
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyurethane–P2S5 composite-based solid-state electrolyte assists low polarization and high stability all-solid-state lithium-ion batteries†
Peng Cui a,
Chun Suna,
Hanqing Daib and
Wei Wei*a
a,
Chun Suna,
Hanqing Daib and
Wei Wei*a
aCollege of Electronic and Optical Engineering, Nanjing University of Posts &Telecommunications, 9 Wenyuan Road, Nanjing, 210023, Jiangsu, China. E-mail: weiwei@njupt.edu.cn
bAcademy for Engineering and Technology Institute for Electric Light Sources School of Information Science and Technology Fudan University Shanghai, 200433, China
First published on 29th September 2022
Abstract
Block copolymer electrolytes represented by polyurethane (PU) have become the forefront field of organic solid-state electrolytes for high-performance lithium-metal batteries due to their superb mechanical properties. However, due to the existence of mechanical hard segments, discontinuous ion transition at the electrolyte–electrode contact is inevitable, which leads to a series of problems such as terrible polarization phenomena, poor cycle stability and inadequate capacity retention. Here, we propose a new strategy to improve the chemical stability and interaction of electrolyte–electrode by modulating soft segments, which successfully reduces the polarization phenomenon. Then a new composite polymer solid electrolyte based on block copolymer PU (abbr. SPE) was prepared by ion-conduct segment modification using P2S5 with high lithium-ion affinity, and the ion conductivity of the SPE reached 7.4 × 10−4 S cm−1 (25 °C) and 4.3 × 10−3 S cm−1 (80 °C) respectively. The assembled LFP|SPE|Li displays a high specific capacity and stable charging/discharging platforms. Besides, an excellent retention capacity of 90% is obtained after 2000 cycles at 5C, and the lithium symmetric battery exhibits no significant polarization over 750 h. This work provides a viable strategy to suppress the polarization phenomenon to develop new block copolymer electrolytes with long cycle stability and high capacity retention.
Introduction
All-solid-state lithium-ion batteries (ASSLBs) are considered one of the most promising alternatives to conventional liquid lithium-ion batteries because of their superior safety and great potential to meet the requirements of high energy and power density.1–3 However, due to the poor solid–solid physical contact between the solid electrolyte and the electrode, the polarization phenomenon is easy to occur, which reduces the capacity retention and working efficiency of the batteries at high C-rates and long cycles, thus reducing the overall performance of the battery.4,5 The polarization phenomenon is primarily caused by high internal impedance, while the high internal resistance is mainly caused by poor solid–solid contact and low ion conductivity of the electrolyte. At present, the primary method to reduce the polarization inside the ASSLBs is to minimize the interface impedance by improving the contact between electrolyte and electrode and to facilitate the kinetics of ion migration under the condition of a specific ion conductivity.6 Therefore, it is essential to develop a solid electrolyte with high conductivity and good spontaneous adsorption to the electrode.Block copolymer electrolytes have many advantages such as good mechanical properties, flexibility and high solubility to lithium salts.7,8 PU elastomers, one of them, can form good contact with electrodes while having high ionic conductivity.9 Different from other solid polymer electrolytes, PU molecules have a unique structure of hard and soft segments. The hard segment offers the molecule mechanical strength to effectively hinder the growth of the lithium dendrite phenomenon, while the soft segment provides a transport path for Li+ ions.10 However, due to the existence of a hard segment, the problem of being unable to transmit ions continuously during electrode contact is inevitable, which leads to a polarization phenomenon. Therefore, we propose a new strategy to improve the chemical stability and interaction of electrolyte–electrode by modulating the soft segment, which successfully reduces the polarization phenomenon.
Sulfur-based electrolytes usually possess high ion conductivity and high stability in which P2S5 is commonly used as a co-admixture in sulfur-based electrolytes.11–14 As the key material used in sulfur electrolytes, P2S5 can not only improve the stability of electrolytes but also modify functional groups to improve the adsorption performance of molecules.15
In this work, we propose a novel composite polymer electrolyte PLAP (PU/LiTFSI–Al2O3–P2S5). Through the reaction between “–OH” and P2S5, P2S5 is introduced into the soft segment (PPG) of PU, which can not only enhance the adsorption of the electrode but also improve the transmission of Li+. LiOH is a functional modifier to enhance the adsorption energy of functional groups. Lithium salts (LiTFSI) and nano-Al2O3 are used as active fillers and inert fillers respectively. Fourier transform infra-red (FTIR) spectra, Raman spectra and X-ray photoelectron spectroscopy (XPS) were used to characterize the structure and properties of PALP. Furthermore, theoretical calculation based on density functional theory (DFT) is used to study the effect of the change of functional groups on the ionic conductivity, and the electrochemical impedance spectroscopy (EIS) test and assembled battery are used to evaluate the specific charge/discharge capacity and electric cycle stability at room temperature.
Results and discussion
Scheme 1 shows the electrolyte PLAP synthesis process. To improve the molecular intrinsic ion conductivity as well as the affinity of electrolyte towards electrode, firstly, P2S5 was grafted to PPG through the reaction of “–OH” and P2S5 due to the role of ions transfer of PU soft segment.16 Then, LiOH was used as a modifier to improve the affinity of functional groups in the soft segment of PU on Li+ and LFP, and to increase the stability between PLAP and electrode. Meanwhile, LiTFSI is the main source of free Li+, and Al2O3 was added at the end mainly to avoid chemical reaction between nano-Al2O3 and strong base (LiOH). Finally, “–SH” segment is polymerized with MDI to form PU, because the “–SH” has higher activity than the “–OH” under acidic conditions.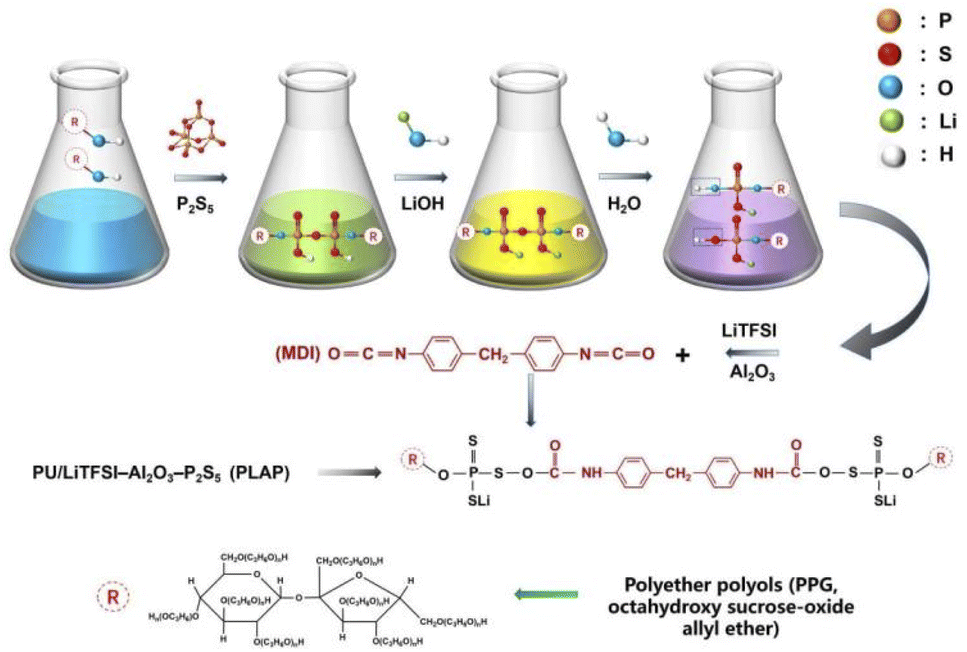 | ||
| Scheme 1 Synthesis and preparation of PLAP. | ||
In order to verify the accuracy of PLAP structure and the effect of intermediate reactant functional groups and chemical bonds on Li+ in each step of synthesis process, we carried out infrared spectrum analysis on each step of PLAP reaction products, and the final product of PLAP was carried out Raman spectrum and infrared spectrum analysis. Fig. 1(a)–(c) are the infrared spectra of the two intermediates in the reaction process and PLAP respectively. Fig. 1(a) is the product in the first step of the reaction. In Fig. 1(a), purple line (-A-), according to infrared spectrum and material properties, “S–S” and “S–C” bonds have no characteristic peak, and “S–H” peak is very weak. The peak located 3416.94 cm−1 is “–OH” stretching vibration signal, 2971.05 cm−1 and 2871.30 cm−1 are typical methyl “–C–H” peaks on the alkane molecular chains, and there are some methylene characteristic peaks on the surface of the multiple peaks included in the middle. The 1452.53 cm−1 is “–CH2” bending vibration signal peak, 1374.36 cm−1 is methyl bending vibration signal peak, 1260.98 cm−1 is “–C–H” bending vibration, the “–C–O” stretching vibration absorption peak of 1083.42 cm−1 is alcohol hydroxyl group, 977.48 cm−1 is the characteristic signal peak of the typical “–C–O–P” group. The red line (-B-) in Fig. 1(a) red line is the infrared spectrum of product after adding lithium salts. By comparing the two spectra, there was no obvious change in the infrared signal of the reaction product from 1500 cm−1 to 3500 cm−1, but the sulfonamide signal on LITFSI appeared near 1320 cm−1. The “S–P” signal peak appeared at 1192.97 cm−1, and the range of “–C–O” stretching vibration absorption peak increased at 1058.34 cm−1, indicating that the system had obvious interaction with Li+.
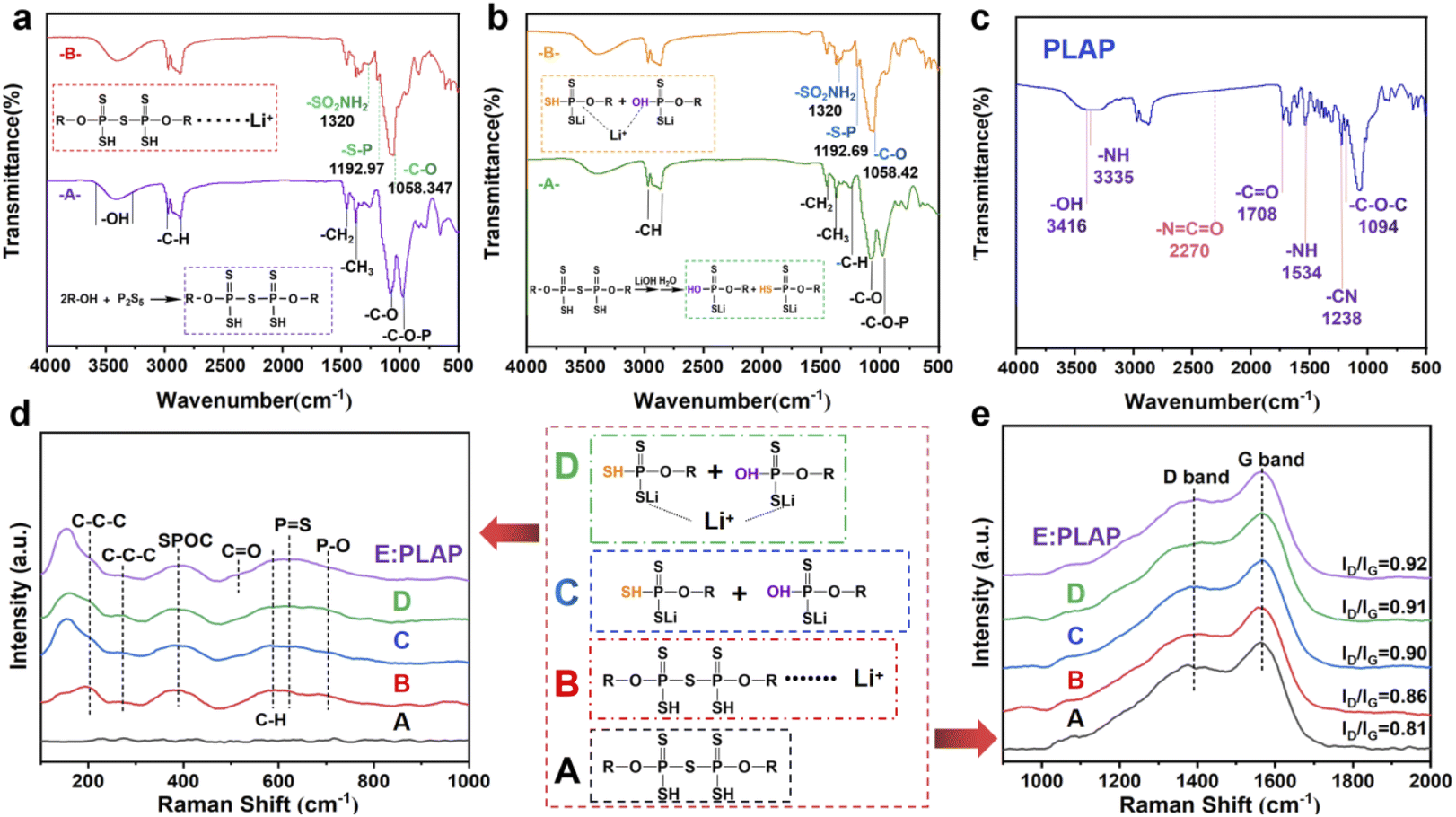 | ||
| Fig. 1 Characterization of electrolyte PLAP. (a) Infrared spectra of the product in the first step of the reaction; (b) infrared spectra of the product in the fourth step of reaction; (c) infrared spectra of PLAP; (d) Raman spectra of reaction intermediates and their addition to Li+ and electrolyte PLAP; (e) D and G peaks in the Raman spectra of five samples. | ||
In Fig. 1(b), the green line (-A-) is the product without Li+. From the figure, we can know that the peak of 3394.83 cm−1 is hydroxyl stretching vibration signal peak, 2970.92 cm−1 and 2871.92 cm−1 represent the multi-peak of the typical methyl and methylene “–C–H” stretching vibration peaks, 1452.80 cm−1 is “–CH2” bending vibration signal peak. The 1374.24 cm−1 is methyl bending vibration signal peak, 1257.31 cm−1 is “–C–H” bending vibration. The “–C–O” stretching vibration absorption peak at 1083.42 cm−1 is belong to “–C–O” alcohol hydroxyl group, 981.76 cm−1 is the characteristic signal peak of the typical “–C–O–P” group. After the lithium salts was added, we can see from Fig. 1(b) yellow line (-B-), at the peak of 1320 cm−1, there is a weak residual sulfonamide (LiTFSI) signal. The “S–P” peak appeared at 1192.69 cm−1, and the range of “–C–O” stretching vibration absorption peak increased at 1058.42 cm−1, indicating that the system had obvious interaction with Li+.
Fig. 1(c) is the infrared spectrum of PLAP. The peaks at 3335, 1708, and 1094 cm−1 correspond to the stretching vibration peaks at “–NH”, “–C–O” and “–C–O–C”, respectively.17 The peaks of 1534 and 1238 cm−1 are bending vibration peaks of “–N–H” and vibration peaks of “–C–N,” respectively. The characteristic peak of “–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO” at 2270 cm−1 disappeared, indicating that the isocyanate has been reacted completely, which means that the PU has been prepared.18 Something to watch out for in the infrared spectrum, as can be seen from Fig. 1(c), the hydroxyl peak still exists, which proves that there are still unreacted “–OH” on the molecules.
CO” at 2270 cm−1 disappeared, indicating that the isocyanate has been reacted completely, which means that the PU has been prepared.18 Something to watch out for in the infrared spectrum, as can be seen from Fig. 1(c), the hydroxyl peak still exists, which proves that there are still unreacted “–OH” on the molecules.
Fig. 1(d) and (e) are Raman spectra of reaction intermediates and their addition to lithium ions and electrolyte PLAP. In Fig. 1(d), we can know that the Raman absorption peaks near 200 cm−1 and 261 cm−1 can be attributed to the vibration peak of carbon chain in PPG group (“–C–C–C”) and the peak near 387 cm−1 is related to the deformation vibration of “–SPOC” in the product, and the Raman absorption peak near 588 cm−1 is related to the “–CH” oscillation vibration in PPG group. Raman absorption peaks near 623 cm−1 and 704 cm−1 can be attributed to stretching vibration peaks of “PS” and “P–O” bonds in the products, respectively. It is noteworthy that “–CO” stretching vibration peak appeared in sample PLAP, which indicates the PU has been synthesized successfully. Meanwhile, the Raman absorption peak near 155 cm−1 increases significantly, which may be related to the hydrolysis of the material. As can be seen from Fig. 1(e), the Raman spectra of all five products showed obvious G and D peaks. Among them, G peak is the main characteristic peak of carbon material graphitization, and is the absorption signal peak formed by aromatic ring vibration, which appears near 1580 cm−1. D peak is generally regarded as the absorption vibration peak of carbon defect and disordered carbon. Therefore, the defect of carbon material will be reflected in its Raman D peak, which can be used to characterize the graphitized structural defect or disorder in the sample. It can also be seen from Fig. 1(e) that the G-peak values of the five products are all high, indicating that the products have good material conductivity and chemical stability.19–23
After demonstrating the structural accuracy and the interaction of functional groups and chemical bonds with Li+, we performed XPS analysis on the stability of PLAP in the electrochemical cycles. Under the condition of 5C, the PLAP was charged/discharged for 500 cycles, and the PLAP before and after the cycles was analyzed by XPS. We used Avantage software to analyze the full spectrum and fine spectrum of XPS respectively. Before the analysis, the system was footnoted with C-spectrum and the “C–C” bond was calibrated to 284.8 eV. Fig. 2(a) is the full spectrum diagram of PLAP. From the Fig. 2(a), we can see that the material contains five elements: C, O, S, P and Li, which are consistent with the actual situation of our material, and there is no change after the charge/discharge cycle. According to the peak fitting of element C–spectrum in Fig. 2(b) and we know that “C1s” contains three main chemical forms. The corresponding proportion “C–C”/“C–H” bonds at 284.8 eV is 34.75%. The “C–O” bond at 286.42 eV, accounting for 62.35%, and “O–CO” bond at 289.01 eV, accounting for 2.9%.24 Fig. 2(c) is the fine spectrum of element “O”. It can be seen from the figure (c) that there are two main forms of “O”, one is “C–O” at 530.95 eV which is belong to “C–O” bond, accounting for 7.58%, and the other is at 532.64 eV which is belong to “CO”/“O–H”, accounting for 92.42%. As the “CO” functional group in the material is actually small, it is likely to correspond to the adsorbed oxygen on the surface of the material, which is belong to “O–H”.25 In Fig. 2(d), we can see that PLAP only has one chemical form of element “P” in XPS test, and the binding energy of 133.22 eV and 134.21 eV correspond to peak 2P3/2 and peak 2P1/2 respectively.26 In Fig. 2(e), after peak fitting of “S2P”, we find that “S” has three chemical forms, which can correspond to “–S–“, “SP” and “OSO” in the material respectively. Among them, the binding energy of 161.5 eV and 162.76 eV correspond to peak 2P3/2 and peak 2P1/2 of “–S–” respectively, accounting for 31%. The binding energy of 163.53 eV and 164.83 eV correspond to peak 2P3/2 and peak 2P1/2 of “SP” respectively, accounting for 29.37%. The binding energy of 168.23 eV and 169.3 eV correspond to peak 2P3/2 and peak 2P1/2 of “OSO” respectively.26–28 Fig. 2(f) shows the peak-splitting fit of “Li1S”, the binding energy of 55.4 eV corresponds to “Li–S” bond. Through the analysis of XPS test results, it can be seen from the Fig. 2(a)–(f) that there is no change before and after the cycles, which proving the stability of PLAP.
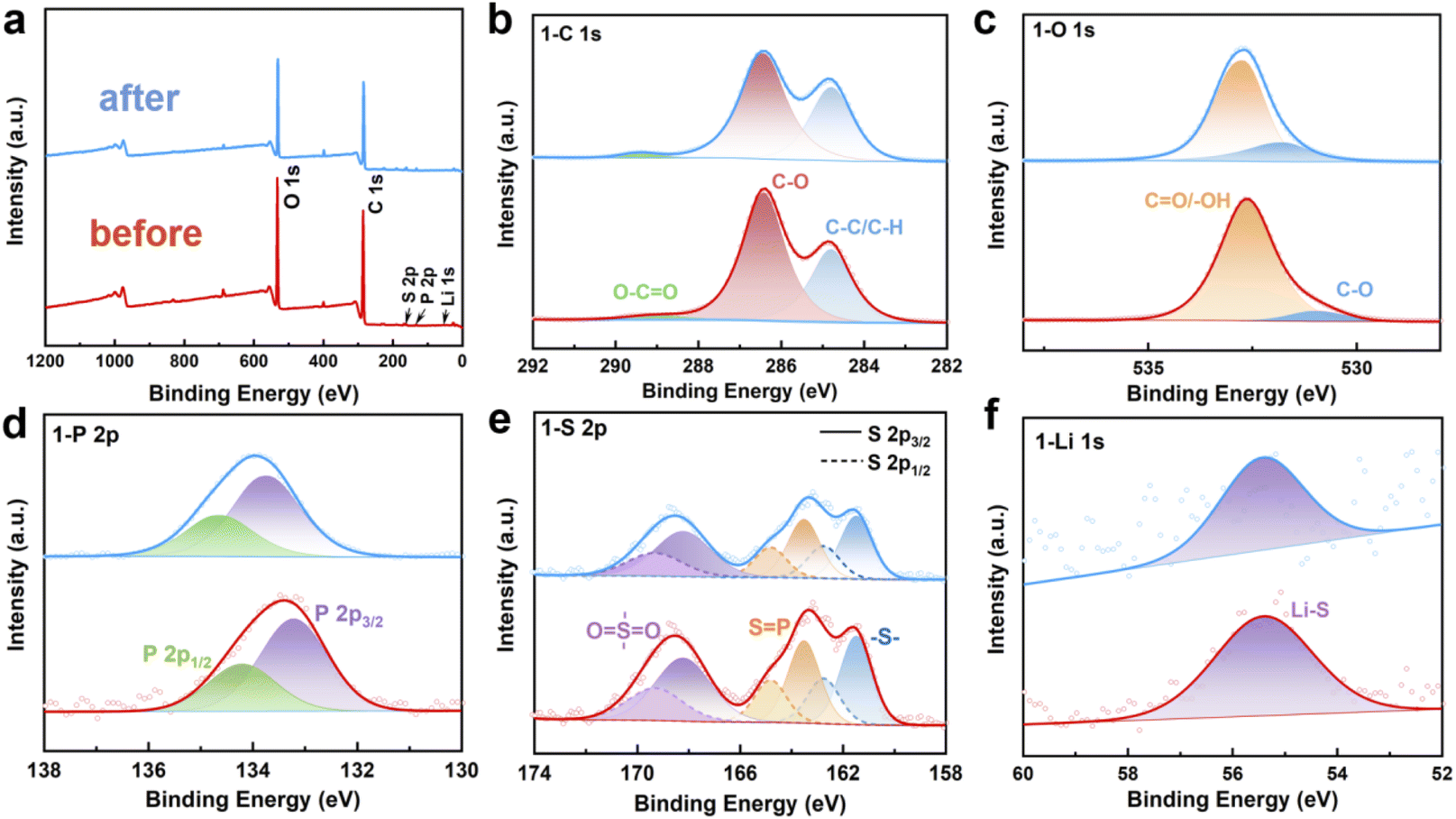 | ||
| Fig. 2 Comparison of XPS spectra of PLAP before and after circulation. (a) Full spectrum diagram; (b) C1S; (c) O1s; (d) P2P; (e) S2P; (f) Li1S. | ||
The physical properties of PLAP are also important, which is related to whether the electrolyte is suitable for high temperature conditions and the effect of tensile strength. Moreover, the surface topography of PLAP can effectively determine the distribution of elements in PLAP and the affinity of PU for various fillers. The thermal properties for the PLAP membranes were characterized by DSC and TGA (SI).† Fig. S1.† shows the degradation temperatures at a 5% weight loss (Td, 5%) of the PLAP is 220 °C, which indicates the electrolyte showed good thermal stability. Fig. S2† shows the value of the glass transition temperature (Tg) is about −43 °C indicating the electrolyte has good flexibility at room temperature. The stress–strain curve of the PLAP is illustrated in Fig. S3.† The stress strength of PLAP is 7 Mpa and the elongation-at-break value was 181.95%, the above characterization indicates PLAP shows good stress strength properties, and their stress strain properties are higher than those of other SPEs reported.29–31 Fig. S4.† shows the surface morphology SEM image and elements mapping of the electrolyte PLAP. We can find that the electrolyte surface is smooth and well distributed. The elements contained in reactant P2S5 are uniformly distributed in the electrolyte system (Fig. S5.†). The Al2O3 nanoparticles and LiTFSI in polymer system are distributed on the system, which proves that the system exhibits good compatibility for the fillers added (Li element can't be mapped out, F element is LiTFSI).
As an electrolyte material, the ions conductivity is an important index of electrolyte, and the change of conductivity with temperature is also an important parameter to measure the SPE. Fig. 3 shows our characterization of the electrical conductivity of electrolyte PLAP and the influence of the interaction between functional groups and Li+ in PLAP on the ions conductivity. As we can see from Fig. 3(a), as the temperature changes from 25 °C to 80 °C, the conductivity goes from 7.4 × 10−4 to 4.3 × 10−3 S cm−1. The reason why ionic conductivity increases with temperature is that the ionic conductivity is determined by the movement of the polymer chain under the interaction or coordination of Li+.32 And from the Fig. 3(b), we know that the Arrhenius plots of the ionic conductivity against the temperature is linear, indicating that the conductivity of the polymer electrolyte obeys Arrhenius law.33
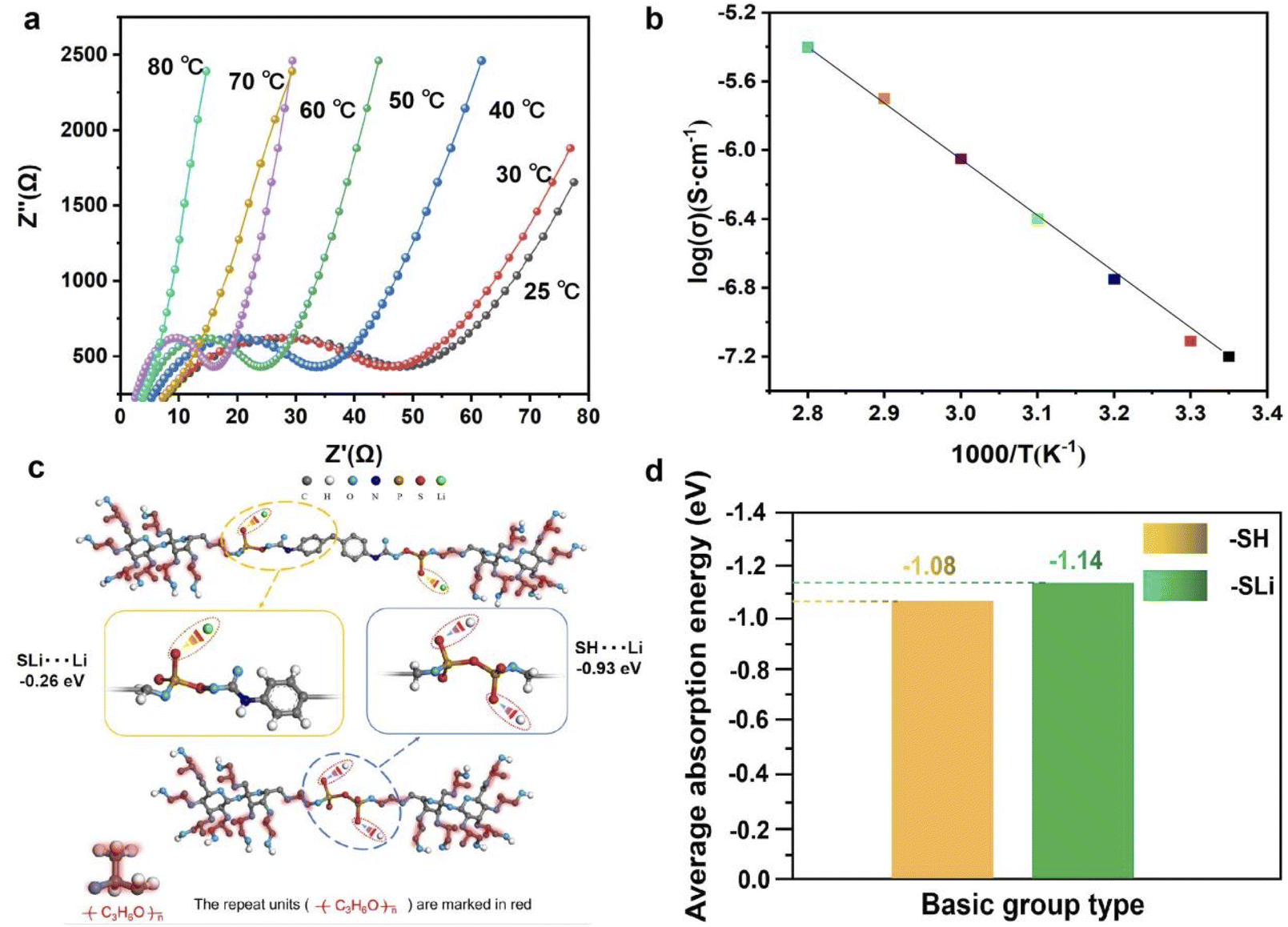 | ||
| Fig. 3 (a) AC impedance of PLAP at different temperatures; (b) the Arrhenius plots for the ionic conductivities of PLAP. (c) Calculation of adsorption energy (DFT) of “–SH” and “–SLi” to Li+; (d) the average adsorption of basic groups of “–SH” and “–SLi”. | ||
We can know that the addition amount of P2S5 has a great influence on the system (ESI Table 1†). The improvement of ionic conductivity is mainly attributed to the reaction between P2S5 and PPG, which makes P2S5 connect to the soft segment of PU through the reaction with “–OH”. To further verify the effect of functional group modification “–SH” into “–SLi” on the coupling effect towards Li+, we performed first principles calculation based on density functional theory (DFT) to analyze the adsorption behavior before and after the modulation. As shown in Fig. 3(c), the absorption energy of “–SLi” towards Li+ is −0.26 eV which is much smaller than that of “–SH” (∼−0.93 eV), which means the coupling effect of Li+ can be conspicuously reduced leading to the improvement of the ion conductivity. Besides, the average adsorption of basic group with/without the functional group modification process on the LFP crystal, as Fig. 3(d) shows, are about −1.08 eV and −1.14 eV respectively which demonstrates a superb contact towards LFP, contributing to the interface ion transport kinetics.
We assemble the LFP|PLAP|Li battery in order to study the application of the electrolyte in the battery device performance. Fig. 4(a) is the structure of the battery that we assembled, and the schematic diagram of ion transport in the battery. Fig. 4(b) exhibits the specific capacity of LFP|SPE|Li batteries with different C-rates. The capacity reached 168 mA h g−1 at 0.2C, which is quite approximate the theoretical specific capacity (170 mA h g−1) in LFP cathode materials. This proves that the prepared electrolyte possesses good ion transport performance and good contact with the electrode of solid-state LFP|SPE|Li batteries. Besides, we find that the battery exhibits a stable charging/discharging platform at different C-rates and no obvious polarization phenomenon, which indicates that the electrolyte prepared by us greatly improves the stability of the battery and enhances the contact between the electrolyte and the electrode, and thus reduces the battery polarization phenomenon.9 Fig. 4(c) shows the charging/discharging performance of the battery at different C-rates. After the changing rate returns to 0.2C, the capacity of the battery can still remain at 99%, which indicates the internal stability of the battery. Fig. 4(d) shows the long-cycle performances of the batteries under three different C-rates. The capacity retention rate is 99% (0.2C) after 500 cycles, and the capacity retention rate are still at 91% (3C) and 90% (5C) after 2000 cycles respectively, indicating the excellent overall performances of the batteries. Fig. 4(e) is a schematic diagram of the battery making the bulb light, which proves that the electrolyte we prepared can be applied in practice.
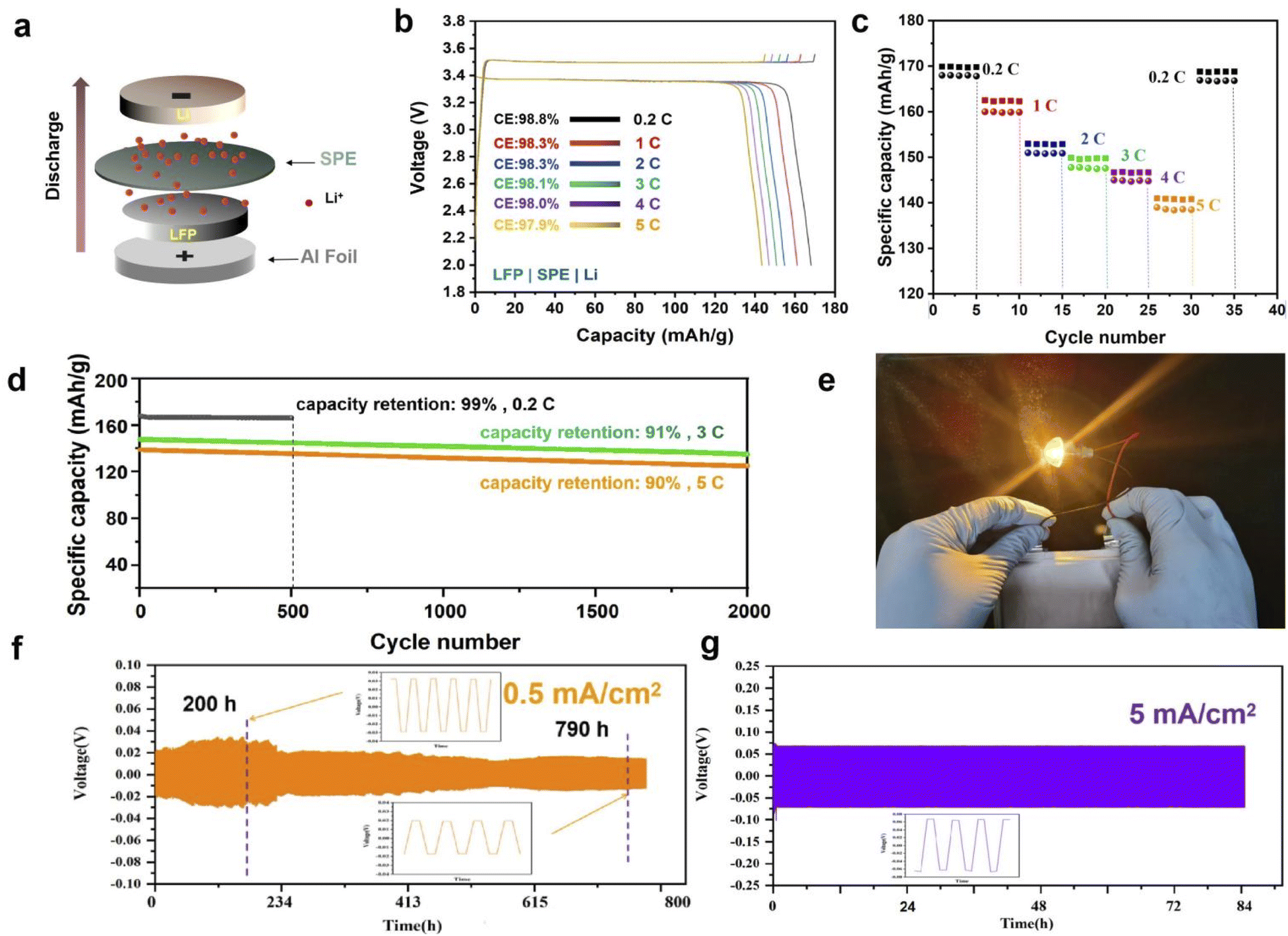 | ||
| Fig. 4 LFP|PLAP|Li batteries. (a) Battery structure diagram; (b) the first charge/discharge curves at ambient temperatures for 0.2C, 1C, 2C, 3C, 4C and 5C; (c) rate capacity with various C-rates; (d) the long-cycling properties provided under different C-rates; (e) battery luminescence picture (LFP|SPE|graphite). Voltage-time curves of the Li|PLAP|Li at the current density of (f) 0.5 mA cm−2 and (g) 5 mA cm−2. | ||
Fig. 4(f) shows the voltage profiles of the symmetric Li|PLAP|Li cells at 0.5 mA cm−2. The cells exhibited stable voltage profiles over 750 h. At the first 200 hours, the polarization phenomenon is more obvious than subsequent time, which may be attributed to the activation process of the interface between electrode and the SPE. Subsequently, the polarization becomes much smaller and the cell keeps stable after 600 h. To further study the evolution of the voltage profiles, those of the symmetric cells at 200 h and 790 h are further enlarged and presented as the inset in Fig. 4(f). The flat voltage plateau during both plating and stripping remains steady throughout long-term cycling. As the current density increased to 5 mA cm−2 (Fig. 4(b)), a stable cycling beyond 80 h with stable hysteresis is attained, which demonstrates the Li|PLAP|Li cells keep excellent stability.
Conclusions
To summarize, a novel PU–P2S5 composite block copolymer electrolyte was prepared. By grafting P2S5 to the PU soft segment, we significantly improved the conductivity of PU and enhanced its stability. The analysis and characterization of the PLAP confirm that PLAP has high conductivity [7.4 × 10−4 S cm−1 (25 °C), 4.3 × 10−3 S cm−1 (80 °C)] and can spontaneously firmly attached to the electrode to form a stable interface to reduce polarization. At the rate of 0.2C, the specific discharge capacity was nearly 168 mA h g−1, which is close to the theoretical specific capacity (170 mA h g−1) of the LFP cathode, yielding ultra-high capacity retention of 99% after 500 cycles at a rate of 0.2C, and a retention of 91% and 90% after 2000 cycles at a rate of 3C and 5C, respectively. The research provides a solid theoretical foundation and experimental support for the preparation of composite polymer electrolytes, resolution for polarization, and strategies of enhancing the performance of solid-state lithium batteries (Table 1).| Ion conductivity (S cm−1) | Specific capacity (mA h g−1) | Polarization (voltage) | Capacity retention | |
|---|---|---|---|---|
| This work | 7.4 × 10−4 (25 °C) | 168 (0.2C) (25 °C) | −0.02–0.02 (0.5 mA cm−2) | 500 cycles ∼99% (0.2C) |
| 4.3 × 10−3 (80 °C) | 143 (5C) (25 °C) | −0.06–0.06 (5 mA cm−2) | 2000 cycles ∼91% (3C) | |
| 2000 cycles ∼90% (5C) | ||||
| Ref | 7.2 × 10−4 (25 °C)39 | 110 (0.1C) (25 °C)36 | −20–20 (1 mA cm−2)34 | 2000 cycles ∼80% (1 mA)35 |
| 20 (5C) (25 °C)36 | −25–25 (0.9 mA cm−2)34 | 200 cycles ∼100% (100 mA)38 | ||
| 100 (1C) (25 °C)37 | −1–1 (1 mA cm−2)9 | 100 cycles ∼85% (0.064 mA)39 | ||
| 55 (0.064 mA cm−1−2)39 |
Experimental section
The main text of the article should appear here with headings as appropriate.Preparation of the PU-based composite electrolyte
Structure characterization
SPE morphology was observed using the field emission scanning electron microscope (S4800). The FTIR spectra were obtained by using the Fourier transform infrared spectrophotometer (PerkinElmer Spectrum Two). The Raman spectra were obtained by using the Japan -Horiba Scientific-LabRAM HR Evolution. The TGA/DSC data were documented by using Mettler DSC3, with temperature range: 10–1000 °C, heating rate of 10 K min−1, under N2. The stress–strain property was evaluated by using the ZQ-990 series universal testing machine. All samples for evaluation have a dimension of 20 mm (W) × 50 mm (L) × 0.035 mm (H). The X-ray photoelectron spectroscopy (XPS) was obtained by using the United States-Thermofisher Nexsa machine.Battery assembly and measurements
Ionic conductivity measurements based on alternating current impedance spectroscopy were performed in the CHI660e electrochemical workstation at a frequency of 100 kHz to 0.1 Hz and an oscillation potential of 10 mV. Composite electrolyte samples about 300 mm thick were sandwiched amid two metal sheet steels for the formation of test cells. The ionic conductivity can be expressed as follows:| σ = L/RbS | (1) |
where σ refers to the ionic conductivity, Rb means the bulk resistance, L represents the thickness of electrolyte membranes, and S denotes the stainless-steel electrode area. All-solid-state lithium batteries adopted LiFePO4 as the cathode and lithium metal as the anode for assembly, and the corresponding charge–discharge and cycling performance were investigated by using the LANHE CT2001A device.
Theoretical calculation
The influence of the functional groups on ion conductivity of the system was calculated and analyzed by using the Vienna abinitio simulation package (VASP). The first-principles calculations under density functional theory (DFT) were carried out with the spin-polarized generalized gradient approximation (GGA). Core electron states were denoted by using the projector augmented-wave technique applied by VASP.40–42 The exchange–correlation interactions were processed by using the GGA parameterized by Perdew, Burke, and Ernzerh (PBE) and represented by a plane wave with a wavefunction cut-off energy of 400 eV. The electronic wave function was converged to a tolerance of 10−5 eV (EDIFF = 10−5), whereas the geometric optimization tolerance was taken as 0.05 eV Å−1 (EDIFFG = −0.05). The calculation absorption energy of Li to the adsorbed C2H5NO3PS3Li and C2H5NO3PS3 is defined as follows:| ΔE = E(C2H5NO3PS3Li/Li) − E(Li) − E(C2H5NO3PS3Li) | (2) |
| ΔE = E(C2H5NO3PS3/Li) − E(Li) − E(C2H5NO3PS3) | (3) |
Author contributions
P. C., C. S., HQ. D and W. W. conceived the idea. P. C and C. S. performed the material synthesis, cell fabrication and battery testing. P. C. performed the electrochemical measurements and cathode characterization. C. S. performed the DFT simulation and provided analysis. P. C and C. S. analyzed the materials characterization data. P. C and C. S. wrote the manuscript. HQ. D and W. W. edited the manuscript. All authors have discussed the results, read the manuscript and agreed with its content.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant numbers: 62075100] and Jiangsu Postgraduate Research Innovation Program [Grant numbers: KYCX20_0701].Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, in Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, World Scientific, 2011, pp. 171–179 Search PubMed.
- J. B. Goodenough, H. D. Abruña and M. V. Buchanan, Basic research needs for electrical energy storage. Report of the basic energy sciences workshop on electrical energy storage, April 2–4, 2007, DOESC (USDOE Office of Science (SC)), 2007 Search PubMed.
- Y. Shen, Y. Zhang, S. Han, J. Wang, Z. Peng and L. Chen, Joule, 2018, 2, 1674–1689 CrossRef CAS.
- X.-B. Cheng, C.-Z. Zhao, Y.-X. Yao, H. Liu and Q. Zhang, Chem, 2019, 5, 74–96 CAS.
- H. Peng, Y. Zhang, Y. Chen, J. Zhang, H. Jiang, X. Chen, Z. Zhang, Y. Zeng, B. Sa and Q. Wei, Mater. Today Energy, 2020, 18, 100519 CrossRef CAS.
- J. R. M. Giles, F. M. Gray, J. R. MacCallum and C. A. Vincent, Polymer, 1987, 28, 1977–1981 CrossRef CAS.
- F. M. Gray, J. R. MacCallum, C. A. Vincent and J. R. M. Giles, Macromolecules, 1988, 21, 392–397 CrossRef CAS.
- P. Cui, Q. Zhang, C. Sun, J. Gu, M. Shu, C. Gao, Q. Zhang and W. Wei, RSC Adv., 2022, 12, 3828–3837 RSC.
- Y.-H. Lee, J.-S. Kim, J. Noh, I. Lee, H. J. Kim, S. Choi, J. Seo, S. Jeon, T.-S. Kim and J.-Y. Lee, Nano Lett., 2013, 13, 5753–5761 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama and K. Kawamoto, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- A. Kuhn, O. Gerbig, C. Zhu, F. Falkenberg, J. Maier and B. V. Lotsch, Phys. Chem. Chem. Phys., 2014, 16, 14669–14674 RSC.
- F. Han, J. Yue, X. Zhu and C. Wang, Adv. Energy Mater., 2018, 8, 1703644 CrossRef.
- N. C. Rosero-Navarro, H. Niwa, A. Miura and K. Tadanaga, Boletín de la Sociedad Española de Cerámica y Vidrio, 2021 Search PubMed.
- S. H. Chung and A. Manthiram, Adv. Energy Mater., 2019, 9, 1901391–1901397 Search PubMed.
- H. Muramatsu, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, Solid State Ionics, 2011, 182, 116–119 CrossRef CAS.
- C. Xu, Y. Huang, L. Tang and Y. Hong, ACS Appl. Mater. Interfaces, 2017, 9, 2169–2180 CrossRef CAS PubMed.
- L. Liu, Y. Wu and Z. Zhu, Polym. Degrad. Stab., 2017, 140, 17–24 CrossRef CAS.
- M. Hagen, P. Schiffels, M. Hammer, S. Dörfler, J. Tübke, M. J. Hoffmann, H. Althues and S. Kaskel, J. Electrochem. Soc., 2013, 160, A1205 CrossRef CAS.
- S. Bhoyate, M. Ionescu, D. Radojcic, P. K. Kahol, J. Chen, S. R. Mishra and R. K. Gupta, J. Appl. Polym. Sci., 2018, 135, 46027 CrossRef.
- K. K. Jena, D. K. Chattopadhyay and K. Raju, Eur. Polym. J., 2007, 43, 1825–1837 CrossRef CAS.
- S. Li, M. Liu, X. Zhou, Y. Dong and J. Li, Macromol. Mater. Eng., 2021, 306, 2000684 CrossRef CAS.
- A. K. Mishra, D. K. Chattopadhyay, B. Sreedhar and K. Raju, Prog. Org. Coat., 2006, 55, 231–243 CrossRef CAS.
- L. Fan, R. Ma, Q. Zhang, X. Jia and B. Lu, Angew. Chem., 2019, 131, 10610–10615 CrossRef.
- S. Zhang, C. Liu, H. Wang, H. Wang, J. Sun, Y. Zhang, X. Han, Y. Cao, S. Liu and J. Sun, ACS Nano, 2021, 15, 3365–3375 CrossRef CAS PubMed.
- Z. Wang, X. Li, W. Guo and Y. Fu, Adv. Funct. Mater., 2021, 31, 2009875 CrossRef CAS.
- J. Zhu, T. Jian, Y. Wu, W. Ma, Y. Lu, L. Sun, F. Meng, B. Wang, F. Cai and J. Gao, Appl. Surf. Sci., 2021, 544, 148882 CrossRef CAS.
- W. Guo, W. Zhang, Y. Si, D. Wang, Y. Fu and A. Manthiram, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- B. Chen, Q. Xu, Z. Huang, Y. Zhao, S. Chen and X. Xu, J. Power Sources, 2016, 331, 322–331 CrossRef CAS.
- Y. Zhao, Z. Huang, S. Chen, B. Chen, J. Yang, Q. Zhang, F. Ding, Y. Chen and X. Xu, Solid State Ionics, 2016, 295, 65–71 CrossRef CAS.
- B. Cong, Y. Song, N. Ren, G. Xie, C. Tao, Y. Huang, G. Xu and J. Bao, Mater. Des., 2018, 142, 221–228 CrossRef CAS.
- J.-H. Baik, D.-G. Kim, J. Shim, J. H. Lee, Y.-S. Choi and J.-C. Lee, Polymer, 2016, 99, 704–712 CrossRef CAS.
- S. Bandyopadhyay, R. F. Marzke, R. K. Singh and N. Newman, Solid State Ionics, 2010, 181, 1727–1731 CrossRef CAS.
- K. Hikima, N. H. Huy Phuc and A. Matsuda, J. Sol-Gel Sci. Technol., 2022, 101, 16–23 CrossRef CAS.
- M. Nagao, H. Kitaura, A. Hayashi and M. Tatsumisago, J. Power Sources, 2009, 189, 672–675 CrossRef CAS.
- T. Ohtomo, A. Hayashi, M. Tatsumisago, Y. Tsuchida, S. Hama and K. Kawamoto, J. Power Sources, 2013, 233, 231–235 CrossRef CAS.
- X. Wu, M. El Kazzi and C. Villevieille, J. Electroceram., 2017, 38, 207–214 CrossRef CAS.
- A. Hayashi, S. Hama, F. Mizuno, K. Tadanaga, T. Minami and M. Tatsumisago, Solid State Ionics, 2004, 175, 683–686 CrossRef CAS.
- K. Hirokazu, H. Akitoshi, T. Kiyoharu and T. Masahiro, J. Electrochem. Soc., 2010, 157, A407–A411 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05330f |
| This journal is © The Royal Society of Chemistry 2022 |