
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
B(C6F5)3-catalyzed oxidation of α-diazoesters using DMF and molecular oxygen as oxygen sources†
Bei Wang‡
abc,
Guo-Min Zhang‡abc,
Hua Zhangabc and
Ji-Yu Wang *ab
*ab
aDepartment of Chemistry, Asymmetric Synthesis and Chiral Technology Key Laboratory of Sichuan Province, Xihua University, Chengdu 610041, P. R. China
bChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 23rd November 2022
Abstract
A metal-free catalytic oxidation of α-diazoesters via a green environmental-friendly route was developed. The α-diazoesters were converted to α-ketoesters using DMF and molecular oxygen as oxygen sources and B(C6F5)3 as the catalyst, without any additives. This protocol has a broad adaptability of substrates and good compatibility with a range of functional groups, and it offers new insight into reactions catalyzed by B(C6F5)3.
Introduction
α-Diazoesters are an important class of organic synthesis synthon.1 Diazo compounds can form metal carbenes by transition metal catalysis, and the reactions of insertion or cyclopropanation based on metal carbines have been developed in the past few decades.2 The oxidation reaction of α-diazoesters can generate α-ketoesters, which usually have biological activity, and can also transform into a variety of functional groups.3 Various methods for this oxidation have been reported, although many require the use of harsh oxidants like dimethyldioxirane (DMDO) or t-BuOCl,4 or expensive transition metal catalysts like Rh.5 In 2016, Stoltz's group found that dimethyl sulfoxide (DMSO) could also serve as an oxidant to achieve oxidation of aryl α-diazoesters (Fig. 1a).6 But the method applied only to the electron-rich diazo compounds. According to green and sustainable chemistry principles, molecular oxygen is considered to be an ideal oxidant due to its natural, inexpensive, and environmentally friendly nature, and therefore it has attractive academic and industrial prospects.7 The reactions of preformed stable metal carbene compounds with oxygen are known.8 In 2021, Xu et al. reported a highly efficient and catalytic procedure for the aerobic oxidation of α-diazoesters to α-ketoesters via a copper carbene intermediate (Fig. 1a).9 In addition, Liu et al. used cheap, readily available Eosin Y as a photocatalyst and O2 (air) as a green oxidant, and achieved the aerobic oxidation of α-diazoesters under visible light in air at room temperature (Fig. 1a).10 Research on the aerobic oxidation of α-diazoesters especially by non-metal catalysts still has space for exploration.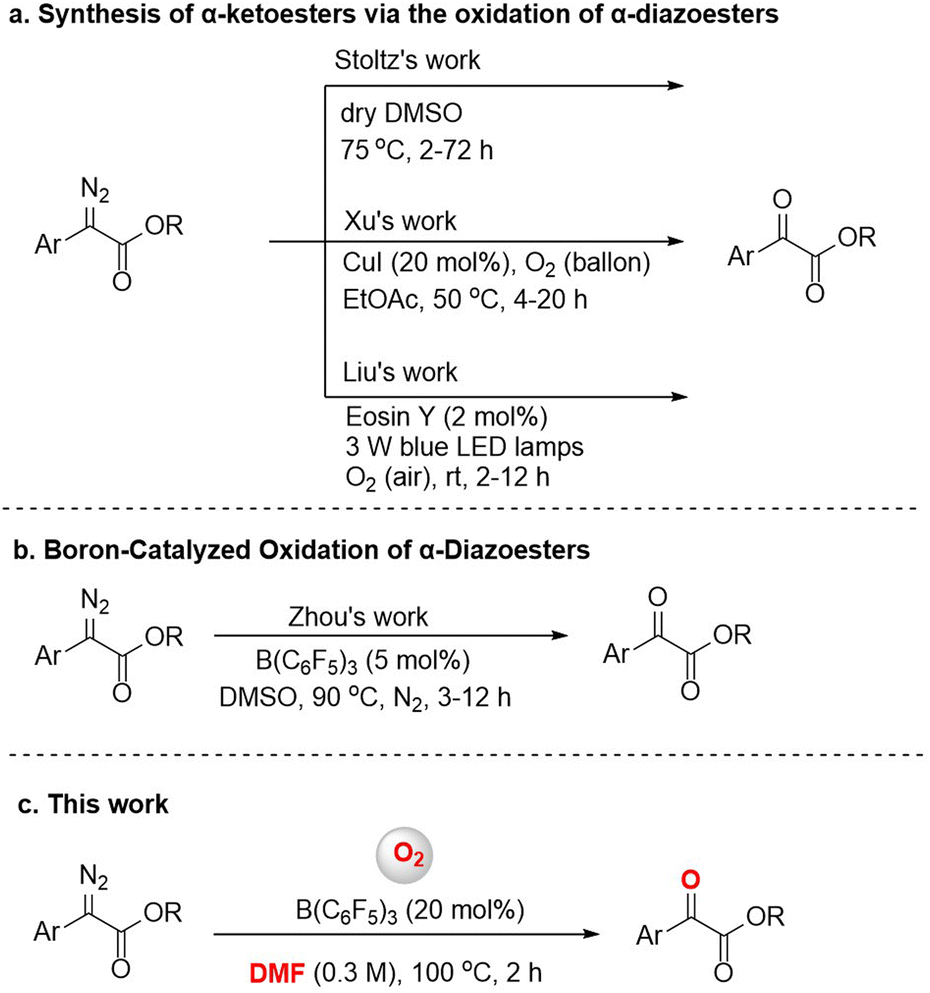 | ||
| Fig. 1 The oxidation reactions of α-diazoesters. | ||
In recent years, tris(pentafluorophenyl)borane has been widely used in the reduction or addition reactions of unsaturated compounds, and in other reactions.11 However, few efforts have been devoted to B(C6F5)3-catalyzed oxidation reactions. For instance, the borane-mediated hydride abstraction of amines results in the generation of reactive iminium hydridoborate salts that participate in a variety of stoichiometric and catalytic processes.12 In 2021, Basak et al. provided a detailed overview of the borane-mediated dehydrogenation functionalization of amine compounds.13 Moreover, in 2017, Babu's group reported an efficient one-pot oxidative esterification and amidation of aldehydes using B(C6F5)3 as the catalyst and TBHP as the oxidant.14 In 2018, Ling et al. reported a B(C6F5)3-catalyzed oxidative deamination/cyclization cascade reaction of benzyl amines and ketones for the synthesis of 2,4,6-triarylpyridines in an oxygen atmosphere.15 In addition, several reports have been published on the B(C6F5)3-catalyzed activation of diazo compounds;16 for example, Tang's group reported a boron-catalyzed O–H bond insertion of α-aryl α-diazoesters in water.17 In previous works, our group has also conducted research on B(C6F5)3-catalyzed reactions and achieved some results.18 During our research, we found that B(C6F5)3 could catalyze the oxidation of α-diazoesters under an O2 atmosphere. Subsequently, Zhou's group described a B(C6F5)3-catalyzed oxidation reaction of α-diazoesters with DMSO as an oxygen source (Fig. 1b).19 But, different to this work, we report a B(C6F5)3-catalyzed oxidation of α-diazoesters to obtain α-ketoesters by activating O2 directly (Fig. 1c). And this protocol features a wide substrate scope including aromatic heterocycle α-diazoesters and good functional group tolerance.
Results and discussion
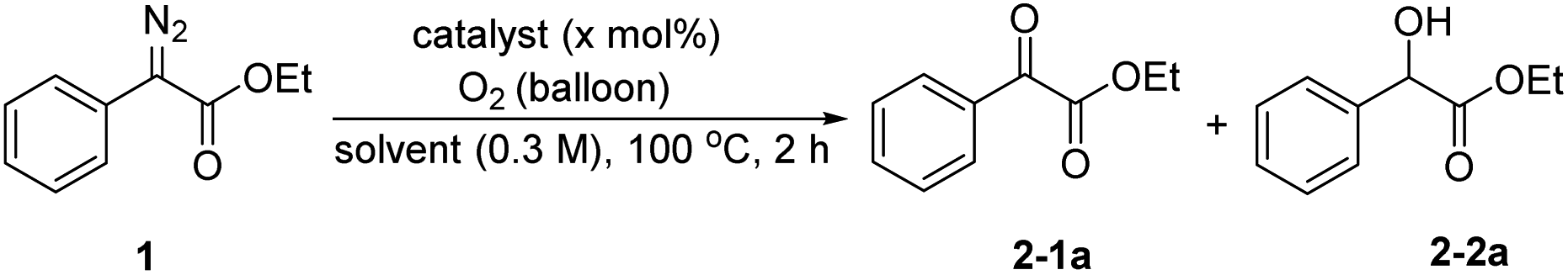
The ethyl phenyldiazoacetate 1a was selected as the model substrate to investigate the oxidation reaction under various reaction conditions (Table 1). Initially, the reaction was carried out using different catalysts at 100 °C for 2 hours in DMF in an oxygen atmosphere. First, only a small amount of oxidation product 2-1a was obtained without any catalyst (entry 1). To our delight, B(C6F5)3 catalyzed the oxidation reaction and the yield of 2-1a increased to 57% (entry 2). 2,4,6-BArF, 3,4,5-BArF, Cu(OAc)2 and CoCl2 also had certain catalytic effects on the reaction, but the results were not as good as those with B(C6F5)3 (entries 3–6). Furthermore, the catalytic effects of metal Lewis acid catalysts with weaker acidity such as La(OTf)3 or Sc(OTf)3 were worse (entries 7 and 8). The byproduct ethyl mandelate was reduced when using dry DMF as solvent (entry 9). Next, we tried to increase the amount of catalyst. When the amount of catalyst was increased to 20 mol%, the yield of 2-1a was 86% (entries 10–12). Then, the temperature was adjusted to 80 °C, and the yield of the expected product decreased significantly (entry 13). Further screening of solvents showed that the type of solvent had effects on the reaction. When the reaction was performed in CH3CN, THF or 1, 4-dioxane, the yields of 2-1a were very low (entries 14–16). Finally, the best conditions for the reaction were determined: the reaction was run with 1a (0.3 mmol) and B(C6F5)3 (20 mol%) under an oxygen atmosphere (balloon) in 1 mL of dry DMF at 100 °C for 2 hours.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Catalyst (x mol%) | 2-1a (%) | 2-2a (%) |
| a Reaction conditions: 1a (54.0 mg, 0.3 mmol) and catalyst (x mol%) in solvent (1 mL) for 2 h at T °C.b Yield of GC.c The dry solvent.d The reaction temperature was 80 °C. | |||||
| 1 | — | DMF | 5 | 5 | Trace |
| 2 | B(C6F5)3 | DMF | 5 | 57 | 20 |
| 3 | 2,4,6-BArF | DMF | 5 | 50 | 18 |
| 4 | 3,4,5-BArF | DMF | 5 | 47 | 22 |
| 5 | Cu(OAc)2 | DMF | 5 | 42 | 18 |
| 6 | CoCl2 | DMF | 5 | 39 | 20 |
| 7 | La(OTf)3 | DMF | 5 | 20 | 25 |
| 8 | Sc(OTf)3 | DMF | 5 | 24 | 23 |
| 9c | B(C6F5)3 | DMF | 5 | 68 | 7 |
| 10c | B(C6F5)3 | DMF | 10 | 63 | 4 |
| 11c | B(C6F5)3 | DMF | 15 | 70 | 6 |
| 12c | B(C6F5)3 | DMF | 20 | 86 | 1 |
| 13c,d | B(C6F5)3 | DMF | 20 | 52 | 7 |
| 14c | B(C6F5)3 | CH3CN | 20 | 15 | 12 |
| 15c | B(C6F5)3 | THF | 20 | 19 | 13 |
| 16c | B(C6F5)3 | 1,4-Dioxane | 20 | 17 | 16 |
Next, using the optimized conditions, the applicability of the protocol for a broad range of substrates was explored, as shown in Scheme 1. Generally, phenyl α-diazoesters with alkyl, benzyl, allyl, or propargyl substituents at R2 all afforded moderate to high yields of the desired α-ketoesters (2-1a–2-1h). The position and electrical properties of the substituents on the benzene ring have a great influence on the reaction. With substituents at the ortho position, the yield of products substantially declined, and the decrease is more obvious when the methyl group was substituted (2-1i–2-1j). This is probably due to steric hindrance and the donating property of the group, because the carbene intermediate that formed during the reaction was unstable. The yield of the products was moderate when a meta substituent was located on the benzene ring (2-1k–2-1l). The yield of the products was lower when the benzene ring had an electron donating substituent at the para position (2-1m–2-1n), and the yield of the products was moderate to good with halogen substituents (2-1o–2-1q). Multisubstituted aryl α-diazoesters such as ethyl 2-diazo-2-(2,3-dichlorophenyl)acetate could be converted to the corresponding oxidation product 2-1r with a yield of 48%. Unsaturated substituents, such as alkyne groups, also tolerated the reaction conditions (2-1s). In the case of polycyclic substrates, such as naphthalene and 2-anthraquinone, the desired products 2-1t and 2-1u could also be formed in good yields; the oxidation product of ethyl-2-(benzo[d][1,3]dioxol-5-yl)-2-diazoacetate could be obtained with a yield of 40% (2-1v). In addition, the substrate bearing a quinoline moiety also gave the corresponding product 2-1w, albeit in a slightly diminished yield. Aryl α-diazoesters containing tocopherol moieties could also smoothly undergo the reaction to give the corresponding product with a yield of 43% (2-1x). Finally, diphenyl–diazomethane was converted to benzophenone in a moderate yield (2-1y) and an oxindole scaffold was compatible with the reaction, allowing for the formation of isatin in 48% yield (2-1z).
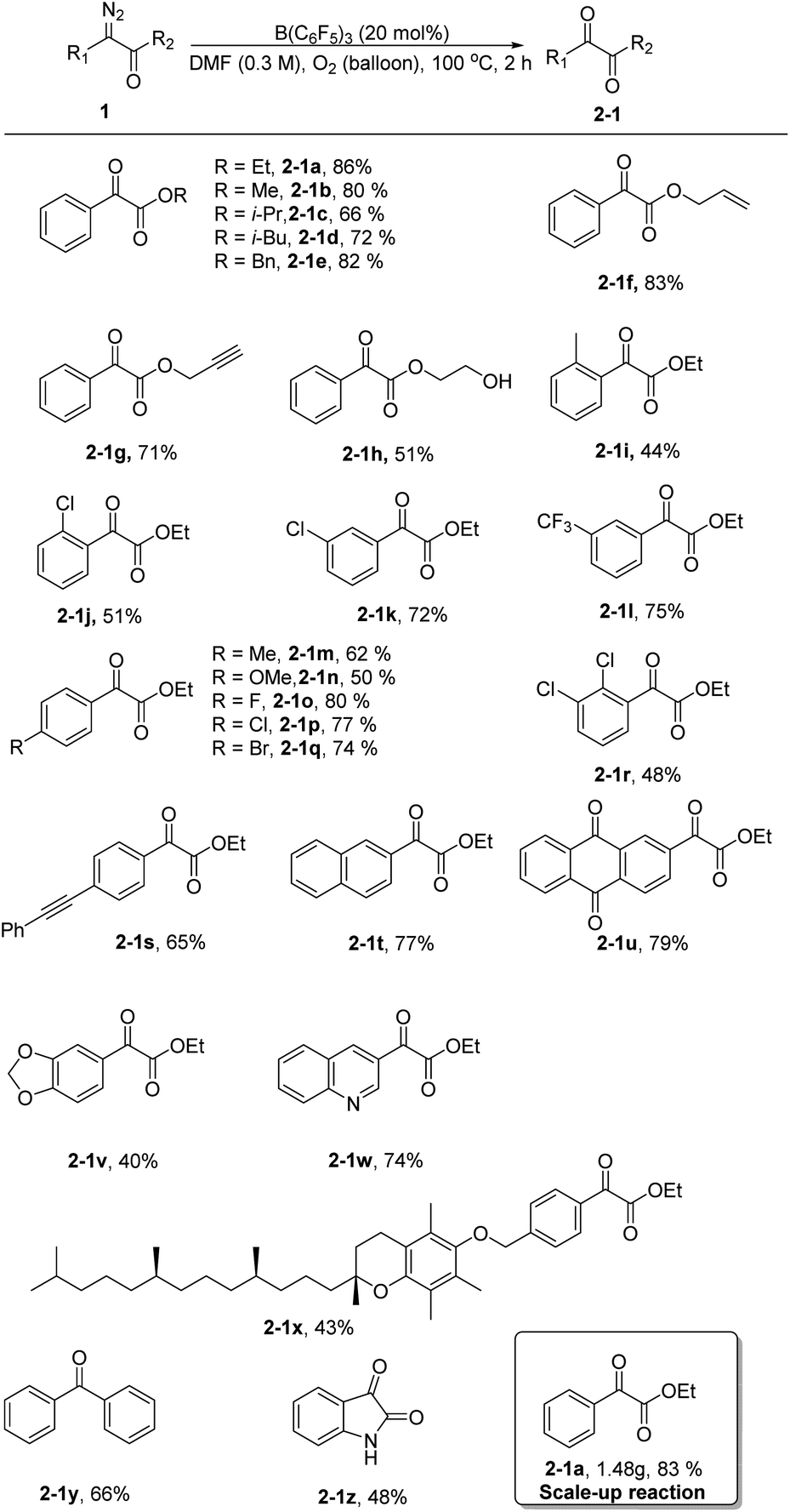 | ||
| Scheme 1 Substrate scope for α-diazoesters. Reaction conditions: 1 (0.3 mmol, 1 equiv.), B(C6F5)3 (20 mol%), and DMF (1 mL) at 100 °C for 2 h. Yield of isolated product. Scale-up reaction conditions: 1 (10 mmol, 1.9 g, 1 equiv.), B(C6F5)3 (20 mol%), and DMF (30 mL) at 100 °C for 2 h. Yield of isolated product. | ||
Subsequently, we increased the amount of ethyl phenyldiazoacetate 1a to 10 mmol and the yield of the product 2-1a did not decrease remarkably, demonstrating that the reaction has potential for industrial scale-up (Scheme 1).
In order to explore the mechanism, we conducted some control experiments. First, we carried out 19F NMR titration experiments using B(C6F5)3 and DMF; when DMF was added to the CDCl3 solution of B(C6F5)3, the 19F NMR spectra of B(C6F5)3 showed two new peaks at chemical shifts of −158 and −165 ppm (Fig. 2), which were caused by B(C6F5)3 coordinating with DMF. Then, the oxidation reaction was carried out in a pure argon atmosphere, and the yield of 2-1a reduced remarkably, while the results of the control experiment with 18O2 confirmed that the oxygen source of the product was mainly DMF, and a small portion of it came from 18O2 (Scheme 2).
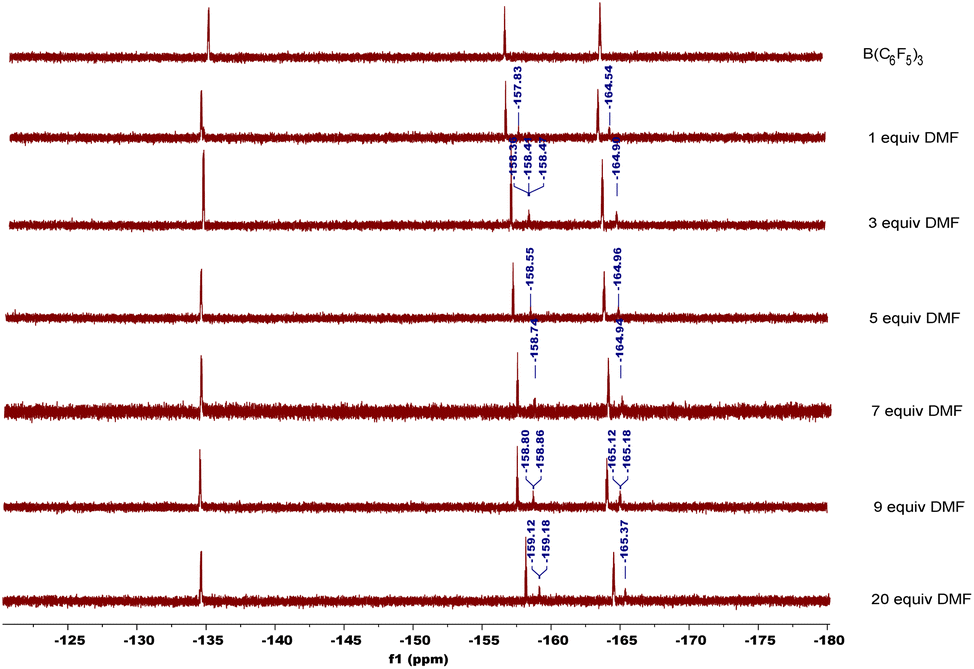 | ||
| Fig. 2 19F NMR titration experiments. | ||
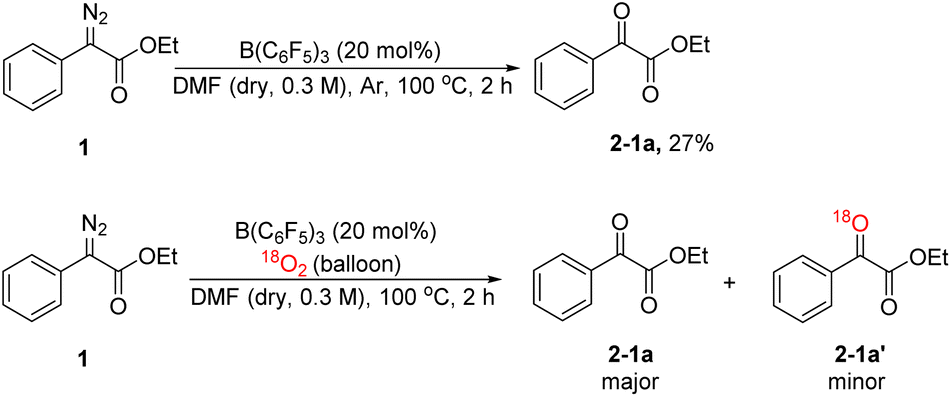 | ||
| Scheme 2 Control experiments. | ||
According to the related literature reports16b,20–22 and the results of control experiments, we can shed light on the reaction mechanism (Scheme 3). On the one hand, the diazoester compound might be activated by B(C6F5)3 and then release N2 to generate carbine intermediate B. Then, the carbene intermediate may directly react with DMF or molecular oxygen to form the corresponding oxidation product 2-1a (path A and path C). On the other hand, B(C6F5)3 might coordinate with DMF, and its coordination intermediate G will then react with the resonance structure F of 1a to form intermediate H. Next, intermediate H will further react to generate the product 2-1a (path B).
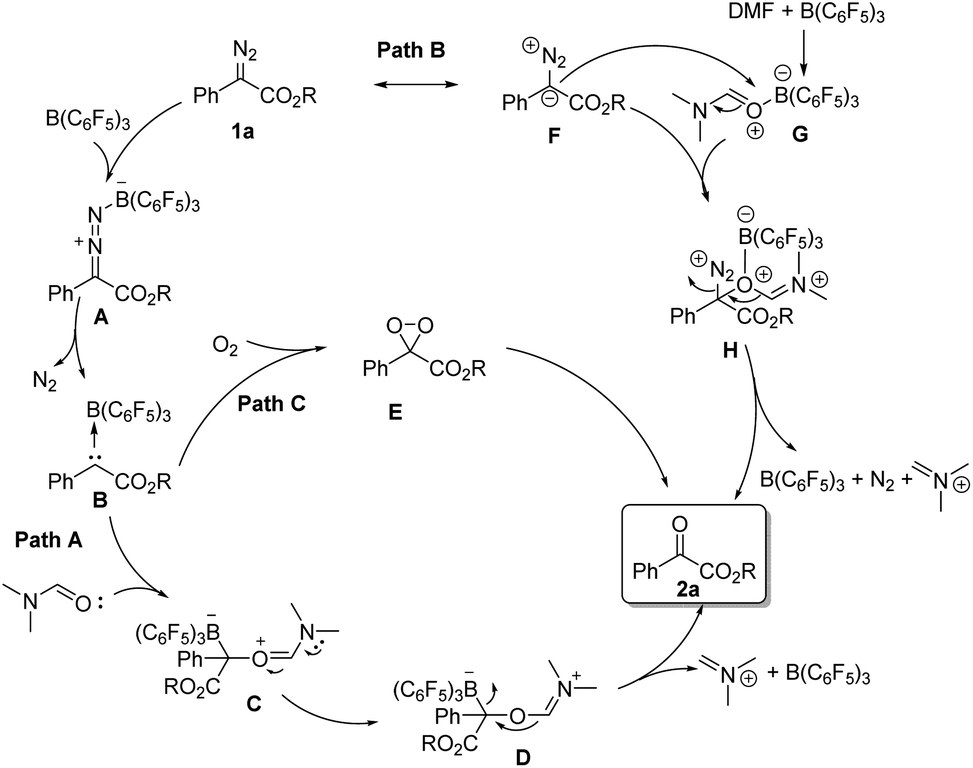 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, a metal-free B(C6F5)3-catalyzed oxidative reaction of α-diazoesters in dry DMF under an oxygen atmosphere was reported. This reaction features a broad substrate scope, good compatibility of functional groups and a green environment-friendly nature. Importantly, the control experiments confirmed that the oxygen sources of the product 2-1a were DMF and molecular oxygen. The protocol offers new insight on reactions catalyzed by B(C6F5)3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from CAS “Light of West China” Program and the Applied Basic Research Programs of Sichuan province, China (No. 20YYJC0492) and Technological Innovation Program of Chengdu, Sichuan province, China (No. 2021-YF05-00776-SN).Notes and references
- (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed; (b) H. M. L. Davies and D. Beckwith, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- (a) Q. Xiao, Y. Zhang and J.-B. Wang, Acc. Chem. Res., 2012, 46, 236–247 CrossRef PubMed; (b) W.-H. Hu, X.-F. Xu, J. Zhou, W.-J. Liu, X.-X. Huang, J. Hu, L.-P. Yang and L.-Z. Gong, J. Am. Chem. Soc., 2008, 130, 7782–7783 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2009, 110, 704–724 CrossRef PubMed; (d) T. Hashimoto and K. Maruoka, Org. Biomol. Chem., 2008, 6, 829–835 RSC; (e) D. C. Cruz, F. Yuste, M. R. Martín, A. Tito and J. L. G. Ruano, J. Org. Chem., 2009, 74, 3820–3826 CrossRef PubMed; (f) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC; (g) Y. Zhang and J.-B. Wang, Chem. Commun., 2009, 36, 5350–5361 RSC; (h) T. Hashimoto, H. Kimura, N. Nakatsu and K. Maruoka, J. Org. Chem., 2011, 76, 6030–6037 CrossRef CAS PubMed.
- For select reviews, see: (a) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed; (b) Y. Kumar, Y. Jaiswl and A. Kumar, J. Org. Chem., 2016, 81, 12247–12257 CrossRef CAS PubMed; (c) P.-J. Zhou, C.-K. Li, S.-F. Zhou, A. Shoberu and J.-P. Zou, Org. Biomol. Chem., 2017, 15, 2629–2637 RSC; (d) P. Saha, S. Kumar Ray and V. K. Singh, Tetrahedron Lett., 2017, 58, 1765–1769 CrossRef CAS; (e) P. Gu, X.-P. Wu, Y. Su, X.-Q. Li, P. Xue and R. Li, Synlett, 2014, 25, 535–538 CrossRef CAS.
- (a) M. Ma, C. Li, L. Peng, F. Xie, X. Zhang and J. Wang, Tetrahedron Lett., 2005, 46, 3927–3929 CrossRef CAS; (b) P. Truong, C. S. Shanahan and M. P. Doyle, Org. Lett., 2012, 14, 3608–3611 CrossRef CAS PubMed; (c) M. B. Rubin and R. Gleiter, Chem. Rev., 2000, 100, 1121–1164 CrossRef CAS PubMed.
- (a) Z. Guo, H. Huang, Q. Fu and W. Hu, Synlett, 2006, 15, 2486–2488 Search PubMed; (b) Y. Yu, Q. Sha, H. Cui, K. S. Chandler and M. P. Doyle, Org. Lett., 2018, 20, 776–779 CrossRef CAS PubMed.
- N. R. O'Connor, P. Bolgar and B. M. Stoltz, Tetrahedron Lett., 2016, 57, 849–851 CrossRef.
- (a) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (b) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- (a) K. P. Kornecki, J. F. Briones, V. Boyarskikh, F. Fullilove, J. Autschbach, K. E. Schrote, K. M. Lancaster, H. M. L. Davies and J. F. Berry, Science, 2013, 342, 351–354 CrossRef CAS PubMed; (b) K. H. Dötz, Angew. Chem., Int. Ed., 1984, 23, 587–608 CrossRef; (c) J. Barluenga, F. Andina, M. A. Fernandez-Rodríguez, P. García-García, I. Merino and E. Aguilar, J. Org. Chem., 2004, 69, 7352–7354 CrossRef CAS PubMed.
- C.-M. Xu, Y.-C. Wang and L. Bai, J. Org. Chem., 2020, 85, 12579–12584 CrossRef CAS PubMed.
- R.-S. Liu, Q.-S. Liu, H.-R. Meng, H.-Y. Ding, J.-D. Hao, Z.-Y. Ji, H.-L. Yue and W. Wei, Org. Chem. Front., 2021, 8, 1970–1975 RSC.
- For select reviews, see: (a) K. Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G. Tárkányi, E. Gábor and S. Tibor, Chem.–Eur. J., 2012, 18, 574–585 CrossRef PubMed; (b) P. Oña-Burgos, A. Kubas, F. C. Falk, F. Breher, K. Fink, G. Lutz and J. Paradies, Dalton Trans., 2012, 41, 9056–9060 RSC; (c) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed; (d) I. Blanca, D. Palomas, S. Holle, S. Steinberg, J. A. Nicasio and M. Alcarazo, Angew. Chem., Int. Ed., 2012, 51, 12367–12369 CrossRef PubMed; (e) Y. Hoshimoto, T. Kinoshita, S. Hazra, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140, 7292–7300 CrossRef CAS PubMed; (f) F. G. M. Alexander, T. Sebastian, T. Schneider, U. Flörke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef PubMed; (g) D. G. Sravanth, R. C. Alpina and B. N. Babu, New J. Chem., 2017, 41, 2328–2332 RSC; (h) Y.-X. Han, S.-T. Zhang, J.-H. He and Y.-T. Zhang, J. Am. Chem. Soc., 2017, 139, 7399–7407 CrossRef CAS PubMed.
- For select reviews, see: (a) M. Cao, A. Yesilcimen and M. Wasa, J. Am. Chem. Soc., 2019, 141, 4199–4203 CrossRef CAS PubMed; (b) M. Hellal, F. C. Falk, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 RSC; (c) M. Dryzhakov, M. Hellal, E. Wolf, F. C. Falk and J. Moran, J. Am. Chem. Soc., 2015, 137, 9555–9558 CrossRef CAS PubMed; (d) G. Ku-maraswamy and M. Gangadhar, ChemistrySelect, 2019, 4, 8973–8977 CrossRef CAS; (e) M. Hatano, K. Hayashi, T. Sakamoto, Y. Makino and K. Ishihara, Synlett, 2016, 27, 1061–1067 CrossRef CAS; (f) M. Hatano, T. Sakamoto, T. Mizuno, Y. Goto and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 16253–16263 CrossRef CAS PubMed; (g) M. Hellal, C. F. Florian, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 RSC; (h) D. Marian and J. Moran, ACS Catal., 2016, 6, 3670–3673 CrossRef.
- S. Basak, L. Winfrey, B. A. Kustiana, R. L. Melen, L. C. Morrill and A. P. Pulis, Chem. Soc. Rev., 2021, 50, 3720–3737 RSC.
- D. G. Sravanthi, A. R. Chari, A. Nagarsenkar, D. K. Sigalapallia and B. N. Babu, New J. Chem., 2017, 41, 2328–2332 RSC.
- F. Ling, L.-X. Shen and W.-H. Zhong, Tetrahedron Lett., 2018, 59, 3678–3682 CrossRef CAS.
- (a) G.-Y. Xu, S.-B. Tang, B. Shao and J.-T. Sun, Chem. Commun., 2019, 55, 9096–9099 RSC; (b) A. Dasgupta, K. Stefkova, R. Babaahmadi, L. Gierlichs, A. Ariafard and R. L. Melen, Angew. Chem., 2020, 59, 15492–15496 CrossRef CAS PubMed; (c) A. Dasgupta, R. Babaahmadi, B. Slater, B. F. Yates, A. Ariafard and R. L. Melen, Chem, 2020, 6, 2364–2381 CrossRef CAS; (d) J. P. Mancinelli and S. M. Wilkerson-Hill, ACS Catal., 2020, 10, 11171–11178 CrossRef CAS; (e) J. Fang and M. Brewer, Org. Lett., 2018, 20, 7384–7387 CrossRef CAS PubMed; (f) M. J. Hensinger, N. J. Dodge and M. Brewer, Org. Lett., 2020, 22, 497–500 CrossRef CAS PubMed; (g) H.-H. San, C.-Y. Wang, H.-P. Zeng, S.-T. Fu, M. Jiang and X.-Y. Tang, J. Org. Chem., 2019, 84, 4478–4485 CrossRef CAS PubMed.
- H.-H. San, S.-J. Wang, M. Jiang and X.-Y. Tang, Org. Lett., 2018, 20, 4672–4676 CrossRef CAS PubMed.
- (a) Y. Dong, H. Zhang, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, ACS Omega, 2019, 4, 21567–21577 CrossRef CAS PubMed; (b) X.-Y. Zhan, H. Zhang, Y. Dong, J. Yang, S. He, Z.-C. Shi, L. Tang and J.-Y. Wang, J. Org. Chem., 2020, 85, 6578–6592 CrossRef CAS PubMed; (c) H. Zhang, X.-Y. Zhan, Y. Dong, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, RSC Adv., 2020, 10, 16942–16948 RSC; (d) G.-M. Zhang, H. Zhang, B. Wang and J.-Y. Wang, RSC Adv., 2021, 11, 17025–17031 RSC; (e) B. Wang, H. Xu, H. Zhang, G.-M. Zhang, F.-Y. Li, S. He, Z.-C. Shi and J.-Y. Wang, Org. Chem. Front., 2021, 8, 6670–6677 RSC.
- X.-Y. Wu, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Adv. Synth. Catal., 2022, 364, 750–754 CrossRef CAS.
- (a) W.-B. Liu, C. Chen and P. N. Zhou, J. Org. Chem., 2017, 82, 2219–2222 CrossRef CAS PubMed; (b) J.-J. Da, H.-Y. Ji Jeon, H.-J. Kim, Y. Kim and S. Lee, Org. Lett., 2014, 16, 2208–2211 CrossRef PubMed; (c) W. Ai, Y. Liu, Q. Wang, Z. Lu and Q. Liu, Org. Lett., 2018, 20, 409–412 CrossRef CAS PubMed.
- D. P. Higley and R. W. Murray, J. Am. Chem. Soc., 1974, 96, 3330–3332 CrossRef CAS.
- (a) M. Santi, D. M. C. Ould, J. Wenz, Y. Soltani, R. L. Melen and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 7861–7865 CrossRef CAS PubMed; (b) S. Rao, R. Kapanaiah and K. R. Prabhu, Adv. Synth. Catal., 2019, 361, 1301–1306 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05739e |
| ‡ B. Wang and G.-M. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |