DOI:
10.1039/D2RA05878B
(Paper)
RSC Adv., 2022,
12, 32652-32658
Room temperature synthesis of pillared-layer metal–organic frameworks (MOFs)
Received
17th September 2022
, Accepted 31st October 2022
First published on 14th November 2022
Abstract
A pillared-layer MOF, {[Cu2(Fu)2(BPY)]·H2O}n, was synthesized at room temperature employing a mixture of water and methanol as the reaction solvent. By using the sodium salts of fumarate, instead of the native acids, the target MOF could be readily prepared in aqueous media. The PXRD of this MOF was found to be in good agreement with the simulated diffractogram from single crystal data of related MOF made at a higher temperature using DMF as the solvent. In addition to characterization by PXRD, TGA, and ESEM, N2-sorption measurements revealed the formation of mesopores in the MOF's structure under study. The electronic band gap and magnetic properties of this pillared-layer MOF were also studied.
Introduction
Metal–organic frameworks (MOFs) denote an attractive class of highly porous materials that have found applications in many varied directions.1,2 The strategy and assembly of metal–organic frameworks (MOFs) are currently of great interest in the fields of coordination chemistry and crystal engineering because they have an interesting variety of topologies, attractive structures, and probable uses.3 In general, the assembly of metal–organic depends on two primary constituents, namely the metal centers, including metal ions, metal clusters, or secondary building units (SBUs), which act as connectors, and the organic ligands, which act as linkers. The remarkable topographies of the connectors and linkers are the number and orientation of the interacting sites.4
Different disciplines such as chemistry, materials science, medicine, and environmental science have shown strong interest in the study and development of MOFs so far. However, most applications of MOFs, including gas separation,5 gas storage,6 chemical sensing,7 catalysis,8 and drug delivery,9–11 are based on exploiting the porosity of materials. Several recent studies have underscored the importance of the framework architecture itself, including the possibility to utilize the structural reinforcement of nitrogen-rich MOFs as energetic materials12 or the ability of MOFs to act as supports for reactive complexes.13,14
Based on the structure of the organic linkers and coordination modes of the metal cations or SBUs, MOF structures can be designed according to targeted properties. Research outputs have shown that metal–organic frameworks have ultra-high porosity and high internal surface areas.15 One class of frameworks that has proven to be particularly popular is pillared-layered MOFs. Such materials are constructed from infinite layers pillared by linear bidentate linkers through dative bonds or supramolecular interactions. Such a topology has greatly advanced recently because of the combined use of bipyridine and polycarboxylate, which allows a high degree of variation and hence the ability to design frameworks and their pores.16 Thus, a variety of different bipyridyl and dicarboxylate ligands have been used in combination with different metal centers, commonly using a bimetallic paddlewheel SBU.17–19 Pillared-layer MOFs have been prepared in many ways typically using solvents with high toxicity and at elevated temperatures. Mechanochemical studies have been done using only small quantities of solvents,20,21 including the demonstration of the synthesis of a pillared MOF that had previously been prepared in DMF at high temperatures.
In this study, we show a simple and sustainable route to the synthesis and characterization of a Cu-pillared MOF. We show that copper(II) nitrate can be reacted with 4, 4′-bipyridine pillar and sodium fumarate (Na2Fu) using a mixture of water and methanol as the solvent rather than DMF. Comparisons of the powder X-ray diffraction (PXRD) patterns of this MOF were made with the pattern calculated from the literature example of analogous MOF prepared in DMF at a higher temperature showing a good match.19,22 Overall, we show a cleaner way to synthesize pillared layer MOFs.
Experimental
Characterization
The synthesized MOF was characterized by PXRD (PANalytical MPD) with Cu Kα radiation (λ = 1.54178 Å) at a scanning rate of 0.001557 s−1. Nitrogen adsorption–desorption isotherm was measured using a Micromeritics-Smart Vac Prep System with a Micromeritics 3Flex Surface Characterization Analyzer. IR spectra were recorded using a PerkinElmer Spectrum 65 FT-IR Spectrometer. The thermogravimetric measurement was carried out using TGA Q500 (TA Instruments) at an Ar flow rate of 10 mL min−1. The elemental analysis measurement of the MOF was carried out using an EA 1112 Flash CHNS/O-analyzer (carrier gas flow rate of 120 mL min−1, reference flow rate 100 mL min−1, oxygen flow rate 250 mL min−1; furnace temperature of 900 °C and oven temperature of 75 °C). UV-visible diffuse reflectance spectra for the band gap determination were recorded using a Cary 5000 UV-vis-NIR spectrometer. PXRD patterns were evaluated using DICVOL06 found in Expo2014 software.23,24 The Scherrer formula (K = 0.94) was applied using Match! 3 Software. The Brunauer–Emmet–Teller (BET) surface area was determined as follows: the MOF sample was degassed under vacuum at 180 °C for 3 h to remove moisture from the pores of the MOF. About 40 mg of the MOF sample was placed in an automatic Surface Area Analyzer (Micromeritics 3Flex Surface Characterization Analyzer), which measures the adsorption/desorption of nitrogen gas at 77 K. SEM micrographs and X-ray microanalysis (SEM/EDX) were recorded by using an FEI Quanta 650 ESEM instrument equipped with Oxford Instruments X-Max 150 mm2 EDS detector using the AZtec software at 20 kV spot 3–4, using low vacuum X-ray cone accessory at 80 Pa.
Synthesis of MOF-T23-RT, {[Cu2(Fu)2(BPY)]·H2O}n
This compound was synthesized by the modification of the procedure22 previously reported (Scheme 1). A clear solution of Na2Fu was prepared by dissolving 8 mmol (0.32 g) of NaOH in 15 mL deionized water and adding 4 mmol (0.4432 g) of fumaric acid while stirring at room temperature. To the above solution, 40 mL of a methanolic solution of BPY (2 mmol, 0.348 g) (BPY = 4,4′-bipyridyl hydrate) was added. The combined solution was stirred for 30 min and Cu(NO3)2·3H2O (4 mmol, 0.9664 g) in water (5 mL) was added forming a cyan-colored suspension. The mixture was stirred at room temperature for 24 hours. The resulting cyan color product was filtered under suction, washed with 3 × 5 mL of deionized water followed by 3 × 5 mL of methanol, and then dried in a vacuum oven at 80 °C for 5 h, yielding 0.95 g (ca. 90% based on BPY) of a cyan color solid {[Cu2(Fu)2(BPY)]·H2O}n. Elemental analyses: Calcd (found)% for C18H14N2O9Cu2: C, 40.80; H, 2.64; N, 5.28%: (C, 40.70); (H, 2.39); (N, 5.04).
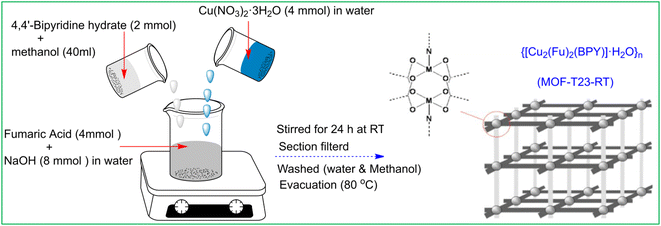 |
| | Scheme 1 Synthetic scheme for MOF-T23-RT, {[Cu2(Fu)2(BPY)]·H2O}n. | |
Results and discussion
PXRD
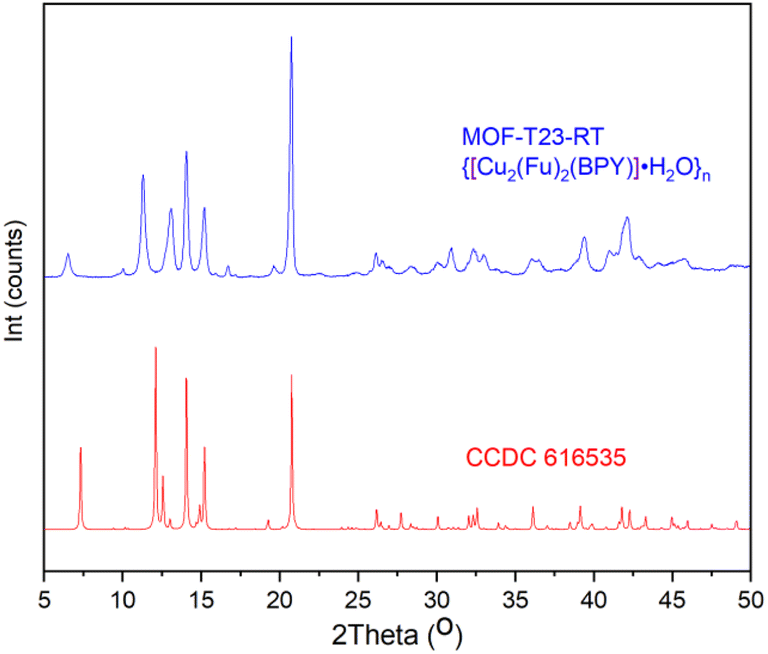
{[Cu2(Fu)2(BPY)]·H2O}n was studied by PXRD and it was found that the patterns of this MOF prepared at room temperature are in good agreement with the corresponding patterns of the similar MOF prepared in DMF at a higher temperature (Fig. 1). Comparison of the PXRD for {[Cu2(Fu)2(BPY)]·H2O}n was made with that simulated from the previously reported CIF (CCDC 616535) of the single crystal of the corresponding pillared MOF prepared in DMF at a higher temperature.22 Both MOFs show similar peak positions at 2θ = 20.85, 15.29, 14.04. The peak positions that are observed at 12.56, 12.12, and 7.3 in the simulation pattern are seen at 11.03, 11.38, and 6.5, respectively, in {[Cu2(Fu)2(BPY)]·H2O}n. Some broadening is observed for these three characteristic peaks, indicating a smaller particle size of the powder obtained using the room temperature synthesis. Overall, a comparison of PXRD of {[Cu2(Fu)2(BPY)]·H2O}n to literature counterparts confirms that it is possible to prepare MOFs at room temperature, replacing DMF with water and methanol as the solvent.
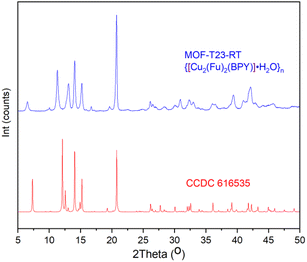 |
| | Fig. 1 PXRD pattern of MOF-T23-RT with its analogue simulated PXRD pattern from the SXRD of pillared MOF CCDC 616535 that was originally made in DMF at higher temperatures. | |
MOF-T23-RT, in this study, is obtained as powder and crystal parameters were determined using DICVOL06 found in Expo2014 software.23 The PXRD pattern of {[Cu2(Fu)2(BPY)]·H2O}n was indexed (Table 1) and a Rietveld refinement was used to confirm that the MOF crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The unit cell volume for the MOF indicates that the system is potentially porous, taking into account the volume occupied by the framework components. The crystallite size of this MOF is estimated using the Scherrer formula (K = 0.94) and calculated as 196 nm (Rp(%) = 11.4). The Match! 3 software automatically selected the peaks in the PXRD patterns that are appropriate for crystallite size estimation between the 2θ values of 30 and 50°.
space group. The unit cell volume for the MOF indicates that the system is potentially porous, taking into account the volume occupied by the framework components. The crystallite size of this MOF is estimated using the Scherrer formula (K = 0.94) and calculated as 196 nm (Rp(%) = 11.4). The Match! 3 software automatically selected the peaks in the PXRD patterns that are appropriate for crystallite size estimation between the 2θ values of 30 and 50°.
Table 1 Crystal data and structural refinement parameters for {[Cu2(Fu)2(BPY)]·H2O}n indexed by DICVOL06
| |
MOF-T23-RT or {[Cu2(Fu)2(BPY)]·H2O}n |
| Empirical formula |
C18H14N2O9Cu2 |
| Formula weight |
527.93 |
| Crystal system |
Triclinic |
| Space group |
P |
| a (Å) |
6.9084 |
| b (Å) |
9.0654 |
| c (Å) |
13.7042 |
| α (°) |
95.93 |
| β (°) |
94.42 |
| γ (°) |
100.60 |
| V (Å3) |
835.04 |
| Z |
1.5 |
| ρcalc (g cm−1) |
1.66 |
| Counts |
2475 |
| λ (nm) |
1.540598 |
FT-IR
The MOF, {[Cu2(Fu)2(BPY)]·H2O}n was also studied by FT-IR spectroscopy. Fig. 2 depicts the FT-IR spectra of this MOF, the linker, and the pillar from which the MOF is derived. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of fumaric acid appears at 1678 cm−1. This peak is shifted to the lower frequency region (1618 cm−1) after the coordination of Cu(II) in {[Cu2(Fu)2(BPY)]·H2O}n. The ring breathing mode observed in BPY in the region between 735–571 cm−1 and 1038–966 cm−1 is seen in the resulting MOF after coordination with the metal with some change in the peak intensity and position. The narrow ring breathing band at 1405 cm−1 in BPY becomes more intense and wider in {[Cu2(Fu)2(BPY)]·H2O}n after coordination. The C–C and C–N stretching frequency of the pillar BPY is observed at 1583 cm−1. This peak is merged to the CO stretching frequency centered at 1618 cm−1 in the MOF. These frequency shifts as well as change in intensity are ascribed to the coordination of nitrogen to the metals in the MOF under study. The symmetric and asymmetric vibrations of the carboxylate (COO–) group coordinated to Cu(II) in {[Cu2(Fu)2(BPY)]·H2O}n are seen at 1618 cm−1 and 1380 cm−1, respectively, but merge with the C–N and ring breathing bands of the pyridyl group. This made the peaks wider and more intense. In general, the shift in FT-IR between the unreacted and reacted linkers and pillar show coordination of oxygen and nitrogen to the metal cation in the MOF.
O stretching frequency of fumaric acid appears at 1678 cm−1. This peak is shifted to the lower frequency region (1618 cm−1) after the coordination of Cu(II) in {[Cu2(Fu)2(BPY)]·H2O}n. The ring breathing mode observed in BPY in the region between 735–571 cm−1 and 1038–966 cm−1 is seen in the resulting MOF after coordination with the metal with some change in the peak intensity and position. The narrow ring breathing band at 1405 cm−1 in BPY becomes more intense and wider in {[Cu2(Fu)2(BPY)]·H2O}n after coordination. The C–C and C–N stretching frequency of the pillar BPY is observed at 1583 cm−1. This peak is merged to the CO stretching frequency centered at 1618 cm−1 in the MOF. These frequency shifts as well as change in intensity are ascribed to the coordination of nitrogen to the metals in the MOF under study. The symmetric and asymmetric vibrations of the carboxylate (COO–) group coordinated to Cu(II) in {[Cu2(Fu)2(BPY)]·H2O}n are seen at 1618 cm−1 and 1380 cm−1, respectively, but merge with the C–N and ring breathing bands of the pyridyl group. This made the peaks wider and more intense. In general, the shift in FT-IR between the unreacted and reacted linkers and pillar show coordination of oxygen and nitrogen to the metal cation in the MOF.
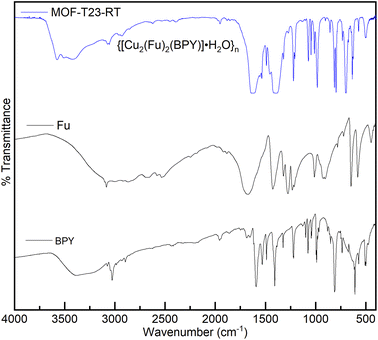 |
| | Fig. 2 FT-IR spectra of MOF-T23-RT, 4,4′-bipyridyl hydrate (BPY) and fumaric acid (Fu). | |
Thermal analysis
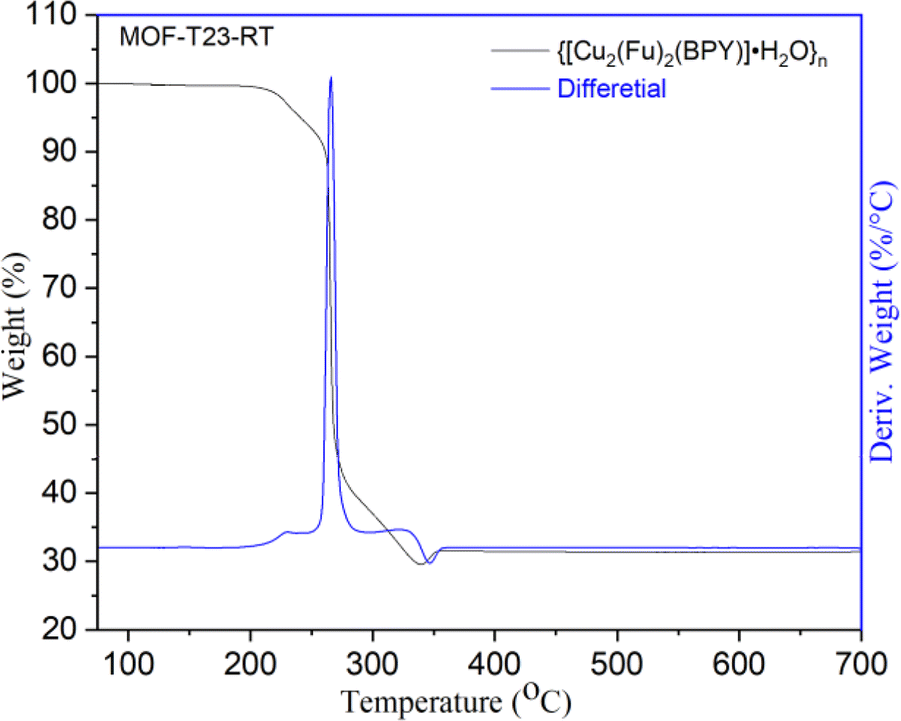
The thermal stability of {[Cu2(Fu)2(BPY)]·H2O}n was examined by TGA (Fig. 3) under argon atmosphere from room temperature to 700 °C at a heating rate of 10 °C min−1 and the differential curves associated with the TGA curve show sharp weight losses at different temperatures. A weight loss below 240 °C (calcd 3.4%) is attributed to the removal of one water molecule from the framework. The second mass loss is witnessed in the range of 240–360 °C, corresponding to the decomposition of the framework. The final 30.9% residual weight for MOF-T23-RT corresponds to two weight equivalents of CuO (calcd 30.05%). This confirms that MOF-T23-RT contains two Cu ions in its empirical formula in the paddle wheel.
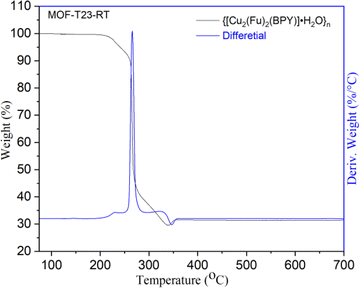 |
| | Fig. 3 Thermogravimetric analysis (TGA) of MOF-T23-RT. | |
Determination of surface area, pore volume and pore size
The Brunauer–Emmet–Teller (BET) surface area of the sample was calculated using N2 adsorption data at 77 K. Pore diameter, BJH pore volume and BET surface area are given in Table 2. It is known that the sample of Cu2(Fu)2(BPY)19,22 prepared from DMF has small pores, ca. 3.6 Å in diameter, probably as a result of doubly interpenetrated arrangements.
Table 2 N2-sorption data for MOF-T23-RT
| MOF |
Pore diameter [nm] |
BJH pore volume [cm3 g−1] |
BET surface area [m2 g−1] |
| MOF-T23-RT |
3.03 |
0.995 |
775.16 |
The pore diameter of over 3 nm observed in this system suggests that the interpenetration or the network effect might be resolved to some extent when the MOF is prepared in water/methanol mixed solvents than in DMF. The shape of the isotherms shown in this study could be ascribed to the fact that dynamic/flexible pillared-layer MOFs can reversibly change their channels by a large magnitude while retaining similar topologies, corresponding to expansion (lp phase) and contraction (np phase) up on the guest N2 adsorption and desorption.25 The structural transformation has an effect26–28 on the shape of the isotherm or hysteresis (Fig. 4a) and could not exactly fit to the models reported by IUPAC.29 This is expected for most real samples; and the models work best for rigid MOFs that do not show flexibility. The pore widths from NLDFT fit or dV dW−1 vs. pore size distribution plot are shown in Fig. 4b for {[Cu2(Fu)2(BPY)]·H2O}n, depicting that the MOF has pore sizes in the lower mesoporous region.
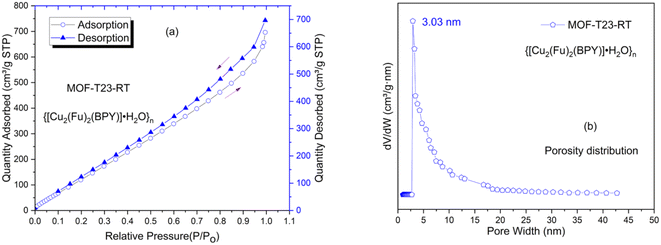 |
| | Fig. 4 Porosity properties of MOF-T23-RT determined by N2 adsorption–desorption method. (a) N2 adsorption–desorption isotherms of MOF-T23-RT, (b) the pore size distribution curves for MOF-T23-RT. | |
Morphological characterization
SEM micrographs and X-ray microanalysis (SEM/EDX) were recorded by using the FEI Quanta 650 ESEM instrument equipped with Oxford Instruments X-Max 150 mm2 EDS detector with the AZtec software at 20 kV spot 3–4, using low vacuum X-ray cone accessory at 80 Pa. SEM images of the MOF are shown in Fig. 5a. The MOF, {[Cu2(Fu)2(BPY)]·H2O}n contains much smaller crystallites giving a cotton-like appearance. SEM/EDX analysis gives information about the sample surfaces with high depth of field and lateral resolutions of around 1–20 nm,30,31 indicating that EDX only provides microanalysis for the surface of particles (Fig. 5b). Importantly, it is remarkable that the SEM/EDX microanalysis confirms the absence of sodium in the MOF material.
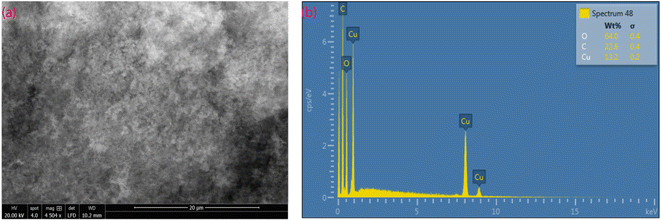 |
| | Fig. 5 SEM image (a) and EDS (b) of MOF-T23-RT. | |
Solid state UV-vis study
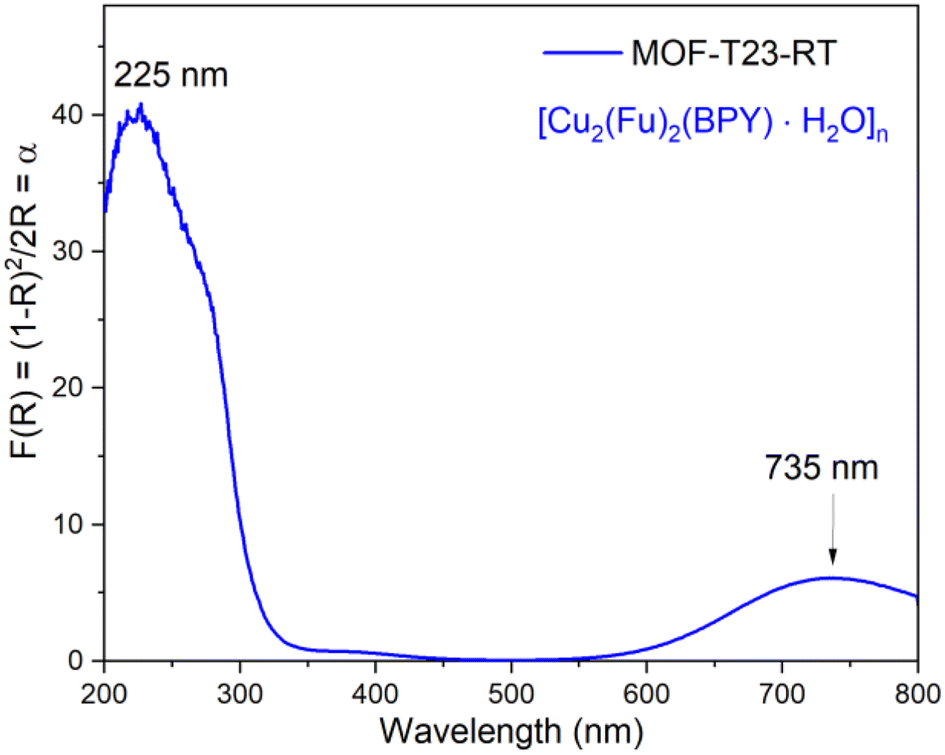
The solid-state UV-vis spectrum shown in Fig. 6 for the powdered microcrystalline {[Cu2(Fu)2(BPY)]·H2O}n depicts the electronic transitions within the linkers. The absorption coefficient (F(R) or α) at 225 nm for {[Cu2(Fu)2(BPY)]·H2O}n is ascribed to the n → σ* electronic transitions within the linker and pillar in the framework. The weak absorption broad band centered at 735 nm for {[Cu2(Fu)2(BPY)]·H2O}n confirms the 2T2 ← 2E electronic transition in the SBU and Cu(II) is in its d9 state. The distorted octahedral geometry around Cu(II) is expected to show three absorptions in this region. The broadband appearing in the spectrum is attributed to be a combination of them.
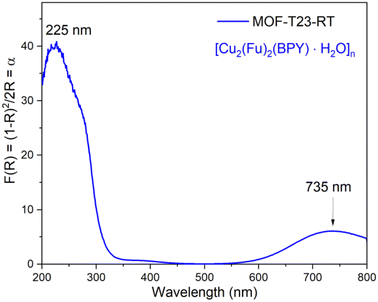 |
| | Fig. 6 Solid state absorption coefficient vs. wavelength spectra for MOF-T23-RT. | |
Band gap determination
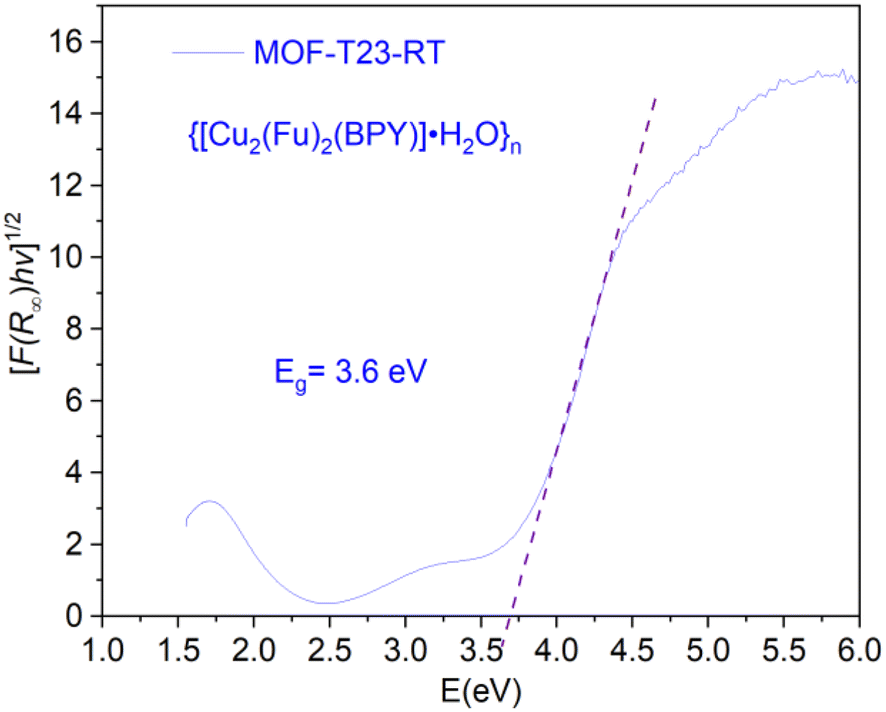
The electronic band gap of the MOF was determined (Fig. 7) from the plot of (αhν)1/2 versus photon energy (hν),32 where α or (F(R∞)) is the absorption coefficient and hν is photon energy. Data for this plot was calculated and obtained from the diffuse reflectance versus wavelength spectral data run using the Cary 5000 UV-vis-NIR spectrometer. The optical edge or gap was inferred by linear extrapolation of the absorbance from the high slope region obtained from the modified Kubelka–Munk ((αhν)1/2) versus hν plot. This results in a band gap of 3.6 eV for {[Cu2(Fu)2(BPY)]·H2O}n. It can be concluded that {[Cu2(Fu)2(BPY)]·H2O}n is active in the UV region for application purposes.
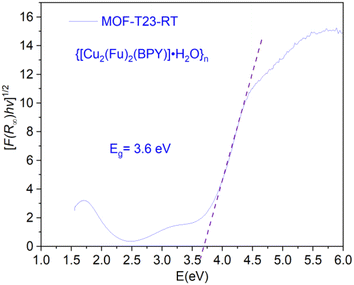 |
| | Fig. 7 The optical band gaps of MOF-T23-RT. | |
Magnetic properties
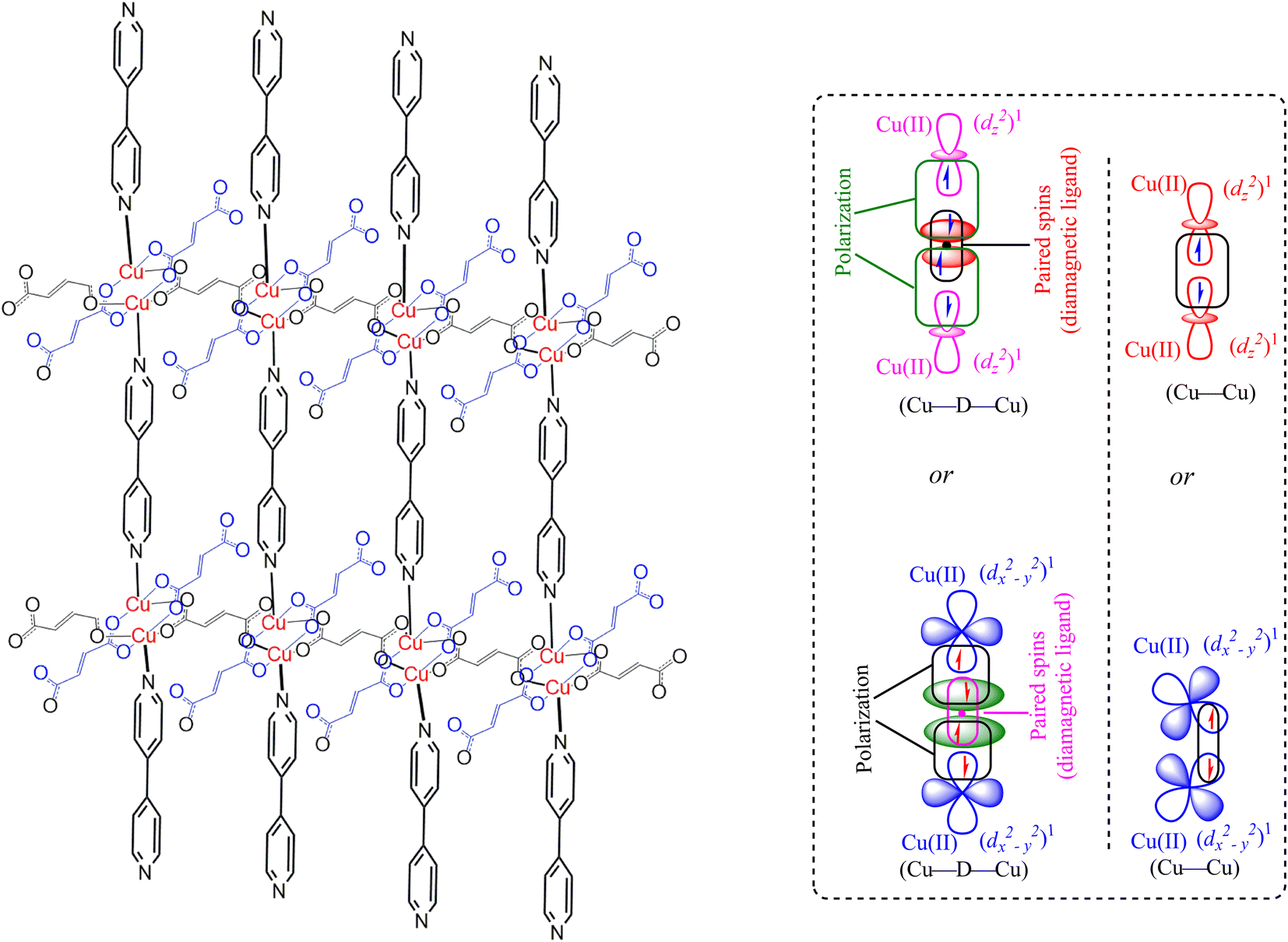
The magnetic properties of the MOF {[Cu2(Fu)2(BPY)]·H2O}n are measured at 294 K using the MSB AUTO (Sherwood Scientific instrument), and shown in Table 3. Magnetic susceptibility measurement of {[Cu2(Fu)2(BPY)]·H2O}n by the inverse Gouy method shows that the gram susceptibility or xg = 3.379 × 10−6 for which the effective magnetic moment, μeff, after calculating the diamagnetic correction is 2.1 BM. This magnetic moment value suggests the presence of 1.37 unpaired electrons (n) in this bimetallic system. This is less than the two unpaired electrons expected in the two Cu(II) centers (one for each, (dz2)1 or (dx2−y2)1) in the paddle wheel. The decrease in the number of unpaired electrons observed is ascribed to the occurrence of metal to metal magnetic communication (Fig. 8) through the diamagnetic pillar (BPY) and/or linker (fumarate). The Cu–Fu–Cu interaction might be extremely strong and dominates the interaction of Cu-BPY-Cu. If the single unpaired electron occupies (dz2) or (dx2−y2) orbital of the first Cu(II) center, it interacts magnetically with another unpaired electron in the (dz2) or (dx2−y2) orbital of the next Cu(II) center through the diamagnetic (D) unit. The interaction might also extend directly between the two metals. This could result in significant antiferromagnetic interaction between the unpaired electrons of the Cu(II) centers through the conjugated bridges.33–36
Table 3 Magnetic susceptibility data of the MOF-T23-RT ({[Cu2(Fu)2(BPY)]·H2O}n) at 294 K
| MOF |
M (g mol−1) |
χg (294 K) |
χM = χg × M |
χdia |
χM′ = χM−χdia |
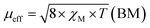
|
n |
| MOF-T23-RT{[Cu2(Fu)2(BPY)]·H2O}n |
527.93 |
3.379 × 10−6 |
1.78 × 10−3 |
−1.84 × 10−4 |
1.96 × 10−3 |
2.1 |
1.37 |
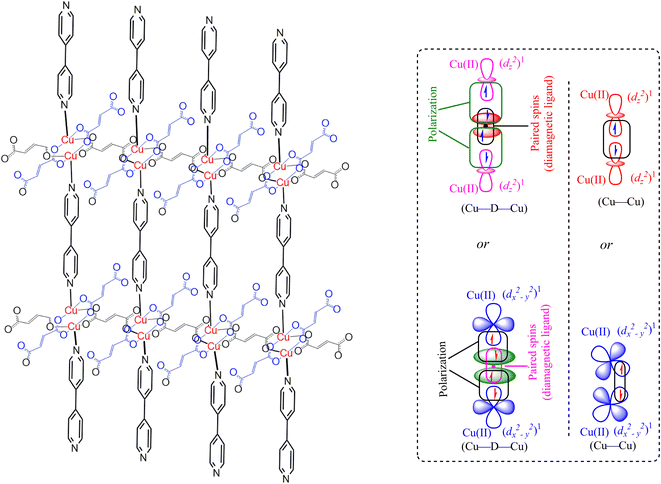 |
| | Fig. 8 Representation of the extended frameworks and magnetic exchange between Cu(II) centers through diamagnetic bridge in MOF-T23-RT ({[Cu2(Fu)2(BPY)]·H2O}n). Note that “D” is a diamagnetic unit (Fumarate or BPY). | |
Conclusion
In summary, the pillared-layer MOF, {[Cu2(Fu)2(BPY)]·H2O}n, was synthesized at room temperature in benign solvents. The PXRD of this MOF was compared to calculated PXRD patterns from literature CIF of the analogous MOF prepared in DMF at higher temperatures, giving close agreement. Determination of the optical band gap revealed that {[Cu2(Fu)2(BPY)]·H2O}n is active in the UV region. Even though single crystals could not be obtained from the current MOF, it is apparent that we are able to prepare quantitative amounts of MOF (compared to the conventional synthesis of MOF in a solvothermal approach using scintillation vials) under simple conditions in a beaker in benign water and methanol solvents. Our approach offers promise for preparing MOF under sustainable conditions and potentially on a large scale. Based on the spectroscopic and non-spectroscopic analysis of data, the extended framework structure of the MOF under study is predicted, as depicted in Fig. 8.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Addis Ababa University for granting financial support, for the research visit to the School of Chemistry, University of Nottingham, UK. NC of the University of Nottingham is also acknowledged for his keenest contribution in allowing his laboratory.
Notes and references
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS
 .
. - K. Sanderson, Nature, 2007, 448, 746–748 CrossRef CAS PubMed .
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC .
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tome, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC .
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés I Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed .
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed .
- M.-Q. Wang, Y. Zhang, S.-J. Bao, Y.-N. Yu and C. Ye, Electrochim. Acta, 2016, 190, 365–370 CrossRef CAS .
- J. Huang, Y. Li, R. K. Huang, C. T. He, L. Gong, Q. Hu, L. Wang, Y. T. Xu, X. Y. Tian, S. Y. Liu, Z. M. Ye, F. Wang, D. D. Zhou, W. X. Zhang and J. P. Zhang, Angew. Chem., Int. Ed., 2018, 57, 4632–4636 CrossRef CAS PubMed .
- H.-X. Zhao, Q. Zou, S.-K. Sun, C. Yu, X. Zhang, R.-J. Li and Y.-Y. Fu, Chem. Sci., 2016, 7, 5294–5301 RSC .
- W. Lin, Q. Hu, K. Jiang, Y. Yang, Y. Yang, Y. Cui and G. Qian, J. Solid State Chem., 2016, 237, 307–312 CrossRef CAS .
- X. Zhao, C. Mao, K. T. Luong, Q. Lin, Q. G. Zhai, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2016, 55, 2768–2772 CrossRef CAS PubMed .
- J. Zhang and J. n. M. Shreeve, Dalton Trans., 2016, 45, 2363–2368 RSC .
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed .
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639–12641 CrossRef CAS PubMed .
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC .
- Z. Yin, Y.-L. Zhou, M.-H. Zeng and M. Kurmoo, Dalton Trans., 2015, 44, 5258–5275 RSC .
- T. Gong, X. Lou, E.-Q. Gao and B. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 21839–21847 CrossRef CAS PubMed .
- A. Pichon, C. M. Fierro, M. Nieuwenhuyzen and S. L. James, CrystEngComm, 2007, 9, 449–451 RSC .
- B.-Q. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912–4914 CrossRef CAS PubMed .
- T. Friscic and L. Fabian, CrystEngComm, 2009, 11, 743–745 RSC .
- H. Sakamoto, R. Matsuda and S. Kitagawa, Dalton Trans., 2012, 41, 3956–3961 RSC .
- B. Chen, S. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H.-C. Zhou, Inorg. Chem., 2007, 46, 1233–1236 CrossRef CAS PubMed .
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231–1235 CrossRef CAS .
- K. Fujii, A. L. Garay, J. Hill, E. Sbircea, Z. Pan, M. Xu, D. C. Apperley, S. L. James and K. D. M. Harris, Chem. Commun., 2010, 46, 7572–7574 RSC .
- Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC .
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC .
- F. ZareKarizi, M. Joharian and A. Morsali, J. Mater. Chem. A, 2018, 6, 19288–19329 RSC .
- I. Senkovska and S. Kaskel, Chem. Commun., 2014, 50, 7089–7098 RSC .
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS .
- P. D. Brown, H. K. Edwards and M. W. Fay, J. Phys.: Conf. Ser., 2010, 241, 012019 CrossRef .
- H. K. Edwards, M. W. Fay, S. I. Anderson, C. A. Scotchford, D. M. Grant and P. D. Brown, J. Microsc., 2009, 234, 16–25 CrossRef CAS PubMed .
- R. López and R. Gómez, J. Sol-Gel Sci. Technol., 2012, 61, 1–7 CrossRef .
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC .
- J. L. Manson, J. A. Schlueter, K. E. Garrett, P. A. Goddard, T. Lancaster, J. S. Möller, S. J. Blundell, A. J. Steele, I. Franke, F. L. Pratt, J. Singleton, J. Bendix, S. H. Lapidus, M. Uhlarz, O. Ayala-Valenzuela, R. D. McDonald, M. Gurak and C. Baines, Chem. Commun., 2016, 52, 12653–12656 RSC .
- S. Höfler, A. Scheja, B. Wolfram and M. Bröring, Z. Anorg. Allg. Chem., 2016, 642, 107–117 CrossRef .
- D. L. Reger, A. Debreczeni, B. Reinecke, V. Rassolov, M. D. Smith and R. F. Semeniuc, Inorg. Chem., 2011, 50, 4669–4670 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
V. J. T. Rajub and
Yonas Chebude
*ab,
V. J. T. Rajub and
Yonas Chebude