DOI:
10.1039/D2RA05514G
(Paper)
RSC Adv., 2022,
12, 29900-29907
Development of Vang Danh anthracite as a cost-effective anode for sodium-ion batteries through a heat-treatment process†
Received
2nd September 2022
, Accepted 3rd October 2022
First published on 19th October 2022
Abstract
This study focuses on the effects of the chemical process and heating time at 900 °C on pristine anthracite coal (provided by Vang Danh coal, Quang Ninh province, Vietnam) and explores its structure and electrochemical performance when used as an anode in Na-ion batteries. After chemical treatment with NaOH and H2SO4, the impurity content in the raw material decreased significantly (e.g., ash content dropped from 4.4% to 0.9%, etc.). The interspacing between the graphene layers in the anthracite structure also increased after the heat treatment. Besides, on extending the heating time, the anthracite structure became more disordered than the samples heated for shorter times. Therefore, the intercalation ability of Na+ ions in the anthracite structure increased, and the sample heated at 900 °C for 6 hours exhibited the highest reversible capacity of up to 160 mA h g−1 with adequate capacity retention after 100 cycles at C/10 rate.
Introduction
Worldwide countries are specifically setting up new energy strategies for the 21st century as non-recoverable fossil resources have been over-used during the last decades owing to the huge energy demand in the industrial and manufacturing sectors. Using fossil energy such as coal and oil unfortunately, releases a large amount of CO2 into the atmosphere, which leads to serious greenhouse effects and climate changes.1 Therefore, finding clean and sustainable energy sources to replace fossil resources has become an essential strategy in every country. It is known that renewable energy sources including wind and solar never get used up or deplete and ensure sustainable development in the future.2–4 However, these sources have not been adequately explored or used due to the lack of efficient systems to store and convert it into electricity for different purposes in a flexible way. As a result, the utilization cost of renewable energy is still unaffordable and cannot be expanded to our daily life. In recent years, the role of energy storage and conversion systems, such as batteries and supercapacitors, has been emphasized to achieve a high yield of energy transition when coupled with a sustainable source. Among them, Li-ion batteries are the most promising devices that enable excellent energy storage and conversion due to their high energy density (100–200 W h kg−1), high voltage (3.8–4.2 V for commercial NMC batteries), low self-discharge (<1% per year), and long cycle life (∼1000 cycles).5,6 However, limited Li reservoirs and uneven Li distribution in the Earth's crust would push up the cost of large-scale batteries in the future, which remains a hard challenge to resolve.7 With the development of Li-ion technology, many rechargeable batteries were also invented earlier but have not been explored much during the last decades, such as Na-ion (NIBs), Mg-ion (MIBs), Ca-ion (CIBs), and Zn-ion batteries (ZIBs). As for CIBs, MIBs, and ZIBs, the higher volumetric and gravimetric capacities, high output voltage, and rich abundance make them promising candidates in the energy storage market. However, the use of these batteries still needs to be considered due to low electrical conductivity and structural degradation.8–10 Therefore, recent research has turned back to studying the Na-ion system and is moving forward quickly through the fruitful achievements of Li-ion technology. In principle, a rechargeable Na-ion battery operating is similar to a Li-ion battery, which is based on ion insertion/extraction between the anode and cathode materials. The larger radius of the Na+ ion than the Li+ ion (∼1.4 times) and the high value of first ionization energy (495.8 kJ mol−1) lead to major differences, such as poor cycling stability and slow kinetics of the intercalation process, and the distance between the layers/holes inside the structure of host materials must be capable of transferring sodium ions in/out.11 In Li-ion batteries, the most common negative electrode is graphite with a layer distance suitable for intercalating the Li+ ions (rLi+ = 0.76 Å). Unfortunately, commercial graphite can hardly store the Na+ ions due to their larger ionic radius (rNa+ = 1.02 Å), and only a small amount of Na+ ions intercalate between the graphene layers of graphite, leading to a low specific capacity.4 Meanwhile, hard carbon materials have been widely investigated as anodes for Na-ion batteries owing to their partially disordered structure. Indeed, this structure not only supports Na+-ion intercalation in the space between the graphene layers but also supports ion absorption in its amorphous region, leading to a higher specific capacity than the graphite anode. Hard carbon materials can be synthesized at different temperatures from various sources, especially biomass-derived carbon materials due to their superior performance, environment-friendliness, abundance, and renewability.12 In particular, Ghimbeu and co-workers have synthesized hard carbon from biomass wastes (coconut shells, walnut shells, and corn silk).2 Hard carbon can also be prepared from isotropic coal tar pitch (CTP) by a simple oxidation–carbonization two-step method.13
To achieve a low-cost carbonaceous anode material, we subjected local anthracite (extracted from the Vang Danh coal mine in the Quang Ninh province) through a heat treatment process. On heating, anthracite coal can produce different crystalline structures of carbon (graphite and hard carbon). Specifically, Kim et al. have reported earlier that the graphitization process evidently occurs at high temperatures, starting from 1600 °C and above,14 to elaborate a highly ordered graphite structure. Such high temperatures achieved by using an inert gas furnace would require typical equipment and incur high manufacturing cost. Interestingly, the carbonization process at a lower temperature (below 1200 °C) can produce a partially ordered structure with some similarities to the hard carbon material and can intercalate Li+/Na+ ions due to abundant micropores along with tiny graphite-like crystals.14–17 Among these materials, anthracite originating from Hon Gai, delivered a high reversible capacity of 370 mA h g−1 when calcined at 1100 °C and used as the anode in a Li-ion battery. In this study, the effects of heating time at a low temperature of 900 °C on the structural and electrochemical properties of anthracite coal were explored for the fundamental understanding of its structure during the intercalation/deintercalation reaction in Na-ion batteries.
Experimental methods
General materials
Raw anthracite was obtained from the Vang Danh coal mine in Quang Ninh province, Vietnam. All the chemicals were purchased from Sigma-Aldrich, Acros Organic, Merck, Imerys, and MTI and used directly, including sodium hydroxide (NaOH, 99%), sulfuric acid (H2SO4, 98%), carboxymethyl cellulose (CMC, Mv = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), carbon Super P (C65), N-methyl pyrrolidone (NMP, 99.5%), sodium perchlorate (NaClO4, 98%), ethylene carbonate (EC, 99%), propylene carbonate (PC, 99.7%), and dimethyl carbonate (DMC, 99%).
000 g mol−1), carbon Super P (C65), N-methyl pyrrolidone (NMP, 99.5%), sodium perchlorate (NaClO4, 98%), ethylene carbonate (EC, 99%), propylene carbonate (PC, 99.7%), and dimethyl carbonate (DMC, 99%).
Chemical and heat treatment process of raw anthracite
Initially, the anthracite coal was thoroughly ground using a ball milling machine, and then it was sieved with a sieve mesh of 53 to eliminate large particles. Secondly, 5 g of the raw material was stirred in 100 mL of a 1 M NaOH solution at 80 °C overnight. The mixture was then filtered and rinsed with deionized water several times until the pH reached 7. Afterward, 5 g of the washed powder was stirred again in 100 mL of a 1 M H2SO4 solution at 80 °C overnight. This mixture was also filtered and rinsed with deionized water until the wash water was completely neutralized (pH = 7). Finally, the sample was dried and heated at 900 °C for 3, 6 and 12 hours with a heating rate of 5 °C min−1 in an Ar atmosphere. All the samples were cooled naturally to room temperature in the argon atmosphere. The sample details are listed in Table 1.
Table 1 The sample abbreviations and explanations along with the treatment conditions (if include)
| Abbreviation |
Sample explanation |
Heating temp. (°C) |
Heating time (hours) |
| RAW |
Raw anthracite from Vang Danh–Quang Ninh |
— |
— |
| CHEM |
RAW sample treated with NaOH and H2SO4, respectively |
— |
— |
| 9003 |
CHEM sample |
900 |
3 |
| 9006 |
CHEM sample |
900 |
6 |
| 90012 |
CHEM sample |
900 |
12 |
| HC |
Commercial hard carbon, from Kuraray Co., Ltd, 9 μm (served as reference sample) |
— |
— |
Material characterization
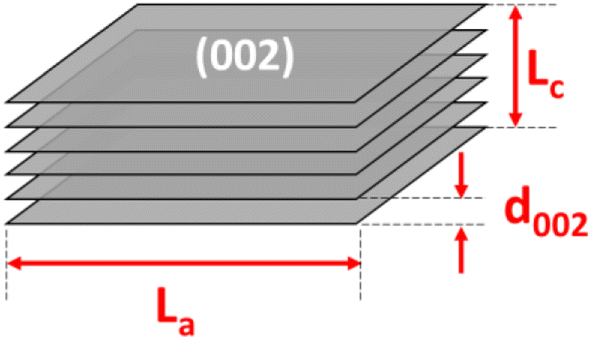
The thermal gravimetric analysis (TGA) was performed on a LABSYS Evo TG-Setaram from room temperature to 1200 °C at a 5 °C min−1 heating rate in dry air or argon gas medium. The phase composition was determined by X-ray diffraction (XRD) obtained on an Empyrean diffractometer using a copper source with λCu Kα1 = 1.540598 Å. The diffraction angle was varied with 2θ from 10° to 70°, and the scanning step was 0.0036° per step (25 seconds per step). The lateral size (La), stacking height (Lc), number of parallel layers (N), and average number of carbon atoms per aromatic lamella (n) were calculated using the below equations:15,18
| La = 1.84λ/β100cosθ |
| Lc = 0.89λ/β002cosθ |
where λ is the wavelength of the X-ray source, β100 and β002 are the full width at half maximum (FWHM) of the (100) and (002) diffraction peaks, respectively, and d002 is the distance between the (002) planes.
Raman spectra were recorded at wavelength numbers from 800 to 2000 cm−1 using a laser source of 532 nm on an XploRA ONE (Horiba). The spectra were fitted to the Lorentzian distribution. The AD/AG ratio was used to evaluate the level of disorder behavior in the structure of anthracite. The material morphology was observed by scanning electron microscopy (SEM) performed on a Hitachi S-4800 with an accelerated voltage of 10.0 kV. The microstructure of all the samples was observed under a high-resolution transmission electron microscope (HR-TEM; JEOL JEM-2010 Electron Microscope) equipped with an Energy Dispersive Spectrometer (EDS) for elemental analysis.
Electrochemical characterization
The active material (HC, RAW, CHEM, 9003, 9006, or 90012) was mixed with conductive carbon C65 and CMC binder at the mass ratio of 90:5:5, respectively, in deionized water to form a uniform slurry. The slurry was cast on an Al foil using the doctor blade technique. The film was dried in a vacuum oven at 100 °C overnight to remove the solvent completely. Then, it was punched into a round shape (d = 12 mm) with a mass loading of around 3 mg cm−2.
All the coin cells were assembled in an Ar-controlled glovebox (H2O and O2 < 0.5 ppm). The counter electrode was a thick Na foil, and the separator was polypropylene immersed in the electrolyte consisting of 1 M NaClO4 dissolved in ethylene carbonate (EC), propylene carbonate (PC), and dimethyl carbonate (DMC) in the volumetric ratio of 3:1:1. The cells were charged/discharged at the current density of C/10 (C = 372 mA h g−1) in the voltage range of 0.01–2.0 V at room temperature on a battery tester (LANHE CT2001A). Cyclic voltammetry was performed on a potentiostat device (Biologic, MPG2) in the same potential window at the scanning rate of 10 μV s−1.
Results and discussions
Material characterization
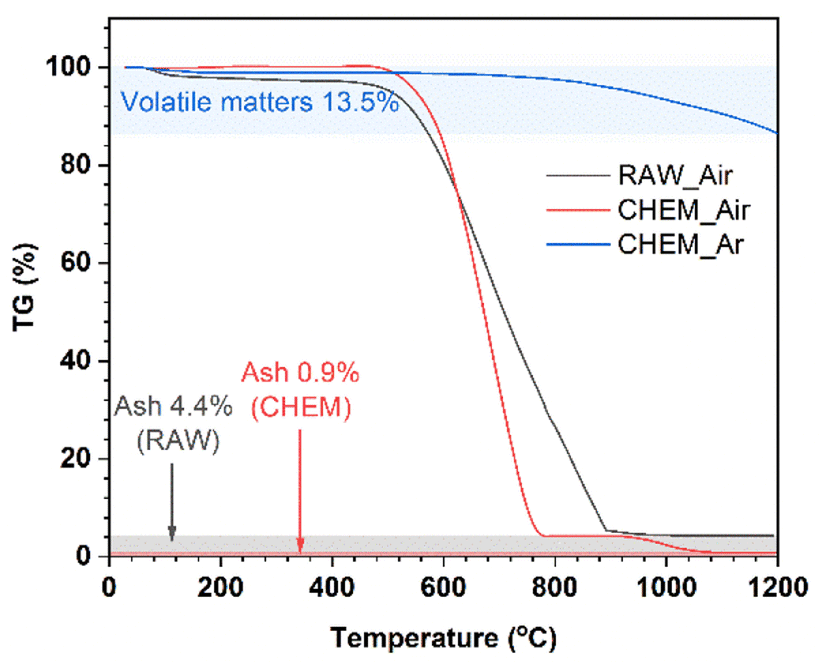
In Fig. 1, thermogravimetric diagrams of the different samples generally consist of three stages: (i) <500 °C; (ii) 500–900 °C (RAW sample) and 500–800 °C (CHEM sample); (iii) the residual part at a very high temperature up to 1200 °C. Generally, the coal samples exhibited a slight weight loss in stage (i), indicating a negligible number of volatile compounds before 500 °C. In stage (ii), a minor difference between the RAW and CHEM samples was observed. The RAW sample was completely burnt in the air at 900 °C (black line), while the combustion temperature of the CHEM sample was lower at about 800 °C (red line). This temperature difference can be explained by the vaporization/decomposition of impurities in the RAW sample under heating, leading to an extended temperature range in stage (ii). The residual mass in stage (iii) can be ascribed to the ash or non-combustible components. The CHEM sample also displayed a lower content of these substances than that of the RAW sample (e.g., 0.9% < 4.4%), confirming that the chemical treatment process using NaOH and H2SO4 helps remove most of the impurities in the RAW sample. The blue line denotes the high pyrolysis temperature of the CHEM sample in the Ar atmosphere. The calculated content of volatile compounds was 13.5%, which might mainly indicate methane, hydrocarbons, hydrogen and carbon monoxide, and non-combustible gases.15 Based on the calculated ash and volatile compound content, the free-state carbon content in the CHEM sample (without involving volatile materials) could be deduced as approximately 85.6%.
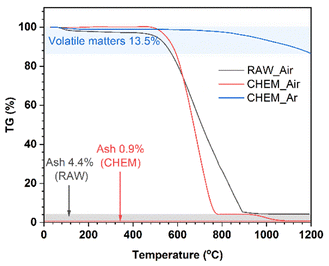 |
| | Fig. 1 The TGA curves of the RAW sample in the air (black line), CHEM sample in the air (red line), and CHEM sample in the Ar atmosphere (blue line). | |
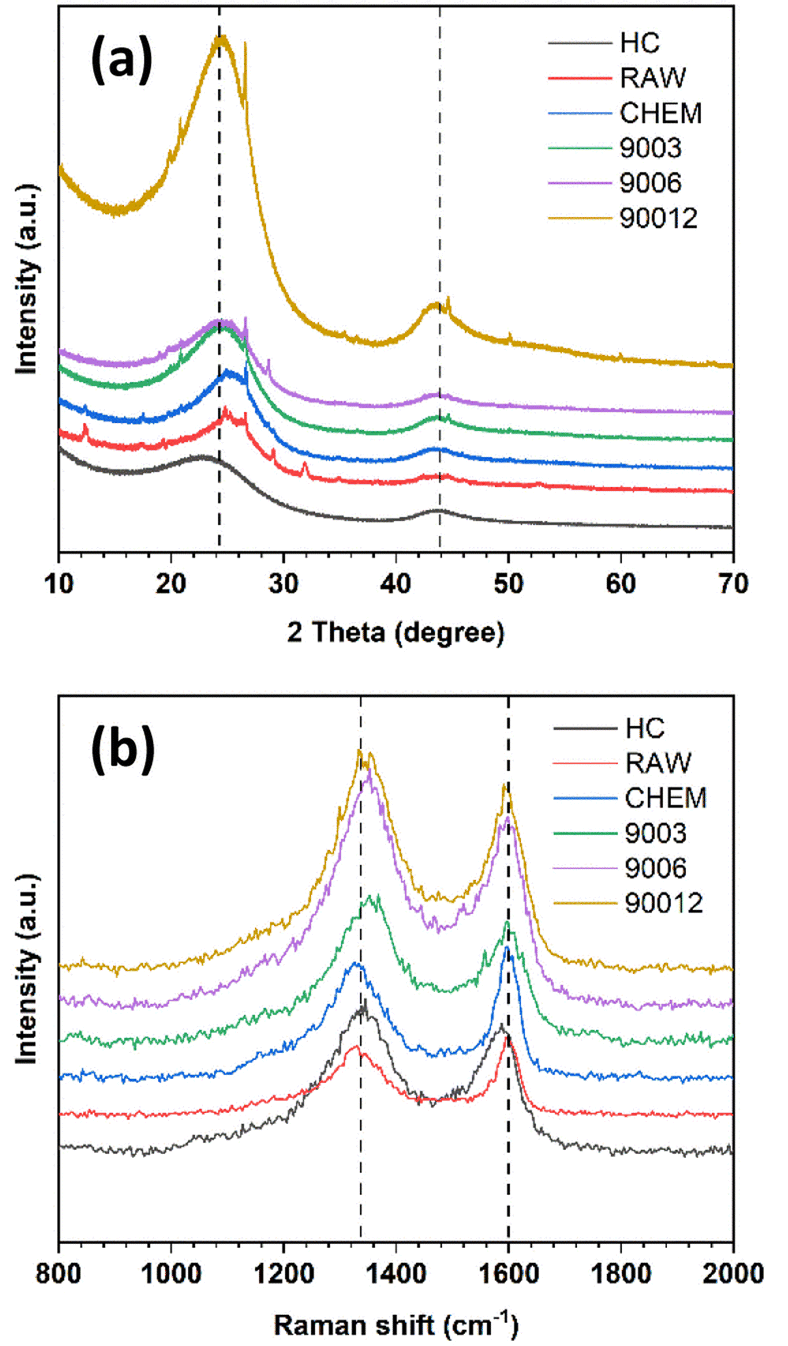
Fig. 2a shows the X-ray diffraction analysis of the coals. The carbon samples displayed two large bands at around 25° and 42°, corresponding to the diffraction patterns of the Miller planes (002) and (100). In the disordered carbon structure, the peak of (002) corresponds to the distance between the interlayers, and the peak of (100) represents the amorphous phase of carbon. In addition, the asymmetric band in the ∼25° region was fitted to two Gaussian peaks at around 22° and 26°, namely the γ-band and the Π-band (d002), ascribed to the reflection of the layers of aromatic rings and the packing distance of the saturated structures, respectively18,19 (Fig. S1†). However, the minor peaks in the RAW sample at 11°, 29.5°, and 31°, corresponded to the foreign impurities present in the original coal sample. After chemical treatment, these diffraction signals vanished, indicating that the chemical treatment could effectively remove some of the impurities, as mentioned. Moreover, the chemical treatment did not change the original structure of the anthracite sample. When pyrolyzed at 900 °C, the pattern peak (002) shifted to the left with the temperature rise, indicating a slight increase in the distance between the (002) planes. This increase in distance is expected to enhance the intercalation ability of Na+ ions inside the material structure. Compared with the reference commercial hard carbon (HC), all calcined anthracite samples exhibited an obvious peak at about 26°, corresponding to the diffraction of the lattice (002) of the graphite structure. However, this pattern of HC was broader than those of the studied anthracite samples, which can be explained by the higher disorder structure of HC. The lattice parameters calculated from the XRD analysis are shown in Table 2 and correspond to the tiny graphite-like crystals in HC and anthracite samples (Fig. 3). The hard carbon electrode with the highest d002 value among the tested materials resulted in a larger extent of Na-ion intercalation between the graphene layers of the crystalline phase. In contrast, the anthracite samples with low d002 values could only accommodate a low Na-ion intercalation capacity. Interestingly, as the heating time was increased, the d002 value slightly increased from 3.50 Å (9003) to 3.54 Å (9006 and 90012). When the treatment temperature was maintained continuously at 900 °C for 3 hours, the d002 value was unchanged. Until 6 hours of heating, d002 constantly remained at 3.54 Å. Indeed, changes in this value might reflect the varied capability of Na+-ion storage in the graphene layers of different samples, as further discussed below. Regarding lateral size (La), commercial hard carbon had the largest value of 54.5 Å, while the three heat-treated samples displayed values around 43.0–44.9 Å. Thus, this confirms that the number of Na+ ions stored between graphene sheets in the studied samples is much less than that in HC.
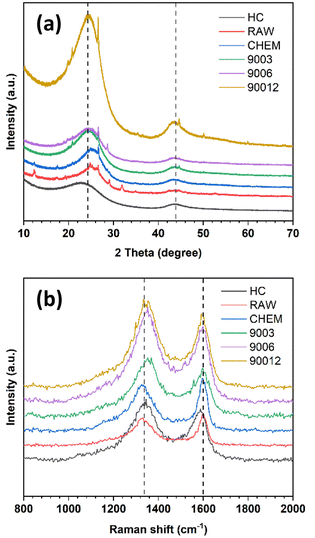 |
| | Fig. 2 (a) X-ray diffraction (XRD) and (b) Raman spectra of raw anthracite, chemical-treated sample, and the heat-treated samples at 900 °C for different durations in comparison with commercial hard carbon. | |
Table 2 Structural parameters of the anthracite samples in comparison with hard carbon
| Sample |
d002 (Å) |
La (Å) |
Lc (Å) |
N |
n |
AD/AG |
| RAW |
3.51 |
47.3 |
17.4 |
6.0 |
11.4 |
2.60 |
| CHEM |
3.49 |
43.6 |
15.9 |
5.6 |
9.9 |
2.67 |
| 9003 |
3.50 |
43.7 |
18.7 |
6.3 |
12.9 |
2.48 |
| 9006 |
3.54 |
43.0 |
14.9 |
5.2 |
8.7 |
2.59 |
| 90012 |
3.54 |
44.9 |
15.5 |
5.4 |
9.3 |
2.67 |
| HC |
3.60 |
54.5 |
15.0 |
5.2 |
8.6 |
2.43 |
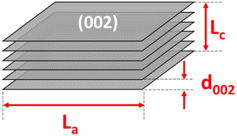 |
| | Fig. 3 The structure parameters of the crystalline phase of the heat-treated anthracite samples. | |
Like other carbonaceous materials, anthracite coal exhibited two clear peaks at wavelength numbers 1250 cm−1 and 1600 cm−1, corresponding to the disordered structure (D-band) and the ordered structure regions (G-band), respectively (Fig. 2b). All the spectra were fitted to four Lorentzian peaks of the D1, D3, D4, and G bands (Fig. S2†), as previously reported by Li et al.20 The calculated AD/AG values are shown in Table 2. The ratio of AD/AG increased with the increase in heating time, indicating higher disorder in the anthracite samples obtained. This disorder originates from the enlargement of the distance between the Miller lattice planes (002) with heating time, as already discussed in the section on XRD analysis. Indeed, heat treatment helped break the small crystalline clusters to rearrange the order level inside the structure. The raw coal sample (RAW) had a smaller AD/AG ratio (2.60) than the treated sample (CHEM) (2.67). This might be because the chemical treatment possibly modifies the anthracite surface via evaporation of some VOC to create some new surface chemical bonding. However, this value decreased from 2.67 to 2.48 in the case of the heat-treated sample (9003). This result indicates the desorption of volatile matter at high temperatures, leading to a decrease in AD/AG correlation.
The morphology of the RAW and CHEM samples, as observed by SEM and TEM, are shown in Fig. 4. The slab-structure of the anthracite samples with a random orientation of the plates represents the clusters of layered structures like graphite (Fig. 4a). As shown in Fig. 4b and d, the ball milling process could break the raw material into some tiny dust surrounding the small particles several micrometers in size. The EDS mapping of RAW anthracite showed some minor impurities (typically the compounds of Fe, S, Si, etc.) unevenly distributed in the original sample (Fig. 4c). Consequently, the Na-ion intercalation ability can be affected if these impurities are not eliminated, which is further discussed below. In contrast to RAW anthracite, the EDS mapping of CHEM anthracite showed a uniform carbon distribution (Fig. 4e). This sample had almost no impurities, which is consistent with the TGA and XRD results discussed above. Therefore, the chemical treatment process effectively removes the impurities in the raw anthracite sample.
 |
| | Fig. 4 (a) The SEM images of the RAW sample. The TEM image and the corresponding EDS mapping of (b and c) RAW anthracite and (d and e) CHEM anthracite. | |
Fig. 5 displays the HRTEM and SAED images of HC and the coal samples. All the samples had three diffraction rings in SAED corresponding to the diffraction of the lattice surfaces (002), (010) and (110), respectively. The (002) diffraction rings showed strong and wide textures related to the amorphous structure of ATC. The raw anthracite (RAW) sample had a spherical bedding corresponding to the aromatic stacking layers. Around these spheres, randomly oriented and anisotropic fringes were observed. This property is similar to the anthracite coal from Hon Gai, Quang Ninh, Vietnam.14 The parallel fringes with only 2–3 layers and short sizes (1–2 nm) are also similar to the anthracite carbon nanostructure reported by Bustin et al.21 and Li et al.20 When carbonized at high temperatures, these spheres tended to flatten and become ordered. This process corresponds with the rearrangement of carbon molecules during high-temperature carbonization, thus changing to the meta-anthracite form. The results of the SAED analysis showed that all five samples displayed three diffraction rings corresponding to the diffraction of the lattice surfaces (002), (010) and (110), respectively, in which the highest noise intensity was observed. The diffraction of the plane (002) was the largest and consistent with the results of the XRD analysis.
 |
| | Fig. 5 The high-resolution TEM lattice fringe images and the corresponding SAED patterns of the (a and b) RAW, (c and d) CHEM, (e and f) 9003, (g and h) 9006, (i and j) and 90012 samples. | |
Electrochemical characterization
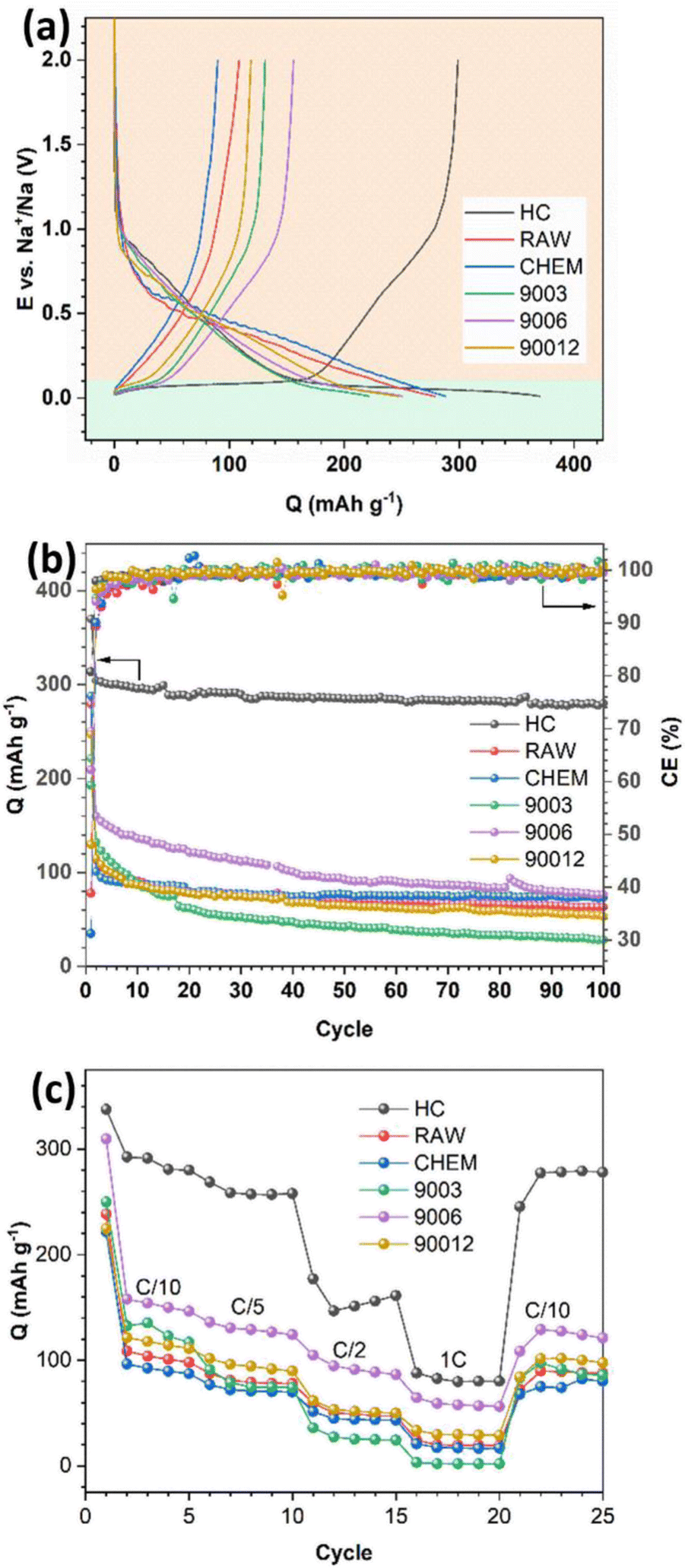
The voltage profiles and long cycling performance of the anthracite samples and hard carbon are presented in Fig. 6. All samples displayed discharge capacities much higher than the respective charge capacities, resulting in low coulombic efficiency (Fig. 6a). The initial irreversible capacity is due to the formation of a passive layer at the electrode interface (SEI) during the discharge process. Generally, the formation of the SEI layer originates from the reduction and decomposition of the electrolyte to create different ionic components depending on the type of electrolyte. In the 1 M NaClO4/EC–PC–DMC (3:1:1) electrolyte, the SEI would mainly consist of Na2CO3 and alkyl carbonate species. Since the initial SEI layer is formed, the coulombic efficiency of the subsequent cycles depends on the stability and physicochemical characteristic of the SEI layer, which can prevent further reduction/oxidation of the electrolytes to minimize the irreversible capacities. In Fig. 6b, as the SEI layer is quickly stabilized after a few cycles, it leads to a charge–discharge efficiency of nearly 100%. In the voltage profiles of the different samples (Fig. 6a), two distinct regions could be observed: a slope with a significant potential drop at potentials > 0.1 V (region I) and a long flat plateau at potentials < 0.1 V (region II). According to Qiu et al.,22 region I mainly refers to the process of Na+ adsorption onto the pores of hard carbon materials, and it represents the intercalation process of the Na+ ions between the graphene layers. Furthermore, no signal in region II was observed for the raw anthracite sample, while both were visible in the heat-treated and hard carbon samples. Indeed, heat treatment eventually increases the disorder in anthracite, and this observation is consistent with the Raman results.
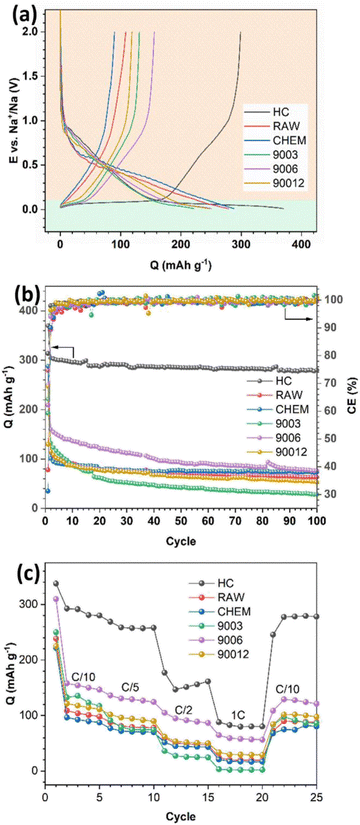 |
| | Fig. 6 (a) The initial charge–discharge profile, (b) long-term cycling performance, and (c) rate capability of the anthracite coal samples and hard carbon. | |
The capacity assigned based on region I was almost similar for all the samples, while a significant difference in the capacity related to region II was observed. In detail, hard carbon had a plateau capacity of 170 mA h g−1, while the anthracite samples showed a short plateau with capacities lower than 50 mA h g−1. Supposing that this plateau represents the adsorption of Na+ ions, it can be concluded that the adsorption process is more favorable in the case of hard carbon because its porous structure and voids accommodate Na+ ions and facilitate their intercalation into the structure. In contrast, both anthracite samples before heat treatment exhibited low capacities but remained really stable in long-term cycling, thus indicating that the reversible intercalation ability of the RAW and CHEM samples was quite good in the voltage region I. However, in the heat-treated samples, although the initial capacity was high (the highest reversible capacity value was observed for sample 9006 at 160 mA h g−1), it decreased rapidly and reached the same value as the RAW and CHEM samples. The result may be explained by the unstable adsorption mechanism of Na+ ions in this voltage plateau region. Among the three heat-treated samples, sample 9003 exhibited the worst discharge capacity, while sample 9006 displayed the best Na+ intercalation ability. Moreover, according to the Raman analysis results, the disorder increases gradually with heating time. As a result, sample 9003 exhibited a largely ordered structure. Since the intercalation ability is mainly related to the disordered region (region II), it unexpectedly decreased in the case of the 9003 sample. However, sample 90012 had the highest disordered structure among the three samples, and the cycling behavior was worse than that of the 9006 sample.
Indeed, a highly disordered structure can eventually impede Na+ ions in some active sites in the adsorption region, negatively affect the discharge reaction and reduce the discharge capacity. Moreover, the 9006 sample also exhibited the most considerable value among the heat-treated anthracite coals by providing more channels for the Na+ ions to be intercalated. Regarding rate capability, the HC electrode exhibited the highest capacity at low current densities of C/10 and C/5 due to the large interlayer spacing (d002) and the nanopores in HC, which can provide more equivalent channels for Na+-ion diffusion.23 However, its capacity rapidly decreased at higher current densities due to the poor conductivity of HC;24 for instance, it retained only 30.1% of the initial capacity at the rate density of 1C. While in the case of the 9006 sample, the capacity retention reached up to 40.9% of the initial capacity at the current density of 1C. This is owing to the increase in electrical conductivity of anthracite upon heating at 900 °C, which leads to the high rate capability of the ATC anode. However, for the other anthracite samples, the low initial capacities resulted in small capacities at high currents.
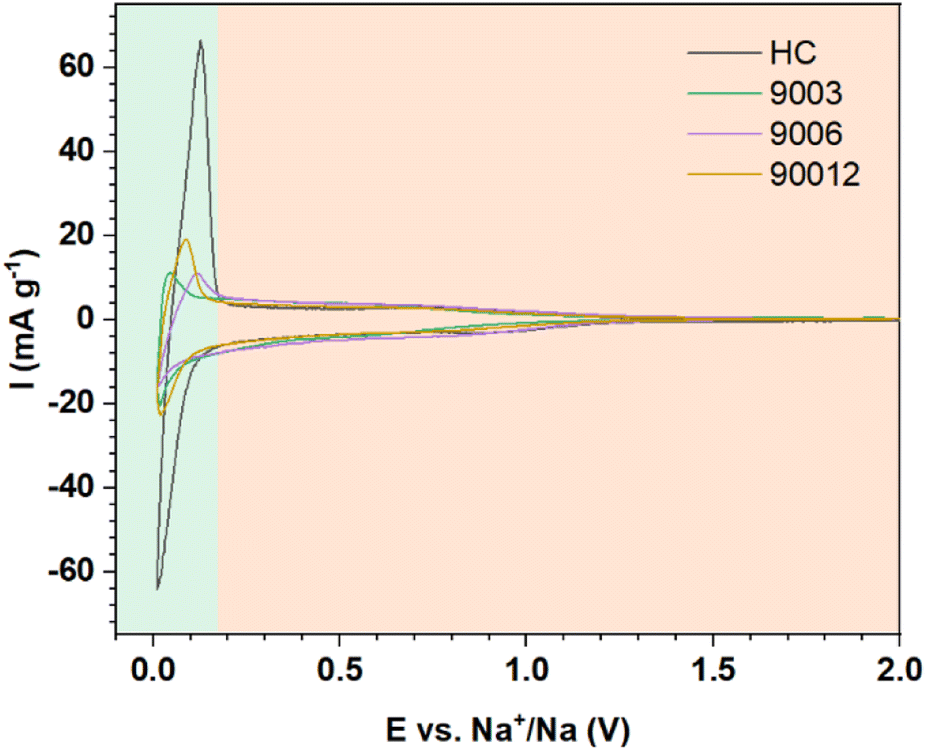
Cyclic voltammetry was performed to compare the hard carbon (HC) and heat-treated anthracite samples (9003, 9006, and 90012) (Fig. 7). Two distinct peak regions were observed, consistent with the proposed mechanisms related to each of the voltage profiles (Fig. 6a). The potential region I and region II in the cycling process correspond to the slope at potentials > 0.1 V and the high-intensity peaks at 0.12 V/0.01 V in the CV results, respectively. For the hard carbon sample, the peak intensity in region I was much higher than that in region II, thus revealing that the charge–discharge capacity obtained in the low-voltage region < 0.1 V contributed mostly to the total capacity of hard carbon. Meanwhile, the anthracite sample displayed a smaller peak, meaning that the contribution of the Na+-ion intercalation ability between the graphene layers to the total capacity of anthracite was lower than hard carbon. Compared with hard carbon, there was no significant difference in current density in the high potential region > 0.1 V in the case of anthracite, indicating that the ability of Na+ adsorption on the surface-active sites in both materials was almost the same.
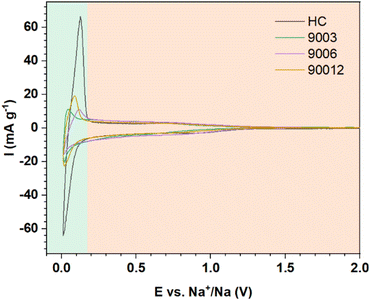 |
| | Fig. 7 The CV curves of hard carbon and the heat-treated anthracite samples. | |
Conclusions
In summary, the heat treatment of the anthracite samples successfully removed the impurities, eventually reducing the ash content and increasing the fixed carbon content. The samples treated at 900 °C were devoid of the volatile components in the original coal sample, resulting in a disordered structure with the formation of voids, which facilitated the absorption and diffusion of Na+ ions. Moreover, the heating time also significantly affected the disorder degree and electrochemical properties of the anthracite samples. The coal sample heated at 900 °C for 6 hours exhibited good discharge capacity (160 mA h g−1) and was stable for 100 cycles (retaining 48% of the original capacity). Even though the performance of anthracite is still inferior to commercial hard carbon, anthracite can be a prospective anode not only for Na-ion batteries but also for Li-ion batteries. Therefore, further work by our team will focus on optimizing the porous structure to enhance Na+-ion adsorption and promote the cycling capacity.
Author contributions
Kha Le Minh: conceived and designed the analysis, collected the data, performed the analysis, wrote, and edited the paper. Tuyen Huynh Thi Kim: contributed analysis tool. Hoang Nguyen Van: conceived and designed the analysis. Thanh Vo Duy: performed the analysis. Nhan Tran Thanh: performed the analysis, wrote the paper. Man Tran Van: contributed to analysis tool. Phung Le My Loan: conceived and designed the analysis, performed the analysis, reviewed the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Viet Nam National University – Ho Chi Minh City (VNUHCM) through the grant number B2020-18-06.
Notes and references
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- C. Nita, B. Zhang, J. Dentzer and C. M. Ghimbeu, J. Energy Chem., 2021, 58, 207–218 CrossRef CAS.
- W. Luo, F. Shen, C. Bommier, H. Zhu, X. Ji and L. Hu, Acc. Chem. Res., 2016, 49, 231–240 CrossRef CAS PubMed.
- Y. Liu, X. Liu, T. Wang, L.-Z. Fan and L. Jiao, Sustain. Energy Fuels, 2017, 1, 986–1006 RSC.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- F. Gao, B. Mei, X. Xu, J. Ren, D. Zhao, Z. Zhang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CrossRef CAS.
- B. Ji, H. He, W. Yao and Y. Tang, Adv. Mater., 2021, 33, 2005501 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, G. Zhao, L. Zhu, Y. Sun, F. Besenbacher and M. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 40451–40459 CrossRef PubMed.
- C. Wang, Z. Wang, D. Zhao, J. Ren, S. Liu, H. Tang, P. Xu, F. Gao, X. Yue and H. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 55020–55028 CrossRef CAS PubMed.
- Y. Chen, X. Guo, A. Liu, H. Zhu and T. Ma, Sustain. Energy Fuels, 2021, 5, 3017–3038 RSC.
- Z. Guo, C. Wang, M. Chen and M. Li, Int. J. Electrochem. Sci., 2013, 8, 2702–2709 CAS.
- Y.-J. Kim, H. Yang, S.-H. Yoon, Y. Korai, I. Mochida and C.-H. Ku, J. Power Sources, 2003, 113, 157–165 CrossRef CAS.
- J. Abou-Rjeily, N. A. Laziz, C. Autret-Lambert, A. Outzourhit, M.-T. Sougrati and F. Ghamouss, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- Y. Li, Y.-S. Hu, X. Qi, X. Rong, H. Li, X. Huang and L. Chen, Energy Storage Mater., 2016, 5, 191–197 CrossRef.
- B.-Y. Wang, J.-L. Xia, X.-L. Dong, X.-S. Wu, L.-J. Jin and W.-C. Li, Energy Fuels, 2020, 34, 16831–16837 CrossRef CAS.
- B. Manoj and A. Kunjomana, Int. J. Electrochem. Sci., 2012, 7, 3127–3134 CAS.
- L. Lu, V. Sahajwalla, C. Kong and D. Harris, Carbon, 2001, 39, 1821–1833 CrossRef CAS.
- K. Li, Q. Liu, H. Cheng, M. Hu and S. Zhang, Spectrochim. Acta, Part A, 2021, 249, 119286 CrossRef CAS PubMed.
- R. Bustin, J.-N. Rouzaud and J. Ross, Carbon, 1995, 33, 679–691 CrossRef CAS.
- S. Qiu, L. Xiao, M. L. Sushko, K. S. Han, Y. Shao, M. Yan, X. Liang, L. Mai, J. Feng and Y. Cao, Adv. Energy Mater., 2017, 7, 1700403 CrossRef.
- J. Yang, X.-y. Zhou, J. Li, Y.-l. Zou and J.-j. Tang, Mater. Chem. Phys., 2012, 135, 445–450 CrossRef CAS.
- C. Ge, Z. Fan, J. Zhang, Y. Qiao, J. Wang and L. Ling, RSC Adv., 2018, 8, 34682–34689 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence acd,
Thanh Nhan Tran
acd,
Thanh Nhan Tran