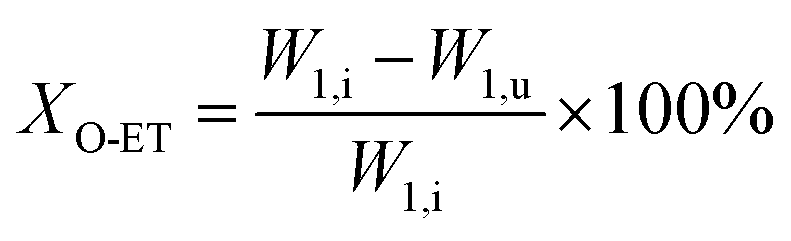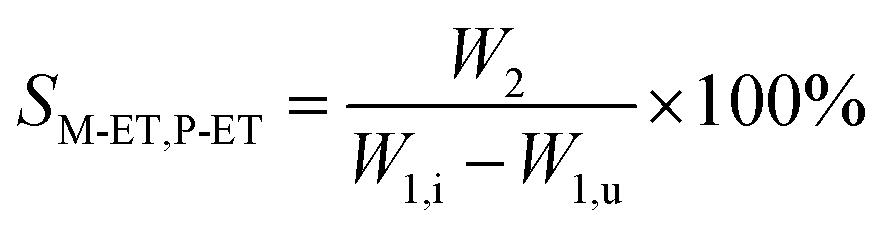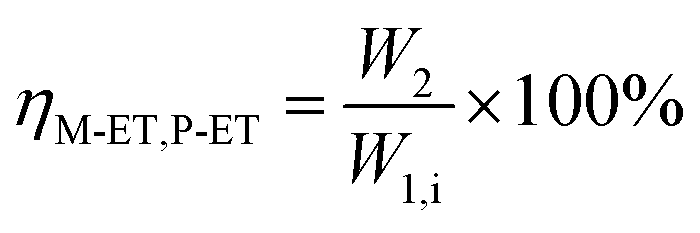DOI:
10.1039/D2RA05390J
(Paper)
RSC Adv., 2022,
12, 31326-31337
Comparison of surface passivation modification of two mordenite zeolites and their application on the isomerisation of o-ethyltoluene†
Received
28th August 2022
, Accepted 18th October 2022
First published on 1st November 2022
Abstract
During the isomerisation of o-ethyltoluene (O-ET) to produce m-ethyltoluene (M-ET) and p-ethyltoluene (P-ET), it is crucial to improve the isomerisation selectivity and reduce side reactions, such as disproportionation, alkyl transfer, and splitting. In this study, in order to improve the selectivities toward M-ET and P-ET during O-ET isomerisation, both the commercial micropore mordenite (HM) and the prepared micro–mesoporous mordenite (HM–M) were treated through chemical liquid deposition using tetraethyl orthosilicate (TEOS) and 3,5-dimethylphenylmagnesium bromide (DPB), respectively. Thereafter, their structure, porosity, and acidity were characterized via X-ray diffraction, transmission electron microscopy, inductively coupled plasma, N2 sorption, temperature-programmed desorption of ammonia, Fourier-transform infrared spectroscopy of pyridine and 2,6-di-tert-butylpyridine, and thermal analysis. The deposition mechanism of DPB was also discussed. The results showed that TEOS could shrink and block the micropores of mordenite. By contrast, DPB passivated the external surface acidity and did not affect the micropore structure. Moreover, HM modified using DPB significantly shortened the self-coking process, improved the product selectivities for M-PT and P-ET as well as their stability, and prolonged the catalytic life. When the amount of magnesium oxide (MgO) deposited on the HM zeolite was 4%, the product selectivities toward M-ET and P-ET increased from 67.27% to 77.54%, and the yields of M-ET and P-ET increased from 47.57% to 52.98%. However, the performance of the catalyst was not significantly enhanced on the HM–M, owing to the passivation of acidic sites in the mesopores by the TEOS and DPB.
Introduction
Poly(methylstyrene) is an essential raw material for manufacturing multifunctional materials and is prepared through dehydrogenation and polymerisation of m-ethyltoluene (M-ET) and p-ethyltoluene (P-ET). However, the commercial application of poly(methylstyrene) in multifunctional materials is limited by the high market price of M-ET and P-ET.1,2 The bottom product of the xylene column in oil refineries contains a variety of C9 aromatic hydrocarbons, including o-ethyltoluene (O-ET), M-ET, P-ET, trimethylbenzene, and tetramethylbenzene. The M-ET and P-ET content in this bottom product can be increased by isomerising O-ET.3,4 Therefore, the development of catalysts with excellent catalytic performance for improving the selectivities toward M-ET and P-ET during O-ET isomerisation is essential.
Recently, crystalline aluminosilicates, such as hydrogen-type mordenite (HM), HZSM-5, and Hβ, and crystalline heteropoly acid salts, such as ammonium silicotungstate and ammonium phosphotungstate, have been widely studied for O-ET isomerisation.4–7 Among these catalysts, the HM zeolite exhibited a higher isomerization conversion rate and selectivity, owing to its suitable acidity and pore structure. However, the initial structural defects of commercial mordenite result in underutilised acid sites in the pores,8,9 the conversion of O-ET was lower at low temperatures. In addition, large-molecule by-products do not readily diffuse out of the pores because of the restriction of pore channels, leading to quick coking and deactivation of the catalyst, thus restricting its industrial application.10–13 To solve these challenges, low-temperature micro–mesoporous mordenite (HM–M) was prepared via alkali-acid post-treatment by our group previously.14 HM–M has a large specific surface area, high accessibility of acid sites in the pores, and short diffusion channels, which can achieve high-efficiency conversion of O-ET at low temperatures and improve catalyst deactivation. Therefore, hierarchical-pore mordenite has broad application prospects in O-ET isomerisation.
Although commercial and hierarchical mordenites exhibit good catalytic performance during the isomerization of O-ET, the acid sites on the outer surface of mordenite are not shape-selective and can induce secondary reactions of isomerisation products.15 Therefore, in order to improve the selectivity of isomerized products and prolong the catalytic life, modification of the acidic sites on the external surface of the zeolites is necessary. Recently, the deposition of chemical silicon and metal oxides on external surfaces of zeolites is often used to modify the acid sites.16–22 Several methods have been used, such as chemical vapour deposition (CVD), chemical liquid deposition (CLD), electrochemical deposition, and impregnation.23–27 Li et al. investigated the deposition of TEOS by CLD to passivate the external surface acidity of Hβ molecular sieves with different grain sizes. Their approach increased the selectivity of the 2-phenyl isomer (2-LAB) by 13% for Hβ with larger grain sizes.28 Zhu et al. deposited TEOS on ZSM-5 zeolites by CVD and CLD methods and discovered that SiO2-CLD/ZSM-5 had a 2% higher EB conversion rate than SiO2-CVD/ZSM-5 with the same p-diethylbenzene (p-DEB) selectivity.22 Consistently, the deposition modification of TEOS not only passivated the acidic sites but also reduced the pore size of the zeolites. Additionally, in the deposition modification of metal oxides such as magnesium oxide (MgO), cerium(IV) oxide (CeO2), and iron(III) oxide (Fe2O3), the precursor nitrate can usually enter the micropores of the molecular sieve. Although this modification can improve selectivity, the catalytic performance also significantly decreases.22 Commonly, metal oxides are not used as a single modifier, but act as additives or auxiliaries. Therefore, it is particularly important to seek a modification that can passivate the acidic sites on the zeolites external surface without affecting their pore size. It was reported that Zhang et al.29 significantly improve the selectivity of p-DEB and found that 3,5-dimethylphenylmagnesium bromide (DPB) does not change its pore size when passivating the external surface acidity of the ZSM-5 zeolite. Therefore, we believe that a nucleophilic macromolecular precursor that can remove the external surface acid sites of mordenite and maintain its pore size may improve the selectivity towards M-ET and P-ET in the O-ET isomerisation process.
In this study, TEOS and DPB macromolecules were chosen to passivate the external surface acidity of mordenite, since the 12-membered ring main channel of mordenite (between 8.2 and 8.6 Å,8) is smaller than that of TEOS and DPB molecules. As a comparison, we modified mordenite zeolites with microporous and micro–mesoporous structures (i.e., HM and HM–M) using TEOS and DPB, respectively, via the CLD method to illustrate the effects of pore size and external surface acidity on the isomerisation process. In addition, the effects of the TEOS and DPB modifiers on the structure, porosity, and acidity of the two mordenite zeolites are discussed in conjunction with their characterisation. Finally, to the best of our knowledge, this study is the first to use the isomerisation reaction of O-ET to evaluate the catalytic properties of surface passivation modified mordenites. This study provides a new idea for shortening the self-coking modification process of mordenites, thus raising the M-ET and P-ET selectivities in the O-ET isomerisation process and improving product stability and catalytic life.
Experimental
Materials
Commercial mordenite was purchased from Nankai Chemist Catalyst Co., Ltd. 3,5-Dimethylphenylmagnesium bromide (DPB) were purchased from Sa en Chemical Technology Co., Ltd. Tetraethyl orthosilicate (TEOS), sodium hydroxide (NaOH), nitric acid (HNO3, 65%), and oxalic acid (C2H2O4) were purchased from Sinopharm Chemical Reagent Co., Ltd. All the chemicals (analytical grade) were used without further purification.
Preparation of micro–mesoporous mordenite
According to previously reported procedures,14 the commercial mordenite (labelled as HM) was refluxed in a 0.2 M sodium hydroxide (NaOH) solution for 1.0 h at 75 °C (25 mL solution per 1 g mordenite). The mordenite was filtered, washed to pH 7, and ion-exchanged with 1.0 M ammonium chloride (NH4Cl). Subsequently, the obtained solid was treated with a 0.1 mol L−1 solution of mixed nitric acid-oxalic acid (HNO3–C2H2O4) (C2H2O4/HNO3 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 1.0 h at 70 °C (25 mL solution per 1 g mordenite). After that, it was filtered, washed to pH 7, dried for 12 h at 110 °C, and calcined in the air for 5 h at 550 °C. This sample was labelled HM–M.
:1) for 1.0 h at 70 °C (25 mL solution per 1 g mordenite). After that, it was filtered, washed to pH 7, dried for 12 h at 110 °C, and calcined in the air for 5 h at 550 °C. This sample was labelled HM–M.
Modification of mordenite zeolite with two structures
The HM zeolite was dispersed in n-hexane (25 mL solution per 1 g mordenite) and stirred thoroughly for 30 min at 300 rpm. Subsequently, TEOS was added to the mixture and stirred for 10 h at 30 °C. After that, n-hexane was removed by vacuum distillation, and the obtained solid was dried for 12 h at 110 °C and calcined in the air for 5 h at 550 °C. The obtained sample was labelled x% SiO2/HM (where x is the SiO2 deposition weight percentage). Similarly, the HM zeolite modified with DPB instead of TEOS was named x% MgO/HM (where x is the MgO deposition weight percentage). HM–M zeolite was modified via the above method to obtain the catalysts x% SiO2/HM–M and x% MgO/HM –M.
Characterisations
The crystallinity of the samples was determined using X-ray powder diffraction (XRD) (Rigaku D/Max 2500VL/PC diffractometer, Rigaku Corporation, Japan) with Cu Kα radiation (λ = 0.1541 nm). Transmission electron microscopy (TEM) images were obtained using a JEM-2100 microscope (JEOL Ltd., Tokyo, Japan). The specific surface area and porosity were measured by N2 sorption (ASAP 2460, Micromeritics Instrument Corporation, USA). The molar ratios of Si/Al and the content of Mg in the zeolite samples were determined via an inductively coupled plasma – optical emission spectrometry (ICP-OES) (IRIS Intrepid II XSP spectrometer, ThermoFisher Scientific Corporation, USA). The number and strength of the acid sites in the zeolite samples were determined using an ammonia temperature-programmed desorption (NH3-TPD) instrument (AutoChem II 2920, Micromeritics Instrument Corporation, USA). The Brønsted and Lewis acid sites of the total and external surfaces were characterised by Fourier-transform infrared spectroscopy (FTIR) with pyridine/2,6-di-tert-butylpyridine adsorption, respectively, on a Vertex 70 (Bruker, Germany) spectrometer. In addition, samples with 2,6-di-tert-butylpyridine adsorption were quantified using Netzsch STA 449F3 thermogravimetric analysis, and samples without 2,6-di-tert-butylpyridine adsorption were used as controls.
Catalytic tests
The catalyst with a 60–80 mesh particle size was placed in a fixed-bed reactor with an inner diameter of 10 mm. The C9 aromatics (Table 1) were fed into the reactor by a metering pump for the isomerisation reactions of O-ET. The isomerisation products were analysed hourly via ThermoFisher Trace 1300 gas chromatography (GC) with a SE-30 capillary column. The products were qualitatively analysed using GC-mass spectrometry (Varian 3800/2200) equipped with a Varian cp-sil-19 column. The experiment was performed under the following conditions: WHSV = 1.0 h−1, H2/HC = 5.0 (mol mol−1) and pressure = 1.5 MPa. According to the preferential isomerisation mechanism of ET,30 the isomerisation process of O-ET is mainly discussed in this work. The conversion of O-ET (XO-ET), selectivities toward M-ET and P-ET (SM-ET,P-ET), and yields of M-ET and P-ET (ηM-ET,P-ET) were calculated using eqn (1), (2) and (3), respectively, where W1,i is the mass of O-ET in the feed, W1,u is the mass of O-ET in the product, and W2 is the mass of generated M-ET and P-ET.| |
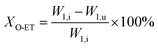 | (1) |
| |
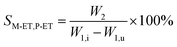 | (2) |
| |
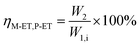 | (3) |
Table 1 Composition of the C9 aromaticsa
| M-ET, P-ET, O-ET, TMB, and others represented m-ethyltoluene, p-ethyltoluene, o-ethyltpoluene, trimethylbenzene and the other components, which were obtained via GC analysis, respectively. |
| Component |
M-ET |
P-ET |
O-ET |
TMB |
Others |
| Content/% |
11.7 |
6.0 |
12.2 |
67.0 |
3.1 |
Results and discussion
Catalyst selection
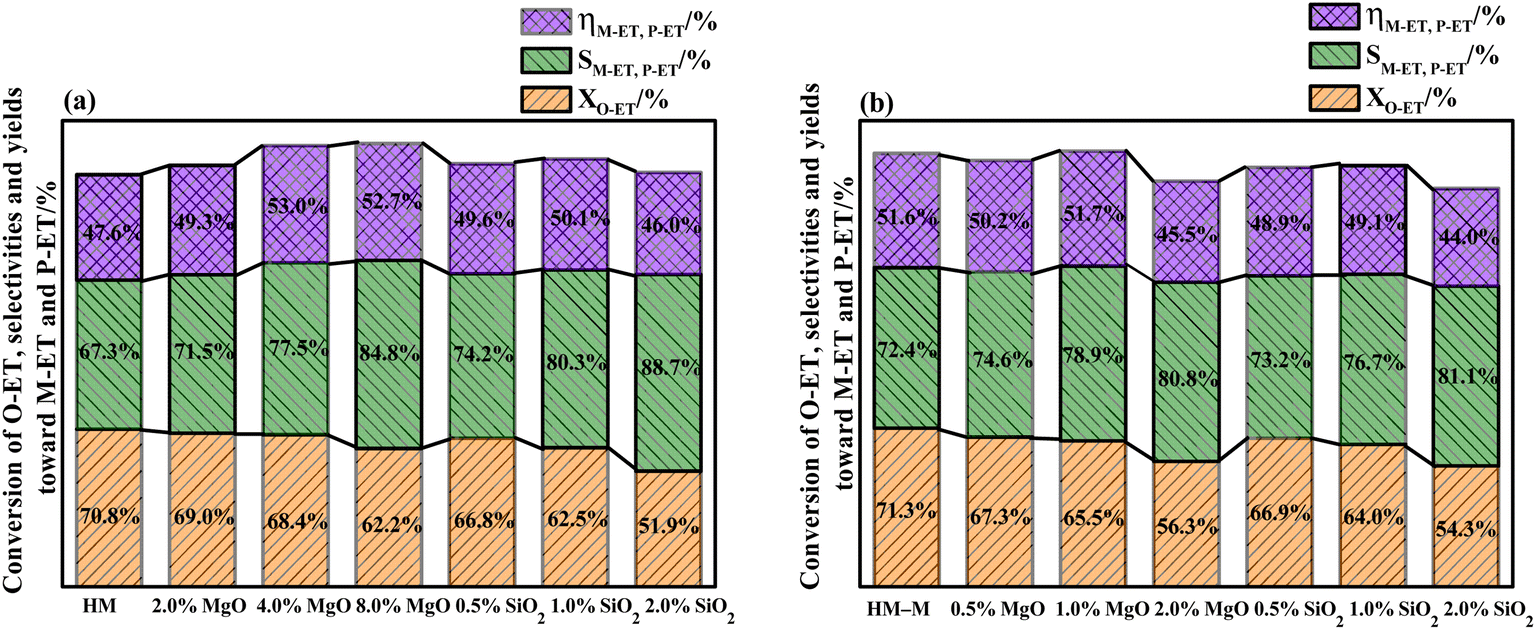
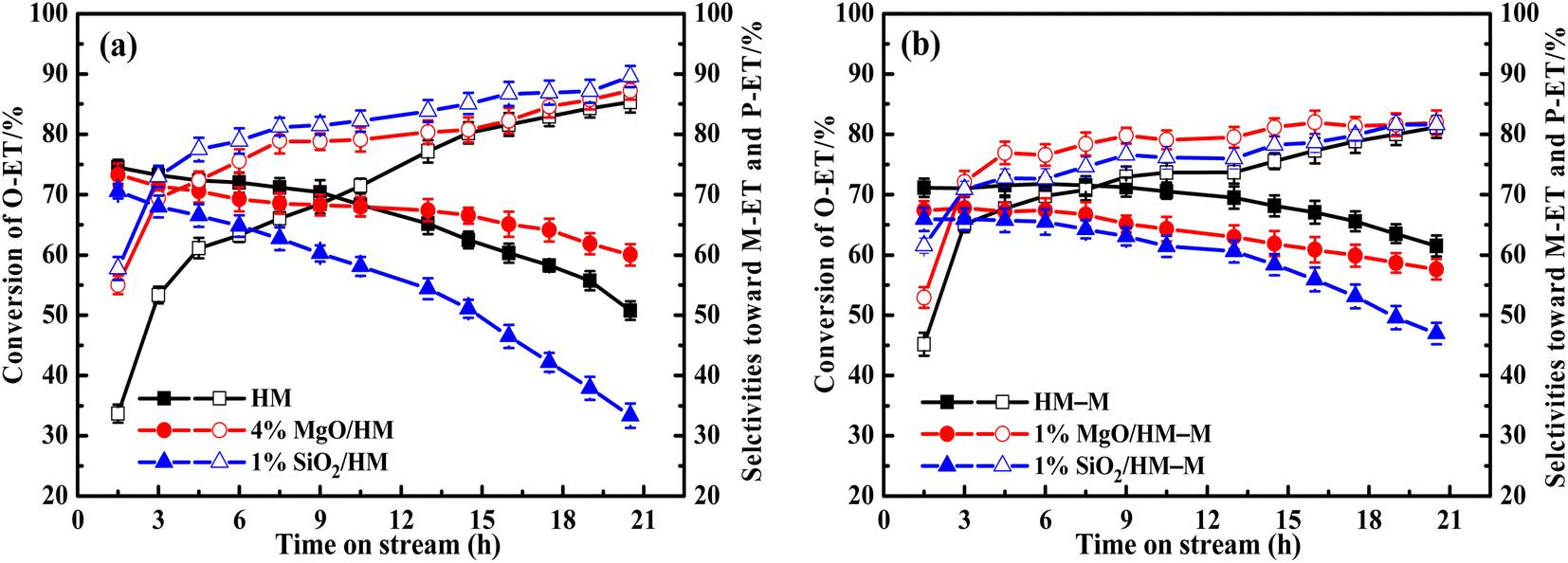
It is common knowledge that during the isomerisation reaction, the external surface acidity of the molecular sieve is very crucial for the selectivity of the target product. Therefore, it is vital to treat the external surface acidity of the molecular sieves. In this study, the influence of the modifier TEOS and DPB amount on the isomerisation performance of O-ET was first evaluated (Fig. 1). These modified zeolites showed higher selectivities for M-ET and P-ET than the parent HM and HM–M. Moreover, increasing the modifier content increased the effect on the conversion rate of O-ET than the selectivities of M-ET and P-ET significantly during isomerisation. Besides, it could be found that the optimal deposition of MgO is 4% for HM and 1% for HM–M. Similarly, the optimal deposition of SiO2 is 1% for both HM and HM–M. Therefore, 1% SiO2/HM, 4% MgO/HM, 1% MgO/HM–M, and 1% SiO2/HM–M were selected as the optimal catalysts for further investigation.
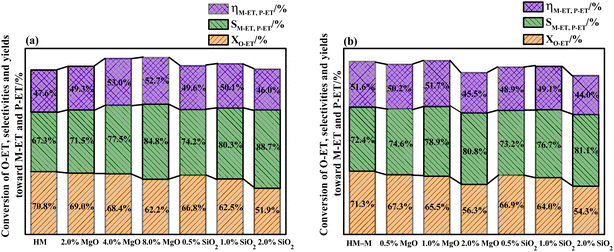 |
| | Fig. 1 Results of O-ET isomerization employing HM, HM–M and their modified zeolites. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. The data were the averages of three experiments, and the maximum standard deviation of each data was <2.0%. | |
Characterisation of catalyst
Physicochemical and structural properties
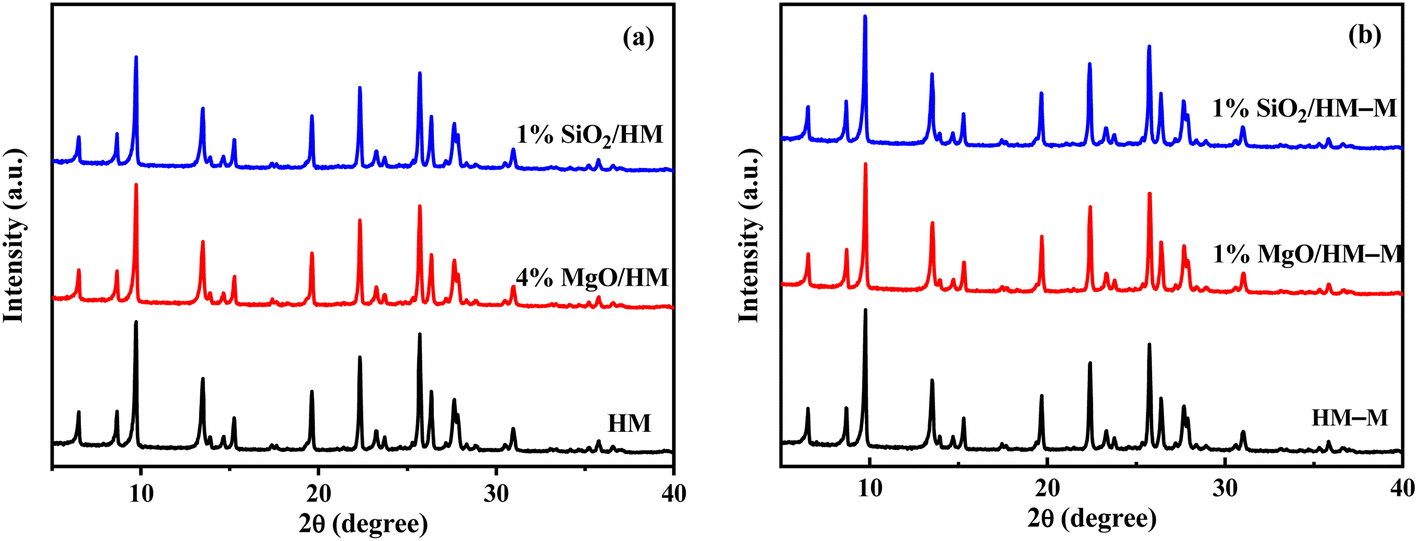
Fig. 2 shows the X-ray diffraction (XRD) patterns of HM, HM–M, and their modified samples. All modified samples exhibited the characteristic diffraction peaks of mordenite zeolite, showing that the CLD treatment with DPB and TEOS did not degrade the crystal integrity of HM and HM–M. Furthermore, the diffraction peaks of SiO2 and MgO are not detected, indicating that SiO2 and MgO particles were minute and highly dispersed on the surface of HM and HM–M zeolites.
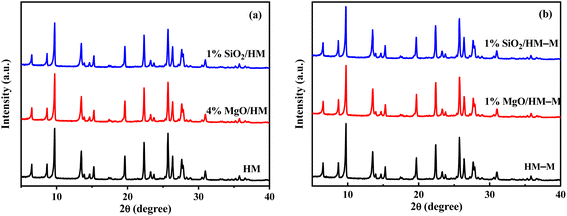 |
| | Fig. 2 XRD patterns of HM, HM–M and their modified samples. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
Fig. 3 presents the transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) images of HM and HM–M. The intergranular mesopores were prominent in HM (Fig. 3a and c), and clear intragranular mesopores were evident in HM–M (Fig. 3b and d). Both the HR-TEM images of HM (Fig. 3c and c′) and HM-M (Fig. 3d and d′) show lattice fringes of microporous channels, indicating that HM–M retained the microporous properties of HM.
 |
| | Fig. 3 TEM images of HM (a, c and c′), HM–M (b, d and d′). | |
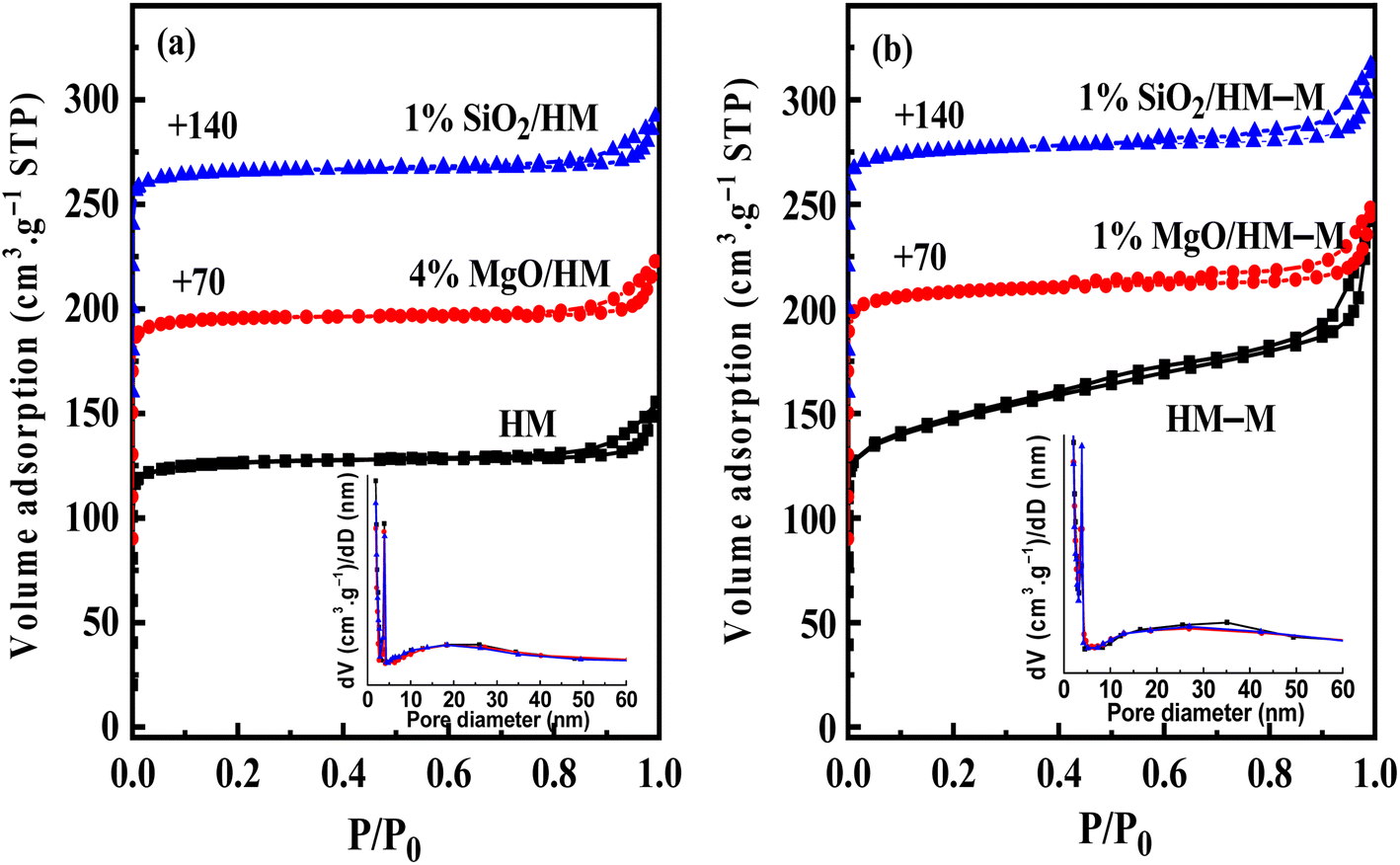
Fig. 4 shows the N2 sorption isotherms and PSDs (inset) of HM, HM–M, and their modified samples. The textural parameters of the parent and modified catalysts are listed in Table 2. From Fig. 4, it can be seen that HM and HM–M both exhibited compound characteristics of type I and type IV. Compared with HM, HM–M had larger specific surface areas (SSA) and more mesopores (Table 2). A small number of intergranular mesopores in HM were formed by the aggregation of particles (Fig. 3a). Many intragranular mesopores in HM–M were formed via acid-alkali treatment (Fig. 3b). For modified HM, the SSA and micropore volume of 4% MgO/HM and 1% MgO/HM–M was almost unchanged, while that of 1% SiO2/HM and 1% SiO2/HM–M decreased, indicating that TEOS deposition had a more significant influence on SSA and micropore volume than DPB. According to the literature,28,29 silanization can block micropores, whereas DPB has little effect on the micropore structure. Consistently, the external specific surface area of all modified samples was reduced. Notably, for 1% SiO2/HM–M, the decrease in SSA and micropore volume was less than that of the 1% SiO2/HM. Moreover, the mesopore volume of the HM-modified samples was unchanged. In contrast, the mesopore volume of the HM–M-modified samples decreased significantly, demonstrating that modification had little effect on the intergranular mesopores. The average mesopore size (PSDs diagram, inset) of HM and its modified samples are almost the same, while those of 1% MgO/HM–M and 1% SiO2/HM–M are both lower than that of HM–M, which further confirmed the above opinion.
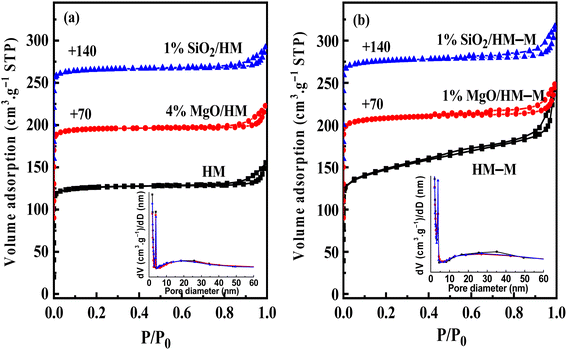 |
| | Fig. 4 N2 adsorption–desorption isotherms and pore-size distributions (PSDs, inset) of HM, HM–M and their modified samples. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
Table 2 Structural properties and chemical compositions of the HM, HM–M and their modified samples
| Sample |
Si/Ald |
Mg content (%) |
SBETa (m2 g−1) |
Sextb (m2 g−1) |
Smicrob (m2 g−1) |
Vmicrob (cm3 g−1) |
Vmesoc (cm3 g−1) |
| Brunauer–Emmett–Teller (BET) method. t-plot method. Barrett–Joyner–Halenda (BJH) method (the adsorption branch). Inductively coupled plasma-optical emission spectroscopy (ICP-OES) method. n. d., not determined. |
| HM |
12.8 |
n. d. |
482 |
25 |
457 |
0.180 |
0.047 |
| 4% MgO/HM |
n. d.e |
2.32 |
476 |
20 |
456 |
0.182 |
0.043 |
| 1% SiO2/HM |
13.5 |
n. d. |
458 |
20 |
438 |
0.162 |
0.043 |
| HM–M |
13.1 |
n. d. |
559 |
130 |
429 |
0.172 |
0.16 |
| 1% MgO/HM–M |
n. d. |
0.62 |
549 |
121 |
428 |
0.171 |
0.10 |
| 1% SiO2/HM–M |
13.7 |
n. d. |
532 |
117 |
415 |
0.174 |
0.10 |
The Si/Al molar ratios and magnesium contents of HM, HM–M, and their modified samples are displayed in Table 2 via ICP measurements. For the TEOS-modified samples, the Si/Al molar ratios increased, which confirmed that SiO2 was effectively deposited on the parent HM and HM–M. In addition, the Mg contents in 4% MgO/HM and 1% MgO/HM–M were 2.32% and 0.62%, respectively, which is consistent with the theoretical deposition of DPB.
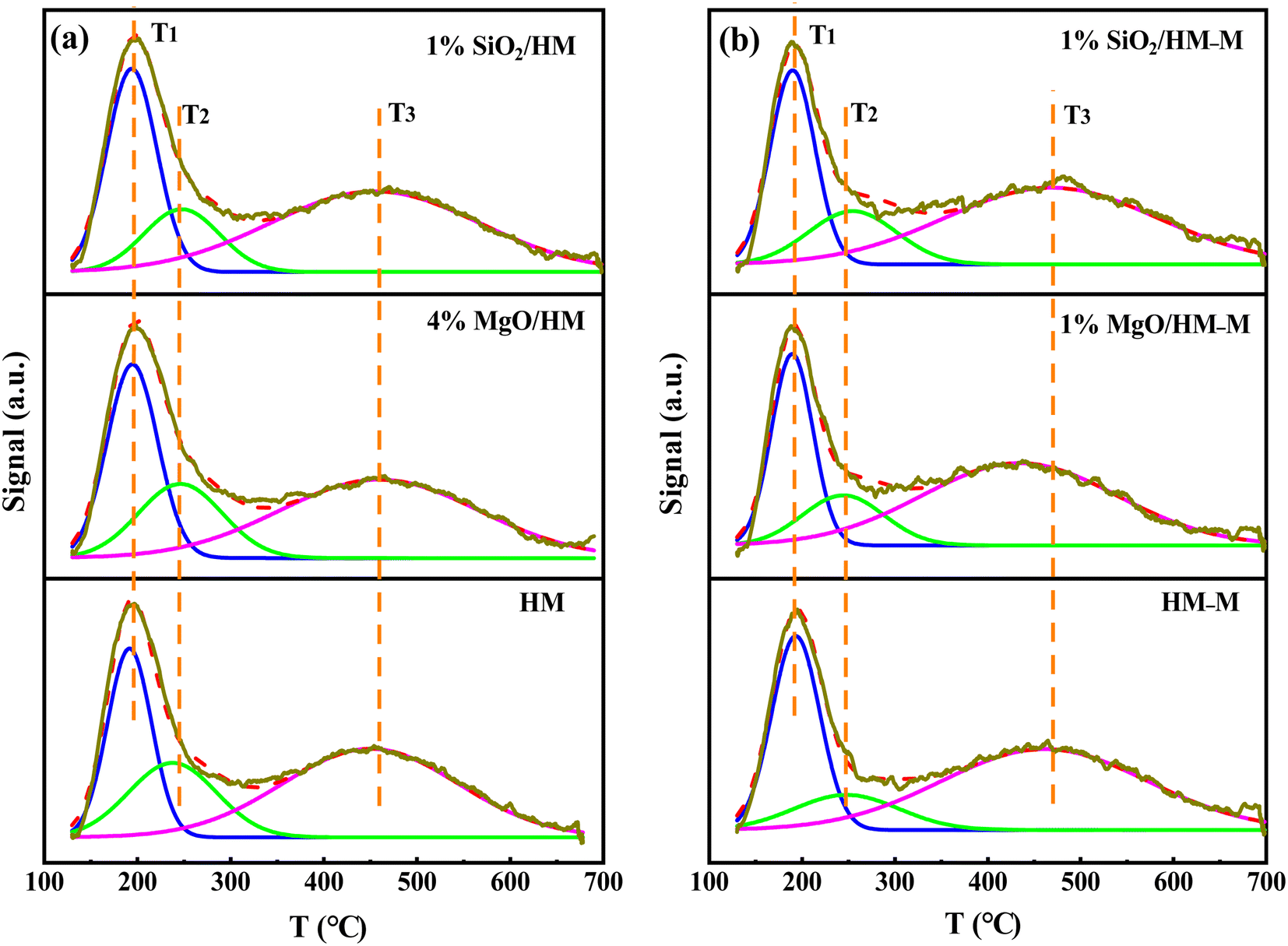
Characterisation of acidity. The total acidity and acid strength of the HM, HM–M, and their modified samples were analysed via temperature-programmed desorption of ammonia (NH3-TPD). Peak fitting of the NH3-TPD curves was performed using Gaussian deconvolution (Fig. 5). The desorption peaks in the ranges of 150–200 °C (T1), 230–260 °C (T2), and 430–470 °C (T3) are attributed to weak, medium, and strong acid sites, respectively.31 According to the literature,32,33 medium and strong acid sites are collectively called strong acid sites. Based on the peak areas and total acid amounts,32 the amounts of weak and strong acid sites were calculated (Table 3). The total acid amount of the micro–mesoporous HM–M sample processed via alkali-acid treatment was lower than that of the commercial HM, consistent with the molar ratio of Si/Al. The total acid amounts of the HM and HM–M samples were reduced to varying degrees after modification. Specifically, the strong acid sites of HM and HM–M showed a significant decrease, whereas the weak acid sites decreased slightly. Notably, the 4% MgO/HM and 1% SiO2/HM have almost the same acid amount, while the acid amount of 1% MgO/HM–M was higher than that of 1% SiO2/HM–M. During the TEOS deposition process, SiO2 deposited on the pores and reduced the pore size or blocked the pores,22,24,28 not just deposited on the external surface of HM and HM–M. Combined with N2 sorption analysis, this outcome can be attributed to some acid sites in the micropores of 1% SiO2/HM and 1% SiO2/HM–M that cannot be detected. Moreover, it was found that the total acid amount of 1% SiO2/HM decreased by 10.3%, while 30.9% of 1% SiO2/HM–M decreased at the same amount of TEOS deposition. The total acid content of 4% MgO/HM and 1% MgO/HM decreased by 9.6% and 10.0%, respectively. These findings demonstrate that TEOS and DPB can access the intragranular mesopores of HM–M and passivate the acidic sites in the pores, which agree with the N2 sorption characterisation.
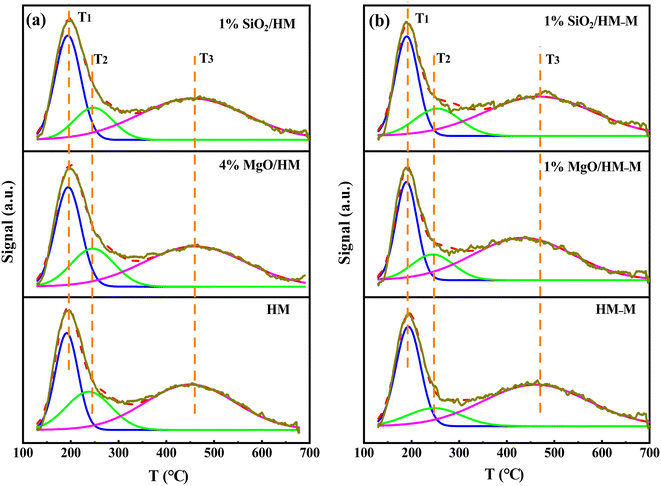 |
| | Fig. 5 NH3-TPD profiles of HM, HM–M and their modified samples. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
Table 3 The acid contents and properties of the parent and modified HM
| Sample |
Peak temperaturea (°C) |
Acid contentsa (cm3 g−1) |
B/Lb |
Brönsted acid sitesc (cm3 g−1) |
Lewis acid sitesc (cm3 g−1) |
| T1 |
T2 |
T3 |
Weak |
Strong |
Total |
200 °C |
350 °C |
200 °C |
350 °C |
200 °C |
350 °C |
| NH3-TPD method. Py-FTIR method. NH3-TPD and Py-FTIR method. |
| HM |
191 |
239 |
449 |
6.7 |
17.6 |
24.3 |
2.0 |
4.9 |
16.2 |
14.6 |
8.1 |
3.0 |
| 4% MgO/HM |
194 |
247 |
457 |
6.8 |
15.2 |
22.0 |
1.8 |
4.0 |
14.1 |
12.1 |
7.8 |
3.0 |
| 1% SiO2/HM |
193 |
249 |
449 |
7.0 |
14.8 |
21.9 |
1.5 |
3.5 |
13.1 |
11.5 |
8.7 |
3.3 |
| HM–M |
190 |
251 |
457 |
4.2 |
10.8 |
15.0 |
2.3 |
6.2 |
10.5 |
9.3 |
4.5 |
1.5 |
| 1% MgO/HM–M |
189 |
248 |
435 |
3.8 |
9.7 |
13.5 |
2.1 |
4.7 |
9.2 |
8.0 |
4.4 |
1.7 |
| 1% SiO2/HM–M |
189 |
253 |
462 |
3.1 |
7.3 |
10.4 |
1.6 |
3.8 |
6.4 |
5.8 |
4.0 |
1.5 |
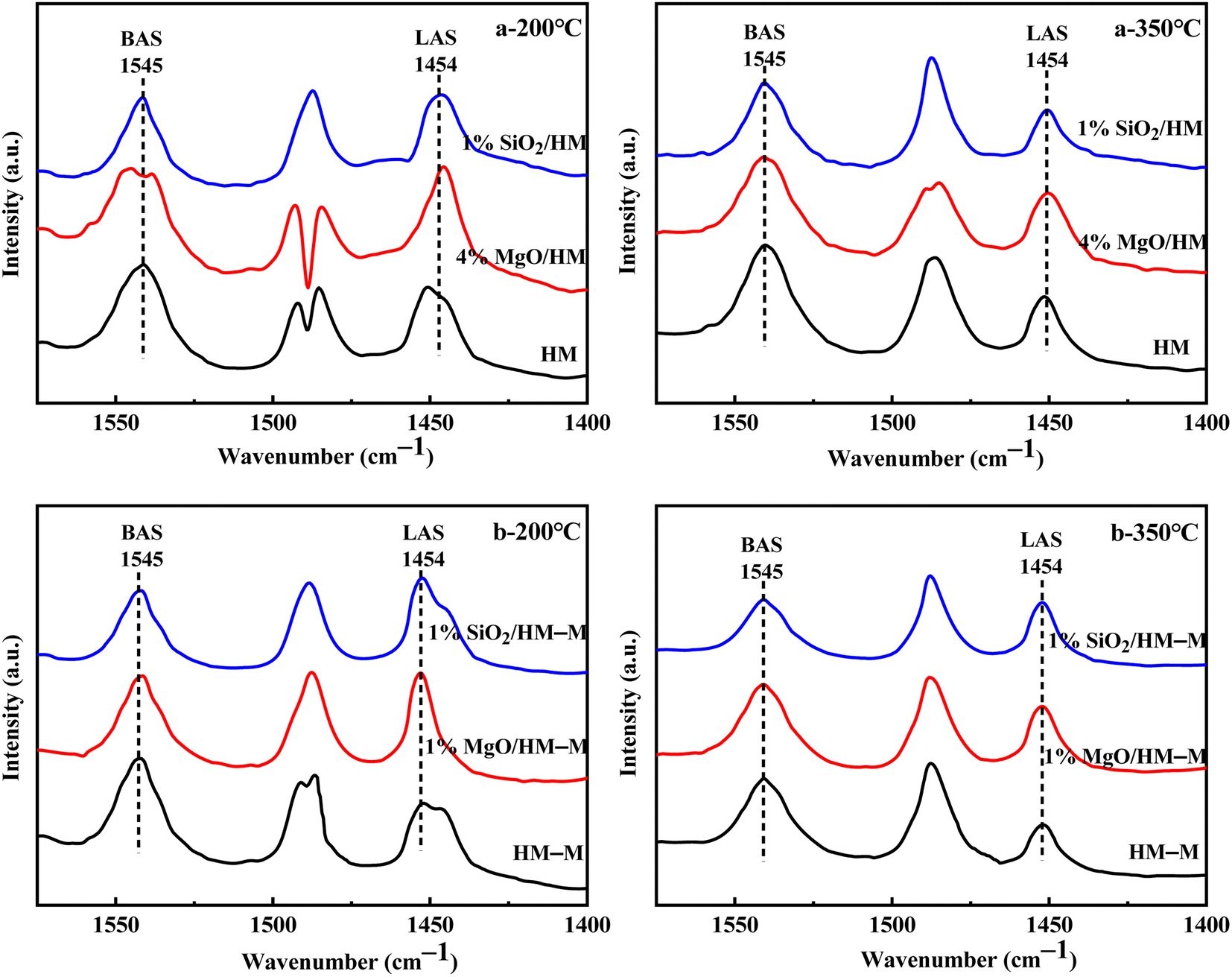
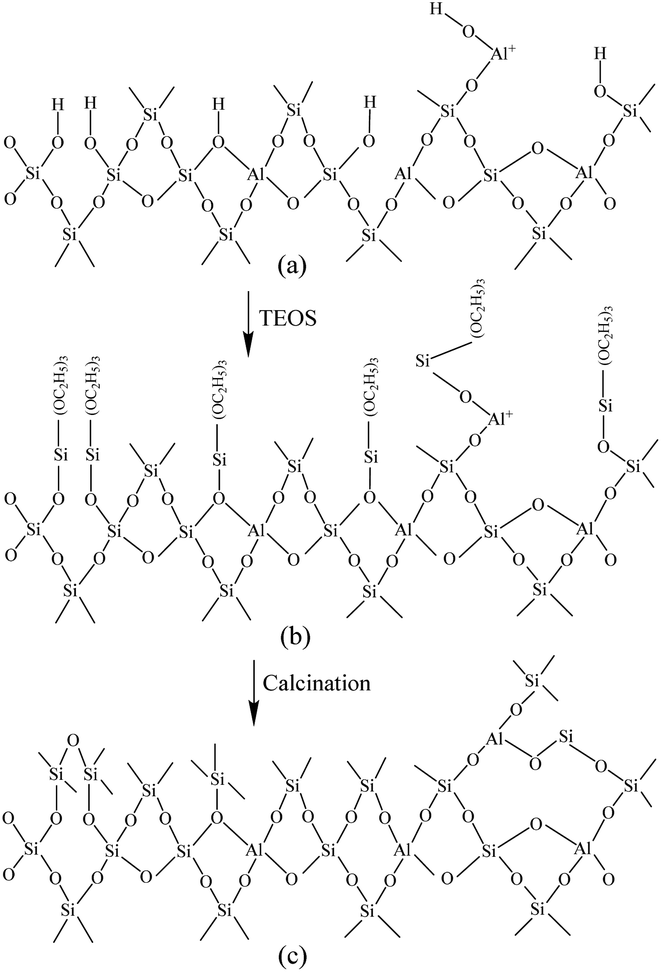
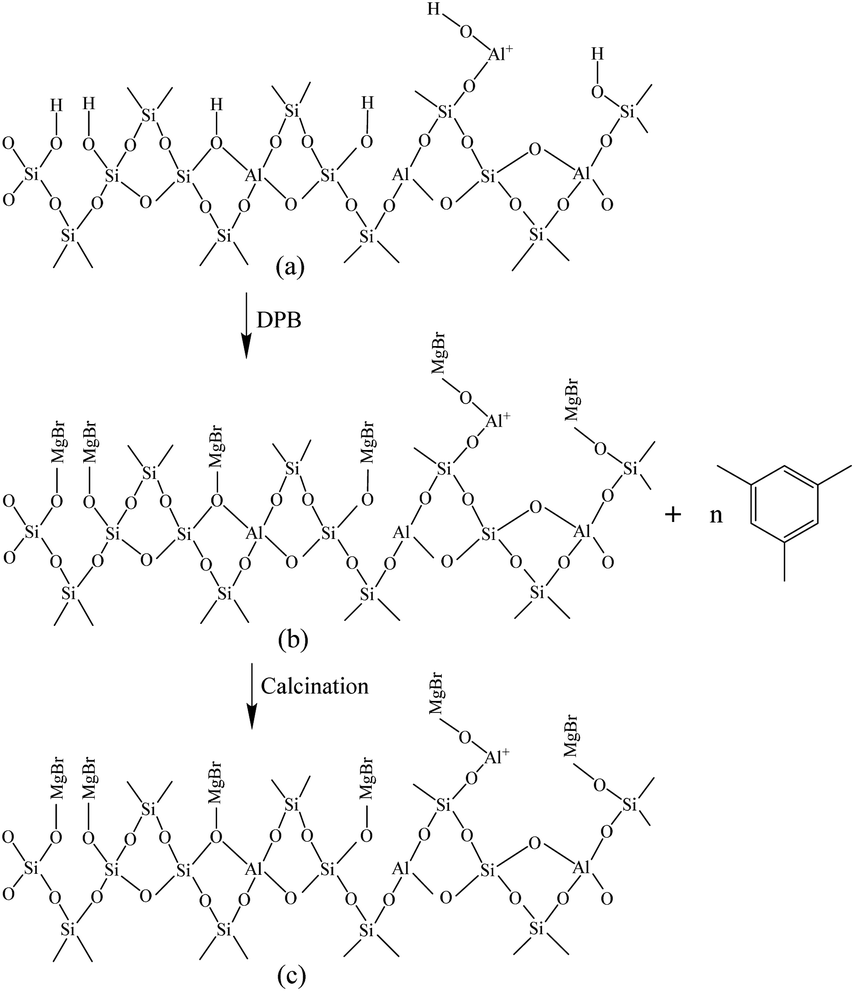
Fig. 6 shows the Fourier-transform infrared spectroscopy of pyridine (Py-FTIR) spectra of the HM, HM–M, and their modified samples in the range of 1575–1400 cm−1 and desorption temperatures of 200 °C and 350 °C. The B/L ratios of HM, HM–M, and their modified samples (Table 3) were calculated using their corresponding molar extinction coefficients34 and integrated areas of the PyH+ and PyL bands. The total acid sites (200 °C) and strong acid sites (350 °C) of Brønsted and Lewis acids were calculated for HM, HM–M, and their modified samples based on the acid amounts measured by NH3-TPD (Table 3). It was found that the deposition of both TEOS and DPB resulted in a reduction in the B/L ratio of the modified zeolites. Furthermore, the decrease of strong Brønsted acid sites was greater than that of weak, indicating that DPB and TEOS had a better passivation effect on the strong Brønsted acid sites on the mordenite external surface. From the deposition mechanism of TEOS (Fig. 7),35 it can be seen that TEOS was mainly adsorbed on the bridging hydroxyl groups (Al–OH–Si) and silica hydroxyl groups (Si–OH) on the zeolite surface (Fig. 7b). TEOS also adsorbed onto the non-framework Al sites. After calcination, a new Al (Si)–O–Si bond formed, which was responsible for the narrowing of the pore size (Fig. 7c). Since the reaction of TEOS with the bridging hydroxyl group dominates,28 it mainly exhibited a decrease in Brønsted acid sites and insignificant changes in Lewis acid sites. In addition, the deposition process of DPB is similar to that of TEOS, except that, combined with the N2 sorption and NH3-TPD characterisation results highlighted earlier, the deposition of DPB had almost no effect on the microporous pore structure. Therefore, it can be inferred that no new Al (Si)–O–Si bonds were formed after calcination (Fig. 8c). Therefore, the deposition mechanism of DPB can be described by the process shown in Fig. 8. The removal of trimethylbenzene mainly occurs during calcination. In addition, TEOS deposition exhibited lower B/L values than DPB deposition in both HM and HM–M. This outcome is due to the TEOS deposition which reduced the pore size or even blocked the micropores, limiting the adsorption of pyridine into internal acid sites of 1% SiO2/HM and 1% SiO2/HM–M.35
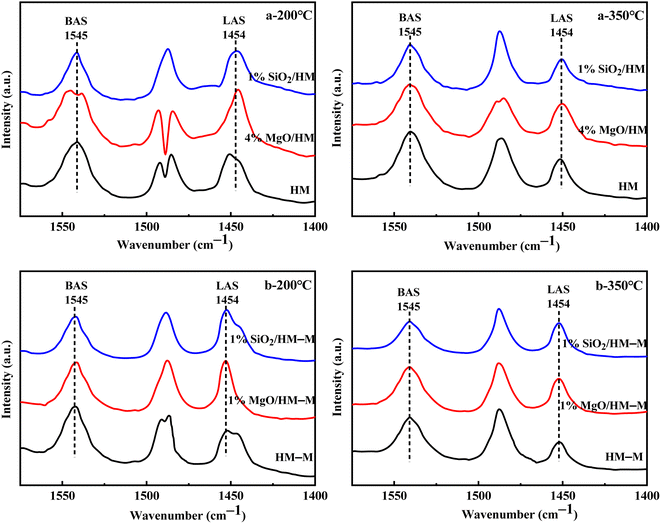 |
| | Fig. 6 FT-IR spectra of pyridine adsorbed on HM, HM–M and their modified samples under different desorption temperature. (a) 200 °C, (a) 350 °C: HM and modified HM samples; (b) 200 °C, (b) 350 °C: HM–M and modified HM–M samples. | |
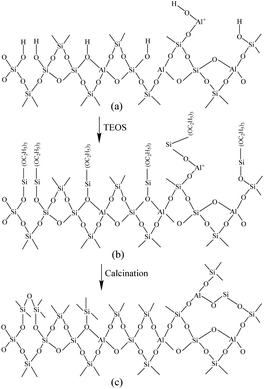 |
| | Fig. 7 Mechanism of TEOS deposition.33 | |
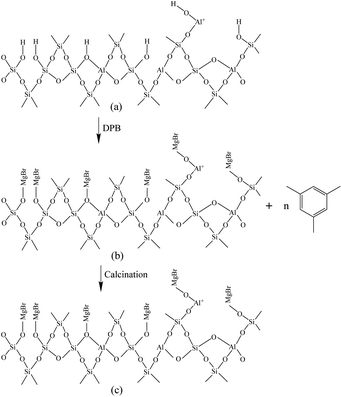 |
| | Fig. 8 Mechanism of DPB deposition. | |
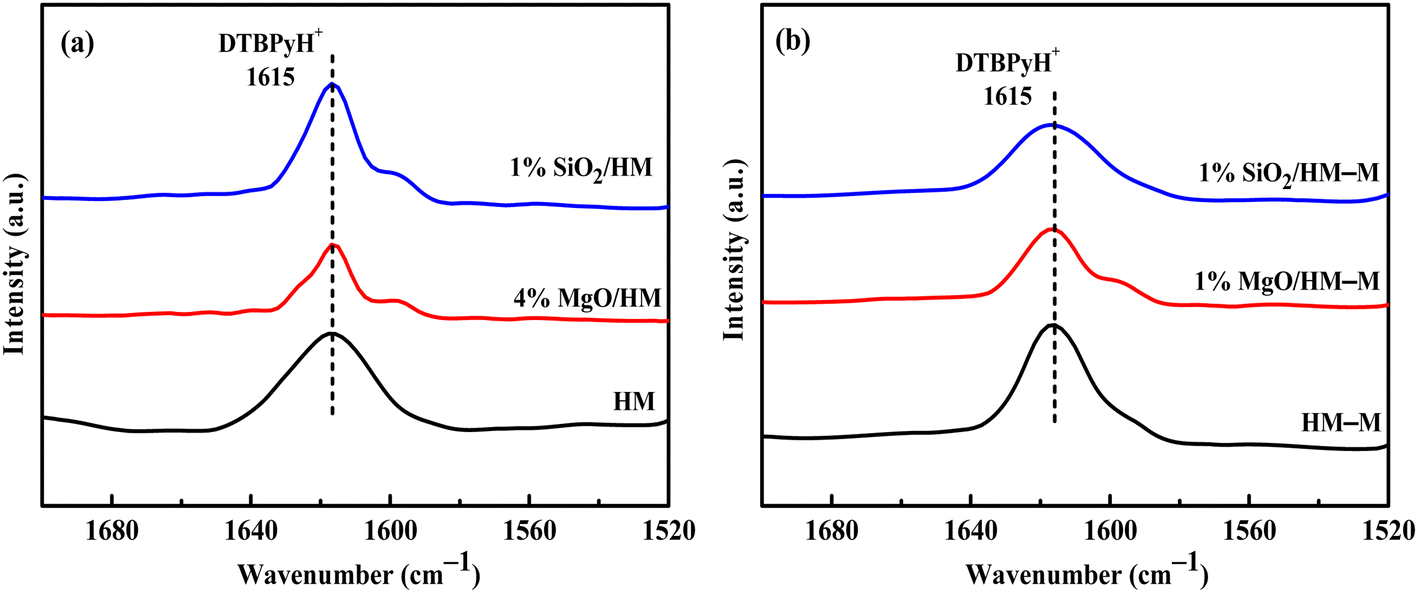
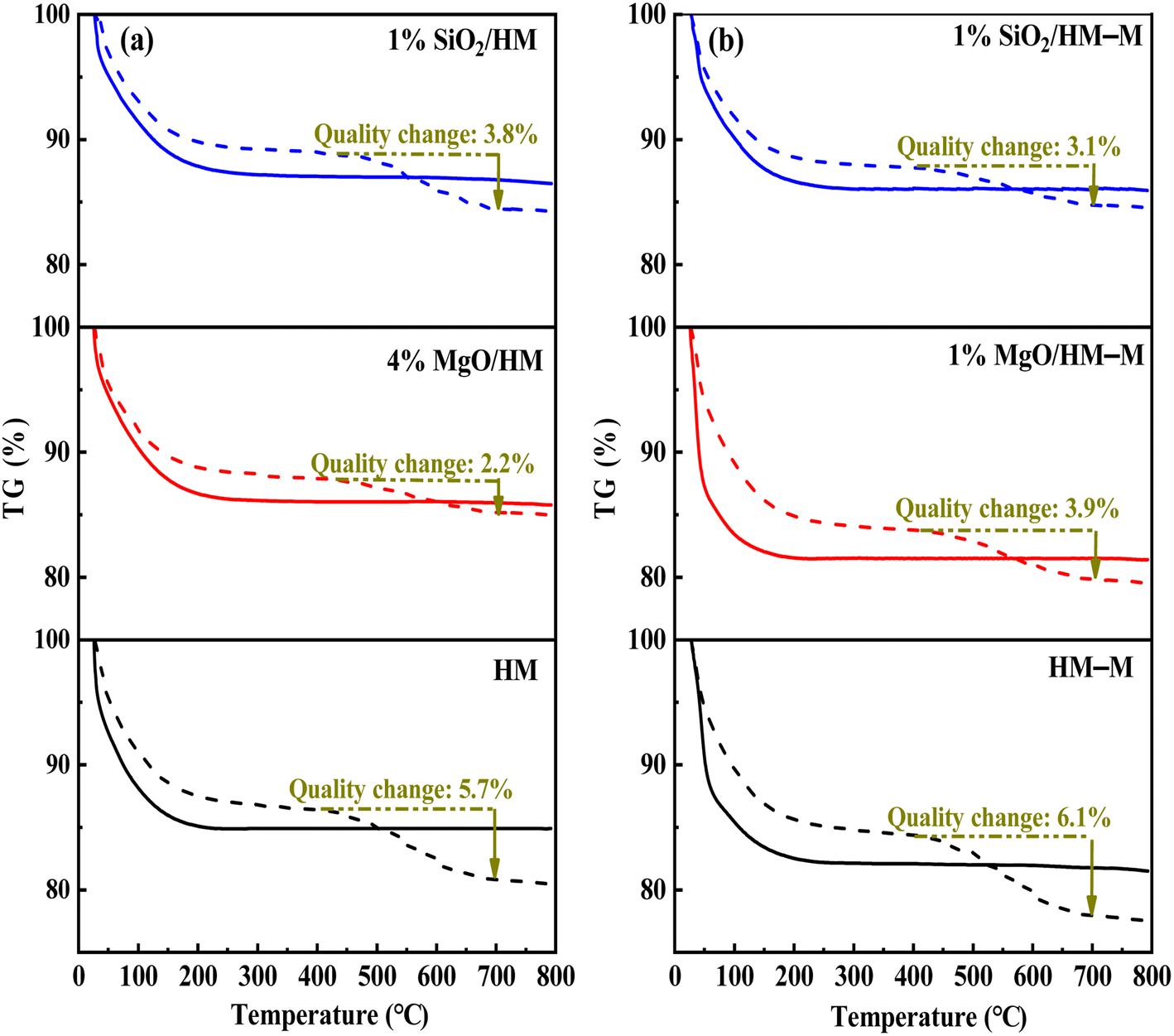
In the microporous structure of mordenite, 2,6-di-tert-butylpyridine (DTBPy) adsorption only occurs at the external surface and pore acid sites.36 Therefore, the external surface acidity of HM, HM-M, and their modified zeolites was qualitatively and quantitatively analysed by infrared spectroscopy of DTBPy adsorption (Fig. 9) combined with synchronous thermal analysis (Fig. 10). The absorption peak at 1615 cm−1 (Fig. 9) is attributed to the adsorption of Brønsted acidic sites by DTBPy,37 indicating that a small number of Brønsted acidic sites remained on the zeolite external surface after modification by TEOS and DPB. The DTBPy adsorption amount was calculated from the changes in thermogravimetric (TG) curves between 400 °C and 700 °C (Fig. 10), and the sample without adsorption of DTBPy was used as a control. It can be found that the samples without DTBPy adsorption exhibit only one stage of weight loss (40–200 °C), while the samples with DTBPy adsorption show two weight loss stages (40–200 °C and 400–700 °C). The first stage at 40–200 °C is due to the release of physical water adsorbed in the pore channel, and the second at 400–700 °C can be attributed to the DTBPy desorption.38 Meanwhile, the modified samples exhibit a significant decrease in the adsorption of DTBPy, indicating that TEOS and DPB mainly neutralised the hydroxyl groups on the zeolite external surface. Combining the N2 sorption results (Table 1) and the deposition mechanism of TEOS and DPB, TEOS leads to the blockage and reduction of the pore size of micropores in HM and HM–M, while DPB has no effect. Therefore, 1% SiO2/HM–M lost more Brønsted acidic sites than 1% MgO/HM–M at the same TEOS and DPB additions (Fig. 10b), which is consistent with the NH3-TPD results. Moreover, more external Brønsted acid sites of 4% MgO/HM were lost compared to those of 1% SiO2/HM (Fig. 10a), while NH3-TPD results showed that they have the same total acid amount, which also confirms that TEOS modification leads to some undetectable acid sites in the micropore channels. In addition, it can be seen from Fig. 3 that obvious intragranular mesopores are present in HM–M. The N2 sorption results showed that both TEOS and DPB could enter the intragranular mesopores of HM–M, reducing the average mesopore size. Thus, HM–M lost more Brønsted acid sites than HM (Fig. 10) with the same amount of TEOS added, identical to the NH3-TPD and Py-FTIR results.
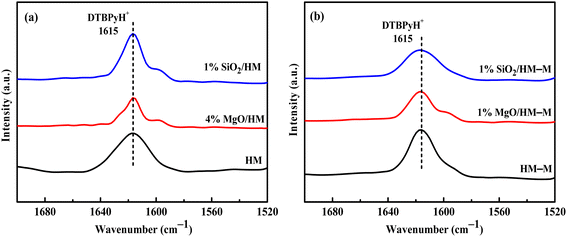 |
| | Fig. 9 FT-IR spectra of 2,6-di-tert-butylpyridine adsorbed on HM, HM–M and their modified samples. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
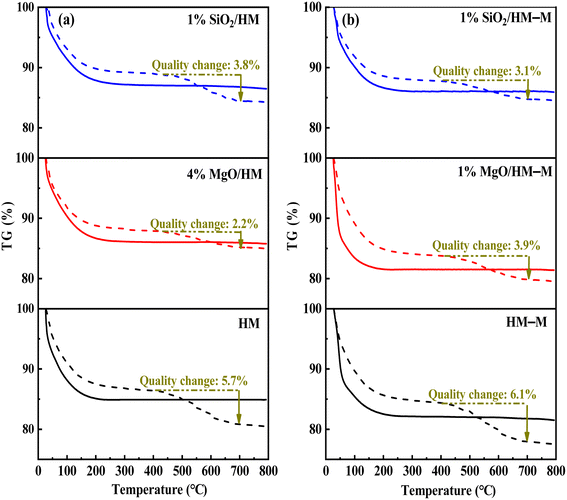 |
| | Fig. 10 The thermogravimetric (TG) curves of HM, HM–M and their modified samples (solid line) and after absorbed 2,6-di-tert-butylpyridine (dash line). (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
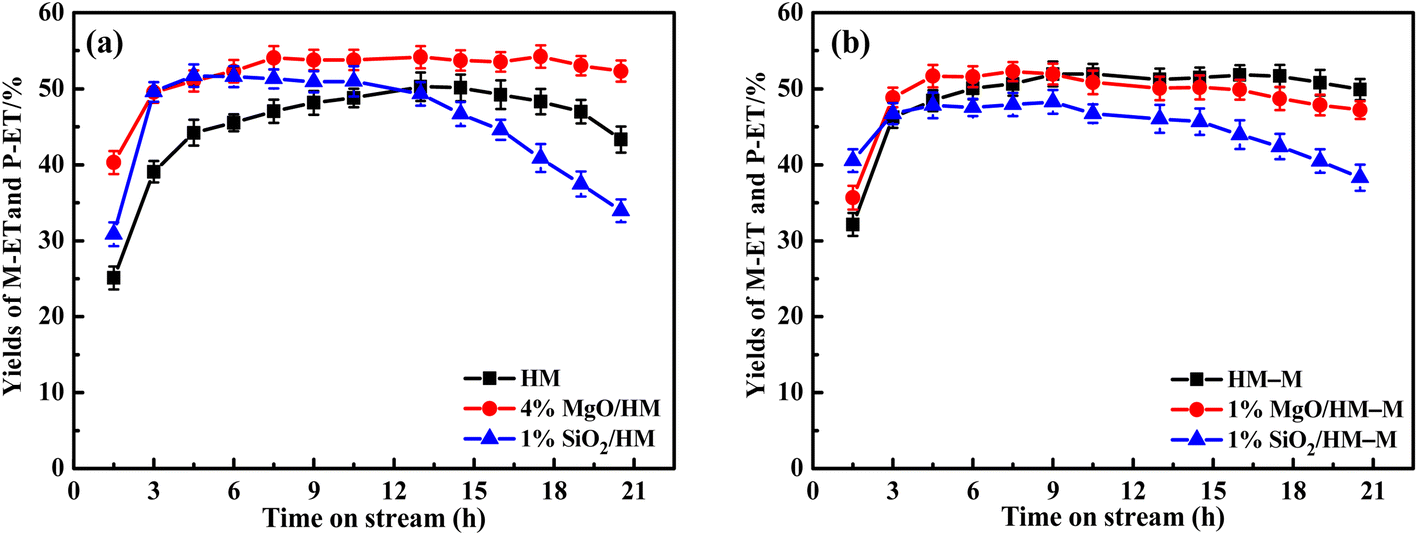
Evaluation of catalytic performance. The isomerisation of O-ET in the C9 aromatic mixture (Table 1) was studied using HM, HM–M, and their modified catalysts. In addition, from Table 1, it can be seen that the content of O-ET in these C9 aromatics is much higher than that in thermodynamic equilibrium,39 so the conversion of O-ET to M-ET and P-ET can be achieved. Fig. 11 displays the influence of the time on stream on the conversion of O-ET and selectivities toward M-ET and P-ET. The yields of M-ET and P-ET are shown in Fig. 12.
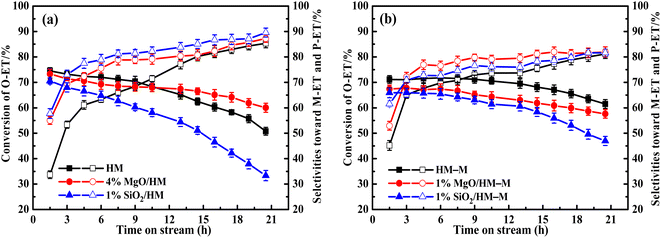 |
| | Fig. 11 Influence of time on stream (TOS) on the catalytic conversion of O-ET (solid symbol) and selectivities toward M-ET and M-ET (hollow symbol) over HM, HM–M and theirs modified zeolites. The maximum standard deviation of each data was <2.0%. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
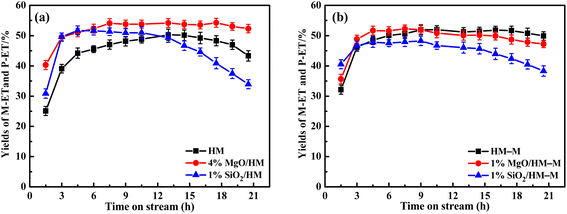 |
| | Fig. 12 Influence of time on stream (TOS) on the catalytic yields of M-ET and P-ET over HM, HM–M and theirs modified zeolites. The maximum standard deviation of each data was <2.0%. (a) HM and modified HM samples, and (b) HM–M and modified HM–M samples. | |
In our previous work,14 the optimal reaction temperatures for HM and HM–M were 235 °C and 165 °C, respectively. Thus, the discussed temperature of HM and modified zeolite is 235 °C, and that of HM–M and modified zeolite is 165 °C in this study. First, regarding the parent HM and HM–M, the conversion of O-ET gradually decreased with increasing reaction time. During the experimental period of 21 h, the conversion of O-ET decreased by 22.8% in HM and 9.6% in HM–M, which can be attributed to the higher mesoporosity of HM–M to increase the accessibility of acidic sites and facilitate the diffusion of macromolecular by-products. In addition, because the Brønsted acid and Lewis acid centres are the active centres for catalysing intramolecular and intermolecular transalkylation, respectively,9 a higher B/L value of HM–M is beneficial for reducing the occurrence of side reactions.
For HM zeolites, a decrease in the O-ET conversion and increased M-ET and P-ET selectivities and yields were observed when TEOS and DPB were deposited (Fig. 11a and 12a). In order to verify the effect of acidic sites on the O-ET isomerization reaction, the parent HM was tested by pyridine poisoning method. From Fig. S1,† it can be found that the conversion of O-ET gradually decreased with the increase of pyridine adsorption, showing that the acidic sites amount was positively correlated with the conversion of O-ET. According to NH3-TPD, Py-IR, and DTBPy-IR analyses, it can be concluded that the decrease in O-ET conversion was mainly caused by the passivation of external surface acidic sites. In contrast, the acidic sites inside the zeolite pores were not affected. Furthermore, 1% SiO2/HM exhibited lower O-ET conversion than 4% MgO/HM, although the NH3-TPD results showed that 4% MgO/HM had the same acid amount as 1% SiO2/HM, which could be attributed to the deposition of TEOS shrinking or blocking some of the micropore channels of HM, thus making the acidic sites in some channels inaccessible for O-ET. Meanwhile, the narrowing or blocking of the micropores restricts the diffusion of products and macromolecular by-products; therefore, 1% SiO2/HM exhibited a distinct decrease in a lifetime. The deposition of DPB passivated the non-shape selective acid sites on the external surface and hardly affected the pore structure, reducing the occurrence of non-shape selective side reactions and improving the catalytic lifetime (Fig. 11a). Notably, the selectivities toward M-ET and P-ET on the parent HM were lower during the first 10 h of the reaction period, gradually increasing with time, and eventually comparable to that of the modified zeolite (Fig. 11a). This process is similar to the coking modification of HM itself, which was also observed by Kaeding et al.40 to improve the selectivity of p-xylene. However, the macromolecular products generated from this self-coking process can block the pores and cover the acid sites, thus showing a fast decrease for the conversion of O-ET with increasing time (Fig. 11a). In addition, the pyridine infrared results showed a decrease in the B/L acid value of 4% MgO/HM and 1% SiO2/HM versus HM. However, the 4% MgO/HM and 1% SiO2/HM samples exhibited high M-ET and P-ET selectivities at the early stage of the reaction and increased slightly with increasing reaction time. This is due to the elimination of many non-selective external surface acidic sites, preventing secondary reaction of the isomerized product and the reaction of macromolecules (TMB) in the feed (Table 1) that almost cannot react in the mordenite channels and produce more by-products. Moreover, the TEOS modification narrowed the pore size. It limited the diffusion of more oversized products, exhibiting higher M-ET and P-ET selectivities than the DPB modification, even though the B/L values of 4% MgO/HM were higher than those of 1% SiO2/HM, which was similar to that reported by Li et al.28 to improve the 2-LAB selectivity of Hβ zeolite. Therefore, we can conclude that the B/L value of acid sites of HM-modified samples is not proportional to the isomerization selectivity.
For HM–M zeolites, TEOS and DPB modification also resulted in a decrease in O-ET conversion and an increase in M-ET and P-ET selectivity (Fig. 11b). Unlike the HM-modified zeolites, the same amount of TEOS deposition did not result in rapid deactivation similar to the 1% SiO2/HM catalytic lifetime, which is due to the access of TEOS to the HM–M mesopores, reducing the effect of TEOS on its micropore structure. The O-ET conversion and M-ET and P-ET selectivities of 1% MgO/HM-M were slightly higher than those of 1% SiO2/HM–M, this results can be attributed to the more acid sites and higher B/L ratio of 1% MgO/HM–M than those of 1% SiO2/HM–M. In addition, from the yields of M-ET and P-ET in Fig. 12, it can be found that, as with HM, DPB showed better results than TEOS in the modification of HM–M. To avoid interference from the self-coking modification of the parent mordenite zeolite, the product distributions of HM, HM–M, and their modified catalysts in Table 4 were derived from the first 9 h of the reaction, and these data indicate the modification effect of TEOS and DPB.
Table 4 Results of the isomerisation of O-ET employing different catalysts at the optimal reaction conditions of each catalyst
| Sample |
Product distributiona (%) |
M-ET, P-ET yield (%) |
Carbon mass balanceb (%) |
| C1–C6 |
Toluene |
Xylene |
M-, P-ET |
O-ET |
Tri-MB |
Tetra-MB |
PTB |
Others |
| Main products of the conversion of O-ET: C1–C6 (alkanes and benzene), xylene (o-, m-, p-xylene), M-, P-ET (m-, p-ethyltoluene), O-ET (o-ethyltoluene), tri-MB (trimethylbenzene isomers), tetra-MB (tetramethylbenzene isomers), PTB (pentamethylbenzene isomers), others (macromolecular aromatics). (Total carbon after the reaction/total carbon before the reaction) × 100%. *Means results in the literature/means not reported in the literature. Reaction conditions: WHSV = 1.0 h−1, H2/HC = 5.0 (mol mol−1), pressure = 1.5 MPa, temperature = 235 °C (HM and its modified ones) and 165 °C (HM–M and its modified ones). |
| HM |
0.25 |
1.10 |
2.30 |
23.46 |
3.60 |
64.10 |
1.99 |
0.98 |
2.22 |
47.6 |
99.5 |
| 4% MgO/HM |
0.23 |
0.77 |
1.40 |
24.16 |
3.87 |
65.59 |
1.59 |
0.56 |
1.76 |
53.0 |
99.3 |
| 1% SiO2/HM |
0.15 |
0.64 |
1.10 |
23.81 |
4.55 |
66.14 |
1.47 |
0.43 |
1.64 |
50.0 |
98.8 |
| HM–M |
0.14 |
1.11 |
1.41 |
24.00 |
3.44 |
65.27 |
1.62 |
0.71 |
2.29 |
51.6 |
99.6 |
| 1% MgO/HM–M |
0.15 |
0.87 |
0.93 |
24.01 |
4.25 |
66.03 |
1.36 |
0.45 |
1.99 |
51.7 |
99.1 |
| 1% SiO2/HM–M |
0.15 |
0.81 |
1.09 |
23.69 |
4.42 |
65.95 |
1.51 |
0.44 |
1.95 |
49.1 |
99.0 |
| *5M-2 composite mordenite |
— |
— |
— |
— |
— |
— |
— |
— |
|
45.0 |
— |
| *7HZSM-5 |
— |
— |
— |
— |
— |
— |
— |
— |
— |
10.0 |
— |
| *7Hβ |
— |
— |
— |
— |
— |
— |
— |
— |
— |
24.0 |
— |
From the above studies, it can be seen that the deposition modification of DPB can significantly improve the selectivity of O-ET isomerisation on commercial HM to generate M-ET and P-ET. In addition, the passivation of non-selective acidic sites on the external surface and the pore size was not affected, allowing the catalytic lifetime to be extended. Importantly, in comparison with the reported catalysts,5,7 4% MgO/HM exhibited the highest yields of M-ET and P-ET in mixed ethyltoluene isomerization process (Table 4). HM–M has a shorter pore channel that can facilitate the diffusion of molecules has a higher catalytic lifetime than the parent HM. The optimal reaction temperature was 70 °C lower than that of HM, allowing for energy savings in industrial applications. The deposition of TEOS and DPB not only passivated the external surface acidic sites of the micropores but also passivated the acidic sites in the mesopores because their sizes were smaller than the mesopore pore diameter. The yields of M-ET and P-ET were unchanged as the selectivity increase for M-ET and P-ET was offset by the decrease in O-ET conversion. Hence, it can be concluded that these two modification methods have no significant effect on HM–M and require further investigation.
Finally, the reaction conditions for the isomerization of O-ET via 4% MgO/HM were optimized, as shown in Fig. S2.† From Fig. S2,† it can be found that the optimal reaction conditions were T = 235 °C, WHSV = 1.0 h−1, pressure = 1.5 MPa, and H2/HC = 5.0 (mol mol−1). Too high reaction temperature, high pressure and low WHSV can affect the selectivities toward M-ET and P-ET. This is because that the severe reaction conditions will aggravate the occurrence of side reactions such as disproportionation and splitting. Under the optimal conditions, the conversion of O-ET reached 68.4% and the selectivities toward M-ET and P-ET reached 77.5%. Furthermore, the renewability study of 4% DPB/HM catalyst was carried out. From the TG results in Fig. S3,† it can be found that the used catalyst has an obvious weight loss peak at 200–600 °C, and the weight loss curve remains unchanged after 600 °C. Hence, the 4% DPB/HM catalyst was cleaned with ethanol to remove residual aromatic compounds on the surface, and then calcined at 600 °C for regeneration. From the renewability results in Fig. S4,† it can be found that the catalytic activity did not decrease significantly, indicating that it has a good regeneration performance and has a good application prospect in industrial catalysis.
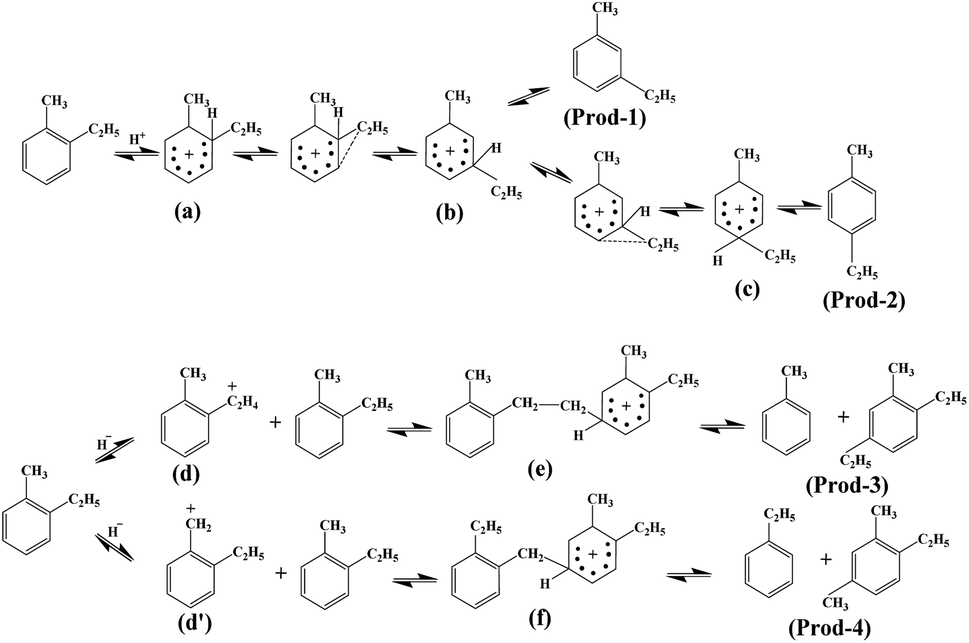
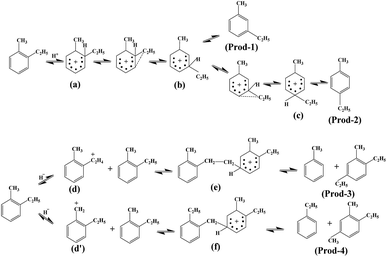
Possible mechanism for the isomerisation of ethyltoluene. According to the characterization above and catalytic performance results in Table 4, as well as our previous work,14 the possible mechanism for the isomerisation of O-ET is presented in Fig. 13. The primary reaction involves the generation of M-ET (Prod-1, Fig. 13) and P-ET (Prod-2, Fig. 13) in the presence of Brønsted acid. First, O-ET was protonated to form the unstable δ-complex (a), then the ethyl group with higher mobility migrates to the ortho position of the methyl group through intramolecular transfer to form the δ-complex (b), and further migration to the para position of the methyl group to form the δ-complex (c). Finally, the target products were obtained by deprotonation. Besides, a bimolecular nucleophilic reaction occurs in the presence of Lewis acid sites. First, O-ET undergoes hydride transfer to form δ-complex (d) or (d′). Afterwards, δ-complex (e) and δ-complex (f) were formed electronically and spatially, and finally by-products (Prod-3 and Prod-4, Fig. 13) are formed through disproportionation. Notably, the by-products contained xylene, tetramethylbenzene and pentamethylbenzene isomers (Table 3), which could be attributed to differences in nucleophilic substitution positions. The macromolecular polycyclic aromatic hydrocarbons (1–2%, Table 4) were generated by the further reaction of by-products, which were also an important factor for catalyst deactivation. Moreover, the by-products could crack to generate small molecular chain alkanes (C1–C6, Table 3).
 |
| | Fig. 13 Mechanism of the isomerization of O-ET. | |
According to the literature,41,42 the high density of acidic sites favors intermolecular transalkylation as well. The deposition of DPB effectively passivated the acid sites on the external surface of HM without changing the pore structure and the acid sites in pore channels. TEOS deposition even blocks some of the pores, reducing the accessibility of acidic sites within the pores. Therefore, the HM-modified zeolites exhibited high selectivities for intramolecular transalkylation products (M-ET, P-ET) (Fig. 11a). Similarly, HM–M modified with DPB and TEOS also showed improved selectivities for M-ET and P-ET (Fig. 11b).
Conclusions
Two mordenite zeolites with different structures were modified with TEOS and DPB using the CLD method, respectively, and their isomerisation property of O-ET in C9 mixed aromatics was evaluated. TEOS and DPB modifications effectively passivated the acidic sites on the external surface of the mordenites. It was found that TEOS caused the pore shrinkage or blockage, while DPB had almost no effect on the pore channels of the mordenite micropores. Therefore, the DPB modifier improved catalytic performance and a longer lifetime than TEOS. Compared with the parent HM, the selectivities toward M-ET and P-ET on the 4% MgO/HM zeolite increased from 67.27% to 77.54%, and the yields of M-ET and P-ET increased from 47.57% to 52.98%. However, for HM–M modified with DPB, the yields of M-ET and P-ET were unchanged as the selectivity increase for the product was offset by the decrease in reactant conversion, owing to the passivation of acidic sites in the mesopores of HM–M. In contrast to the external surface acid passivation of HM with micropores, TEOS and DPB were not effective on that of HM–M with a micro–mesoporous structure, and the modification for this zeolite will be further investigated in future studies. Finally, this study provides a new approval to improve the product stability and catalytic life in the industrial isomerisation of O-ET with C9 mixed aromatics as raw materials using mordenite as a catalyst.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (21973045), China Petroleum & Chemical Corporation (J418013-3) and Jiangsu scientific and technological transformative project (SBA2018030374). The related measure and analysis instrument for this work were supported by Centre for Analysis, Nanjing Normal University. We would like to thank Editage (https://www.editage.cn) for English language editing.
References
- J. Lin and Z. Gu, Chem. Technol., 2002, 10, 4 Search PubMed.
- Z. Zeng, Y. Li and B. Zhan, Guangdong Chem. Ind., 1986, 3–8 Search PubMed.
- M. J. Firth, Regul. Toxicol. Pharmacol., 2008, 52, 248–256 CrossRef CAS PubMed.
- L. Liu, F. Jin, G. Xiong, H. Long and X. Wang, J. Porous Mater., 2013, 20, 637–645 CrossRef CAS.
- I. Wang, T. C. Tsai and S. T. Huang, Ind. Eng. Chem. Res., 1990, 29, 2005–2012 CrossRef CAS.
- I. Wang, T. C. Tsai and S. T. Huang, Ind. Eng. Chem. Res., 1990, 29, 2005–2012 CrossRef CAS.
- X. Cheng, X. Wang and H. Long, Microporous Mesoporous Mater., 2009, 119, 171–175 CrossRef CAS.
- W. Μ. Meier, Zeitschrift fur Kristallographie, 1961, 115, 439–450 CrossRef CAS.
- S. M. Csicsery, J. Catal., 1970, 19, 394–397 CrossRef CAS.
- M. Tromp, J. A. van Bokhoven, M. T. Garriga Oostenbrink, J. H. Bitter, K. P. de Jong and D. C. Koningsberger, J. Catal., 2000, 190, 209–214 CrossRef CAS.
- J. S. Jung, J. W. Park and G. Seo, Appl. Catal., A, 2005, 288, 149–157 CrossRef CAS.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS.
- Y. Li, S. Liu, Z. Zhang, S. Xie, X. Zhu and L. Xu, Appl. Catal., A, 2008, 338, 100–113 CrossRef CAS.
- X. Cao, R. Wang, K. Wang, Z. Gu and F. Wang, ACS Omega, 2021, 6, 22688–22699 CrossRef CAS.
- H.-T. Yen, J.-J. Wang, S.-H. Siao, S. H. Cha, S. B. Hong, S. S. Al-Khattaf, I. Wang and T.-C. Tsai, Catal. Sci. Technol., 2016, 6, 2715–2724 RSC.
- J. Qian, G. Xiong, J. Liu, C. Liu and H. Guo, Ind. Eng. Chem. Res., 2019, 58, 9006–9016 CrossRef CAS.
- S. Akiyama, H. Mochizuki, H. Yamazaki, T. Yokoi, T. Tatsumi and J. N. Kondo, Mol. Catal., 2017, 433, 48–54 CrossRef CAS.
- U. Khalil, O. Muraza and A. Al-Amer, Adv. Powder Technol., 2016, 27, 1404–1410 CrossRef CAS.
- E. Kiliç and S. Yilmaz, React. Kinet., Mech. Catal., 2014, 112, 283–294 CrossRef.
- J. Lv, Z. Hua, J. Zhou, Z. Liu, H. Guo and J. Shi, ChemCatChem, 2018, 10, 2278–2284 CrossRef CAS.
- C. Zhang, X. Guo, C. Song, S. Zhao and X. Wang, Catal. Today, 2010, 149, 196–201 CrossRef CAS.
- Z. Zhu, Q. Chen, Z. Xie, W. Yang, D. Kong and C. Li, J. Mol. Catal. A: Chem., 2006, 248, 152–158 CrossRef CAS.
- X. Li, S. Liu, X. Zhu, Y. Wang, S. Xie, W. Xin, L. Zhang and L. Xu, Catal. Lett., 2011, 141, 1498 CrossRef CAS.
- R. W. Weber, K. P. Möller and C. T. O'Connor, Microporous Mesoporous Mater., 2000, 35–36, 533–543 CrossRef CAS.
- C. Gründling, G. Eder-Mirth and J. A. Lercher, J. Catal., 1996, 160, 299–308 CrossRef.
- M. J. Zheng, L. D. Zhang, G. H. Li and W. Z. Shen, Chem. Phys. Lett., 2002, 363, 123–128 CrossRef CAS.
- S. P. Jiang, J. Mater. Sci. Eng. A, 2006, 418, 199–210 CrossRef.
- R. Li, S. Xing, S. Zhang and M. Han, RSC Adv., 2020, 10, 10006–10016 RSC.
- Y. Zhang, J. W. Li, J. W. Wu and B. L. Xu, Chin. J. Inorg. Chem., 2017, 33, 2351–2356 Search PubMed.
- Y. Wang, M. Wang and X. Zhang, Chem. Ind. Eng., 2016, 33, 35–39 CAS.
- Z. Huang, J. Zhang, Q. Han, X. Zhang, P. Lu, L. Xu, Y. Yuan and L. J. A. C. A. G. Xu, Appl. Catal., A, 2018, 572, 80–89 CrossRef.
- D. Esquivel, A. J. Cruz-Cabeza, C. Jimenez-Sanchidrian and F. J. Romero-Salguero, Catal. Lett., 2012, 142, 112–117 CrossRef CAS.
- D. Jin, B. Zhu, Z. Hou, J. Fei, H. Lou and X. Zheng, Fuel, 2007, 86, 2707–2713 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- R. A. Shaikh, S. G. Hegde, A. A. Behlekar and B. S. Rao, Catal. Today, 1999, 49, 201–209 CrossRef CAS.
- A. Corma, V. Fornés, L. Forni, F. Márquez, J. Martínez-Triguero and D. Moscotti, J. Catal., 1998, 179, 451–458 CrossRef CAS.
- K. Góra-Marek, K. Tarach and M. Choi, J. Phys. Chem. C, 2014, 118, 12266–12274 CrossRef.
- Z. Huang, J. Zhang, Q. Han, X. Zhang, P. Lu, L. Xu, Y. Yuan and L. Xu, Appl. Catal., A, 2019, 572, 80–89 CrossRef CAS.
- S. Csicsery, J. Chem. Eng. Data, 1967, 12, 118–122 CrossRef CAS.
- W. W. Kaeding, L. B. Young and C. C. Chu, Chem. Informationsdienst, 1984, 89, 267–273 CAS.
- S. M. Csicsery and D. A. Hickson, J. Catal., 1970, 19, 386–393 CrossRef CAS.
- K. M. Wang and J. H. Lunsford, J. Catal., 1972, 24, 262–271 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Kaijun Wang
ab,
Kaijun Wang