DOI:
10.1039/D2RA05565A
(Paper)
RSC Adv., 2022,
12, 34843-34850
Novel pharmaceutical salts of cephalexin with organic counterions: structural analysis and properties†
Received
4th September 2022
, Accepted 10th November 2022
First published on 6th December 2022
Abstract
Three novel pharmaceutical salts of cephalexin (CPX) with 2,6-dihydroxybenzoic acid (DHBA), 5-chlorosalicylic acid (CSA) and 5-sulfosalicylic acid (SSA), which were obtained and thoroughly explored by various analytical techniques, were found to be crystallized invariably in hydrated forms. It is the proton transfer from carboxylic or sulfonic counterions to the CPX molecules that results in the salt formation. Crystal structure analyses reveal that the N–H⋯O and O–H⋯O hydrogen bonding interactions among the CPX, acidic guest molecules and water molecules play a crucial role in the packing motifs of crystal stabilization. All the salts exhibit higher solubility compared with the parent drug. These salts offer an alternative way of increasing the number of solid forms for CPX, which facilitates selection of a suitable form in the context of drug formulation development for further repurposing investigations.
1. Introduction
The active pharmaceutical ingredient (API) is a drug or a chemical liable for the pharmacological and therapeutic activities to treat a disease or disorder. The poor physicochemical properties of APIs which prohibit their utilization in formulation development are nonnegligible disadvantages in the pharmaceutical industry.1–6 In recent years, crystal engineering facilitating the design and synthesis of novel solid forms of APIs with optimized physicochemical properties has emerged into the fields of the pharmaceutical industry with paramount importance in the process of drug development.7–12 Such various solid forms of APIs including amorphous, polymorphs, hydrates, solvates, cocrystals and salts formulation can enable the improvement of solubility, dissolution, stability and bioavailability, without affecting the pharmacological activities of APIs.13–19 In the pharmaceutical industry, salt formation, which shows a dramatic enhancement of the water solubility, is of first priority among the different solid-state formulation methods and more than 50% of drug APIs are marketed in the form of salts.20–23 The preparation of salts demands that APIs have ionizable functional groups and the acid–base differences between the drug substance and guest molecules meet the ΔpKa rule of 3.24–26 However, the design of salts with a suitable selection of counterions and synthon compatibility, which would render expected properties of a new solid form, is still a challenging area of research.
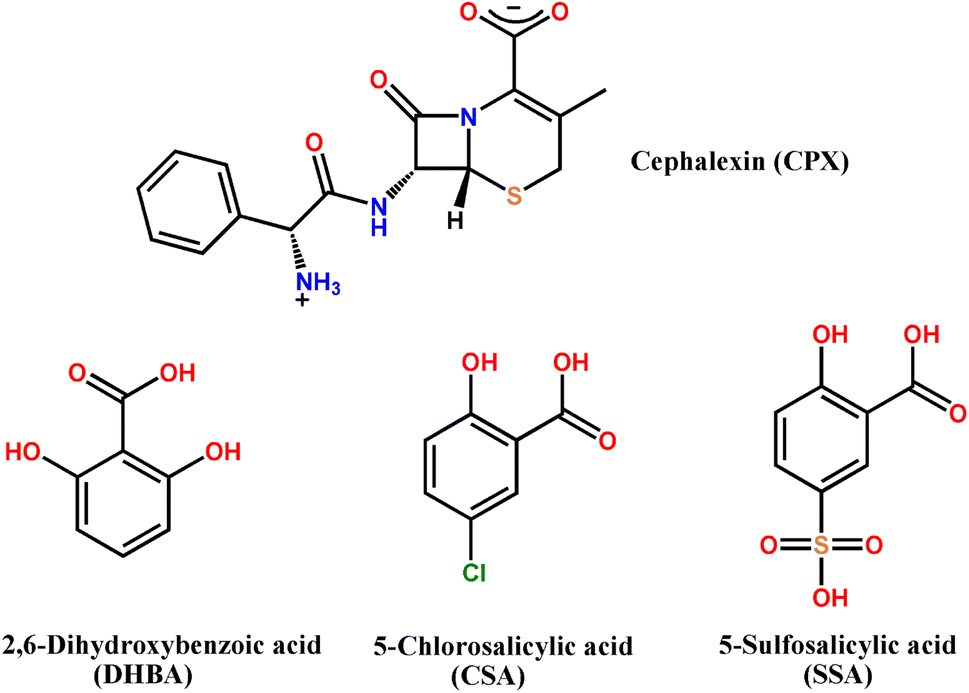
Cephalexin (CPX), ((6R,7R)-7-[(R)-2-amino-2-phenylacetamido]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylic acid), is the first generation of β-lactam cephalosporin antibiotics, which exhibits excellent activities in killing Gram-positive and some Gram-negative bacteria through inhibition of cell wall synthesis. It is effectively used for the treatment of infections caused by bacteria, including skin infections, upper respiratory infections, bone infections and urinary tract infections, with the characteristics of a broad antibacterial spectrum, strong antibacterial activity and less prone to cause allergic reactions. In recent years, it has maintained a high growth rate of usage and is a kind of medicine with research significance and functionality. CPX exhibits slight solubility in water. However, CPX usually exists in the form of a hydrate and emerges in a zwitterionic form with the acid/base interaction between the basic primary amine (pKa = 7.23) and the carboxylic acid group (pKa = 3.26), showing better aqueous solubility. Except the hydrates, salt formation is of great interest as well to improve the aqueous solubility of CPX for the design of dosage forms to be taken orally. Furthermore, although a tremendous amount of study has been carried out on the CPX molecule, the crystal structure data of CPX in either the pure or the complexed state have rarely been reported. Only two kinds of hydrates, CPX·1.9H2O and CPX·H2O, and one hydrated cocrystal with the coformer of β-naphthol have been recorded.27–30 Recently, Braga et al. synthesized a cocrystal hydrate of CPX with thymol, which exists in two polymorphic forms.31 Nevertheless, there is no structural evidence for any other crystal forms, especially the salt formulation of CPX.
The main objective of this work was to explore the salts preparation of CPX. Herein, we report three novel pharmaceutical salts involving CPX in the presence of organic counterions of 2,6-dihydroxybenzoic acid (DHBA), 5-chlorosalicylic acid (CSA) and 5-sulfosalicylic acid (SSA) (Scheme 1), namely CPX·DHBA·H2O (salt 1), 2CPX·2CSA·3H2O (salt 2) and CPX·SSA·4H2O (salt 3). Interestingly, three salts were all obtained in hydrate forms. The crystal structures of CPX salts were elucidated by single crystal X-ray diffraction data. A broad range of analytical techniques, consisting of powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), thermogravimetry (TGA) analyses and infrared (IR) spectroscopy, have been employed to characterize the new solid forms of CPX. UV-vis spectral measurements were conducted to check the improvement of CPX salts. Moreover, the Hirshfeld analyses were utilized to shed light on the significance of intermolecular interactions like hydrogen bonding in structure stabilization of pharmaceutical salts.
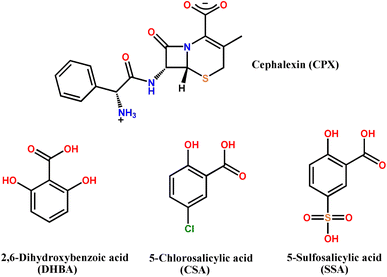 |
| | Scheme 1 Molecular diagrams of CPX and the salt formers used in this work. CPX was used in its zwitterionic form. | |
2. Experimental section
2.1 Materials
The equired reagents and solvents for the synthesis are commercially available and were used without further purification. Cephalexin monohydrate (CPX, C16H19N3O5S, 98%) and 5-sulfosalicylic acid dihydrate (SSA, C7H10O8S, 99%) were purchased from Macklin reagent. 2,6-Dihydroxybenzoic acid (DHBA, C7H6O4, 98%) and 5-chlorosalicylic acid (CSA, C7H5O3Cl, 99%) were obtained from the Aladdin reagent company. Analytical grade solvents were used for the crystallization experiments.
2.2 Preparation of salts 1–3
Preliminary screening experiments were employed to validate new solid forms of cephalexin. Several pharmaceutically acceptable carboxylic and sulfonic acids were chosen in light of their solubility and a range of functional groups that show great potential to form hydrogen bonds with complementary sites in the cephalexin molecule. Among them, three single crystals of CPX salts were obtained with organic counterions of DHBA, CSA and SSA as coformers, respectively. A mixture of equimolar CPX (109.6 mg, 0.3 mmol) and DHBA (46.2 mg, 0.3 mmol) was dissolved in 15 mL of aqueous solution under stirring. The solution was heated to 50 °C and 2 mL of ethanol solvent was added until all the solids were completely dissolved. The resulting homogeneous solution was filtered and evaporated slowly under ambient conditions. After two weeks, yellow strip-like crystals of salt 1 (yield 68%, based on DHBA) were obtained.
The same method was exploited for the synthesis of salts 2 and 3 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio of CPX and corresponding salt formers. Then yellow strip-like crystals of salts 2 (yield 75%, based on CSA) and 3 (yield 61%, based on SSA) were harvested by slow evaporation at room temperature for more than one month.
:1 stoichiometric ratio of CPX and corresponding salt formers. Then yellow strip-like crystals of salts 2 (yield 75%, based on CSA) and 3 (yield 61%, based on SSA) were harvested by slow evaporation at room temperature for more than one month.
2.3 Single crystal X-ray diffraction
Good quality single crystals crystallized from the solution were chosen for the structure determination. X-ray reflection data of salt 1 was collected on a Rigaku XtalAB mini II diffractometer equipped with a graphite monochromator and Mo Kα radiation (λ = 0.71073 Å) operating at 293 K. CrysAlisPro 1.171.39.46e (Rigaku OD, 2018) was used to perform such data reduction and multi-scan absorption correction. The data collection for salts 2 and 3 was performed on a Bruker APEX-II CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) at 150 K. The data reduction, scaling and absorption corrections were processed using SAINT (Bruker, V8.40B, after 2013). The crystal structures for the three compounds were all solved by direct methods and refined by full-matric least-squares on F2 implemented in the SHELXTL-2014 program. The non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms bonded to the C atoms were generated geometrically and refined using the riding model with Uiso(H) = 1.2 Ueq(C) or 1.5 Ueq(C). The H atoms on the N and O atoms were located from electron density difference-Fourier maps and freely refined isotropically. The crystallographic data are presented in Table 1 and can be obtained free of charge from Cambridge Crystallographic Centre with CCDC numbers 2117239–2117241†.
Table 1 Crystallographic data of salts 1–3
| Compound |
CPX·DHBA·H2O (salt 1) |
2CPX·2CSA·3H2O (salt 2) |
CPX·SSA·4H2O (salt 3) |
| Empirical formula |
C23H25N3O9S |
C46H50N6O17S2Cl2 |
C23H31N3O14S2 |
| Formula weight |
519.52 |
1093.94 |
637.63 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21 |
C2 |
P21 |
| T (K) |
293 |
150 |
150 |
| a (Å) |
11.3188(13) |
30.1649(13) |
14.7579(9) |
| b (Å) |
7.2613(9) |
7.7132(3) |
7.0785(3) |
| c (Å) |
14.983(2) |
10.9391(4) |
27.0868(16) |
| α (°) |
90 |
90 |
90 |
| β (°) |
95.858(12) |
95.377(1) |
97.464(2) |
| γ (°) |
90 |
90 |
90 |
| V (Å3) |
1225.0(3) |
2533.98(17) |
2805.6(3) |
| Z |
2 |
2 |
4 |
| Dx (g cm−3) |
1.408 |
1.434 |
1.510 |
| F(000) |
544.0 |
1140.0 |
1336.0 |
| Unique reflections |
3942 |
5090 |
11413 |
| Parameters refined |
330 |
334 |
767 |
| GOF |
0.998 |
1.091 |
1.149 |
| R1 |
0.0436 |
0.0472 |
0.0567 |
| wR2 |
0.1080 |
0.1164 |
0.1280 |
| CCDC |
2117239 |
2117240 |
2117241 |
2.4 PXRD measurements
PXRD data were recorded on the Rigaku Ultima IV diffractometer equipped with Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA) at room temperature. Powder samples obtained by grinding were measured in the reflection mode in the 2θ range of 5–45° at a scan rate of 5° min−1. Mercury 2020.3.0 was used to simulate diffraction patterns from the single-crystal X-ray diffraction data.
2.5 IR spectroscopy
IR spectra were collected on a Nicolet 5700 FT-IR Spectrometer. All of the samples were dispersed in KBr pellets over the scanning range of 4000–400 cm−1 with the resolution of 4 cm−1.
2.6 Thermal analysis
TGA and DSC analyses were carried out on a NETZSCH STA 449F3 instrument. The samples (5–6 mg) were kept under nitrogen atmosphere in the temperature range of 30–600 °C at a heating rate of 10 °C min−1.
2.7 Hirshfeld surface analysis and fingerprint plot
The three-dimensional (3D) Hirshfeld surface analysis was carried out on the CrystalExplorer21 (ref. 32) based on the crystal structure to clarify the nature of intermolecular close contacts in a crystal. A two-dimensional (2D) fingerprint plot generated from these reduced Hirshfeld surfaces enables quantitative comparison of diverse intermolecular interactions to the packing arrangement.
2.8 Phase solubility study
Bulk crystals used in the solubility studies were gently ground up into powder samples. Solubility experiments in distilled water were performed using UV-vis spectroscopy on a SHIMADZU UV-2550 UV-vis spectrometer after appropriate dilution. CPX showed absorbance maxima at 260 nm in the UV-vis spectra. Prior to the solubility measurements, calibration curves of every sample were constructed (Fig. S9†). To determine the solubility of CPX and its salts, saturated aqueous solutions were prepared and filtered with a 0.22 μm nylon filter and analysed from the corresponding calibration curve.
3. Results and discussion
3.1 PXRD analysis
Powder X-ray diffraction is frequently used to verify that new crystalline materials are established. All the multi-component salts display unique crystalline PXRD patterns in comparison to the initial components of CPX (Fig. 1), which indicate the formation of new solid forms. The PXRD patterns of cephalexin monohydrate are in good accordance with the literature data.27 The experimental PXRD patterns of salts 1–3 also match well with those calculated from the crystal structures, confirming the phase purities of the powder samples. The powder diffraction characteristic peaks of salts 1–3 remained unchanged under ambient temperature, pressure over time, indicating that the three pharmaceutical salts had good physical stability (Fig. S1–S3†).
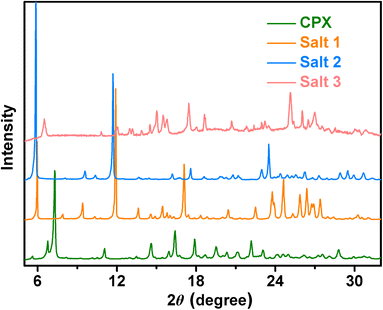 |
| | Fig. 1 Experimental PXRD patterns of parent CPX and salts 1–3. | |
3.2 ΔpKa rule
It is widely known that whether an API and a guest molecule can form a salt or cocrystal can be forecasted in light of the ΔpKa rule. When the ΔpKa is greater than 3, a salt is usually expected to form. If the ΔpKa is less than 0, the reactant constituents tend to form a cocrystal. However, in the range of 0 < ΔpKa < 3, the formation of a salt or cocrystal is unpredictable in such a situation. Considering the first ionization constants of the acid molecules employed in this study, the ΔpKa values of CPX salts are estimated to be 5.59 for DHBA (pKa = 1.64), 4.64 for CSA (pKa = 2.59), and 10.04 for SSA (pKa = −2.81), respectively. Based on the ΔpKa rule of 3, it is evident that the crystallization of CPX in the presence of such counterions probably results in the formation of corresponding salts.
3.3 Crystal structure analysis
3.3.1 Salt 1 of cephalexin with 2,6-dihydroxybenzoic acid (CPX·DHBA·H2O). Salt 1 crystallized in the monoclinic space group of P21 with one CPX cation, one DHBA anion and one water molecule in the asymmetric unit. For the DHBA molecules, it is worthwhile to note that the difference between the two C–O bond distances ΔDC–O in the carboxylate group is capable of illustrating the proton transfer (Table 2). The small ΔDC–O value of 0.01 Å provides evidence for such a transfer of proton and the formation of a salt.33,34 A carboxylic acid proton is transferred from DHBA to the terminal N1 atom of the CPX molecule resulting in a strong ionic hydrogen bond N1+–H1A⋯O6− (Fig. 2a). In the deprotonated DHBA molecule with the hydroxyl groups distributed in adjacent locations on both sides of the carboxyl group, two intramolecular hydrogen bonds of O7–H7⋯O5− and O8–H8⋯O6− are distinctly found. Meanwhile, as shown in Fig. 2a and b, the protonated CPX molecule links with one DHBA and another two CPX molecules through the hydrogen bonds of N1+–H1A⋯O6−, N1+–H1B⋯O2 and N1+–H1C⋯O1, respectively. In particular, the water molecules are located in the interstitial sites and act as bridges connecting the CPX and DHBA molecules via accepting one hydrogen bond of O1–H1⋯O9 from one CPX molecule and donating the two hydrogen bonds of O9–H9A⋯O5− and O9–H9B⋯O3 to one DHBA molecule and the other CPX molecule, respectively. The units are self-assembled in the crystal structure via such above mentioned hydrogen bonds and form a layered network in the crystallographic ac-plane (Fig. 2c).
Table 2 Distribution of C–O bond lengths in the anions for salts 1–3
| Complex |
DC–O (Å) |
ΔDC–O (Å) |
| Salt 1 |
C23–O6: 1.266(4), C23–O5: 1.256(4) |
0.01 |
| Salt 2 |
C23–O6: 1.272(5), C23–O7: 1.253(4) |
0.019 |
| Salt 3 |
C23–O7: 1.311(6), C23–O6: 1.225(6) |
0.086 |
| C46–O17: 1.307(6), C46–O16: 1.226(6) |
0.081 |
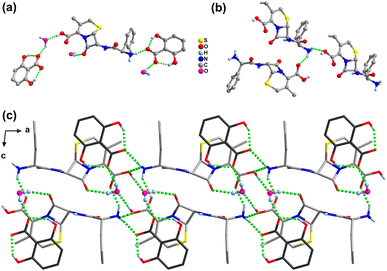 |
| | Fig. 2 The molecular structure of salt 1. (a) Hydrogen bonding interactions among CPX, DHBA and water molecules. (b) Hydrogen bonding interactions among CPX molecules. (c) The two-dimensional packing arrangement in the ac-plane (C atoms in DHBA molecules in black). For the sake of clarity, the O atoms of water molecules are drawn in purple and H atoms on C atoms have been omitted. Hydrogen bonds are shown as green dashed lines. | |
3.3.2 Salt 2 of cephalexin with 5-chlorosalicylic acid (2CPX·2CSA·3H2O). The crystal structure of molecular salt 2 was solved in the monoclinic C2 space group, involving one and a half water molecules, one CPX cation and one CSA anion in the asymmetric unit. It is the proton transfer from 5-chlorosalicylic acid to the CPX molecule that results in the salt formation, which is further corroborated by the ΔDC–O (0.019 Å) of the carboxylate group in the CSA molecule (Table 2). The proton transfer may be promoted by the water molecule from the carboxylic acids to the carboxylate group of CPX due to the zwitterionic form in the hydrate form. In the asymmetric unit of salt 2, the CPX, CSA and one water molecule all occupy the normal positions, while a half water molecule is located at a special position with a crystallographic 2-fold rotation axis along the b direction. The salt structure is primarily stabilized by hydrogen bonding interactions between the CPX, CSA and water molecules. Among them, a CSA anion is connected with CPX via the N3+–H3B⋯O5 hydrogen bond between the protonated amino donor of CPX and the phenolic hydroxyl of CSA molecule, as shown in Fig. 3a and b, linking with the other CPX as well via N2–H2A⋯O7− hydrogen bond between the non-protonated imino donor of CPX and the carboxylate acceptor of CSA to form a trimeric unit. As expected, a conventional intramolecular hydrogen bond O5–H5⋯O6− between a phenolic hydroxyl donor and carboxylate accepter is also involved in the CSA anion. Meanwhile, the N3+–H3A⋯O1 hydrogen bonds between the protonated amino donor of CPX with the carboxylate accepter of the other drug assemble the trimeric units into a chain expanded along the c-axis. Such a protonated amino donor of CPX further links with the carbonyl acceptor of another drug via the N3+–H3C⋯O3 hydrogen bond. In addition, the water molecules are also involved in strong hydrogen bonding interactions and presented as bridges between the CPX cations and CSA anions, manipulating the one-dimensional chains into the two-dimensional layered framework in the crystallographic bc-plane (Fig. 3c).
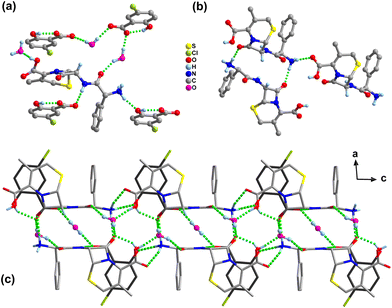 |
| | Fig. 3 The molecular structure of salt 2. (a) Hydrogen bonding interactions among CPX, CSA and water molecules. (b) Hydrogen bonding interactions among CPX molecules. (c) The two-dimensional packing arrangement in the ac-plane (C atoms in CSA molecules in black). For the sake of clarity, the O atoms of water molecules are drawn in purple and H atoms on C atoms have been omitted. Hydrogen bonds are shown as green dashed lines. | |
3.3.3 Salt 3 of cephalexin with 5-sulfosalicylic acid (CPX·SSA·4H2O). Single crystal X-ray diffraction analysis revealed that salt 3 crystallized in the monoclinic crystal system with the P21 space group. The asymmetric unit is comprised of two CPX cations (A and B), two SSA anions (C and D) and eight water molecules, and no higher symmetry could be identified in the monoclinic cell. On account of the presence of the eight water molecules of solvation in salt 3, the hydrogen bonding interactions are much more extensive than those in salts 1 and 2. In this case, only one proton of every 5-sulfosalicylic acid has transferred to the CPX molecule, while the COOH and phenol OH of CSA molecule remain unprotonated (Fig. 4a). The reason may be that the sulfonic acid is much stronger than the carboxylic acid. Similar to salt 2, the proton may be delivered through the water molecule from the sulfonic acids to the carboxylate group of CPX. The S–O bond distances in the SO3− groups of SSA-C and SSA-D molecules are in the range of 1.444(4)–1.458(4) Å and 1.450(4)–1.454(4) Å, respectively. The corresponding O–S–O bond angles vary from 111.3(3)° to 113.2(3)° and 111.6(2)° to 112.8(3)°. Such bond lengths and angles for SSA-C and SSA-D are both in the range of the monodeprotonated SO3− moiety.35 In addition, according to the relatively large ΔDC–O values of 0.086 and 0.081 Å for the SSA-C and SSA-D molecules (Table 2), respectively, the carboxylic acid groups in the two SSA molecules are evidenced to remain unionized.
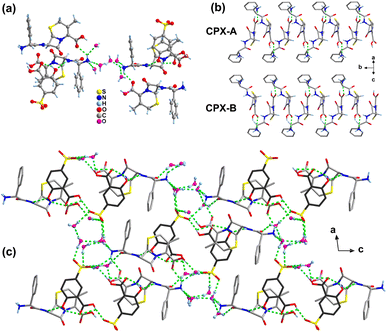 |
| | Fig. 4 The molecular structure of salt 3. (a) Part of hydrogen bonding interactions linking CPX, SSA and water molecules. (b) One-dimensional cationic chains of CPX-A and CPX-B along the b-axis. (c) Packing diagram of salt 3 in the ac-plane (C atoms in SSA molecules in black). For the sake of clarity, the O atoms of water molecules are drawn in purple and H atoms on C atoms have been omitted. Hydrogen bonds are shown as green dashed lines. | |
In the structure of salt 3, the protonated N3 atom of the CPX-A cation as the donor forms four hydrogen bonds with three water molecules (N3+–H3A⋯O24, N3+–H3B⋯O23, N3+–H3B⋯O25) and one COOH group (N3+–H3C⋯O2) of the adjacent CPX-A molecule, resulting in a one-dimensional cationic chain along the b-axis (Fig. 4b). In the meantime, the unprotonated N2 atom of the CPX-A cation also forms a hydrogen bond N2–H2A⋯O5 as donation with the phenol OH group of the SSA-C molecule. With regard to the CPX-B cation, there appears to demonstrate similar hydrogen bonding interactions as CPX-A with adjacent molecules. For the SSA anions in salt 3, like salts 1 and 2, the usual intramolecular hydrogen bond is found between the phenol OH group and the O atom of carboxylic acid group as O5–H5⋯O6 (SSA-C) and O15–H15A⋯O16 (SSA-D). The O7 atom of the carboxylic acid group in SSA-C serves as a donor link sulfonate O18 atom with the hydrogen bond of O7–H7⋯O18−, while the carboxylic O17 atom in SSA-D serving as a donor is connected with the water molecule by the O17–H17A⋯O21 hydrogen bond. It is also of particular interest that the three O atoms of the sulfonate group in the SSA-C molecule form five hydrogen bond associations with five water molecules, as listed in Table S3,† yet sulfonate O atoms in SSA-D molecules form six hydrogen bonds with five water molecules and one COOH group of the SSA-C molecule. The water molecules also act as bridges between CPX and SSA molecules, allowing propagation of the structure through hydrogen bonding interactions and resulting in the three-dimensional framework structure (Fig. 4c).
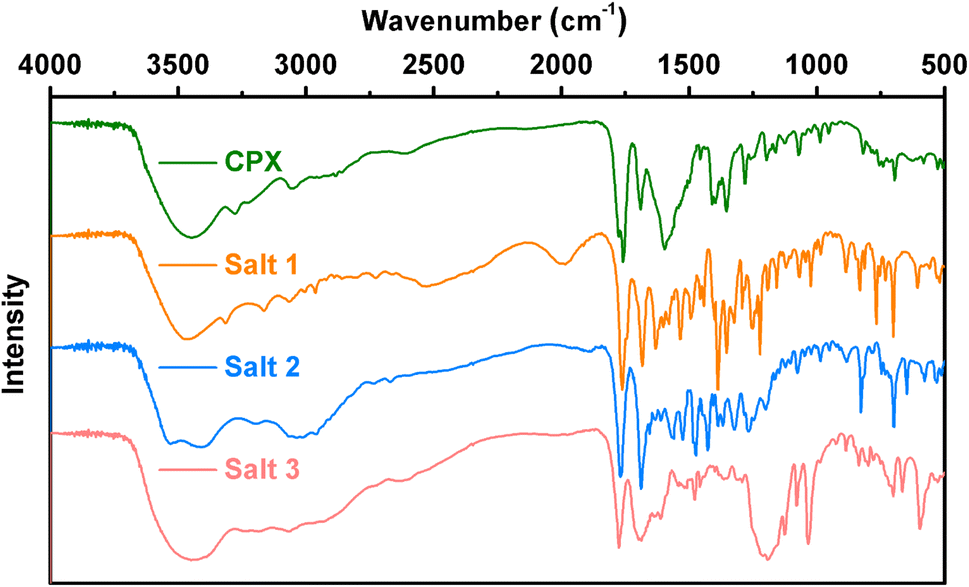
3.4 Infrared spectroscopy analysis
Additional insights into the structural features of salts 1–3 are supported by vibrational spectroscopy analysis, associated with the identification of the formation of novel multicomponent crystals. As shown in Fig. 5, intense absorption bands for salts 1–3 appearing around 1760 cm−1 indicate that the COOH group of the CPX molecule is unionized in the three salts. The peak at approximately 1680 cm−1 in the spectrum of CPX can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the ketone carbonyl. In the meantime, the two characteristic carboxylate peaks around 1550 and 1400 cm−1 in salts 1 and 2 are due to asymmetric and symmetric O–C–O stretching, respectively, while the characteristic IR absorption peaks at 1033 and 1190 cm−1 for salt 3 are ascribed to the SO stretching vibration of the sulfonate moiety. Furthermore, the moderate absorption band around 1500 cm−1 in all of the spectra confirms the protonation (–NH3+) of the primary amine group, of which the protons are transferred from the zwitterionic CPX molecules or salt formers.
O stretch of the ketone carbonyl. In the meantime, the two characteristic carboxylate peaks around 1550 and 1400 cm−1 in salts 1 and 2 are due to asymmetric and symmetric O–C–O stretching, respectively, while the characteristic IR absorption peaks at 1033 and 1190 cm−1 for salt 3 are ascribed to the SO stretching vibration of the sulfonate moiety. Furthermore, the moderate absorption band around 1500 cm−1 in all of the spectra confirms the protonation (–NH3+) of the primary amine group, of which the protons are transferred from the zwitterionic CPX molecules or salt formers.
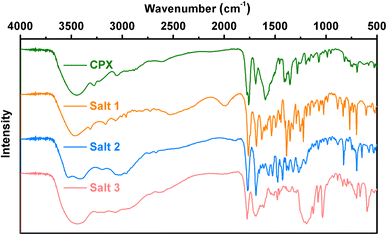 |
| | Fig. 5 FT-IR spectra of CPX and salts 1–3. | |
3.5 Thermal analysis
The thermal stability of cephalexin and its salts was investigated by DSC and TGA analysis (Fig. S4–S7†). The DSC thermograms of salts 1–3 and CPX confirm their melting points as endothermic peaks observed at 191, 152, 202 and 195 °C, respectively. As expected, the melting properties of the three multicomponent crystals are obviously different from that of CPX, signifying the formation of novel phases. The TGA curves display an early weight loss (60–120 °C) of 3.9%, 4.4%, 12.9% and 5.3% in salts 1–3 and CPX, respectively, denoting the presence of water molecules in the lattice, as is confirmed by the structural and PXRD analysis as well. After the early loss, CPX remains stable up to 190 °C, while salts 1 and 2 exhibit inferior stability up to approximately 180 and 150 °C, respectively. Fortunately, salt 3 shows a relatively higher stability up to 210 °C, after which the organic components start decomposing.
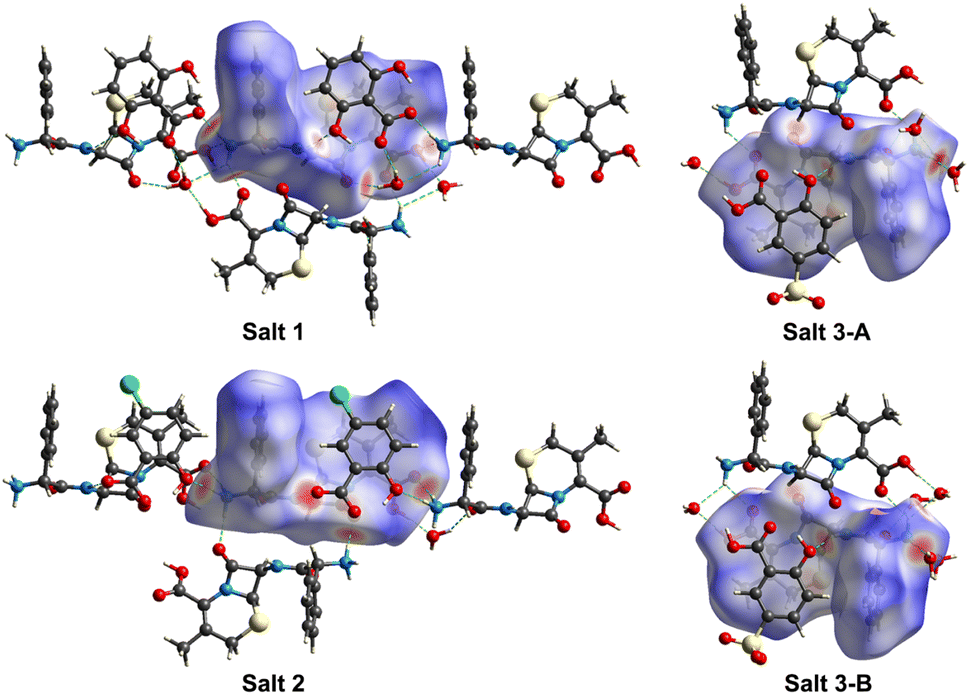
3.6 Hirshfeld surfaces and 2D fingerprint plots analyses
3D Hirshfeld surfaces analysis is a valuable tool for the visualization and exploration of intermolecular interactions towards the stability and arrangement of molecule in the crystal lattice of pharmaceutical salts 1–3. The distances from the Hirshfeld surface to the nearest nucleus outside and inside the surface are designated as de and di, respectively. The normalized contact distance dnorm is based on de, di and the van der Waals radii of the atoms,36,37 which can be mapped onto the Hirshfeld surface for easy comparison of intermolecular contacts. As shown in Fig. 6, the Hirshfeld surfaces of CPX molecules in pharmaceutical salts 1–3 denote the area encoded with different colours, in which the white regions imply the contacts with a distance close to the sum of the van der Waals radii and the contacts shorter and longer than the van der Waals radii are visualized as red and blue colours in the Hirshfeld surface, respectively. Considering the two CPX molecules in the asymmetric unit of salt 3, both CPX molecules were explored by Hirshfeld surfaces analyses individually and taken as salts 3-A and 3-B, respectively. According to the 3D Hirshfeld surface figures, similarities and differences of the influences of the three different co-formers are demonstrated on the intermolecular interactions of CPX molecules in salts 1–3. The large red circular regions on the dnorm surfaces indicate the presence of strong hydrogen bonding interactions, especially when there is a close contact caused by proton transfer. From the 3D Hirshfeld surface figures, the N atom of the ammonium group of CPX molecule displayed in the largest red area suggests strong N–H⋯O hydrogen bonds. The red area also appears around the O atom of the carboxylic group, representing the O–H⋯O hydrogen bonds. The protonated amine group and carboxylic group act as the hydrogen-bonding site, which interacts with organic counterions, water and other CPX molecules.
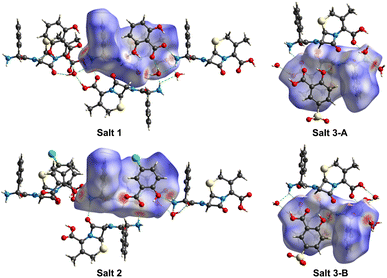 |
| | Fig. 6 Hirshfeld surfaces of CPX molecules in salts 1–3 generated on dnorm parameters with di and de. Two CPX molecules in the asymmetric unit of salts 3 were explored individually and recorded as salts 3-A and 3-B, respectively. | |
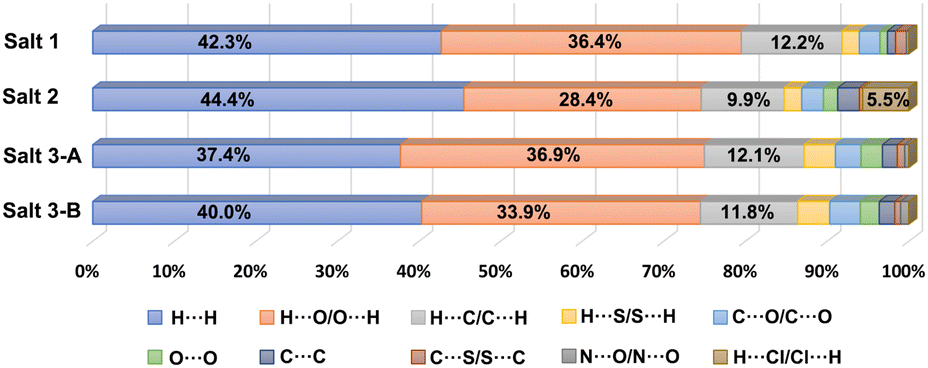
The 2D fingerprint plots derived from the Hirshfeld surface, which are unique for a crystal structure, were employed to identify and compare various kinds of intermolecular contacts in pharmaceutical salts 1–3. As illustrated in Fig. S8,† the main body of the fingerprint plots for the three salts was associated with the H⋯H contacts. The two sharp spikes projecting along the diagonal of the plots were derived from the H⋯O/O⋯H contacts relative to O–H⋯O and N–H⋯O hydrogen bonding interactions, where the upper and lower spikes are associated with the hydrogen bond donor and acceptor, respectively. The H⋯C/C⋯H contacts contributed to the wings of the fingerprint plots. The relative contributions of the different intermolecular interactions to the Hirshfeld area of the CPX molecules in the crystal structures of three pharmaceutical salts are summarized in Fig. 7. It can be seen that H⋯H contacts which represent interactions to the Hirshfeld area of the CPX molecules in the crystal structures of three pharmaceutical salts are summarized as follows: the van der Waals forces exhibit the dominant contributions (37.4%–44.4%) in all the crystals, followed by H⋯O/O⋯H (28.4–36.9%) and H⋯C/C⋯H (9.9%–12.2%) interactions. In salts 1–3, the H⋯O/O⋯H contacts are the strongest intermolecular interaction and the interaction of H⋯H is the main force on the Hirshfeld surface. It is the strength and directional feature that make hydrogen bonds play an essential role in the construction of crystals that are responsible for the stability. Notably, another type of weak H⋯Cl/Cl⋯H contacts was also found to make contributions of 5.5% for the surface of salt 2 in the crystal structure formation. The distinctive percentages occupied by these contacts change along with variation in the crystal structures.
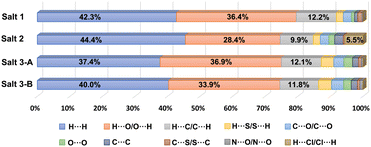 |
| | Fig. 7 Relative contributions of various intermolecular contacts to the Hirshfeld surface area for CPX molecules in salts 1–3. Salts 3-A and 3-B denote the two CPX molecules in the asymmetric unit of salt 3. | |
3.7 Aqueous solubility
Solubility is one of the key factors for drug oral activity and pharmaceutical preparation. Salt formation is a useful strategy to fine-tune the solubility properties of the parent API. The solubility of cephalexin salts was determined in water at room temperature and compared with that of the parent cephalexin monohydrate (Table 3). The solubility value obtained for cephalexin monohydrate (5.6 mg mL−1) is in good agreement with that reported in the literature.31 The three pharmaceutical salts all show improved solubility compared to pure CPX. Salt 3 with the coformer of SSA is found to be 6.32 times more soluble than the parent drug. Salt 1 shows a relatively moderate 3.09 times improvement in the solubility of CPX. Salt 2 is the least soluble salt, whose solubility is 2.98 times higher than CPX.
Table 3 Solubility in water of CPX and its salts
| Complex |
Solubility (mg mL−1) |
Solubility enhancement compared with parent CPX |
| CPX |
5.60 ± 0.003 |
|
| Salt 1 |
17.30 ± 0.001 |
3.09 |
| Salt 2 |
16.68 ± 0.003 |
2.98 |
| Salt 3 |
35.39 ± 0.004 |
6.32 |
4. Conclusions
In summary, we have obtained three hydrated pharmaceutical salts 1–3 of CPX with acidic organic counterions of DHBA, CSA and SSA, respectively, for the first time, which were structurally characterized by single crystal and powder X-ray diffraction. The salts formation and proton transfer were also demonstrated by the pKa difference between CPX and the carboxylic and sulfonic acids and spectroscopy analyses. The crystal structures of all the salts were stabilized by the N–H⋯O and O–H⋯O hydrogen bonding interactions formed by the protonated CPX species, carboxylic or sulfonic counterions and water molecules. The donations of various intermolecular interactions that facilitate the crystal packing of three pharmaceutical salts were further studied by Hirshfeld surfaces and 2D fingerprint plots analyses. Solubility measurements in water confirmed that the three salts exhibit higher solubility that the parent CPX. The novel solid forms provide a better understanding of the structural landscape of cephalexin.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support of the National Nature Science Foundation of China (grant number 22201134) and the Natural Science Foundation for Colleges and Universities of Jiangsu Province (grant number 22KJB150028).
Notes and references
- D. Guan, B. Xuan, C. Wang, R. Long, Y. Jiang, L. Mao, J. Kang, Z. Wang, S. F. Chow and Q. Zhou, Pharmaceutics, 2021, 13, 2160 CrossRef CAS
 .
. - L. Li, X.-H. Yin and K.-S. Diao, RSC Adv., 2020, 10, 36125–36134 RSC .
- P. Sanphui, M. K. Mishra, U. Ramamurty and G. R. Desiraju, Mol. Pharmaceutics, 2015, 12, 889–897 CrossRef CAS .
- M. Guo, X. Sun, J. Chen and T. Cai, Acta Pharm. Sin. B, 2021, 11, 2537–2564 CrossRef CAS PubMed .
- S. G. Khare, S. K. Jena, A. T. Sangamwar, S. Khullar and S. K. Mandal, Cryst. Growth Des., 2017, 17, 1589–1599 Search PubMed .
- S. SeethaLekshmi, T. S. Thakur and S. Varughese, J. Photochem. Photobiol., C, 2021, 49, 100455 CrossRef CAS .
- P. Vishweshwar, J. A. McMahon, M. L. Peterson, M. B. Hickey, T. R. Shattock and M. J. Zaworotko, Chem. Commun., 2005, 36, 4601–4603 RSC .
- X. Fu, J. Li, L. Wang, B. Wu, X. Xu, Z. Deng and H. Zhang, RSC Adv., 2016, 6, 26474–26478 Search PubMed .
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CrossRef CAS PubMed .
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017, 117, 3–24 Search PubMed .
- D. P. Elder, R. Holm and H. L. de Diego, Int. J. Pharm., 2013, 453, 88–100 Search PubMed .
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS .
- L. Yu, Adv. Drug Delivery Rev., 2001, 48, 27–42 CrossRef CAS .
- B. C. Hancock and G. Zograf, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS .
- A. Sokal, E. Pindelska, L. Szeleszczuk and W. Kolodziejski, Int. J. Pharm., 2017, 522, 80–89 CrossRef CAS PubMed .
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Adv. Drug Delivery Rev., 2017, 117, 25–46 CrossRef CAS PubMed .
- A. O. Surov, A. N. Manin, A. P. Voronin, K. V. Drozd, A. A. Simagina, A. V. Churakov and G. L. Perlovich, Eur. J. Pharm. Sci., 2015, 77, 112–121 CrossRef CAS .
- S. K. Nechipadappu and D. R. Trivedi, Eur. J. Pharm. Sci., 2017, 96, 578–589 CrossRef CAS .
- M. Zhang, X. Xiong, Z. Suo, Q. Hou, N. Gan, P. Tang, X. Ding and H. Li, RSC Adv., 2019, 9, 3946–3955 RSC .
- K. K. Sarmah, A. Sarma, K. Roy, D. R. Rao and R. Thakuria, Cryst. Growth Des., 2016, 16, 1047–1055 Search PubMed .
- R. Du, J. Xu, J. Li, L. Zhang, L. Ning and S. Li, New J. Chem., 2021, 45, 1474–1481 RSC .
- S. S. Bharate, Drug Discovery Today, 2021, 26, 384–398 CrossRef CAS .
- Y. Wu, X. Hao, J. Li, A. Guan, Z. Zhou and F. Guo, CrystEngComm, 2021, 23, 6191–6198 RSC .
- B. Sarma, N. K. Nath, B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2009, 9, 1546–1557 CrossRef CAS .
- A. O. Surov, N. A. Vasilev, M. V. Vener, O. D. Parashchuk, A. V. Churakov, O. V. Magdysyuk and G. L. Perlovich, Cryst. Growth Des., 2021, 21, 4516–4530 CrossRef CAS .
- S.-Y. Yang, F.-K. Zhao, H. Pang, L.-Z. Chen, R.-B. Shi and B.-H. Fang, J. Mol. Struct., 2022, 1265, 133335 Search PubMed .
- J. A. Kaduk, A. M. Gindhart and T. N. Blanton, Powder Diffr., 2020, 35, 293–300 CrossRef CAS .
- A. R. Kennedy, M. O. Okoth, D. B. Sheen, J. N. Sherwood, S. J. Teat and R. M. Vrcelj, Acta Crystallogr., Sect. C: Struct. Chem., 2003, 59, O650–O652 Search PubMed .
- G. J. Kemperman, R. de Gelder, F. J. Dommerholt, P. C. Raemakers-Franken, A. J. H. Klunder and B. Zwanenburg, Chem.–Eur. J., 1999, 5, 2163–2168 CrossRef CAS .
- G. J. Kemperman, R. d. Gelder, F. J. Dommerholt, P. C. Raemakers-Franken, A. J. H. Klunder and B. Zwanenburg, Eur. J. Org. Chem., 2001, 2001, 3641–3650 CrossRef .
- C. Fiore, A. Baraghini, O. Shemchuk, V. Sambri, M. Morotti, F. Grepioni and D. Braga, Cryst. Growth Des., 2022, 22, 1467–1475 CrossRef CAS .
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed .
- L. Orola and M. V. Veidis, CrystEngComm, 2009, 11, 415–417 Search PubMed .
- K. A. Solomon, O. Blacque and R. Venkatnarayan, J. Mol. Struct., 2017, 1134, 190–198 Search PubMed .
- G. Smith, U. D. Wermuth and J. M. White, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o105–o109 CrossRef PubMed .
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC .
- Lalhruaizela, B. N. Marak, D. Gogoi, J. Dowarah, B. S. Sran, Z. Pachuau and V. P. Singh, J. Mol. Struct., 2021, 1235, 130214 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Xia Pana,
Yang Zhuab,
Zhuoer Caib,
Qi Songa,
Yaozhenhui Lia,
Wenbin Fenga,
Xin Chena,
Hui Zhang
*a,
Xia Pana,
Yang Zhuab,
Zhuoer Caib,
Qi Songa,
Yaozhenhui Lia,
Wenbin Fenga,
Xin Chena,
Hui Zhang