars, 30 μm. | ||
Next, we tested the inhibitory activity of 11d in tumor cells using a cell-based in vivo biosensor assay. Our biosensor fusion protein (TASPBS) consists of a red fluorescent (mCherry) protein coupled to the simian virus 40 (SV40) nuclear localization sequence (NLS) and a dominant, strong nuclear export signal (NES) from the human immunodeficiency virus 1 (HIV-1) Rev protein separated by the bona fide MLL CS2 Taspase 1 cleavage site (Fig. 8A). As illustrated, TASPBS shuttles continuously between the nucleus and the cytoplasm. As the NES is dominant over the NLS, the uncleaved biosensor's steady-state localization is predominantly cytoplasmic. Cleavage by Taspase 1 however results in the loss of the NES, thereby triggering TASPBS’ nuclear accumulation (Fig. 8A). Experimentally, the red fluorescent TASPBS accumulated in the nucleus in Taspase 1 co-expressing HeLa Kyoto cells (Fig. 8B, lower panel). In contrast, treatment with 11d effectively inhibited Taspase 1-mediated biosensor cleavage (>10-fold; Fig. 8C and D) as demonstrated by TASPBS's exclusive cytoplasmic localization in 11d-treated cells (Fig. 8B, upper panel, and Fig. 8C).
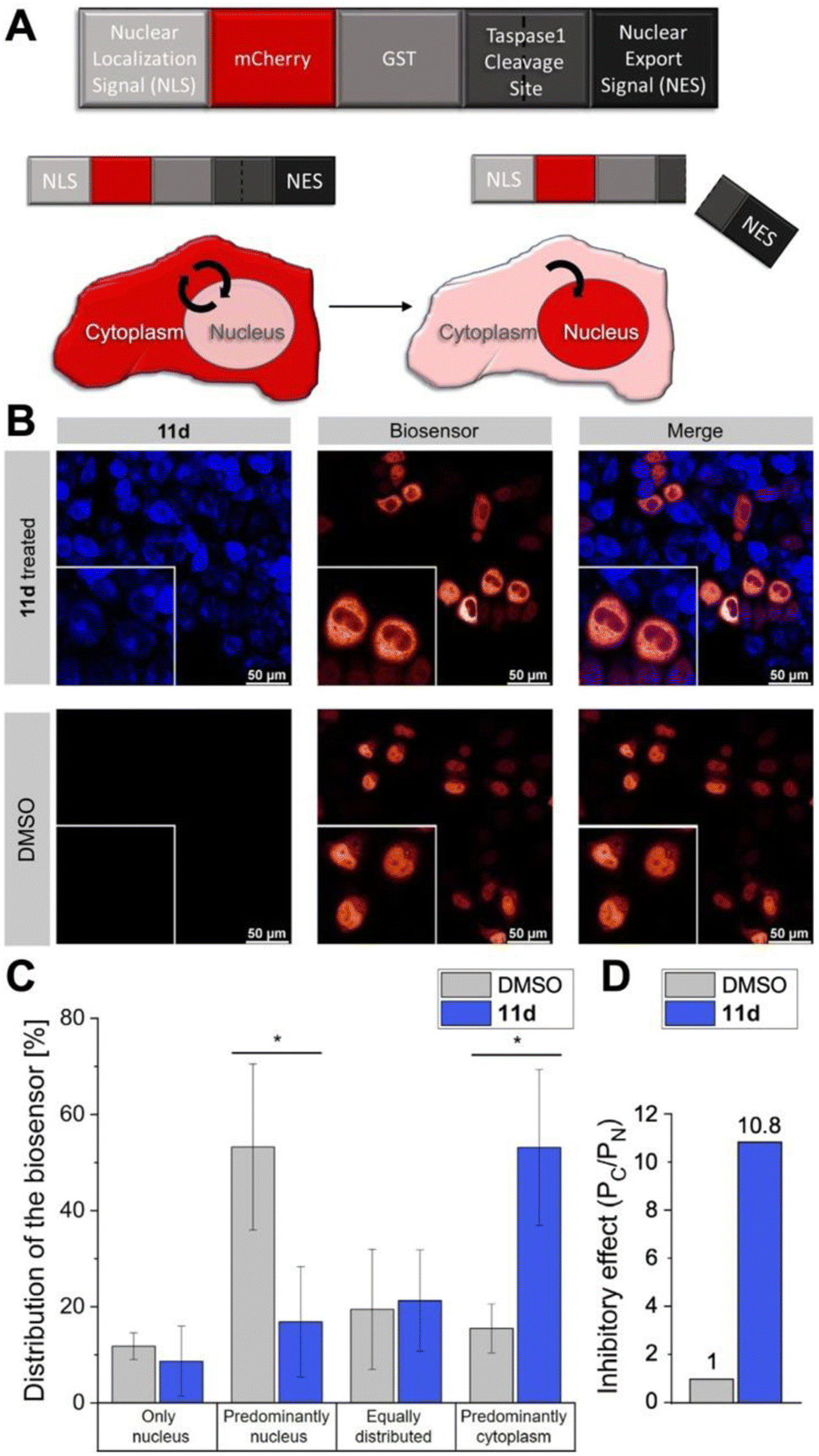 | ||
| Fig. 8 11d interferes with Taspase 1 proteolytic activity in living cancer cells. (A) Modular structure of the autofluorescent Taspase 1-biosensor (TASPBS). Taspase 1-mediated cleavage results in the biosensor's nuclear accumulation. (B) Confocal microscopy of TASPBS-expressing HeLa Kyoto cells, treated with 50 μM 11d for 24 h (upper panel) or DMSO control (lower panel). Scale bars, 50 μm; frames indicate representative close-ups. (C) Quantification of TASPBS's intracellular localization (see ESI, Table S5†). Microscopic images were acquired in at least 100 cells and randomized for localization assignment (see ESI, Table S5†). Results are the mean of triplicates ± standard deviation (*p < 0.5; ***p < 0.001). (D) Inhibitory effect of 11d as represented by the ratio of the mean percentage of cells in the category ‘Predominantly cytoplasm’ (PC) vs. ‘Predominately nucleus' (PN) normalized to 1 for the DMSO control. | ||
Results were additionally quantified by randomized image quantification (see ESI, Fig. S20 and Table S5†), characterizing 11d as a unique highly potent cell-permeable inhibitor of this protease. To exclude the formal possibility that 11d unspecifically interferes with the import of the biosensor itself, Taspase 1-expressing HeLa Kyoto cells were transfected with TASPBS and successively treated with 11d in combination with the nuclear export inhibitor LMB.64 Blocking export allowed nuclear import and thus nuclear accumulation of the biosensor even in the presence of 11d, excluding an unspecific inhibition with the TASPBS NLS or import in general (see ESI, Fig. S21†). Although our cell studies did not reveal any changes in cellular morphology, we additionally tested ligand 11d in a cell viability assay where it showed no toxicity (see ESI, Fig. S22†).
Clearly, future work needs to thoroughly investigate 11d's effectivity and bioavailability in tumour models with improved complexity. Although compounds such as 11d have a relatively low solubility in aqueous environments, our data obtained in biochemical as well as cancer cell culture condition clearly demonstrate their applicability. Nevertheless, one may seek to improve their solubility by additional chemical modifications, such as the introduction of peripheral solubilizing groups or a downsizing of the aromatic backbones (whilst maintaining the distance between the two phosphate groups). Moreover, the rational engineering of pharmacophores and delivery by nano-based drug formulations might be explored.32,65 However, convincing Taspase 1 specific small animal tumour models are not yet available.23 Thus, we do not wish to postulate that 11d is ‘ready’ for clinical translation, which may be considered a potential limitation of our work. We are also aware that our approach may not be generalized to any disease-relevant protein as it relies on the surface presentation of relevant basic amino acids in a distinct orientation of a bipartite NLS on a flexible loop structure. Although the monopartite SV40 NLS present in our biosensor was not affected by 11d (see ESI, Fig. S21†), a plethora of NLSs have been identified in many cancer-/disease-relevant proteins, such as p53, NPM, TFIIA, or the superfamily of nuclear receptors, with the vast majority classified as monopartite signals.19,30,66–68
However, recent studies revealed that the traditional definition of bipartite NLSs might be too restrictive and linker length can vary within a broader range.69 As such, extension of the surface-exposed, importin-interacting amino acid residues of NLSs initially classified as monopartite might render them amenable to analogous interference strategies.
Moreover, as enzymatic/functional relevant domains are often localized in close proximity, we feel that our ‘proof of principle’ study is not limited to Taspase 1 but similar approaches might be successfully translated to other targets.
Conclusions
Based on a comprehensive in silico, analytical, biochemical, and live cell approach, we demonstrated that the bisphosphate ligand 11d is a potent dual active inhibitor for Taspase 1. Our conclusion is based on independent experimental evidence: for one, molecular modeling combined with NMR and binding experiments confirmed 11d's selectivity for the basic cluster (220KKRR223) in Taspase 1's NLS. Second, 11d's high NLS binding affinity prevented Taspase 1's interaction with Importin α interfering with nuclear entry and protease activation (activity #1). Third, biochemical cell-free assays showed that 11d additionally affected the protease's catalytic cleavage function, most likely by restricting the molecular flexibility of the bound loop (activity #2; Fig. 1). Fourth, 11d efficiently inhibited Taspase 1 also in cancer-relevant cell models.We demonstrate that by binding to a single site, compound 11d simultaneously inhibits Taspase 1's protein-protein-interaction-based activation and its enzymatic function also in a physiological environment. This study thereby introduces the concept that dual activity inhibition through a single ligand by binding to a single site is possible with high efficiency also inside cells. Hence, ‘targeting two for the price of one’ by addressing multiple independent functions in a disease-relevant cellular protein via a single inhibitor should no longer be considered a ‘too ambitious’ goal, thereby stimulating the design of similar inhibitors for other targets.
Author contributions
AH purified the proteins and performed all biological experiments. RS synthesized and purified the compounds. CB performed the NMR experiments and interpreted the data together with PB. NR carried out the computational studies and analyzed the results together with DH. SK and JN designed the study and supervised the experiments. AH assembled the figures and wrote the manuscript together with SK, RHS and JN. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the CRC 1093 funded by the ‘Deutsche Forschungsgemeinschaft’. We acknowledge the use of the imaging equipment and the support of the “Imaging Center Campus Essen” (ICCE). Instrument Leica TCS SP8X FALCON was obtained through DFG funding (Major Research Instrumentation Program, INST 20876/294-1 FUGG).References
- K. Baby, S. Maity, C. H. Mehta, A. Suresh, U. Y. Nayak and Y. Nayak, Eur. J. Pharmacol., 2021, 896, 173922 CrossRef CAS PubMed.
- T. Stankovic, J. Dinic, A. Podolski-Renic, L. Musso, S. S. Buric, S. Dallavalle and M. Pesic, Curr. Med. Chem., 2019, 26, 6074–6106 CrossRef CAS PubMed.
- M. Szumilak, A. Wiktorowska-Owczarek and A. Stanczak, Molecules, 2021, 26, 2601 CrossRef CAS PubMed.
- A. Flemming, Nat. Rev. Drug Discovery, 2016, 15, 455 CrossRef PubMed.
- C. Harrison, Nat. Rev. Drug Discovery, 2010, 9, 271 CrossRef PubMed.
- P. Cohen, D. Cross and P. A. Jänne, Nat. Rev. Drug Discovery, 2021, 20, 551–569 CrossRef CAS PubMed.
- D. Romero, Nat. Rev. Clin. Oncol., 2017, 14, 328 CrossRef.
- M. Brazil, Nat. Rev. Drug Discovery, 2003, 2, 170–171 CrossRef CAS.
- J. Sohn, T. I. Kim, Y. H. Yoon, J. Y. Kim and S. Y. Kim, J. Clin. Invest., 2003, 111, 121–128 CrossRef CAS PubMed.
- C. Vallet, D. Aschmann, C. Beuck, M. Killa, A. Meiners, M. Mertel, M. Ehlers, P. Bayer, C. Schmuck, M. Giese and S. K. Knauer, Angew. Chem., Int. Ed., 2020, 59, 5567–5571 CrossRef CAS PubMed.
- A. Meiners, S. Backer, I. Hadrovic, C. Heid, C. Beuck, Y. B. Ruiz-Blanco, J. Mieres-Perez, M. Porschke, J. N. Grad, C. Vallet, D. Hoffmann, P. Bayer, E. Sanchez-Garcia, T. Schrader and S. K. Knauer, Nat. Commun., 2021, 12, 1505 CrossRef CAS PubMed.
- D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- S. K. Knauer, V. Fetz, J. Rabenstein, S. Friedl, B. Hofmann, S. Sabiani, E. Schröder, L. Kunst, E. Proschak, E. Thines, T. Kindler, G. Schneider, R. Marschalek, R. H. Stauber and C. Bier, PLoS One, 2011, 6, e18253 CrossRef CAS PubMed.
- A. Höing, A. Zimmermann, L. Moews, M. Killa, M. Heimann, A. Hensel, J. Voskuhl and S. K. Knauer, ChemMedChem, 2022, 17, e202100640 CrossRef PubMed.
- A. Gribko, A. Hahlbrock, S. Strieth, S. Becker, J. Hagemann, M. Deichelbohrer, A. Hildebrandt, N. Habtemichael and D. Wünsch, Sci. Rep., 2017, 7, 14937, DOI:10.1038/s41598-017-14814-x.
- J. v. d. Boom, M. Mamić, D. Baccelliere, S. Zweerink, F. Kaschani, S. Knauer, P. Bayer and M. Kaiser, ChemBioChem, 2014, 15, 2233–2237 CrossRef PubMed.
- C. Bier, S. K. Knauer, D. Wünsch, L. Kunst, S. Scheiding, M. Kaiser, C. Ottmann, O. H. Krämer and R. H. Stauber, FASEB J., 2012, 26, 3421–3429 CrossRef CAS PubMed.
- C. Bier, S. K. Knauer, A. Klapthor, A. Schweitzer, A. Rekik, O. H. Krämer, R. Marschalek and R. H. Stauber, J. Biol. Chem., 2011, 286, 3007–3017 CrossRef CAS PubMed.
- C. Bier, S. K. Knauer, D. Docter, G. Schneider, O. H. Kramer and R. H. Stauber, Traffic, 2011, 12, 703–714 CrossRef CAS PubMed.
- C. Bier, R. Hecht, L. Kunst, S. Scheiding, D. Wunsch, D. Goesswein, G. Schneider, O. H. Kramer, S. K. Knauer and R. H. Stauber, PLoS One, 2012, 7, e34142 CrossRef CAS PubMed.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283, 30433–30437 CrossRef PubMed.
- J. S. Bond, J. Biol. Chem., 2019, 294, 1643–1651 CrossRef CAS.
- D. Wünsch, A. Hahlbrock, S. Jung, T. Schirmeister, J. v. d. Boom, O. Schilling, S. K. Knauer and R. H. Stauber, Oncogene, 2016, 35, 3351–3364 CrossRef PubMed.
- D. Y. Chen, H. Liu, S. Takeda, H.-C. Tu, S. Sasagawa, B. A. van Tine, D. Lu, E. H.-Y. Cheng and J. J.-D. Hsieh, Cancer Res., 2010, 70, 5358–5367 CrossRef CAS PubMed.
- J. J.-D. Hsieh, E. H.-Y. Cheng and S. J. Korsmeyer, Cell, 2003, 115, 293–303 CrossRef CAS PubMed.
- J. A. Khan, B. M. Dunn and L. Tong, Structure, 2005, 13, 1443–1452 CrossRef CAS PubMed.
- S. Takeda, D. Y. Chen, T. D. Westergard, J. K. Fisher, J. A. Rubens, S. Sasagawa, J. T. Kan, S. J. Korsmeyer, E. H.-Y. Cheng and J. J.-D. Hsieh, Genes Dev., 2006, 20, 2397–2409 CrossRef CAS PubMed.
- H. Zhou, S. Spicuglia, J. J.-D. Hsieh, D. J. Mitsiou, T. Høiby, G. J. C. Veenstra, S. J. Korsmeyer and H. G. Stunnenberg, Mol. Cell. Biol., 2006, 26, 2728–2735 CrossRef CAS PubMed.
- S. Takeda, S. Sasagawa, T. Oyama, A. C. Searleman, T. D. Westergard, E. H. Cheng and J. J. Hsieh, J. Clin. Invest., 2015, 125, 1203–1214 CrossRef PubMed.
- C. Schrenk, V. Fetz, C. Vallet, C. Heiselmayer, E. Schroder, A. Hensel, A. Hahlbrock, D. Wunsch, D. Goesswein, C. Bier, N. Habtemichael, G. Schneider, R. H. Stauber and S. K. Knauer, J. Mol. Cell Biol., 2018, 10, 33–47 CrossRef CAS PubMed.
- D. Y. Chen, Y. Lee, B. A. van Tine, A. C. Searleman, T. D. Westergard, H. Liu, H.-C. Tu, S. Takeda, Y. Dong, D. R. Piwnica-Worms, K. J. Oh, S. J. Korsmeyer, A. Hermone, R. Gussio, R. H. Shoemaker, E. H.-Y. Cheng and J. J.-D. Hsieh, Cancer Res., 2012, 72, 736–746 CrossRef CAS PubMed.
- S. Siemer, T. A. Bauer, P. Scholz, C. Breder, F. Fenaroli, G. Harms, D. Dietrich, J. Dietrich, C. Rosenauer, M. Barz, S. Becker, S. Strieth, C. Reinhardt, T. Fauth, J. Hagemann and R. H. Stauber, ACS Nano, 2021, 15, 18541–18556 CrossRef CAS PubMed.
- S. Siemer, T. Fauth, P. Scholz, Y. Al-Zamel, A. Khamis, D. Gül, L. Freudelsperger, B. Wollenberg, S. Becker, R. H. Stauber and J. Hagemann, Cancers, 2021, 13, 4831, DOI:10.3390/cancers13194831.
- J. van den Boom, A. Hensel, F. Trusch, A. Matena, S. Siemer, D. Guel, D. Docter, A. Höing, P. Bayer, R. H. Stauber and S. K. Knauer, Nanoscale, 2020, 12, 19093–19103 RSC.
- P. Pasch, A. Höing, S. Ueclue, M. Killa, J. Voskuhl, S. K. Knauer and L. Hartmann, Chem. Commun., 2021, 57, 3091–3094 RSC.
- J. van den Boom, F. Trusch, L. Hoppstock, C. Beuck and P. Bayer, PLoS One, 2016, 11, e0151431 CrossRef PubMed.
- N. Nagaratnam, S. L. Delker, R. Jernigan, T. E. Edwards, J. Snider, D. Thifault, D. Williams, B. L. Nannenga, M. Stofega, L. Sambucetti, J. J. Hsieh, A. J. Flint, P. Fromme and J. M. Martin-Garcia, Structure, 2021, 29, 873–885 CrossRef CAS PubMed.
- M. Giese, J. Niemeyer and J. Voskuhl, ChemPlusChem, 2020, 85, 985–997 CrossRef CAS PubMed.
- D. Aschmann, C. Vallet, S. K. Tripathi, Y. B. Ruiz-Blanco, M. Brabender, C. Schmuck, E. Sanchez-Garcia, S. K. Knauer and M. Giese, ChemBioChem, 2022, 23, e202100618 CrossRef CAS PubMed.
- L. Bartsch, M. Bartel, A. Gigante, J. Iglesias-Fernández, Y. B. Ruiz-Blanco, C. Beuck, J. Briels, N. Toetsch, P. Bayer, E. Sanchez-Garcia, C. Ottmann and C. Schmuck, ChemBioChem, 2019, 20, 2921–2926 CrossRef CAS PubMed.
- A. Gigante, E. Sijbesma, P. A. Sanchez-Murcia, X. Hu, D. Bier, S. Backer, S. Knauer, F. Gago, C. Ottmann and C. Schmuck, Angew. Chem., Int. Ed., 2020, 59, 5284–5287 CrossRef CAS.
- D. Maity, A. Gigante, P. A. Sanchez-Murcia, E. Sijbesma, M. Li, D. Bier, S. Mosel, S. Knauer, C. Ottmann and C. Schmuck, Org. Biomol. Chem., 2019, 17, 4359–4363 RSC.
- N. Rafieiolhosseini, M. Killa, T. Neumann, N. Tötsch, J.-N. Grad, A. Höing, T. Dirksmeyer, J. Niemeyer, C. Ottmann, S. K. Knauer, M. Giese, J. Voskuhl and D. Hoffmann, Beilstein J. Org. Chem., 2022, 18, 1322–1331 CrossRef CAS PubMed.
- T. Dirksmeyer, P. Stahl, C. Vallet, S. Knauer, M. Giese, C. Schmuck and C. Hirschhauser, Chem. –Eur. J., 2022, 28, e202104618 CrossRef CAS PubMed.
- X. Y. Hu, M. Ehlers, T. Wang, E. Zellermann, S. Mosel, H. Jiang, J. E. Ostwaldt, S. K. Knauer, L. Wang and C. Schmuck, Chem. –Eur. J., 2018, 24, 9754–9759 CrossRef CAS PubMed.
- P. Jana, K. Samanta, S. Bäcker, E. Zellermann, S. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2017, 56, 8083–8088 CrossRef CAS PubMed.
- H. Jiang, X. Y. Hu, S. Mosel, S. K. Knauer, C. Hirschhauser and C. Schmuck, ChemBioChem, 2019, 20, 1410–1416 CrossRef CAS.
- S. Junghanel, S. Karczewski, S. Backer, S. K. Knauer and C. Schmuck, ChemBioChem, 2017, 18, 2268–2279 CrossRef.
- H. Y. Kuchelmeister, A. Gutschmidt, S. Tillmann, S. Knauer and C. Schmuck, Chem. Sci., 2012, 3, 996–1002 RSC.
- H. Y. Kuchelmeister, S. Karczewski, A. Gutschmidt, S. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2013, 52, 14016–14020 CrossRef CAS PubMed.
- M. Li, M. Ehlers, S. Schlesiger, E. Zellermann, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2016, 55, 598–601 CrossRef CAS PubMed.
- M. Li, S. Schlesiger, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2015, 54, 2941–2944 CrossRef CAS PubMed.
- K. Samanta, P. Jana, S. Backer, S. Knauer and C. Schmuck, Chem. Commun., 2016, 52, 12446–12449 RSC.
- A. Höing, A. Zimmermann, L. Moews, M. Killa, M. Heimann, A. Hensel, J. Voskuhl and S. K. Knauer, ChemMedChem, 2022, 17, e202100640 CrossRef.
- M. Fokkens, T. Schrader and F. G. Klarner, J. Am. Chem. Soc., 2005, 127, 14415–14421 CrossRef CAS.
- A. Höing, A. Kirupakaran, C. Beuck, M. Pörschke, F. C. Niemeyer, T. Seiler, L. Hartmann, P. Bayer, T. Schrader and S. K. Knauer, Biomacromolecules, 2022, 23, 4504–4518 CrossRef.
- F. Octa-Smolin, R. Mitra, M. Thiele, C. G. Daniliuc, L. Stegemann, C. Strassert and J. Niemeyer, Chem. –Eur. J., 2017, 23, 10058–10067 CrossRef CAS PubMed.
- F. Octa-Smolin, F. van der Vight, R. Yadav, J. Bhangu, K. Soloviova, C. Wolper, C. G. Daniliuc, C. A. Strassert, H. Somnitz, G. Jansen and J. Niemeyer, J. Org. Chem., 2018, 83, 14568–14587 CrossRef CAS PubMed.
- R. Yadav, C. Kwamen and J. Niemeyer, Isr. J. Chem., 2021, 61, 231–233 CrossRef CAS.
- F. Octa-Smolin, M. Thiele, R. Yadav, A. Platzek, G. H. Clever and J. Niemeyer, Org. Lett., 2018, 20, 6153–6156 CrossRef CAS PubMed.
- M. K. Goshisht, G. K. Patra and N. Tripathi, Mater. Adv., 2022, 3, 2612–2669 RSC.
- M. K. Goshisht and N. Tripathi, J. Mat. Chem. C, 2021, 9, 9820–9850 RSC.
- J. T. Lee, D. Y. Chen, Z. Yang, A. D. Ramos, J. J.-D. Hsieh and M. Bogyo, Bioorg. Med. Chem. Lett., 2009, 19, 5086–5090 CrossRef CAS PubMed.
- S. K. Knauer, W. Mann and R. H. Stauber, Cell Cycle, 2007, 6, 518–521 CrossRef CAS.
- S. B. van der Meer, I. Hadrovic, A. Meiners, K. Loza, M. Heggen, S. K. Knauer, P. Bayer, T. Schrader, C. Beuck and M. Epple, J. Phys. Chem. B, 2021, 125, 115–127 CrossRef CAS PubMed.
- A. Brandl, T. Wagner, K. M. Uhlig, S. K. Knauer, R. H. Stauber, F. Melchior, G. Schneider, T. Heinzel and O. H. Kramer, J. Mol. Cell Biol., 2012, 4, 284–293 CrossRef CAS PubMed.
- S. K. Knauer and R. H. Stauber, Anal. Chem., 2005, 77, 4815–4820 CrossRef CAS PubMed.
- A. Schweitzer, S. K. Knauer and R. H. Stauber, Int. J. Cancer, 2010, 126, 801–809 CAS.
- A. Lange, L. M. McLane, R. E. Mills, S. E. Devine and A. H. Corbett, Traffic, 2010, 11, 311–323 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical and biological assays, computational studies and microscopy, supporting figures and tables. See DOI: https://doi.org/10.1039/d2ra06019a |
| This journal is © The Royal Society of Chemistry 2022 |