
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effects of Ni–Fe alloy catalysts on dry reforming of methane at low temperatures in an electric field†
Ayaka Motomuraa,
Yuki Nakaya b,
Clarence Sampsona,
Takuma Higoa,
Maki Torimotoa,
Hideaki Tsunekia,
Shinya Furukawab and
Yasushi Sekine*a
b,
Clarence Sampsona,
Takuma Higoa,
Maki Torimotoa,
Hideaki Tsunekia,
Shinya Furukawab and
Yasushi Sekine*a
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo, 169-8555, Japan. E-mail: ysekine@waseda.jp
bInstitute for Catalysts, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, 001-0021, Japan
First published on 5th October 2022
Abstract
Dry reforming of methane (DRM) is a promising reaction able to convert greenhouse gases (CO2 and CH4) into syngas: an important chemical feedstock. Several difficulties limit the applicability of DRM in conventional thermal catalytic reactions; it is an endothermic reaction that requires high temperatures, resulting in high carbon deposition and a low H2/CO ratio. Catalysis with the application of an electric field (EF) at low temperatures can resolve these difficulties. Synergistic effects with alloys have also been reported for reactions promoted by the application of EF. Therefore, the synergistic effects of low-temperature DRM and Ni–Fe bimetallic catalysts were investigated using various methods and several characterisations (XRD, XPS, FE-STEM, etc.), which revealed that Ni–Fe binary catalysts show high performance in low-temperature DRM. In particular, the Ni0.8Fe0.2 catalyst supported on CeO2 was found to carry out DRM in EF effectively and selectively by virtue of its bimetallic characteristics.
1. Introduction
In recent years, environmental burdens such as global warming caused by greenhouse gases and extreme weather events have become important issues, and their resolution is essential for the long-term well-being of humankind. Therefore, attention is focused on reducing CO2 and CH4 emissions, which have a high environmental impact, and on the effective use of these resources.1 Dry reforming of methane (DRM, as shown in eqn (1)) is an environmentally favorable reaction that can convert two greenhouse gases into syngas: a gas mixture of H2 and CO which is a raw material for various industrial processes.| CH4 + CO2 → 2H2 + 2CO ΔH0298 = 247.2 kJ mol−1 | (1) |
In addition, DRM can directly convert biogas resulting from methane fermentation from renewable energy sources.2–5 For these benefits, DRM has attracted significant attention in recent years. However, due to its high endothermicity, DRM requires temperatures higher than 800 K.6 Moreover, this reaction is well-known to deposit great amounts of carbon on the catalyst. It causes deactivation of catalyst and blockage of reactor, especially on base metal catalysts such as Ni, which are industrially desirable by virtue of their low cost and catalytic activity. Carbon deposition occurs most frequently at temperatures between 800 and 1000 K through methane decomposition and CO disproportionation.7,8 Consequently, conventional thermal catalytic processes for DRM have been conducted at temperatures higher than 1000 K. Better handling features for low-temperature reactions along with less carbon deposition, are highly anticipated.
A possible approach is the application of an electric field (EF) to a semiconductor-based catalyst. Compared with the conventional thermal processes, EF-assisted process can promote DRM even at low temperatures under 500 K. Notably, the assistance of EF can alter the reaction mechanism differs from the conventionals,9–11 thereby enabling the enhancement of DRM activity even at low temperatures. The reaction mechanism of DRM with EF has already been clarified in the previous reports, which showed hopping proton on the catalyst surface via adsorbed water or hydroxyl groups (the Grotthuss mechanism) in collision with physisorbed CH4 at the metal–support interface. Simultaneously, adsorbed CO2 reacts with lattice oxygen defects that are produced by reaction between physisorbed CH4 and lattice oxygen.11 This specific mechanism enables activation of CH4 and CO2 molecules, even at low temperatures. Furthermore, because reactions proceed at temperatures less than 500 K, carbon deposition is suppressed drastically. Inhibition of the side reaction, reverse water gas shift (RWGS, as shown in eqn (2)), causes production with a high H2/CO ratio.
| CO2 + H2 → CO + H2O ΔH0298 = 41.2 kJ mol−1 | (2) |
One widely used method of decreasing carbon deposition amounts is the use of alloy catalysts.7,12,13 Two major effects of alloy catalysts are ligand and ensemble effects.14–17 Ligand effects, which lead to change of the surface electronic state because of electronic contributions from other surface atoms, affect catalysis activity and selectivity. Ensemble effects occur by the dilution of active metal ensembles. In general, dilution of active metal ensembles typically lowers the bond breaking ability of the active metal, thus suppressing carbon deposition in DRM,18,19 whereas decreasing the catalytic activity at the same time. Therefore, applications of alloys are limited because of the tradeoff relationship.
A promising strategy to overcome this challenge is the combination of alloying effects and the use of an electric field.20,21 Large active metal ensembles are not required for reaction with the electric field because it proceeds at the metal–support interface. Furthermore, with increasing dilution, catalytic activity increased by virtue of enhancement of the electron density of the active metal by charge transfer from the dilution metal. For this study, we specifically examined the synergistic effects between alloy catalysts and reactions with the electric field on DRM to develop a highly active catalytic process with less carbon deposition.
2. Experimental
2.1 Catalyst preparation
Catalyst samples used for this study were prepared by a deposition precipitation method using urea and CeO2(JRC-CEO-1: 156.9 m2 g−1) as a support. Also, Ni(NO3)2·6H2O, Co(NO3)2·6H2O, Fe(NO3)3·9H2O, and Zn(NO3)2·6H2O were used as metal precursors and were mixed to obtain the following Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M molar ratios of 4:1 (M = Co, Fe, Zn), 9:1 (M = Fe) and 7:3 (M = Fe). Every catalyst was prepared as containing 10 wt% metal in total. First, metal precursors were dissolved in distilled water. Then the urea solution (5 times the molar amount of metal) was added dropwise into the solution. After that, it was stirred at 368 K overnight. Then, the precipitate was filtered and washed with distilled water, and dried at 393 K for 6 h. Finally, it was calcined at 773 K for 1 h and reduced at 873 K for 1 h (H2:Ar = 1:4, total flow rate: 100 mL min−1).
:M molar ratios of 4:1 (M = Co, Fe, Zn), 9:1 (M = Fe) and 7:3 (M = Fe). Every catalyst was prepared as containing 10 wt% metal in total. First, metal precursors were dissolved in distilled water. Then the urea solution (5 times the molar amount of metal) was added dropwise into the solution. After that, it was stirred at 368 K overnight. Then, the precipitate was filtered and washed with distilled water, and dried at 393 K for 6 h. Finally, it was calcined at 773 K for 1 h and reduced at 873 K for 1 h (H2:Ar = 1:4, total flow rate: 100 mL min−1).
2.2 Catalytic activity test
Catalytic activity tests were conducted using a fixed-bed flow reactor at atmospheric pressure. A quartz tube (8.0 mm o.d., 6.0 mm i.d.) was used as the reactor tube. Catalyst of 100 mg was used for each activity test. Two stainless steel electrodes (2 mmϕ) were inserted into the reactor as contacting the top and bottom sides of the catalyst bed. Direct current (DC) was applied through these electrodes. The response voltage was recorded using a digital phosphor oscilloscope. A thermocouple was inserted to monitor the actual catalyst bed temperature. Reaction pre-treatment was conducted before each test at 673 K for 30 min (H2:Ar = 1:4 total flow, rate: 100 mL min−1). The reaction gas composition was CH4:CO2:Ar = 1:1:1 (total flow rate: 30 mL min−1) for activity tests and CH4:CO2:Ar = 1:1:6 (total flow rate: 80 mL min−1) for temperature-changing tests. The catalyst bed temperature was controlled to be at 673 and 473 K for reactions with and without EF. The product gases were analysed using GC-TCD and GC-FID with a methanizer. Reaction rate of CH4, CO2 and H2/CO ratio were calculated according to the following equations.
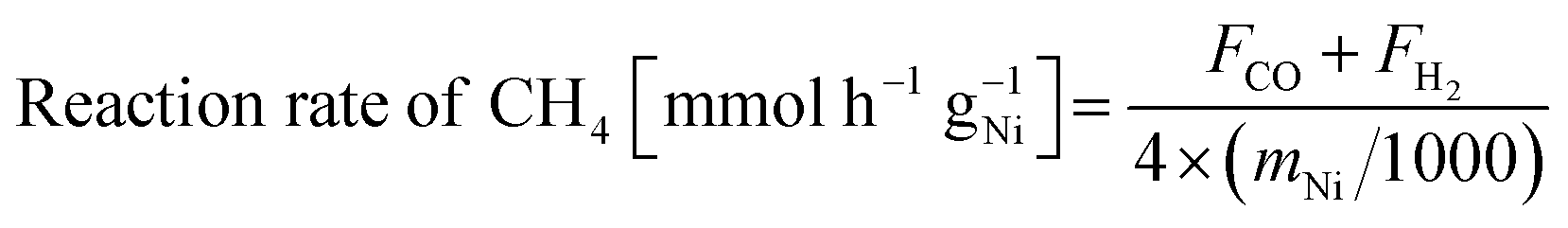 | (3) |
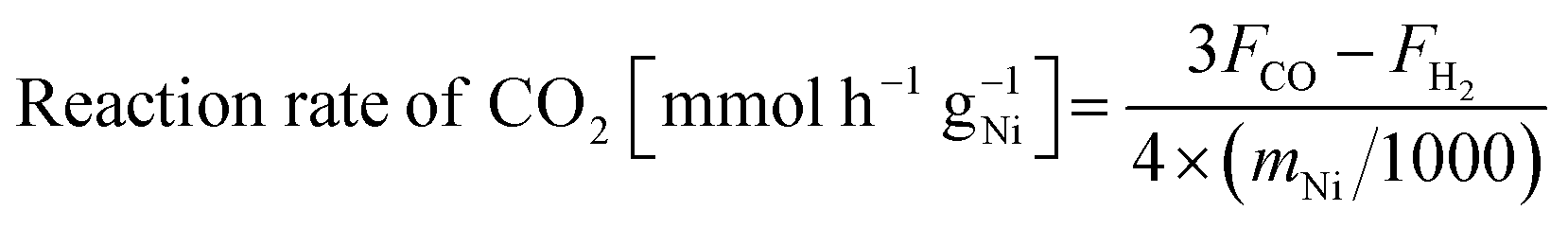 | (4) |
| H2/CO[−] = FH2/FCO | (5) |
2.3 Characterizations
For characterization of the catalysts, we used powder X-ray diffraction (XRD, SmartLab III; Rigaku Corp.) at 40 kV and 40 mA with Cu Kα radiation to measure each catalyst structure.The structure of catalysts was observed using field emission scanning transmission electron microscopy (FE-STEM; JEM-2100F; JEOL) at 200 kV. Energy dispersive X-ray spectroscopy (EDS) mapping was performed for all samples to observe the distribution state of the elements.
The diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs) spectra of adsorbed CO were measured by an FT-IR spectrophotometer (FT/IR-6200; Jasco Corp.) equipped with a ZnSe window and MCT-M detector cooled with liquid nitrogen. Spectra were recorded with 2 cm−1 resolution and 25 times of scans. Before measurement, the powder catalyst (ca. 80 mg) was reduced at 673 K for 30 min (H2:Ar = 1:4, total flow rate: 50 mL min−1). The catalyst was purged with Ar for 30 min and exposed to CO:Ar = 1:199 (total flow rate: 50 mL min−1) at 323 K.
The electronic state of Ni over catalyst was investigated using X-ray photoelectron spectroscopy (XPS, VersaProbe 2; Ulvac-Phi Inc.). The measurements were taken with an Al Kα X-ray source. For the fresh Ni0.8Fe0.2/CeO2 catalyst, the sample was reduced at 673 K. For the spent Ni0.8Fe0.2/CeO2 catalyst, the catalyst was operated in the DRM condition for 2 h at 473 K with applying 10 mA of EF. Afterwards, the catalysts were loaded on a carbon tape using a transfer vessel, and transferred into the spectrometer from a glove box. The obtained binding energies were calibrated with the Si 2s emission (154.8 eV) of the internal standard SiO2 (JRC–SIO–7). The distributions of Ni0, Ni2+, and the satellite peak were estimated by the respective peak areas of 2p3/2.
The carbon amount deposited on the spent catalyst was measured using temperature programmed oxidation (TPO, O2:Ar = 1:9, total flow rate: 100 mL min−1) with an infrared analyser (CGT-7100; Shimadzu Corp.).
3. Results and discussion
3.1 Structural analysis of Ni1−xFex/CeO2
A series of Ni1−xFex/CeO2 (x = 0, 0.1, 0.2, and 0.3) were prepared by a deposition precipitation method using CeO2 as a support. Fig. 1a and b shows the elemental mappings, acquired by EDX analysis, of the representative Ni0.8Fe0.2/CeO2 catalyst, which showing that Ni and Fe co-existed in each nanoparticle. Further, no segregation of Ni and Fe phase was observed. Fig. 1c shows the powder XRD patterns of the Ni/CeO2 and Ni0.8Fe0.2/CeO2 catalysts. Upon the introduction of Fe, the diffraction angles, which can be assigned to fcc-type structure, were shifted to lower values, indicating the alloying Ni with Fe.22,23 The surfaces of the Ni and Ni0.8Fe0.2 catalysts were analysed by DRIFTs measurement using CO as a probe molecule (Fig. 1d). Apparently, a peak assigned to linearly adsorbed CO on Ni atoms was shifted from 2051 cm−1 to 2038 cm−1 upon the formation of Ni–Fe alloys, demonstrating the electron transfer from Fe to Ni, as previously reported in other Ni–Fe systems.24 We also prepared the Ni0.8Co0.2/CeO2 and Ni0.8Zn0.2/CeO2 catalysts and ascertained their alloy structures (see Fig. S1 for the corresponding XRD patterns†).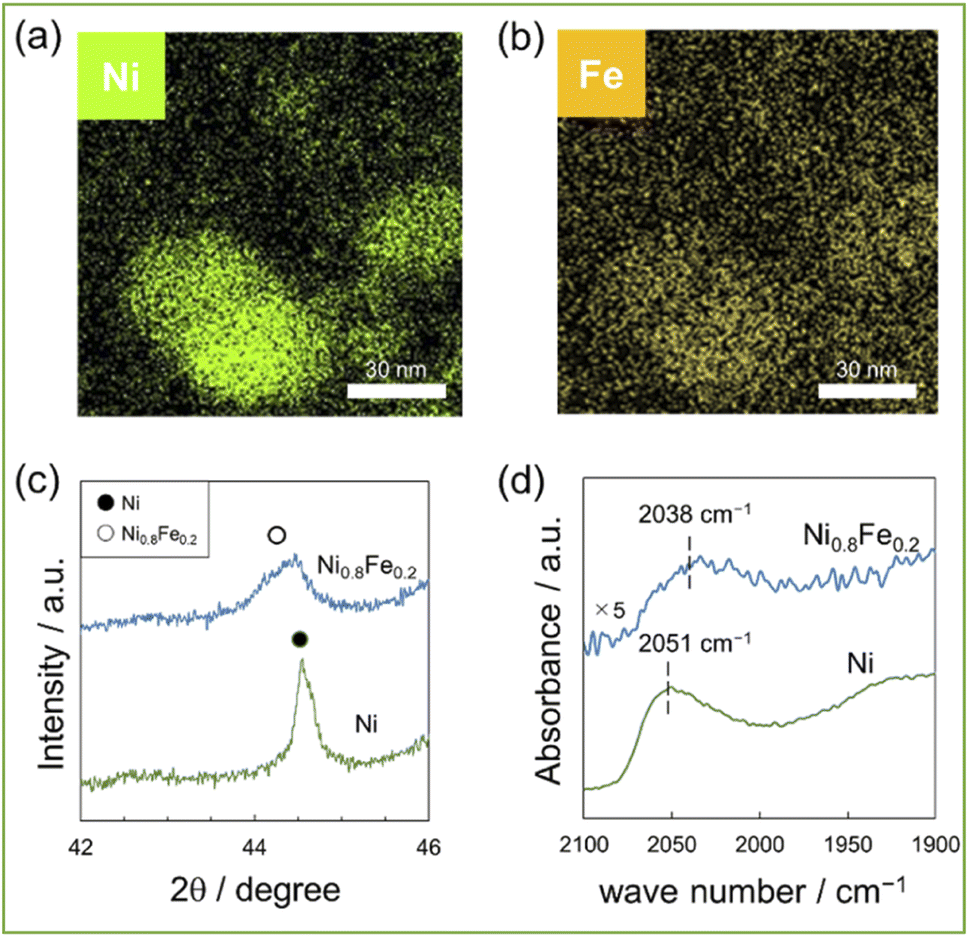 | ||
| Fig. 1 (a) and (b) EDX mappings for 10wt%(Ni0.8Fe0.2)/CeO2, (c) XRD patterns of 10wt%Ni, (Ni0.8Fe0.2)/CeO2, (d)DRIFTs spectra of CO adsorbed on 10wt%Ni, (Ni0.8Fe0.2)/CeO2. | ||
3.2 Catalytic activity
Fig. 2a shows the catalytic performance of Ni0.8M0.2/CeO2 catalysts (M = Fe, Co, and Zn) with EF and without EF (i.e. conventional thermal catalysis) at 673 K. The catalytic performance at 673 K retained unchanged regardless of EF addition. This similarity of activity might be attributable to the reduced number of adsorbed OH at higher temperatures, which suppresses surface proton conduction.25 So these two reaction systems work with the conventional thermal reaction, i.e. by the Langmuir–Hinshelwood mechanism. The addition of second metals reduced the amounts of accumulated coke (as shown in Table S1†), while reducing their catalytic activities. It is well-known that the dilution of the Ni–Ni ensembles by the addition of second metals results in the coke deposition at the sacrifice of catalytic activity.26,27 Therefore, the conventional catalysis inevitably compromises on this trade-off relationship. Besides, the H2/CO ratios of all catalysts were below 0.5, indicating the considerable contribution of RWGS at this high reaction temperature (673 K). However, the catalytic performance at 473 K considerably differs from those at 673 K.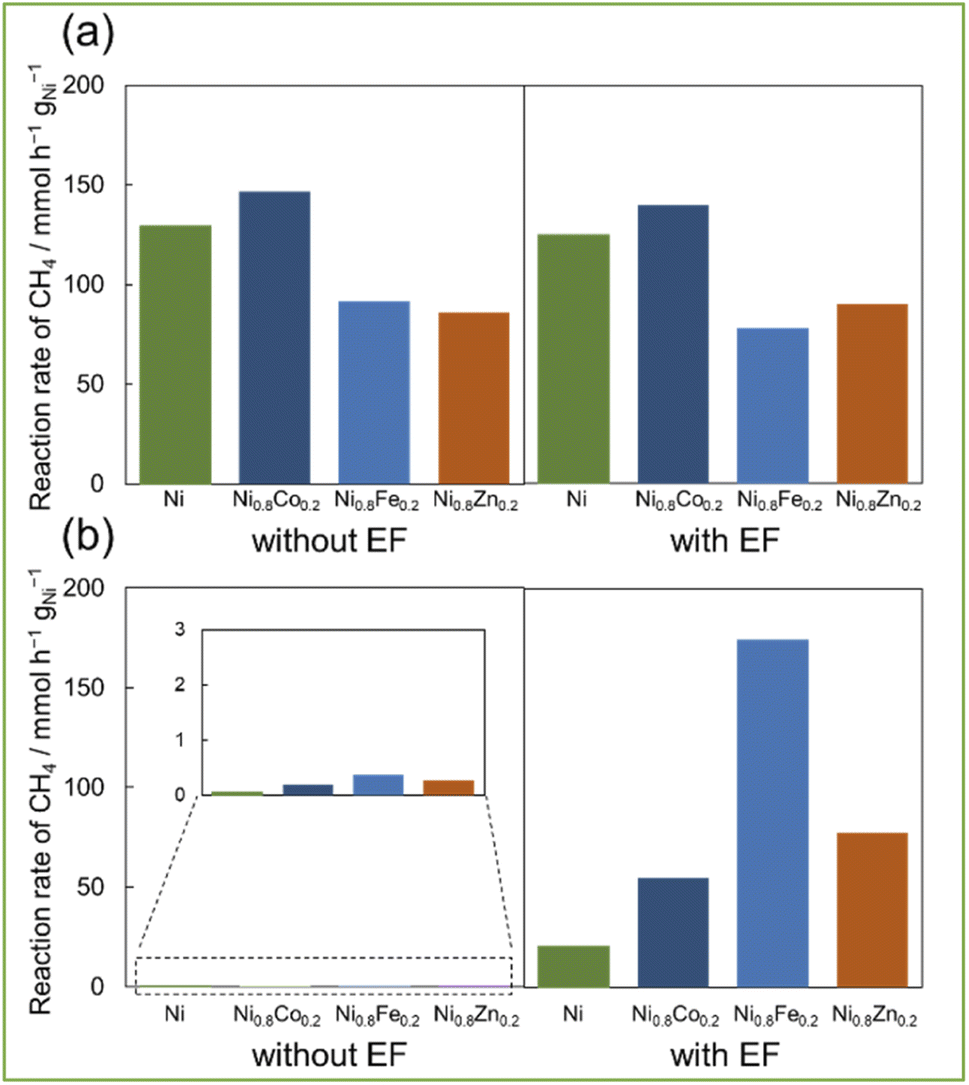 | ||
| Fig. 2 Catalytic activities of 10wt%Ni, (Ni0.8M0.2)/CeO2 (M = Co, Fe, Zn) (a) 673 K without EF and with EF (10 mA) and (b) 473 K without EF and with EF (10 mA); 30 SCCM total flow rate (CH4:CO2:Ar = 1:1:1). | ||
Surprisingly, the catalytic activities of Ni0.8M0.2/CeO2 resulted in the drastic increase by applying EF at low temperature (473 K) (Fig. 2b). All catalysts showed negligible catalytic activities in the case of the conventional thermal catalysis (Fig. 2b: left), whereas yielding much higher catalytic activities with EF assistance and all bimetallic catalysts showed higher activity than the monometallic Ni catalyst. Among them, Ni0.8Fe0.2/CeO2 exhibited the highest catalytic activity with EF, which was 8 times higher than the monometallic Ni/CeO2. The order of catalytic activities is as follows: Ni < Ni0.8Co0.2 < Ni0.8Zn0.2 ≪ Ni0.8Fe0.2. Compared with the thermal catalysis, EF-assisted catalysis resulted in the increased catalytic activity upon the alloy formation. Okada et al.28 reported that the methane dissociation mechanism, the rate-limiting step of DRM, proceeds through the formation of an intermediate [CH3–H–H]+ by colliding a proton with physisorbed CH4. It is therefore considered that the increased electron density of the active metal contributes to stabilize the intermediates, resulting in the stable formation of certain intermediates. So, it is assumed that electronic state of Ni is a dominant factor in DRM by EF. Thus, because of ligand effect, the existence of electron-enriched Ni (as presented in Fig. 1d) in Ni0.8Fe0.2 alloy catalyst contributed high activity. Notably, the H2/CO ratios were greater than 0.6 which is much higher than the thermal reaction. In addition, alloying also suppressed the coke deposition (as shown in Table S1†) by the ensemble effect. Considering the previous studies, the advantage of low carbon deposition on the catalyst in the current EF-assisted reaction is not only due to the ensemble effect of the alloy and the fact that the reaction is proceeding at low temperature, but also the oxidation of carbon by lattice oxygen is thought to contribute at the same time. Ogo et al.29 showed that the application of EF reduced Ce4+ to Ce3+ and facilitated the release of lattice oxygen on the catalyst support. Thus, this easy release of lattice oxygen at low temperatures is presumably responsible for the carbon suppression. These results showed that applying EF greatly enhance the DRM catalysis at even low temperature of 473 K and coke suppression.
Based on these results, Fe was selected as the most suitable second metal among the three metals in the EF-assisted DRM for additional investigation. So, we tested the catalytic performance of Ni–Fe alloys with different compositions (Fig. 3, XRD for these catalysts are shown in ESI Fig. S2†). Without EF at 673 K (Fig. 3a), catalytic activities decreased concomitantly with the increasing Fe ratio. Furthermore, the H2/CO ratio decreased corresponding with the increasing Fe ratio. On the other hand, activities with EF at 473 K (Fig. 3b) increased along with the Fe ratio up to Ni0.8Fe0.2. Additionally, because of the ensemble effect, carbon deposition amount decreased along with increasing Fe ratio while there was no decrease in activity (ESI Table S1(b)†). So, it can be said that the synergy between alloy catalysts and applying EF enables to achieve both suppression of side reactions and high activity. In fact, when the Ni/Fe ratio is greater than 0.8/0.2 (i.e., Ni0.7Fe0.3), it showed much lower activity because Ni and Fe are not uniformly mixed (Fig. S3 in ESI†). The catalyst of Ni0.8Fe0.2/CeO2 showed the highest reaction rate of CH4 at 473 K with EF, 174.4 mmol h−1 gNi−1 (Table S1(b) in ESI†), and the highest conversion of CH4; 6.0% and of CO2; 6.5%, note that these reactions were conducted in a kinetic region (i.e. high space velocity). These activity tests showed that both high activity and less carbon deposition with Ni0.8Fe0.2 catalyst improved by application of EF, even at low temperatures.
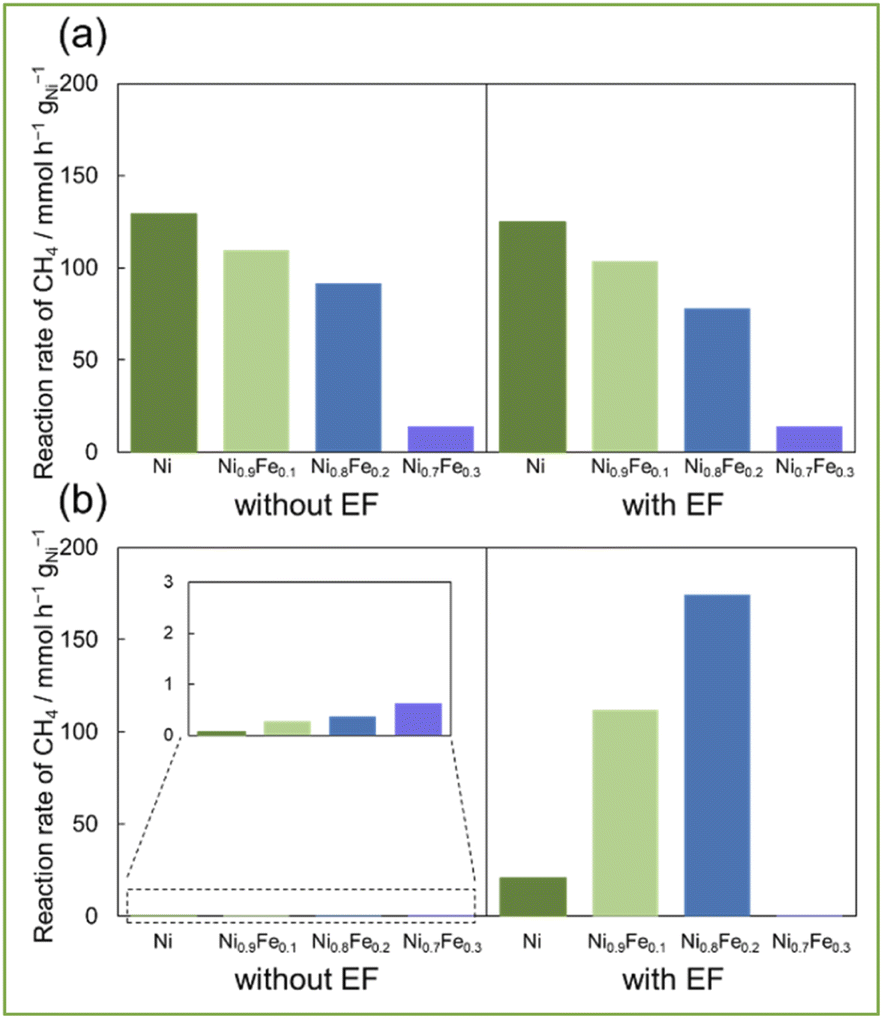 | ||
| Fig. 3 Catalytic activities of 10wt%Ni, (Ni1−xFex)/CeO2 (x = 0.1, 0.2, 0.3) (a) 673 K without EF and with EF (10 mA) and (b) 473 K without EF and with EF (10 mA); 30 SCCM total flow rate (CH4:CO2:Ar = 1:1:1). | ||
3.3 Structural comparison between before and after activity tests of Ni, Ni0.8Fe0.2/CeO2
To compare the structure of catalysts between before and after activity tests, some characterizations were performed: Fig. 4a and b show the elemental distribution using EDX mapping of Ni0.8Fe0.2/CeO2 after activity tests with EF; Fig. 4c shows XRD patterns of Ni, Ni0.8Fe0.2/CeO2, which are as made and after activity tests with EF one; and Fig. 4d shows XPS spectra of Ni0.8Fe0.2/CeO2, which are as made and after activity test with EF one. Fig. 4a and b show the distribution of Ni and Fe was consistent and no Ni and Fe segregation was occurred through the activity test with EF. Additionally, in Fig. 4c, the peak assigned to Ni–Fe alloy was observed in XRD patterns of Ni0.8Fe0.2/CeO2 after the activity test with EF. Also, each catalyst after activity test with other conditions maintained its original structure. Fig. 4d shows the electronic state of Ni in Ni0.8Fe0.2 after the reaction, and the structure was unchanged compared to the as-made catalyst. These results show alloying Ni with Fe and the electronic state of Ni were remained, even applying EF for 2 h.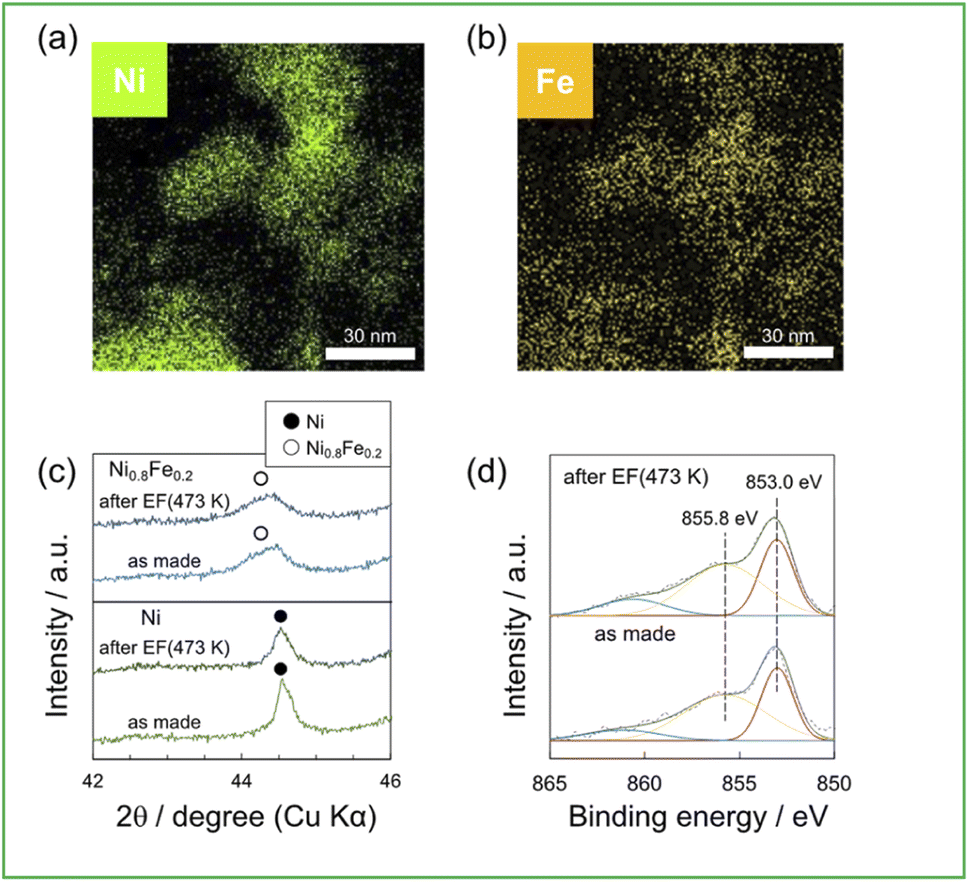 | ||
| Fig. 4 (a) and (b) EDX mappings for 10wt%(Ni0.8Fe0.2)/CeO2 after activity test with EF, (c) XRD patterns of 10wt%Ni, (Ni0.8Fe0.2)/CeO2 (as made and after activity test with EF), (d) XPS spectra of 10wt%(Ni0.8Fe0.2)/CeO2 (as made and after activity test with EF). | ||
4. Conclusion
An Ni–Fe alloy catalyst showed excellent performance for methane dry reforming in an electric field, even at temperatures as low as 473 K, at which the conventional thermal reaction showed negligible low activity. Because of ensemble effects, the alloy catalyst showed reduced activity and less carbon deposition in the reaction with conventional heating. For the EF-assisted catalysis at low temperatures, the catalyst was able to achieve high activity with low carbon deposition by virtue of the influence of surface ionics without decreasing activity thanks to the ensemble effect. Thus, it was confirmed that Ni0.8Fe0.2/CeO2 can improve its activity by applying EF without expressing the disadvantage of alloy catalysts. Investigations into the Fe substitution ratio indicated that the activity increased to 20% substitution, but at higher iron substitutions, the activity decreased because of non-uniform bi-metal formation. The alloying of iron and nickel is considered to have affected the electronic state of nickel, which led to high performance. These results, produced by the synergy between the alloy catalyst and the application of the electric field, are expected to lead to the use of widely various applications.Conflicts of interest
The authors have no conflict to declare.Acknowledgements
This work was the result of using research equipment (JEM-2100F and Versa Probe II: Material Characterization Central Laboratory in Waseda University) shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for sup-porting construction of core facilities) Grant Number JPMXS0440500021. This work was supported by JSPS KAKENHI Grant Number 19K22090. The authors thank S. Enomoto and N. Hanzawa at Kagami Memorial Research Institute for Materials Science and Technology, Waseda University, for TEM observation.References
- IPCC, AR6 WG1, 2021 Search PubMed.
- M. F. Demirbas, M. Balat and H. Balat, Energy Convers. Manage., 2011, 52, 1815–1828 CrossRef.
- N. J. Themelis and P. A. Ulloa, Renewable Energy, 2007, 32, 1243–1257 CrossRef CAS.
- J. M. Lavoie, Front. Chem., 2014, 2, 81 Search PubMed.
- R. Parsapur, S. Chatterjee and K. Huang, ACS Energy Lett., 2020, 5, 2881–2885 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- J. Zhang, H. Wang and A. K. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668–108688 RSC.
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, Fuel Process. Technol., 2017, 158, 96–103 CrossRef CAS.
- T. Yabe, K. Yamada, K. Murakami, K. Toko, K. Ito, T. Higo, S. Ogo and Y. Sekine, ACS Sustainable Chem. Eng., 2019, 7, 5690–5697 CrossRef CAS.
- N. Nakano, M. Torimoto, H. Sampei, R. Yamashita, R. Yamano, K. Saegusa, A. Motomura, K. Nagakawa, H. Tsuneki, S. Ogo and Y. Sekine, RSC Adv., 2022, 12, 9036–9043 RSC.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Coperet and C. Müller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS PubMed.
- M. Torimoto and Y. Sekine, Catal. Sci. Technol., 2022, 12, 3387–3411 RSC.
- J. W. A. Sachtler and G. A. Somorjai, J. Catal., 1983, 81, 77–94 CrossRef CAS.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
- S. Sakong, C. Mosch and A. Groß, Phys. Chem. Chem. Phys., 2007, 9, 2216–2225 RSC.
- P. Liu and J. K. Nørskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 RSC.
- J. R. Rostrup-Nielsen, J. Catal., 1984, 85, 31–43 CrossRef CAS.
- T. Osaki and T. Mori, J. Catal., 2001, 204, 89–97 CrossRef CAS.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J. G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
- J. Zhang, R. Ma, H. Ham, K. Shimizu and S. Furukawa, JACS Au, 2021, 1, 1688–1693 CrossRef CAS PubMed.
- L. Wang, D. Li, M. Koike, S. Koso, Y. Nakagawa, Y. Xu and K. Tomishige, Appl. Catal., A, 2011, 392, 248–255 CrossRef CAS.
- J. Ashok and S. Kawi, ACS Catal., 2014, 4, 289–301 CrossRef CAS.
- D. Li, M. Koike, L. Wang, Y. Nakagawa, Y. Xu and K. Tomishige, ChemSusChem, 2014, 7, 510–522 CrossRef CAS PubMed.
- K. Murakami, Y. Tanaka, R. Sakai, Y. Hisai, S. Hayashi, Y. Mizutani, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki and Y. Sekine, Chem. Commun., 2020, 56, 3365–3368 RSC.
- K. Nagaoka, K. Takanabe and K. Aika, Appl. Catal., A, 2004, 268, 151–158 CrossRef CAS.
- G. Valderrama, A. Kiennemann and M. R. Goldwasser, J. Power Sources, 2010, 195, 1765–1771 CrossRef CAS.
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS.
- S. Ogo, H. Nakatsubo, K. Iwasaki, A. Sato, K. Murakami, T. Yabe, A. Ishikawa, H. Nakai and Y. Sekine, J. Phys. Chem. C, 2018, 122, 2089–2096 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05946k |
| This journal is © The Royal Society of Chemistry 2022 |