
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlack phosphorous/palladium functionalized carbon aerogel nanocomposite for highly efficient ethanol electrooxidation†
Ibrahim Abdelwahab*a and
Abdalla Abdelwahab *bc
*bc
aDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore. E-mail: chmiaa@nus.edu.sg
bFaculty of Science, Galala University, Sokhna, Suez 43511, Egypt
cMaterials Science and Nanotechnology Department, Faculty of Postgraduate Studies for Advanced Sciences, Beni-Suef University, Beni-Suef 62511, Egypt. E-mail: aabdelwahab@psas.bsu.edu.eg
First published on 1st November 2022
Abstract
Direct ethanol fuel cells have great potential for practical power applications due to their easy operation, high energy density, and low toxicity. However, the slow and incomplete ethanol electrooxidation (EEO) reaction is a major drawback that hinders the development of this type of fuel cell. Here, we report a facile approach for the preparation of highly active, low cost and stable electrocatalysts based on palladium (Pd) nanoparticles and black phosphorus/palladium (BP/Pd) nanohybrids supported on a carbon aerogel (CA). The nanocomposites show remarkable catalytic performance and stability as anode electrocatalysts for EEO in an alkaline medium. A mass peak current density of 8376 mA mgPd−1 is attained for EEO on the BP/Pd/CA catalyst, which is 11.4 times higher than that of the commercial Pd/C catalyst. To gain deep insight into the structure–property relationship associated with superior electroactivity, the catalysts are well characterized in terms of morphology, surface chemistry, and catalytic activity. It is found that the BP-doped CA support provides high catalyst dispersibility, protection against leaching, and modification of the electronic and catalytic properties of Pd, while the catalyst modifies CA into a more open and conductive structure. This synergistic interaction between the support and the catalyst improves the transport of active species and electrons at the electrode/electrolyte interface, leading to rapid EEO reaction kinetics.
1. Introduction
Fossil fuel combustion is the main contributor to greenhouse gases that drive global warming. The search for clean, sustainable, and ecological energy sources has become a global, yet challenging, demand in recent years. Fuel cells are devices that convert chemical energy into electrical energy in a clean manner. If the supplied fuel is hydrogen, electricity, heat, and water are the only products.1,2 Among different types of fuel cells, liquid fuel cells (LFCs) have considerable advantages in terms of easy handling, low operating temperatures, and the availability of a wide range of liquid fuels such as ethanol, methanol and formic acid.3–5 The ease of producing ethanol from the fermentation of biomass makes its use as a fuel in LFCs an attractive choice. Despite these appealing features, the realization of efficient, cost-effective ethanol LFCs is still hampered by some issues such as the slow and inefficient ethanol electrooxidation (EEO) reaction, the poisoning of the catalysts by the intermediate species especially in acidic media, and the high cost of the catalysts, of which platinum (Pt) is the most famous.4,6–9 Palladium (Pd) based catalysts have become competitive with the Pt ones due to their relatively low cost and high resistance to poisoning species such as carbon monoxide (CO).5,10,11 In addition, Pd electrocatalysts have shown high catalytic activities for EEO in the less corrosive alkaline media,3,12 which can be attributed to the formation of ethoxy intermediates rather than CO-like species.7The electrocatalytic performance of Pd can be further enhanced by modifying its electronic and/or morphological properties. It has been found that combining Pd with other transition metals13,14 or two-dimensional (2D) materials15,16 can modulate the electronic structure of Pd, boosting its electrochemical activities. Owing to the accumulation characteristics of naked Pd nanoparticles (NPs), different catalyst support materials such as carbon nanomaterials, conductive polymers, and metal oxides have been utilized to achieve uniform Pd particle dispersion, size control and thus enhanced catalytic properties. For example, hollow nitrogen-doped graphene frameworks17 and polypyrrole–carbon composite4 were used as Pd support materials, and the results demonstrated that efficient Pd catalyst NPs with an average size of about 5.5 nm could be well dispersed on the surface of the support material. However, many of these support materials are morphologically flat where the incorporated catalyst tends to dissolute in the electrolyte and gets separated from the support. Moreover, the poor conductivity of some support materials (e.g., metal oxides) limits their use in high-performance electrocatalysis.
Carbon gels, in both of their forms; xerogels (CXs) and aerogels (CAs), have attracted extensive interest as electrode materials for wide-ranging applications such as catalysis,18,19 fuel cells20,21 and energy storage.22,23 The appeal of carbon gels stems from their low mass density, high surface area, large mesopore volume, high electrical and thermal conductivities, and inexpensive preparation methods.24,25 More specifically, CAs can be readily prepared from resorcinol and formaldehyde monomers by the sol–gel method in the presence of a catalyst. Functionalized metal doped CAs can also be obtained by adding a soluble metal salt to the resorcinol/formaldehyde mixture or by post-synthesis modifications. The doped metal is typically trapped and chelated within the gel matrix with high tolerance against leakage. As a result, carbon aerogel-supported catalysts have shown great promise in many liquid-phase catalytic applications.
Herein, highly efficient anodic Pd catalysts supported on CA are prepared and tested for EEO in an alkaline medium. The Pd/CA catalyst exhibits high EEO performance which can be ascribed to different factors: (1) high electroactive area owing to the better dispersion of Pd NPs; (2) enhanced electrolyte penetration through the porous structure of the CA support; and (3) improved catalyst stability and durability. The EEO reaction is further boosted by doping the Pd/CA catalyst with back phosphorus (BP); a layered 2D material with high electronic mobility in which each phosphorus atom covalently bonded to three adjacent phosphorus atoms in a puckered honeycomb structure.26 It has been shown that the Pd/BP interaction can trigger a Pd–P coordination bond of a covalent nature.27 The electron transfer from BP to Pd increases the Pd charge density and upshifts its d-band center, promoting the catalytic performance of the hybrid BP/Pd/CA catalyst.
2. Materials and methods
2.1. Materials
Analytical grade resorcinol, formaldehyde, palladium(II) chloride (PdCl2), cobalt acetate (Co(Ac)2·6H2O), ethylene glycol, urea, N,N-dimethylformamide (DMF), Nafion solution (5 wt%), isopropanol, potassium hydroxide (KOH), and ethanol (C2H5OH) were purchased from Sigma-Aldrich. The black phosphorus (BP) single crystal was supplied by HQ Graphene.2.2. Carbon aerogel (CA) synthesis
CA samples were synthesized by a polymerization reaction between resorcinol and formaldehyde with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Cobalt acetate was used as a polymerization catalyst and water as a solvent. The molar ratio between resorcinol and water was adjusted to be 1:23, and the cobalt (Co) content was calculated to be 1 wt% of the final carbon structure. All the reactants were mixed using a magnetic stirrer, filled into glass molds, and heated at 40 °C for a day and then at 80 °C for five days. The resulting organic gels were placed in acetone for three days for solvent exchange, and then super-critically dried to afford their corresponding organic aerogels. The CA samples were obtained by carbonizing the organic aerogels at 900 °C for two hours under nitrogen (N2) atmosphere.
:2. Cobalt acetate was used as a polymerization catalyst and water as a solvent. The molar ratio between resorcinol and water was adjusted to be 1:23, and the cobalt (Co) content was calculated to be 1 wt% of the final carbon structure. All the reactants were mixed using a magnetic stirrer, filled into glass molds, and heated at 40 °C for a day and then at 80 °C for five days. The resulting organic gels were placed in acetone for three days for solvent exchange, and then super-critically dried to afford their corresponding organic aerogels. The CA samples were obtained by carbonizing the organic aerogels at 900 °C for two hours under nitrogen (N2) atmosphere.
2.3. Preparation of palladium doped carbon aerogel (Pd/CA)
Pd/CA was prepared through a solvothermal process, following a previous protocol21 with little modification. Explicitly, a solution containing 120 mg of CA dispersed in 40 ml DMF, was added to a solution containing 1.2 mmole of PdCl2 dissolved in 30 ml of HCl (0.1 M) and ethylene glycol with a volumetric ratio of 1:2. The mixture was sonicated and transferred into 100 ml Teflon lined stainless steel autoclave, and thereafter the solvothermal process was carried out at 180 °C for 12 hours. After cooling to room temperature, the product was collected with centrifugation and washed several times with absolute ethanol then dried at 60 °C for another 12 hours.
2.4. Preparation of black phosphorus/palladium doped carbon aerogel (BP/Pd/CA)
The same preparation method of Pd/CA was followed in preparing BP/Pd/CA, except for the addition of BP. BP nanosheets were prepared by ball-milling the BP single crystal under an argon (Ar) atmosphere for 24 hours at 250 rpm. The molar ratio between Pd and BP was adjusted to be 2:1.
2.5. Physicochemical characterization
Surface area and porosity analysis was performed by N2 adsorption/desorption at 77 K using surface area analyzer (TriStar II 3020, Micromeritics, USA). The samples were first outgassed under Ar gas flow for 3 hours at 110 °C and then the isotherm was recorded over a relative pressure range (P/P0) from 0.01 to 0.99 and back. The specific surface area (SBET) was determined using the Brunauer–Emmett–Teller (BET) equation. The pore volume versus diameter distribution was evaluated by analyzing the desorption branch of the isotherms using the Barrett–Joyner–Halenda (BJH) method. Powder X-ray diffraction (PXRD) patterns of the samples were recorded on a Bruker D8 Focus Powder X-ray diffractometer using Cu Kα radiation (40 kV, 40 mA) at room temperature. The zeta potential measurements were performed using a Zetasizer Nano ZS (Malvern Instruments, United Kingdom) with a particle size range from 0.3 nm to 10 μm. The powder sample was dispersed in ethanol, and each measurement was repeated three times. The final zeta potential vs. pH curves were averaged from the three measurements.Scanning electron microscopy (SEM) was performed using a JEOL system (JEOL JSM-6701F SEM, USA) installed with a field-emission gun and a specimen stage with three motorized axes. The SEM accelerating voltage was adjustable from 0.5 to 30 kV. High Resolution Transmission Electron Microscopy (HRTEM) was performed using a JEOL JEM-3011 system installed with a LaB6 emitter and a Gatan Orius SC200 CCD camera. The TEM system operated at an accelerating voltage of 300 kV. The system was also equipped with an Oxford Instruments X-Max 80T Energy Dispersive Spectrometer (EDS) for elemental analysis. X-ray Photoemission Spectroscopy (XPS) was performed with a SPECS XR-50 X-ray Mg Kα (1253.7 eV) source with a pass energy of 30 eV and a spot size of 5 mm.
2.6. Electrode preparation and electrochemical techniques
5 mg of CA, Pd/CA or BP/Pd/CA was dispersed in 400 μL of isopropanol and 30 μL Nafion solution (5 wt%). The suspension was sonicated for 10 minutes at room temperature to form a homogeneous ink, and then 10 μL of the ink was deposited onto the active area of a glassy carbon electrode (3 mm in diameter) served as the working electrode. The electrochemical measurements were carried out in a three-electrode system with a potentiostat (VMP, BioLogic Science Instruments) in which silver/silver chloride (Ag/AgCl) and platinum (Pt) electrodes acted as reference and counter electrodes, respectively. 1.0 M KOH was used as the electrolyte and all experiments were carried out at room temperature. Electrochemical impedance spectroscopy (EIS) was performed by applying a sinusoidal AC excitation signal of 10 mV to the working electrode in the 100 kHz to 1 mHz frequency range. The EIS data is fitted to a mathematical model (i.e., an equivalent electrical circuit) for interpretation and analysis. The impedance components are calculated as the complex ratio between the applied potential sinewave and the response current. The chronoamperometric analysis was conducted at −1 V for 1 hour in 1.0 M KOH + 0.5 M C2H5OH electrolyte.3. Results and discussion
3.1. Materials characterization
The textural properties of the CA, Pd/CA and BP/Pd/CA samples are analyzed by N2 physisorption at 77 K. Fig. 1a shows the N2 adsorption/desorption isotherms of the samples. The isotherms are of type IV, which is characteristic of mesoporous materials with monolayer–multilayer adsorption at low P/P0 and capillary condensation at high P/P0. The capillary condensation transition of the isotherms occurs at relatively low P/P0 values, indicating that the pore size lies in the upper micropore–lower mesopore size range. The wide hysteresis loops of the isotherms can be classified as type H2, reflecting a complex pore system with an ill-defined shape and wide pore size distribution. The sharpness of the capillary condensation steep rise recorded for the Pd and Pd/BP doped samples is attributed to a narrower pore size distribution and higher uniformity, relative to CA. The textural parameters deduced from the isotherms using the BET equation and the BJH method are listed in Table S1.† The BET surface areas (SBET) of Pd/CA and BP/Pd/CA are lower than that of CA due to the pore filling effect caused by the introduction of the dopants.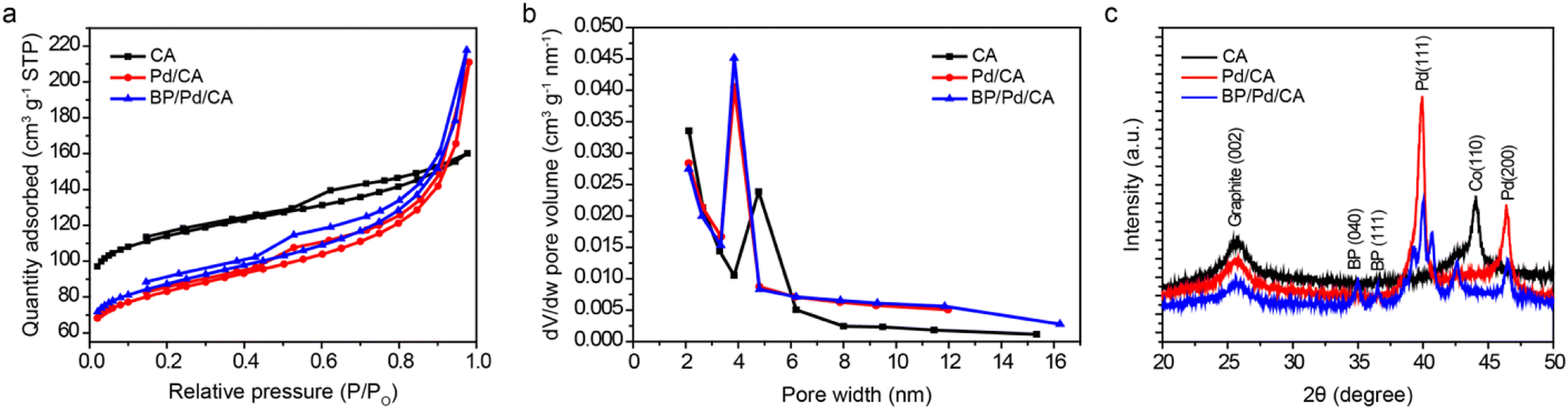 | ||
| Fig. 1 (a) N2 adsorption/desorption isotherms, (b) Barrett–Joyner–Halanda (BJH) pore size distribution, and (c) X-ray diffraction (XRD) patterns of the synthesized samples. | ||
Despite their lower SBET values, the amount of liquid N2 condensate within the pores of Pd/CA and BP/Pd/CA in the high relative pressure range is higher than that of CA, implying more developed mesoporous carbon structures in these modified samples. The well-developed mesoporosity of the carbon matrices upon Pd and Pd/BP doping is also confirmed by their larger mesopore volumes and sizes when compared with pristine CA, as shown in Fig. 1b. It is worth mentioning that several studies28–31 have established a clear correlation between the extent of mesoporosity in a material system and the adsorption capacity on its surface. For example, Bjelopavlic et al.30 demonstrated that the adsorption capacity of activated carbon for natural organic matter (NOM) significantly increased when the mesoporosity of activated carbon was developed into a more open structure. In our work, the largest pore volume/average pore diameter is registered for BP/Pd/CA, revealing that the sample has the most open porous structure. From a structural point of view, this denotes that the BP doping of the Pd/CA catalyst increases the number of adsorption sites for ethanol and suggests a higher EEO efficiency.
XRD patterns of the samples are shown in Fig. 1c. The patterns show a broad peak at 2θ ≃ 26°, which corresponds to the (002) diffraction peak of graphite. The average distance between the (002) planes (i.e., d(002)-spacing) is found to be larger than that of an ideal graphite crystal, indicating that the samples have an amorphous carbon structure with a low degree of graphitization.32 It should be emphasized that the CA graphitic structures are mainly present around the Co particles that are used as a catalyst for the preparation of carbon aerogels.23 The presence of Co NPs in the aerogel matrix is confirmed by a peak at 44.22° assigned to the (111) peak of metallic cobalt.18 The diffractogram of the Pd/CA sample displays two additional peaks at 39.9° and 46.4° which can be indexed to the (111) and (200) crystallographic planes of fcc-Pd, respectively. In addition to the graphite and Pd peaks, the BP/Pd/CA sample shows peaks at 34.9°, 36.5°, 39.5°, 41.3° and 42.5°, which can be related to the (040), (111), (041), (002) and (131) planes of BP, respectively. This demonstrates that the crystalline structure of BP is preserved after ball-milling. The slight shift of the Pd peaks towards higher 2θ values in the BP/Pd/CA sample suggests lattice contraction upon doping with BP. The lattice contraction is likely due to the formation of Pd–P bonds (confirmed by XPS) as well as size effects.27 The decreased intensity of the (002) graphite peak in the cases of Pd/CA and BP/Pd/CA is attributed to turbostratic carbon with disordered graphitic planes, corroborating the textural observations that more developed porous structures are formed after the Pd and BP/Pd doping.33 The absence of the Co peak in Pd/CA and BP/Pd/CA could be ascribed to the formation of Co solid solutions with the dopants as well as graphitic shielding after the solvothermal process. The mean particle size (dXRD) of the metal NPs in the different samples was estimated using Scherrer's equation; the results are summarized in Table S2.† The dXRD values of Pd NPs in Pd/CA and BP/Pd/CA are found to be 17.5 nm and 19.2 nm, respectively, giving an indication that carbon aerogel is an excellent support material for pristine and alloyed Pd NPs.
The exterior and interior surface morphologies of the samples are investigated using SEM and TEM, respectively. Fig. 2a–c shows typical SEM images of the three different samples. It is noticeable that the introduction of Pd and Pd/BP leads to less compact aggregates with slightly larger gel particles (i.e. larger pores between them) (Fig. 2b and c), consistent with the textural data. The amorphous carbon structure of CA is clearly observed by TEM (Fig. 2d–f), together with a small fraction of graphitic clusters. The TEM images also reveal that the Pd NPs (Fig. 2e) are round and uniformly dispersed in the carbon matrix with a narrow particle size distribution. The intimate linkage between the dopants and the CA support makes the Pd/CA and BP/Pd/CA catalysts excellent candidates for liquid phase electrocatalytic applications due to their low leaching tendency. Fig. S1† represents the EDS spectrum and elemental mapping for the BP/Pd/CA catalyst. Uniform and homogeneous elemental (C, N, O, Pd and P) distribution is registered throughout the entire structure. In the case of BP/Pd/CA, the HRTEM imaging (Fig. 2f) reveals some relatively large dark regions, probably due to the presence of thick BP nanosheets. Moreover, the high-resolution TEM images of BP/Pd/CA at different magnifications (Fig. S2†) reveal that the catalyst nanoparticles have diameters in the range of 3.89 nm to 19.3 nm.
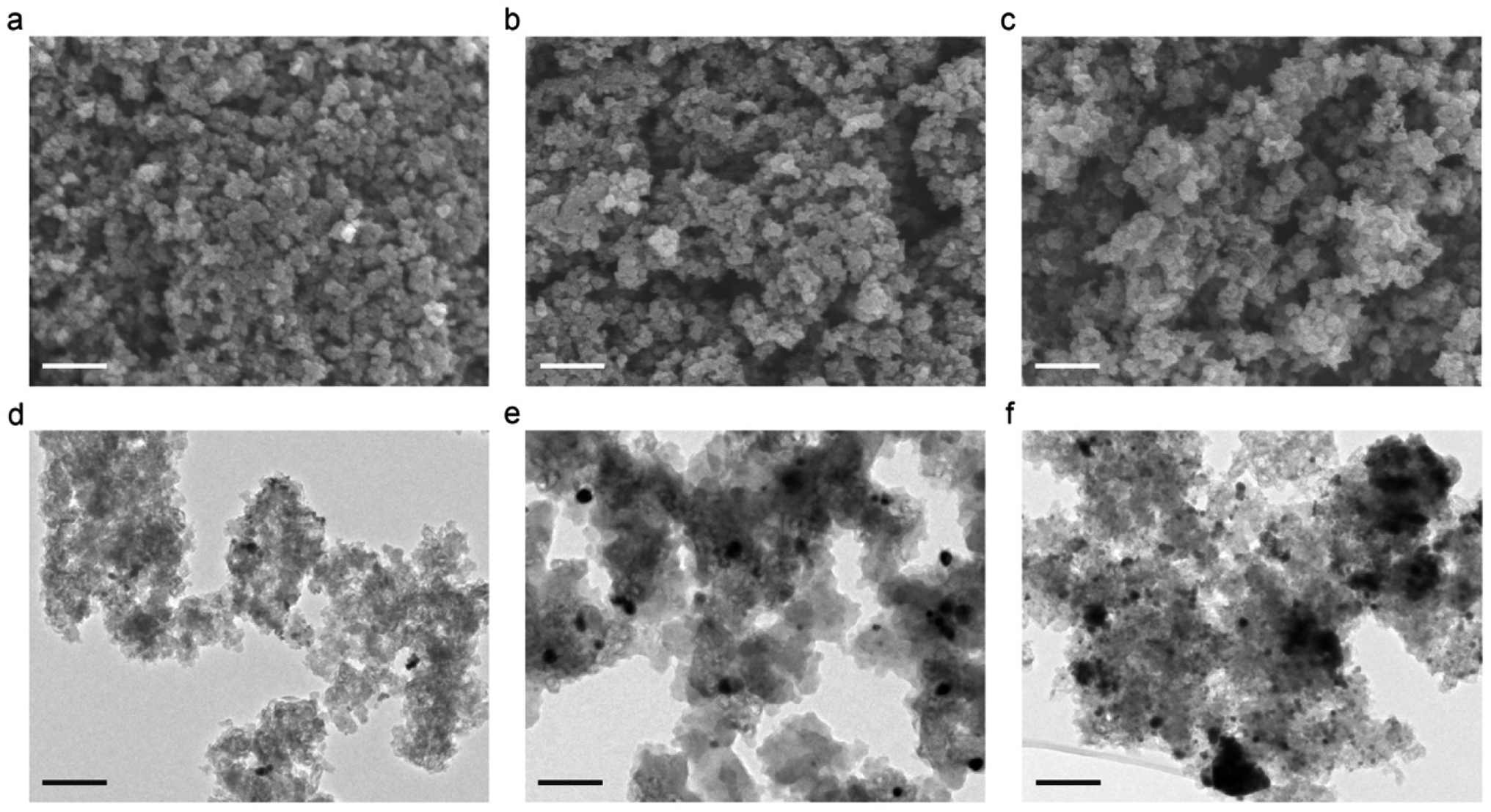 | ||
| Fig. 2 STM (a–c) and TEM (d–f) images of the different samples. (a and d) CA. (b and e) Pd/CA. (c and f) BP/Pd/CA. Scale bars: 100 nm. | ||
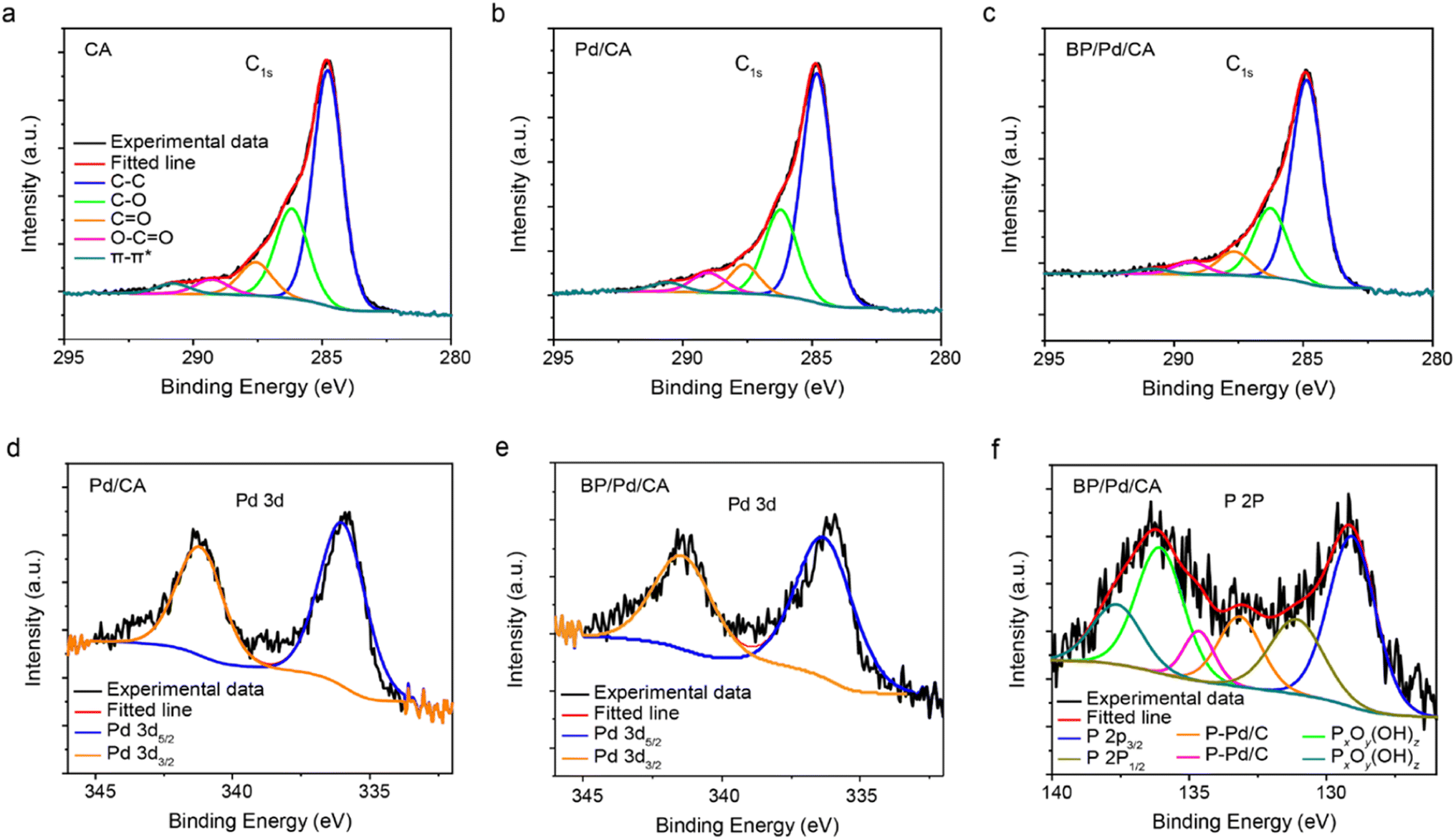
The chemical states of the near-surface part of the samples are determined using XPS (Fig. 3). The XPS spectra of the pristine and doped CA samples show broad, asymmetric C1s peaks that tail toward high binding energies as depicted in Fig. 3a–c. The peak asymmetry is due to the presence of several oxygen groups in the aerogels that contribute to the carbon XPS spectra. The C1s peak can be fitted by five component peaks assigned to aromatic and aliphatic carbon C–C (sp2) or carbon bonded to hydrogen at 284.8 eV (red), C–O at 286.1 eV (green), carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 288.5 eV, carboxyl O–CO at 290.2 eV, and C π–π* shake-up satellite or ester group at 292.3 eV.34,35 As shown in Fig. 3b and c, the relative intensity of the C–O peak increases whereas those of the double-bond functional groups decrease, indicating that the original CA structure is partially reduced during the solvothermal process. A comparison of the C–C peaks shows that the Pd/CA and BP/Pd/CA samples possess a high crystalline carbon content in the amorphous carbon matrix in comparison to CA, as reflected in their narrower C–C peaks (refer to the FWHM values in Table S2†).
O at 288.5 eV, carboxyl O–CO at 290.2 eV, and C π–π* shake-up satellite or ester group at 292.3 eV.34,35 As shown in Fig. 3b and c, the relative intensity of the C–O peak increases whereas those of the double-bond functional groups decrease, indicating that the original CA structure is partially reduced during the solvothermal process. A comparison of the C–C peaks shows that the Pd/CA and BP/Pd/CA samples possess a high crystalline carbon content in the amorphous carbon matrix in comparison to CA, as reflected in their narrower C–C peaks (refer to the FWHM values in Table S2†).
 | ||
| Fig. 3 (a–c) C1s core-level XPS spectra of CA (a), Pd/CA (b), BP/Pd/CA (c). (d) Pd 3d core-level XPS spectrum of Pd/CA, Pd 3d core-level XPS spectrum of BP/Pd/CA (e) and P 2p core-level XPS spectrum of BP/Pd/CA (f). | ||
This further confirms that the dopants serve as catalysts for promoting the formation of graphitic structures during the solvothermal process, which is in agreement with the XRD and TEM data. The catalytic graphitization of the carbon gel boosts its electrical conductivity and thus, enhances its performance in electrochemical and electro-catalytic processes. The Pd 3d core-level XPS spectra of Pd/CA and BP/Pd/CA are composed of two-component peaks at 335.9 eV and 341.2 eV (Fig. 3d and e). The peak at 335.9 eV accounts for Pd 3d5/2, while the peak at 341.2 eV accounts for Pd 3d3/2.36 Our Pd 3d5/2 peak position is slightly higher than that of elemental Pd (334.87 eV) yet lower than that of PbO (336.7 eV), which suggests that the Pd NPs are partially oxidized. The P 2p core-level spectrum of BP/Pd/CA shows two peaks at 129.1 eV and 131.1 eV which can be assigned to P 2p3/2 and P 2p1/2 of elemental phosphorus, respectively.37 The high binding energy peaks (>135 eV) are attributed to phosphorus oxides.38 It has been reported that BP oxidizes in ambient conditions, forming several phosphorus oxide species, PxOy(OH)z, including native stable oxides such as P2O5.39 The oxidized phosphorus species in BP/Pd/CA could be due to air oxidation and/or interaction between elemental phosphorus and the gel's oxygen-containing groups. In addition, components at intermediate binding energy values (around 134 eV) are also present. These peaks provide evidence for the presence of phosphorus in different chemical environments and are likely due to P–Pd and P–C bonds.27,40 The XPS results confirm that BP is well incorporated into the CA support through various chemical bonds such as P–C, P–Pd, P–OH, PO, P–O–P, etc.
3.2. Electrochemical performance
The background electrochemical performance of CA, Pd/CA, and BP/Pd/CA is tested in a 1.0 M KOH electrolyte at room temperature using a three-electrode configuration. Fig. S3† depicts the cyclic voltammograms (CVs) of pristine and doped carbon aerogels between −0.8 and 0.3 V (vs. Ag/AgCl) at scan rates (current loadings) from 5 to 60 mV s−1. The CV curves show rectangle-like shapes, indicating good capacitive behavior of the electrode materials. No apparent redox peaks are detected for the CA support, as shown in Fig. S3a.† In contrast, the reduction peaks of Pd oxide are observed in the reverse scan at 0.34 V and 0.38 V for Pd/CA and BP/Pd/CA, respectively (Fig. S3b and c†).41 The positive shift of the Pd oxide reduction peak of BP/Pd/CA indicates that the reduction of Pd oxide is enhanced by the charge transfer between BP/CA and Pd. In addition, the charge associated with the Pd oxide reduction of BP/Pd/CA is slightly higher than that of Pd/CA. This feature suggests that the presence of black phosphorus increases the electrochemically active surface area of the Pd/CA catalyst and therefore enhances its electrochemical activity.413.3. Electrooxidation of ethanol
The catalytic performance of CA, Pd/CA, and BP/Pd/CA as anode catalysts for EEO is examined in 1.0 M KOH/0.5 M C2H5OH electrolyte at a scan rate of 60 mV s−1 (Fig. 4). The electro-oxidation of ethanol on the catalyst surface exhibits two oxidation peaks; the first is at 0.026 V for Pd/CA or at 0.06 V for BP/Pd/CA on the forward scan under anodic conditions and the second is at −0.30 V for Pd/CA or at −0.34 V for BP/Pd/CA on the reverse scan under cathodic conditions. The anodic peak is attributed to the oxidation of the freshly chemisorbed ethanol species on the doped electrode while the cathodic peak is associated with the adsorption and oxidation of the intermediate carbonaceous species that are not completely oxidized in the forward scan. On the forward scan, the onset potential of ethanol oxidation is around −0.60 V for both Pd/CA and BP/Pd/CA. The current densities at the anodic peaks of CA, Pd/CA and BP/Pd/CA are 0.7, 27.7 and 35.9 mA cm−2, respectively (Fig. 4a–c). To better compare the electrochemical performance of our catalysts with the results of previous studies, we normalized the current density by the total amount of Pd (0.125 mg) loaded on the catalyst to obtain the catalytic mass activity (Fig. 4d). The mass peak current densities are 6520 mA mgPd−1 for Pd/CA and 8376 mA mgPd−1 for BP/Pd/CA. These extremely high peak current density values surpass those of previously reported Pd- and Pt-based catalysts (Table 1). Furthermore, the BP/Pd nanohybrids on carbon aerogel support catalytically outperform Pd NPs on BP and BP/graphene supports.42,43 By further increasing the positive anode potential, the Pd oxide formation takes place and inhibits EEO, resulting in a drop in the faradaic current. On the reverse scan, the EEO reaction is inactive until the reduction of Pd oxides that reactivates the process and, as a consequence, the cathodic peaks around −0.30 are observed.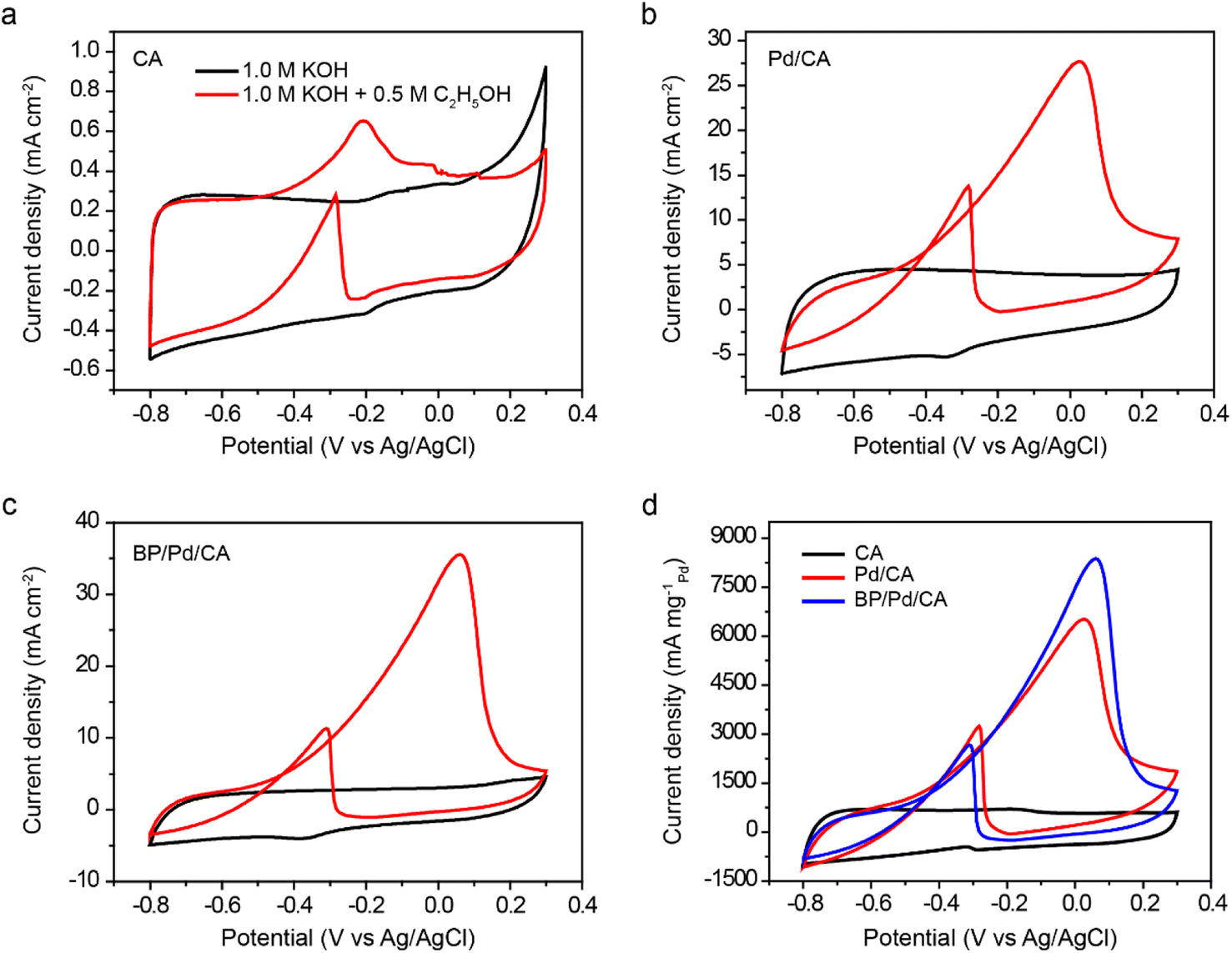 | ||
| Fig. 4 Room-temperature cyclic voltammograms (red curves) of (a) CA, (b) Pd/CA, and (c) BP/Pd/CA in 1.0 M KOH + 0.5 M C2H5OH solution at a scan rate of 60 mV s−1. The background electrochemical performance (black curves) of the samples in 1.0 M KOH is also shown for comparison. (d) The mass current densities of the samples. | ||
| Catalyst | Mass peak current density (mA mgPd−1) | Solution conditions | Ref. |
|---|---|---|---|
| BP/Pd/CA | 8376 | 1.0 M KOH + 0.5 M C2H5OH | This work |
| Pd/CA | 6520 | 1.0 M KOH + 0.5 M C2H5OH | This work |
| Pd/ATN-BP-30% | 5023.8 | 1.0 M NaOH + 1.0 M C2H5OH | 15 |
| Pd–Ni–P nanocatalysts (Pd40Ni43P17) | 4950 | 1.0 M NaOH + 1.0 M C2H5OH | 13 |
| NiAuPt nanoparticles/rGO (Ni40Au33Pt27-NGs) | 4938 | 0.5 M NaOH + 1.0 M C2H5OH | 44 |
| PdNi hollow nanospheres aerogels (Pd83Ni17 HNS) | 3630 | 1.0 M NaOH + 1.0 M C2H5OH | 45 |
| Pd77Cu23 aerogel | 3472 | 1.0 M KOH + 1.0 M C2H5OH | 46 |
| Pd/B,N-codoped graphene nanoribbons (BN-GNRs) | 2156 | 1.0 M NaOH + 1.0 M C2H5OH | 47 |
| PdCo nanotube arrays (NTAs) supported on carbon fibre cloth (CFC) (PdCo NTAs/CFC) | 1500 | 1.0 M KOH + 1.0 M C2H5OH | 48 |
| Pd nanoparticles/CoP nanosheets attached to the cloth of carbon fibres (Pd@CoP NSs/CFC) | 1413 | 1.0 M KOH + 1.0 M C2H5OH | 49 |
| Commercial Pd/C | 730.7 | 1.0 M NaOH + 1.0 M C2H5OH | 15 |
The ratio of the forward oxidation peak current density (If) to the backward oxidation peak current density (Ib), If/Ib, is an important indicator of the catalyst tolerance to poisoning by the reaction intermediates. A higher If/Ib value implies more efficient oxidation of ethanol and less accumulation of carbonaceous oxidative intermediates on the catalyst. As listed in Table 2, the If/Ib ratio of the Pd/CA catalyst is 2.0, remarkably higher than those of commercial Pd/C catalyst (If/Ib = 0.54) and other nanocomposites.15 The anti-poisoning effect is further enhanced by the addition of BP (If/Ib ∼ 3.1). The results affirm that the Pd/CA and BP/Pd/CA catalysts have both high catalytic activity for alcohol electrooxidation and high tolerance to poisoning species.
| Sample | J (mA cm−2) | If/Ib | Zeta-potential (mV) | ESR (Ω) |
|---|---|---|---|---|
| a J = If. | ||||
| CA | 0.7 | 1.6 | −6.9 | 3.58 |
| Pd/CA | 27.6 | 2.0 | −4.1 | 1.47 |
| BP/Pd/CA | 35.9 | 3.1 | −0.8 | 0.49 |
The catalytic current densities of BP/Pd/CA at different scan rates ranging from 5 to 60 mV s−1 are represented in Fig. 5a. The forward and backward oxidation peak potentials (Ep) shift towards more positive values with a rising scan rate, with a linear correlation between Ep and the natural logarithm of scan rate (lnv). This suggests that the electrooxidation of ethanol is an irreversible process in which the reaction products diffuse away from the active sites of the electrode. The relationship between the anodic peak current density and the square root of the scan rate for BP/Pd/CA is shown in Fig. 5b. The increase in the peak current density is linearly proportional to the square root of the scan rate, which indicates that the EEO process is a diffusion-controlled process.
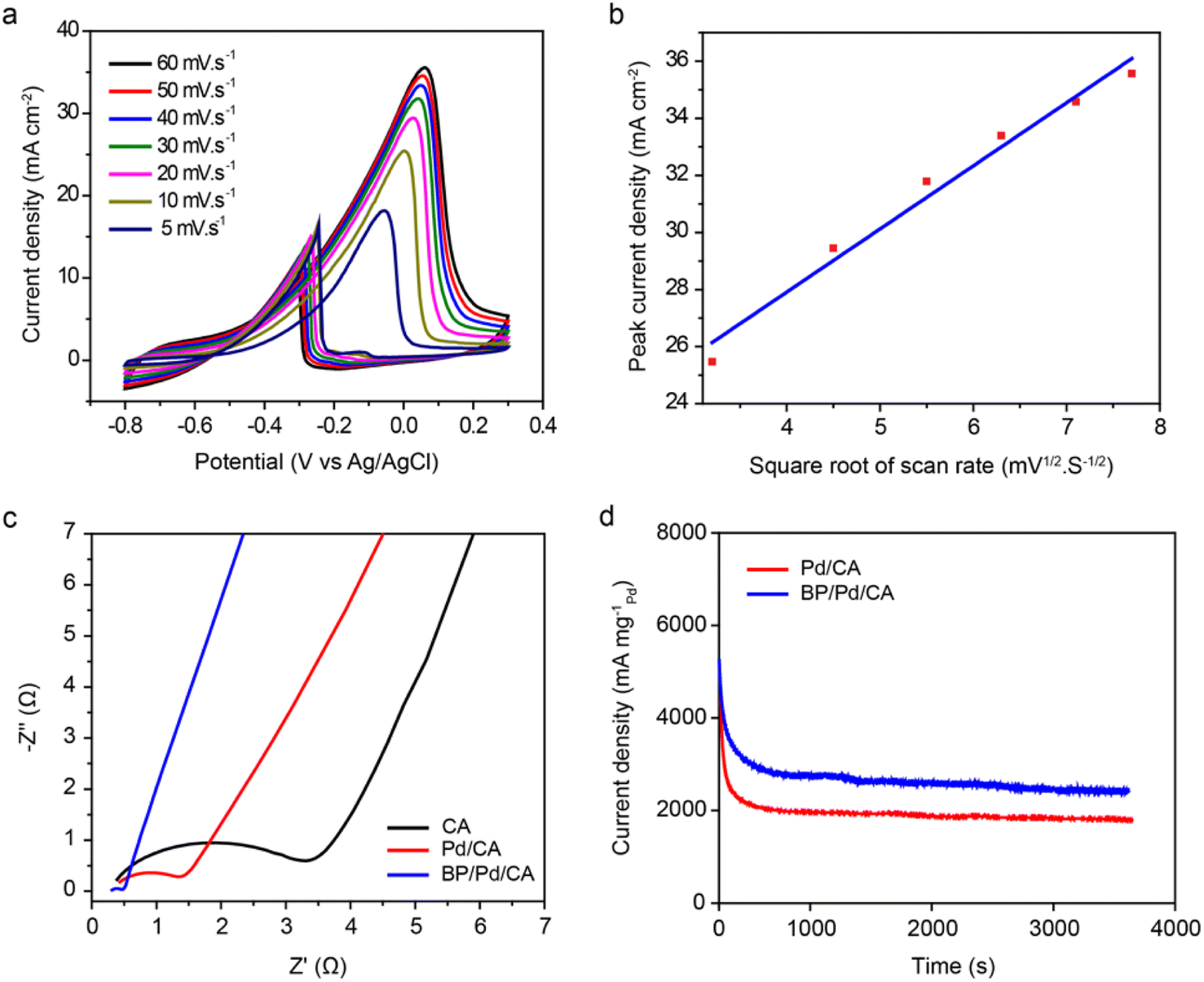 | ||
| Fig. 5 (a) Cyclic voltammograms of BP/Pd/CA in 1.0 M KOH + 0.5 M C2H5OH solution at various scan rates. (b) Plot of anodic current density versus the square root of the scan rate for BP/Pd/CA. (c) Nyquist plots obtained from EIS for CA (black), Pd/CA (red), and BP/Pd/CA (blue) in 1.0 M KOH + 0.5 M C2H5OH solution with a potential of 10 mV. (d) Chronoamperograms of the Pd/CA and BP/Pd/CA catalysts in 1.0 M KOH + 0.5 M C2H5OH solution at −1 V. | ||
The reaction mechanism of EEO has pH-dependent complex pathways that involve several reactive intermediates. The EEO on Pd-based electrocatalysts in alkaline media is proposed to occur according to the following reactions:50–53
| CH3CH2OH + 3OH− ↔ CH3COads + 3H2O + e | (1) |
| OH− ↔ OHads + e | (2) |
| CH3COads + OHads → CH3COOHads | (3) |
| CH3COOHads + OH− → CH3COO− + H2O | (4) |
The third stage is the slowest step during EEO and is reported as the rate-determining step. Moreover, CH3COO− may be oxidized at higher potentials to CO2.54 The high catalytic activities of Pd/CA and BP/Pd/CA are attributed to the synergistic interaction between the support and catalyst. The Pd and Pd-BP catalysts modify CA into a more open and conductive structure, while the 3D network structure of CA uniformly distributes the Pd NPs and BP/Pd nanohybrids and protects them from leaching. It has been demonstrated that the catalytic performance of Pd has a strong dependence on its surface structure with the highest catalytic activity achieved for Pd {111} facets.12 As evidenced by the XRD analysis, our facile preparation method produces Pd NPs with mainly (111) surface arrangement, which is ideal for EEO. The electrocatalytic reactivity and charge density of the Pd NPs are further improved through BP doping.55 This in turn leads to fast dissociative adsorption of alcohol on the Pd surface (eqn (1)) and high coverage of the adsorbed acyl and hydroxyl species (eqn (3)).
To further characterize the interfacial behavior of the electrode/electrolyte interface, we performed EIS measurements. EIS measures the impedance of an electrochemical system in dependence of an applied AC potentials frequency. Fig. 5c displays the Nyquist impedance spectra of ethanol oxidation on the CA, Pd/CA and BP/Pd/CA electrodes, in which the imaginary impedance component (Z′′(Ω)) is plotted against the real impedance component (Z′(Ω)) at each excitation frequency. The catalysts exhibit a semicircle in the high frequency region, a diagonal line (Warburg impedance) at middle frequencies, and an approximate vertical line at low frequencies. The semicircle corresponds to the faradaic charge-transfer resistance at the electrode/electrolyte interface.56,57 We noticed that the charge-transfer resistance (i.e., the diameter of the semicircle) of EEO decreases in the following order: CA > Pd/CA > BP/Pd/CA, demonstrating the enhanced charge-transfer kinetics of the BP/Pd/CA catalyst, in good agreement with the voltammetric results. The length of the Warburg-type lines is the shortest for BP/Pd/CA, which indicates high ion diffusion efficiency for ion transport. In addition, the straight line of BP/Pd/CA in the low-frequency region is the steepest. This reveals that BP/Pd/CA has the best capacitive behavior.
The intersection of the capacitive line with the Z′ axis indicates the equivalent series resistance (ESR) of the electrode, defined as the sum of the resistances of the electrode, the electrolyte, and the interface. The lowest ESR value (0.49 Ω) is attained for BP/Pd/CA (Table 2), manifesting the superior EEO performance of the BP/Pd/CA catalyst. Table 2 also shows the zeta-potential values of the three electro-catalysts. The effective electric charge on the CA surface is about −6.9 mV which drops to −0.8 mV for BP/Pd/CA. The lower zeta-potential value of BP/Pd/CA is an inductive of higher electrode activity towards ethanol oxidation. The stability of the electrocatalysts for EEO is further investigated by performing chronoamperometry measurements at a constant potential of −1 V for 3600 seconds. The chronoamperic responses of Pd/CA and BP/Pd/CA are shown in Fig. 5d. The EEO current density drops quickly in the initial period and then reaches a pseudo-steady state. The initial current decay is due to the formation of some Pd oxides/hydroxides and strongly adsorbed intermediates on the surface of reactive sites. The quasi-steady current densities of the catalysts are quite stable and attest to high electrode durability and long-term stability. As expected, the initial and final current densities of BP/Pd/CA are higher than those of Pd/CA. BP/Pd/CA retains 92% of its initial activity, while Pd/CA preserves 88% of its initial activity. In addition, the initial current of BP/Pd/CA declines more slowly than that of Pd/CA which signifies that BP/Pd/CA has superior electrochemical catalytic stability for the oxidation of alcohols.
4. Conclusions
In summary, we have utilized carbon aerogel prepared by the sol–gel method as a support material for palladium nanocatalysts. The CA support influences the catalyst dispersion, size, and morphology, resulting in improved catalyst stability and reactivity. The catalyst increases the graphitic character and pore volume of the support, facilitating electron transportation and electrolyte penetration. The hybrid system, therefore, shows remarkable catalytic activity as an anode electrocatalyst for EEO in an alkaline medium. The electroactivity of the catalyst is further enhanced by BP doping. BP modifies the electronic properties of the Pd catalyst, increasing the adsorption strength of ethanol and reactive intermediates onto the catalyst surface. Our work suggests that electrode materials that combine carbon gels and electroactive metals enable high catalytic performance with potential applications in catalysis and fuel cells.Conflicts of interest
There are no conflicts to declare.References
- S. Wasmus and A. Küver, Methanol oxidation and direct methanol fuel cells: a selective review, J. Electroanal. Chem., 1999, 461(1–2), 14–31 CrossRef.
- R. Parsons and T. VanderNoot, The oxidation of small organic molecules: a survey of recent fuel cell related research, J. Electroanal. Chem. Interfacial Electrochem., 1988, 257(1–2), 9–45 CrossRef.
- J. C. Ferreira, et al., Palladium nanoparticles supported on mesoporous biocarbon from coconut shell for ethanol electro-oxidation in alkaline media, Mater Renew Sustain Energy, 2018, 7(4), 9 Search PubMed.
- H. Han, et al., Electrocatalytic Oxidations of Formic Acid and Ethanol over Pd Catalysts Supported on a Doped Polypyrrole-Carbon Composite, ChemistrySelect, 2017, 2(22), 6260–6268 CrossRef.
- Y. Cui, et al., Ultrasmall AuPd nanoclusters on amine-functionalized carbon blacks as high-performance bi-functional catalysts for ethanol electrooxidation and formic acid dehydrogenation, J. Energy Chem., 2022, 68, 556–563 CrossRef.
- P. Kanninen, et al., Highly active platinum nanoparticles supported by nitrogen/sulfur functionalized graphene composite for ethanol electro-oxidation, Electrochim. Acta, 2017, 242, 315–326 CrossRef.
- R. Kiyani, M. J. Parnian and S. Rowshanzamir, Investigation of the effect of carbonaceous supports on the activity and stability of supported palladium catalysts for methanol electro-oxidation reaction, Int. J. Hydrogen Energy, 2017, 42(36), 23070–23084 CrossRef.
- C. S. Yellatur, et al., Facile electrooxidation of ethanol on reduced graphene oxide supported Pt-Pd bimetallic nanocomposite surfaces in acidic media, Nanotechnology, 2022, 33(33), 335401 CrossRef.
- S. Li, et al., PtNiCu nanowires with advantageous lattice-plane boundary for enhanced ethanol electrooxidation, Nano Res., 2022, 15(4), 2877–2886 CrossRef.
- Y. Xiang, et al., Construction of PdCo catalysts on Ni bowl-like micro/nano array films for efficient methanol and ethanol electrooxidation, J. Alloys Compd., 2022, 924, 166483 CrossRef.
- S. Mohammad Mostashari, et al., Engineering three-dimensional superstructure of Pd aerogel with enhanced performance for ethanol electrooxidation, J. Mol. Liq., 2022, 360, 119363 CrossRef.
- M. López-Atalaya, et al., Electrochemical oxidation of ethanol on Pt (hkl) basal surfaces in NaOH and Na2CO3 media, J. Power Sources, 1994, 52(1), 109–117 CrossRef.
- L. Chen, et al., Improved ethanol electrooxidation performance by shortening Pd–Ni active site distance in Pd–Ni–P nanocatalysts, Nat. Commun., 2017, 8(1), 14136 CrossRef PubMed.
- Y.-C. Wei, C.-W. Liu and K.-W. Wang, Improvement of oxygen reduction reaction and methanol tolerance characteristics for PdCo electrocatalysts by Au alloying and CO treatment, Chem. Commun., 2011, 47(43), 11927–11929 RSC.
- T. Wu, et al., Palladium Nanoparticles Anchored on Anatase Titanium Dioxide-Black Phosphorus Hybrids with Heterointerfaces: Highly Electroactive and Durable Catalysts for Ethanol Electrooxidation, Adv. Energy Mater., 2018, 8(1), 1701799 CrossRef.
- S. Li, et al., Mxene coupled over nitrogen-doped graphene anchoring palladium nanocrystals as an advanced electrocatalyst for the ethanol electrooxidation, J. Colloid Interface Sci., 2022, 610, 944–952 CrossRef PubMed.
- Q. Liu, et al., Well-dispersed palladium nanoparticles on three-dimensional hollow N-doped graphene frameworks for enhancement of methanol electro-oxidation, Electrochem. Commun., 2016, 73, 75–79 CrossRef.
- A. Abdelwahab, et al., Cobalt-Doped Carbon Gels as Electro-Catalysts for the Reduction of CO2 to Hydrocarbons, Catalysts, 2017, 7(1), 25 CrossRef.
- A. F. Pérez-Cadenas, et al., Metal-doped carbon xerogels for the electro-catalytic conversion of CO2 to hydrocarbons, Carbon, 2013, 56, 324–331 CrossRef.
- A. Abdelwahab, et al., Electrodes Based on Carbon Aerogels Partially Graphitized by Doping with Transition Metals for Oxygen Reduction Reaction, Nanomaterials, 2018, 8(4), 266 CrossRef PubMed.
- M. M. EL-Deeb, et al., Effect of pore geometry on the electrocatalytic performance of nickel cobaltite/carbon xerogel nanocomposite for methanol oxidation, Electrochim. Acta, 2018, 259, 77–85 CrossRef.
- A. Elmouwahidi, et al., Electrochemical performances of supercapacitors from carbon-ZrO2 composites, Electrochim. Acta, 2018, 259, 803–814 CrossRef.
- A. Abdelwahab, et al., Insight of the effect of graphitic cluster in the performance of carbon aerogels doped with nickel as electrodes for supercapacitors, Carbon, 2018, 139, 888–895 CrossRef CAS.
- R. Pekala, Organic aerogels from the polycondensation of resorcinol with formaldehyde, J. Mater. Sci., 1989, 24(9), 3221–3227 CrossRef CAS.
- R. Pekala, et al., Aerogels derived from multifunctional organic monomers, J. Non-Cryst. Solids, 1992, 145, 90–98 CrossRef CAS.
- C. M. Park and H. J. Sohn, Black phosphorus and its composite for lithium rechargeable batteries, Adv. Mater., 2007, 19(18), 2465–2468 CrossRef CAS.
- M. Vanni, et al., Black Phosphorus/Palladium Nanohybrid: Unraveling the Nature of P–Pd Interaction and Application in Selective Hydrogenation, Chem. Mater., 2019, 31(14), 5075–5080 CrossRef CAS.
- G. Gryglewicz, et al., Effect of pore size distribution of coal-based activated carbons on double layer capacitance, Electrochim. Acta, 2005, 50(5), 1197–1206 CrossRef CAS.
- H.-K. Song, et al., Electrochemical impedance spectroscopy of porous electrodes: the effect of pore size distribution, Electrochim. Acta, 1999, 44(20), 3513–3519 CrossRef CAS.
- M. Bjelopavlic, G. Newcombe and R. Hayes, Adsorption of NOM onto activated carbon: effect of surface charge, ionic strength, and pore volume distribution, J. Colloid Interface Sci., 1999, 210(2), 271–280 CrossRef CAS PubMed.
- G. Newcombe, M. Drikas and R. Hayes, Influence of characterised natural organic material on activated carbon adsorption: II. Effect on pore volume distribution and adsorption of 2-methylisoborneol, Water Res., 1997, 31(5), 1065–1073 CrossRef CAS.
- K. Wang, et al., Carbon nanocages with nanographene shell for high-rate lithium ion batteries, J. Mater. Chem., 2010, 20(43), 9748–9753 RSC.
- F. Xu, et al., A facile approach for tailoring carbon frameworks from microporous to nonporous for nanocarbons, J. Mater. Chem. A, 2013, 1(16), 5001–5005 RSC.
- R. Fu, et al., XPS Study of Copper-Doped Carbon Aerogels, Langmuir, 2002, 18(26), 10100–10104 CrossRef CAS.
- A. L. Smirnova, et al., Synthesis of Novel Electrode Materials Using Supercritical Fluids, ECS Trans., 2019, 19(21), 9–21 CrossRef.
- Z. Jin, et al., Decoration, Migration, and Aggregation of Palladium Nanoparticles on Graphene Sheets, Chem. Mater., 2010, 22(20), 5695–5699 CrossRef.
- S. J. R. Tan, et al., Quasi-Monolayer Black Phosphorus with High Mobility and Air Stability, Adv. Mater., 2018, 30(6), 1704619 CrossRef.
- Y. Wu, et al., Preparation of a graphene–phosphorene composite by pressure quenching and its ferromagnetism, Chem. Commun., 2020, 56(13), 2016–2019 RSC.
- K. L. Kuntz, et al., Control of Surface and Edge Oxidation on Phosphorene, ACS Appl. Mater. Interfaces, 2017, 9(10), 9126–9135 CrossRef.
- X. Zhu, et al., Stabilizing black phosphorus nanosheets via edge-selective bonding of sacrificial C60 molecules, Nat. Commun., 2018, 9(1), 4177 CrossRef PubMed.
- M. Farsadrooh, et al., Two-dimensional Pd-nanosheets as efficient electrocatalysts for ethanol electrooxidation. Evidences of the CC scission at low potentials, Appl. Catal., B, 2018, 237, 866–875 CrossRef CAS.
- Q. Zhang, J. Weng and J. Xu, Palladium Phosphide/Black Phosphorus Heterostructures with Enhanced Ethanol Oxidation Activity and Stability, J. Phys. Chem. C, 2021, 125(34), 18717–18724 CrossRef CAS.
- T. Wu, et al., Black Phosphorus–Graphene Heterostructure-Supported Pd Nanoparticles with Superior Activity and Stability for Ethanol Electro-oxidation, ACS Appl. Mater. Interfaces, 2019, 11(5), 5136–5145 CrossRef CAS.
- A. Dutta and J. Ouyang, Ternary NiAuPt Nanoparticles on Reduced Graphene Oxide as Catalysts toward the Electrochemical Oxidation Reaction of Ethanol, ACS Catal., 2015, 5(2), 1371–1380 CrossRef CAS.
- B. Cai, et al., Function-Led Design of Aerogels: Self-Assembly of Alloyed PdNi Hollow Nanospheres for Efficient Electrocatalysis, Angew. Chem., Int. Ed., 2015, 54(44), 13101–13105 CrossRef CAS PubMed.
- C. Zhu, et al., Efficient Synthesis of MCu (M = Pd, Pt, and Au) Aerogels with Accelerated Gelation Kinetics and their High Electrocatalytic Activity, Adv. Mater., 2016, 28(39), 8779–8783 CrossRef CAS PubMed.
- Q. Liu, et al., B,N-codoped graphene nanoribbons supported Pd nanoparticles for ethanol electrooxidation enhancement, J. Mater. Chem. A, 2016, 4(13), 4929–4933 RSC.
- A.-L. Wang, et al., Palladium–Cobalt Nanotube Arrays Supported on Carbon Fiber Cloth as High-Performance Flexible Electrocatalysts for Ethanol Oxidation, Angew. Chem., Int. Ed., 2015, 54(12), 3669–3673 CrossRef CAS PubMed.
- S.-H. Ye, J.-X. Feng and G.-R. Li, Pd Nanoparticle/CoP Nanosheet Hybrids: Highly Electroactive and Durable Catalysts for Ethanol Electrooxidation, ACS Catal., 2016, 6(11), 7962–7969 CrossRef CAS.
- J. Liu, et al., Kinetics of ethanol electrooxidation at Pd electrodeposited on Ti, Electrochem. Commun., 2007, 9(9), 2334–2339 CrossRef.
- Z. X. Liang, et al., Mechanism study of the ethanol oxidation reaction on palladium in alkaline media, Electrochim. Acta, 2009, 54(8), 2203–2208 CrossRef.
- J. Wang, et al., Comparative study to understand the intrinsic properties of Pt and Pd catalysts for methanol and ethanol oxidation in alkaline media, Electrochim. Acta, 2015, 185, 267–275 CrossRef.
- A. Sadiki, et al., Increased Electrochemical Oxidation Rate of Alcohols in Alkaline Media on Palladium Surfaces Electrochemically Modified by Antimony, Lead, and Tin, Electrochim. Acta, 2014, 139, 302–307 CrossRef.
- W. Du, et al., Palladium–Tin Alloyed Catalysts for the Ethanol Oxidation Reaction in an Alkaline Medium, ACS Catal., 2012, 2(2), 287–297 CrossRef CAS.
- J. K. Nørskov, et al., Density functional theory in surface chemistry and catalysis, Proc. Natl. Acad. Sci., 2011, 108(3), 937 CrossRef.
- Z. Wang, et al., Hybrids of NiCo2O4 nanorods and nanobundles with graphene as promising electrode materials for supercapacitors, J. Colloid Interface Sci., 2015, 460, 303–309 CrossRef CAS PubMed.
- S. Biswas and L. T. Drzal, Multilayered nanoarchitecture of graphene nanosheets and polypyrrole nanowires for high performance supercapacitor electrodes, Chem. Mater., 2010, 22(20), 5667–5671 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05452c |
| This journal is © The Royal Society of Chemistry 2022 |