
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple protocol for determination of enantiopurity of amines using BINOL derivatives as chiral solvating agents via 1H- and 19F-NMR spectroscopic analysis†
Pooja Chaudharya,
Geeta Devi Yadavb and
Surendra Singh *a
*a
aDepartment of Chemistry, University of Delhi, Delhi-110007, India. E-mail: ssingh1@chemistry.du.ac.in
bDepartment of Chemistry, Swami Shraddhanand College, University of Delhi, Delhi-110036, India
First published on 8th September 2022
Abstract
A rapid and simple protocol for the determination of enantiopurity of primary and secondary amines was developed by using (S)-BINOL/(S)-BINOL derivatives/(R)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate as chiral solvating agents via 1H- and 19F-NMR spectroscopic analysis. In this protocol, the analyte and chiral solvating agent were directly mixed in an NMR tube in chloroform-d and after shaking for 30 seconds the 1H- and 19F-NMR spectra were recorded, which affords well-resolved resonance peaks for both the enantiomers present in an analyte. The enantiomeric excess of 1,2-diphenylethylenediamine was determined and linear relationship with coefficient of R2 = 0.9995 was observed. The binding constant and associated ΔG values were also calculated for diastereomeric complexes formed between both the enantiomers of analyte 5 with CSA (S)-3a by using UV-visible spectroscopy.
Introduction
Extraordinary development in asymmetric synthesis provided easy entrance to various classes of chiral compounds. Chiral compounds are important building blocks in the biochemistry and pharmaceutical industry. Therefore, the enantiodifferentiation and awareness of enantiomeric excess are extremely essential in several areas such as catalysis, pharmacology, asymmetric synthesis, and biochemistry.1–5 Among all organic compounds used in the pharmaceutical industry, chiral amines are in high demand because of their application as chiral building blocks in drug discovery and their broad range of biological activities.6,7 The most extensively used methods to determine the enantiopurity of chiral compounds are circular dichroism (CD), optical rotation, high-performance liquid chromatography (HPLC), and gas chromatography (GC).8–13 The measurement of optical purity by optical rotation and circular dichroism (CD) is easy and inexpensive but the results obtained are dependent on various parameters like amount, temperature, solvent, and the presence of impurities in the sample. HPLC and GC provided the accurate measurement of enantiomeric purity of chiral compounds but they required expensive chiral columns and HPLC solvents.The fast HPLC methods14,15 and supercritical fluid chromatography (SFC)16 already reported as efficient techniques for rapid enantiopurity determination of chiral compounds but it has been always in demand to develop a simple and rapid protocol to determine the enantiopurity and absolute configuration of synthesized chiral products. In the last few decades, the measurement of enantiomeric excess by nuclear magnetic resonance (NMR) spectroscopy has already been reported as a rapid and reliable protocol in the literature.17–37 The determination of enantiopurity via NMR spectroscopy required the presence of a chiral auxiliary to convert the enantiomers into diastereomers. The most commonly used chiral auxiliaries are chiral solvating agents {CSAs like tyrosine-modified pillar[5]arenes, R-VAPOL-phosphoric acid, organic-soluble acids, benzene tricarboxamide-based hydrogelators, BINOL phosphoric acid, Kagan's amides, roof-shape amines, BINOL and its derivatives},17–31 chiral derivatizing agents {CDAs like Δ-[Ir(ppy)2(MeCN)2](PF6) (ppy is 2-phenylpyridine), α-methoxy-α-phenylacetic acid (MPA), α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) and BINOL},31–36 chiral lanthanide shift reagents {CLSRs like [Eu(tfc)3], [Eu(hfc)3], [Sm(tfc)3], and [(R)-Pr(tfc)3]}.37–39 Among all the chiral auxiliaries reported for NMR chiral recognition, BINOL and its derivatives represent a useful auxiliary because of their direct utilization as a CSA. The BINOL and its derivatives are reported as a chiral solvating agent for NMR enantiodifferentiation of amines,26 promethazine,27 alkaloid crispine A,28 isoflavones,40 sulfinimines,41 omeprazole,42 and flavones.43
The nature of solvent is also important for this type of chiral analysis because the anisochrony observed between diastereomeric host–guest complexes is preeminent in non-polar deuterated solvents like CCl4, CDCl3, and C6D6. While polar solvents like acetonitrile-d3, acetone-d6, and methanol-d4 preferentially solvate the analyte (guest) and the enantiodifferentiation (ΔδR/S) of analyte falls to zero.44 This explains the importance of intermolecular hydrogen bond formation between CSA and analyte to form the diastereomeric complexes.
Bull and James reported the enantiopurity determination of amine 5 using (R)-BINOL as an effective chiral derivatizing agent with 2-formylphenylboronic acid. In that case, chiral BINOL undergoes the enantiodifferentiation of amine 5 by forming diastereomeric iminoboronate ester complexes via covalent (strong) interactions.35 The chemical shift difference value obtained with Bull and James protocol is higher but it is not easy to recovered and purify the BINOL from the system for reusing it in next experiment.
In this manuscript, we report a complete assessment of the effectiveness of (S)-BINOL (1), 3,3′-disubstituted (S)-BINOLs (2 and 3a–d), and (R)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (4) as chiral solvating agents for the enantiodifferentiation of various types of amines (5-11) via 1H- and 19F-NMR spectroscopic analysis (Fig. 1). In this protocol, the enantiodifferentiation of amine 5 is based on the principle that enantiomers of amine 5 (guest) complexed with CSA (S)-3a (host) through non-covalent (weak) interactions30 such as hydrogen bonding, π–π interactions to form two diastereomeric host–guest complexes, as a result different chemical shift values observed for both the enantiomers of amine 5 in 1H-NMR spectrum. Herein, the chiral solvating agent (S)-3a can be easily purified by column chromatography and reused for the next experiments.
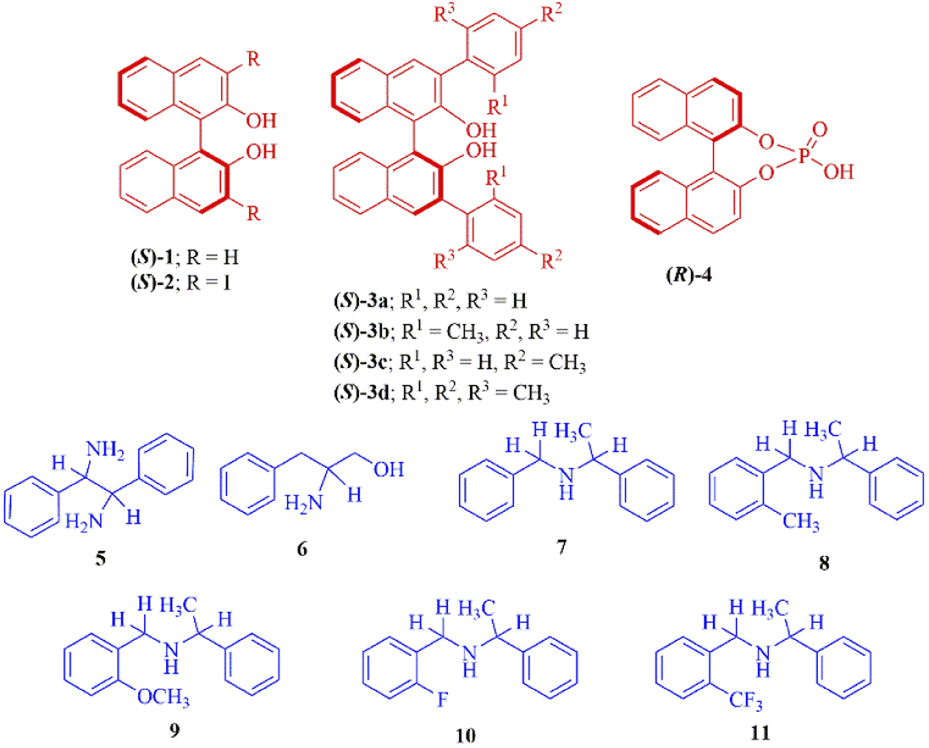 | ||
| Fig. 1 The (S)-BINOL (1), 3,3′-disubstituted (S)-BINOLs (2 and 3a–d) and (R)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (4) as a chiral solvating agents (CSAs) (red colour) and different types of amines as analytes (blue colour). | ||
Results and discussion
The 3,3′-disubstituted (S)-BINOLs (2 and 3a–d) were synthesized from (S)-BINOL (>99% ee) according to Scheme 1. The hydroxy group of (S)-BINOL was protected as methoxymethyl (MOM) ether using sodium hydride and methoxymethyl chloride by following the reported procedure.45 The direct ortho-lithiation of MOM-protected (S)-BINOL (12) using n-BuLi followed by quenching with iodine afforded the 3,3′-diiodo substituted MOM-protected (S)-BINOL (13) in 75% yield.46 The Suzuki coupling reaction of diiodide (13) with various boronic acids such as phenylboronic acid, ortho-tolylboronic acid, para-tolylboronic acid, and 2,4,6-trimethylphenylboronic acid, catalyzed by Pd(PPh3)4, afforded the corresponding 3,3′-disubstituted MOM-protected (S)-BINOLs (14a–d) in 76–85% yields.47 Further, deprotection of 3,3′-disubstituted MOM-protected (S)-BINOLs (14a–d) under acidic conditions afforded the 3,3-disubstituted (S)-BINOLs (3a–d) in 96–98% yields.48 3,3′-Diiodo (S)-BINOL (2) was also obtained in 98% yield after deprotection of 3,3′-diiodo MOM-protected (S)-BINOL (13) under acidic conditions.48 All of the reported 3,3′-disubstituted (S)-BINOLs (2, 3a–d) were characterized by 1H- and 13C-NMR spectroscopy. The optical purity of these 3,3′-disubstituted (S)-BINOLs was confirmed by comparing their optical rotation values with the reported literature.49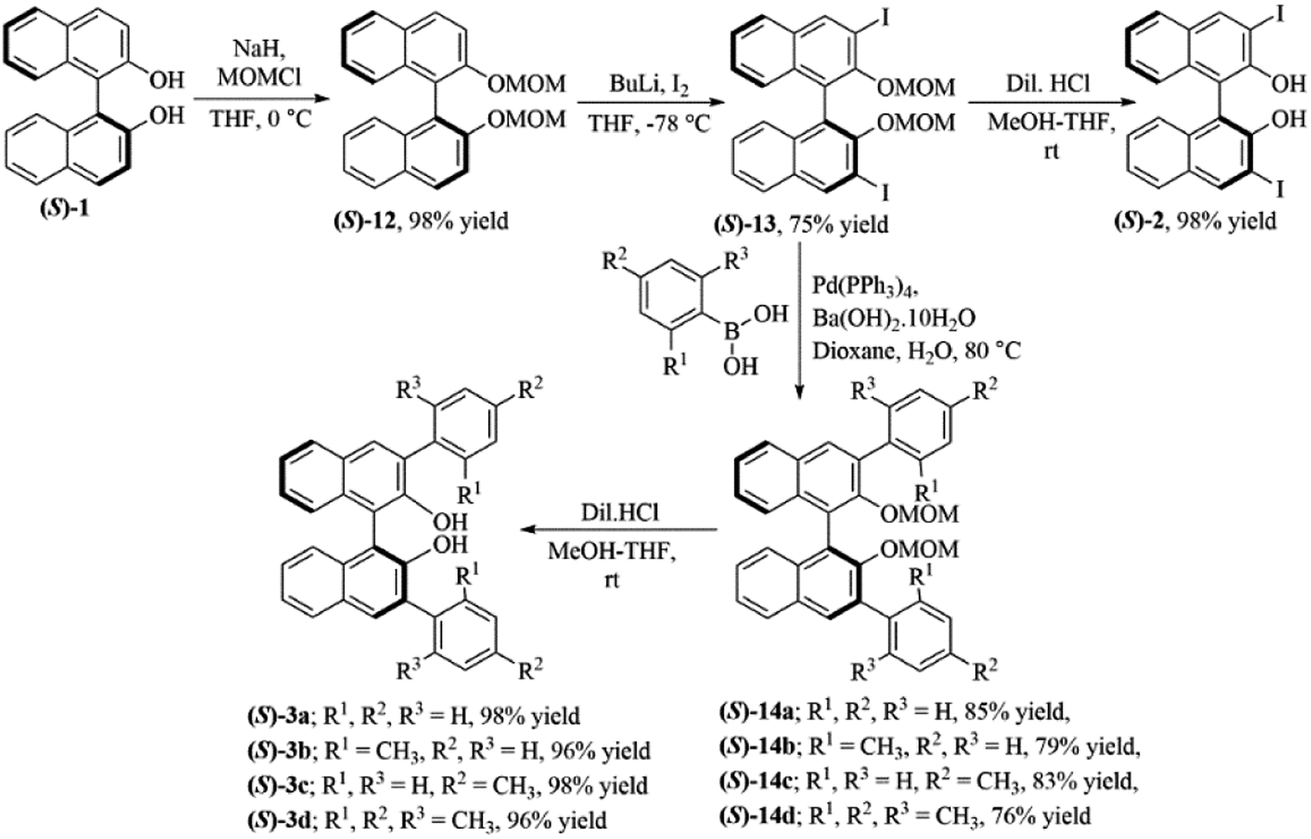 | ||
| Scheme 1 Synthesis of 3,3′-disubstituted (S)-BINOLs (2 and 3a–d) from (S)-BINOL (1). | ||
The enantiopure amine 5 is a valuable chiral source for the synthesis of chiral ligands and organocatalysts.50–52 The enantiopurity of chiral amine 5 can be determined by HPLC after its derivatization but it takes a lot of time, whereas by using this protocol it can be determined in five minutes (including the sample preparation time). Initially, enantiodifferentiation of rac-5 was carried by using 0.1 mmol of MOM-protected (S)-BINOL (12)/3,3′-diiodo-MOM-protected (S)-BINOL (13) in 0.6 mL chloroform-d at 25 °C. In the absence of CSAs, the C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) resonance of rac-5 appeared at δ = 4.10 ppm (singlet signal) and with MOM-protected (S)-BINOLs 12 and 13 no change was observed in singlet signal of rac-5, this may be due to the absence of hydrogen bonding interaction between CSAs (12 and 13) and analyte 5 (Table 1, entries 1 and 2 and Fig. 2). Later, the (S)-BINOL and its derivatives 1, 2, and 3a–d were tested as CSAs and it was observed that the singlet signal for C proton of rac-5 split into two discrete resonance peaks with a chemical shift difference (ΔδR/S) values of 0.02 to 0.06 ppm (Table 1, entries 3–8 and Fig. 2). This may be due to intermolecular hydrogen bond formation between OH and NH2 groups of (S)-BINOLs and rac-5, respectively. It was also observed that the proton signal for O group of (S)-BINOLs was upfield shifted while for N2 group of rac-5 downfield shifted and merged as a broad singlet signal, which also confirms the intermolecular hydrogen bonding interactions between (S)-BINOLs and rac-5.53
resonance of rac-5 appeared at δ = 4.10 ppm (singlet signal) and with MOM-protected (S)-BINOLs 12 and 13 no change was observed in singlet signal of rac-5, this may be due to the absence of hydrogen bonding interaction between CSAs (12 and 13) and analyte 5 (Table 1, entries 1 and 2 and Fig. 2). Later, the (S)-BINOL and its derivatives 1, 2, and 3a–d were tested as CSAs and it was observed that the singlet signal for C proton of rac-5 split into two discrete resonance peaks with a chemical shift difference (ΔδR/S) values of 0.02 to 0.06 ppm (Table 1, entries 3–8 and Fig. 2). This may be due to intermolecular hydrogen bond formation between OH and NH2 groups of (S)-BINOLs and rac-5, respectively. It was also observed that the proton signal for O group of (S)-BINOLs was upfield shifted while for N2 group of rac-5 downfield shifted and merged as a broad singlet signal, which also confirms the intermolecular hydrogen bonding interactions between (S)-BINOLs and rac-5.53
| Entry | (CSAs) | Chemical shift difference (ΔδR/S)b (ppm) |
|---|---|---|
| a In NMR tube, the CSAs (0.1 mmol) and rac-5 (0.05 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at 25 °C.b Calculated from the 1H-NMR spectrum. | ||
| 1 | (S)-12 | No resol. |
| 2 | (S)-13 | No resol. |
| 3 | (S)-1 | 0.02 |
| 4 | (S)-2 | 0.03 |
| 5 | (S)-3a | 0.06 |
| 6 | (S)-3b | 0.03 |
| 7 | (S)-3c | 0.04 |
| 8 | (S)-3d | 0.02 |
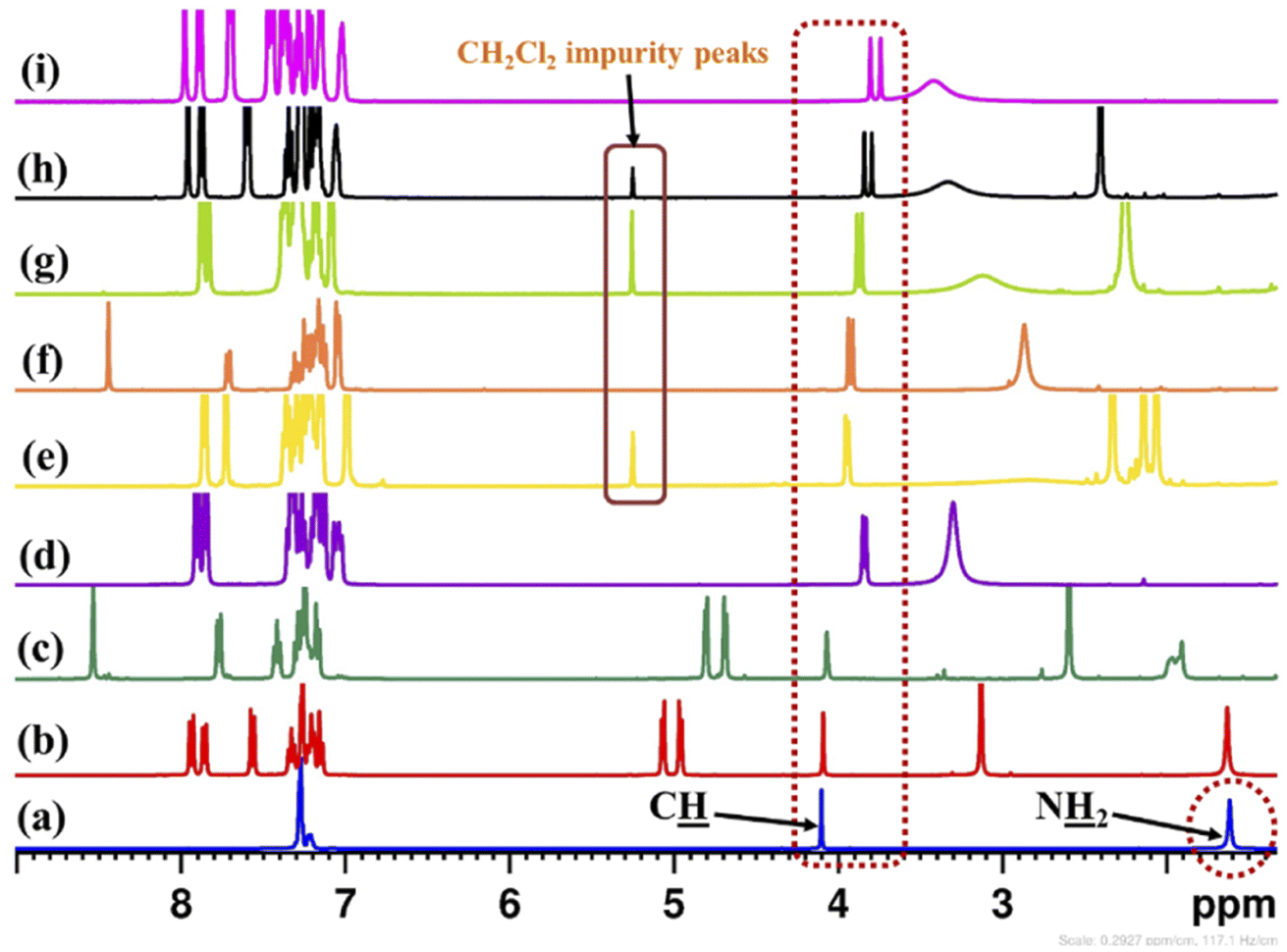 | ||
| Fig. 2 Partial stacked plot of 1H-NMR spectrum (400 MHz, CDCl3) of (a) neat rac-5, (b) rac-5 with (S)-12, (c) rac-5 with (S)-13, (d) rac-5 with (S)-1, (e) rac-5 with (S)-3d, (f) rac-5 with (S)-2, (g) rac-5 with (S)-3b, (h) rac-5 with (S)-3c, and (i) rac-5 with (S)-3a in 0.6 mL of chloroform-d at 25 °C. C resonances of rac-5 are highlighted in wine red colour box. | ||
The maximum enantiodifferentiation observed with (S)-3a, indicates the stronger hydrogen bonding interactions between NH2 and OH group of rac-5 and (S)-3a, respectively. The better results obtained with (S)-3a as compared to other 3,3′-disubstituted (S)-BINOLs may be due to extra stabilization of hydrogen bonding interactions by π–π stacking interactions between phenyl rings of rac-5 and (S)-3a. While for other 3,3′-disubstituted (S)-BINOLs (2 and 3b–d), no π–π stacking interactions possible due to the presence of bulky methyl group(s) on phenyl ring and iodide group on these BINOLs. These results reveal that the chemical shift difference (ΔδR/S) value is dependent on optimal host–guest complexation, which arise from the appropriate substitution in the chiral solvating agents (CSAs).
After assessing the various chiral solvating agents (CSAs), the typical experiments were conducted to optimize the amounts of (S)-3a and rac-5. 1H-NMR spectra of rac-5 (0.05 mmol) were recorded in 0.6 mL of chloroform-d by gradually increasing the amount of (S)-3a as a chiral solvating agent (Table 2, entries 1–4 and see ESI, Fig. 1A†). The amount of (S)-3a was varied from 0.0125 mmol to 0.1 mmol with fixed amount (0.05 mmol) of rac-5 and 0.1 mmol was found to be sufficient amount to get the clear baseline separation (ΔδR/S = 0.06 ppm) of C resonance of rac-5. Experiments were also conducted by varying the amount of rac-5 from 0.0125 mmol to 0.1 mmol with fixed amount (0.1 mmol) of (S)-3a (Table 2, entries 4–7 and see ESI, Fig. 1B†). The 0.05 mmol of rac-5 and 0.1 mmol of (S)-3a in 0.6 mL of chloroform-d was found to be the optimum amounts to get a clear baseline separation of C resonance of rac-5. Fig. 2 (ESI†) shows that, as the amount of rac-5 is increased from 0.0125 mmol to 0.1 mmol then observed broad singlet signal is upfield shifted from δ = 4.35 to 2.94 ppm. This clearly confirm the intramolecular hydrogen bond formation between OH and NH2 groups of (S)-3a and rac-5, respectively.
| Entry | Amount of (S)-3a (mmol) | Amount of rac-5 (mmol) | (ΔδR/S)b (ppm) |
|---|---|---|---|
| a In NMR tube, the CSA (S)-3a (0.0125–0.1 mmol) and rac-5 (0.0125–0.1 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at 25 °C.b Calculated from the 1H-NMR spectrum. | |||
| 1 | 0.0125 | 0.05 | 0.02 |
| 2 | 0.025 | 0.05 | 0.03 |
| 3 | 0.05 | 0.05 | 0.04 |
| 4 | 0.1 | 0.05 | 0.06 |
| 5 | 0.1 | 0.0125 | 0.06 |
| 6 | 0.1 | 0.025 | 0.06 |
| 7 | 0.1 | 0.1 | 0.06 |
To know the effect of temperature on enantiodifferentiation of 1,2-diphenylethylenediamine (5) using (S)-3a as a chiral solvating agent, the 1H-NMR experiments were conducted at variable temperature (VT) in 0.6 mL of chloroform-d. The temperature was varied from 25 to −50 °C and at −50 °C very high chemical shift difference value (ΔδR/S = 0.52 ppm) was observed for C1 resonance of rac-5, because at low temperature the two enantiomers of 1,2-diphenylethylenediamine (5) reside in different local chemical environment for a longer time. At this temperature, even the aromatic proton signals for C2 and C3 of rac-5 were also split into two distinct resonance peaks. The clear baseline separation was observed for C2 resonance at −40 °C (ΔδR/S = 0.11 ppm) and −50 °C (ΔδR/S = 0.15 ppm) but C3 resonance was overlapped in the aromatic 1H-NMR region of (S)-3a (Table 3, and Fig. 3).
| Entry | Temp. (°C) | Chemical shift difference (ΔδR/S)b (ppm) | |
|---|---|---|---|
| C1 |
C2 |
||
| a In NMR tube, the CSA (S)-3a (0.1 mmol) and rac-5 (0.05 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at variable temperatures.b Calculated from the 1H-NMR spectrum. | |||
| 1 | −50 | 0.52 | 0.15 |
| 2 | −40 | 0.40 | 0.11 |
| 3 | −20 | 0.19 | 0.05 |
| 4 | −10 | 0.14 | No resol. |
| 5 | 0 | 0.11 | No resol. |
| 6 | 10 | 0.09 | No resol. |
| 7 | 25 | 0.06 | No resol. |
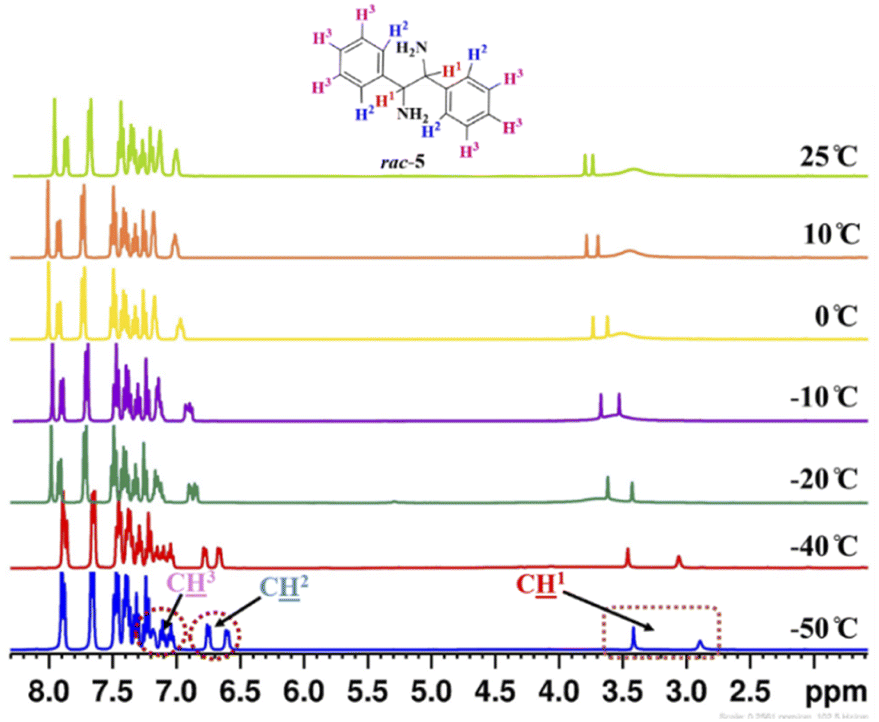 | ||
| Fig. 3 Partial stacked plot of 1H-NMR spectrum (400 MHz, CDCl3) of rac-5 (0.05 mmol) with (S)-3a (0.1 mmol) at variable temperatures in chloroform-d (0.6 mL). C1, C2, and C3 resonances are highlighted in wine red colour box. | ||
The reliability of the proposed protocol was evaluated by performing the experiments to measure the enantiomeric excess of 1,2-diphenylethylenediamine (5). The six different scalemic mixtures of enantiopure (1R,2R) and (1S,2S)-1,2-diphenylethylenediamine (5) (0.05 mmol) were prepared and their enantiomeric excess was determined by using (S)-3a (0.1 mmol) as a CSA in chloroform-d (0.6 mL) at 25 °C. The enantiomeric excess was calculated by integrating the C resonance peaks observed for each of the enantiomers of 1,2-diphenylethylenediamine (5) from their respective 1H-NMR spectrum (Fig. 4A). A linear relationship with a coefficient of R2 = 0.9995 was observed between the theoretically calculated ee values and the ee values determined by 1H-NMR spectra (Fig. 4B). These outcomes indicate, (S)-3a is an effective chiral solvating agent to determine the enantiomeric excess of 1,2-diphenylethylenediamine (5).
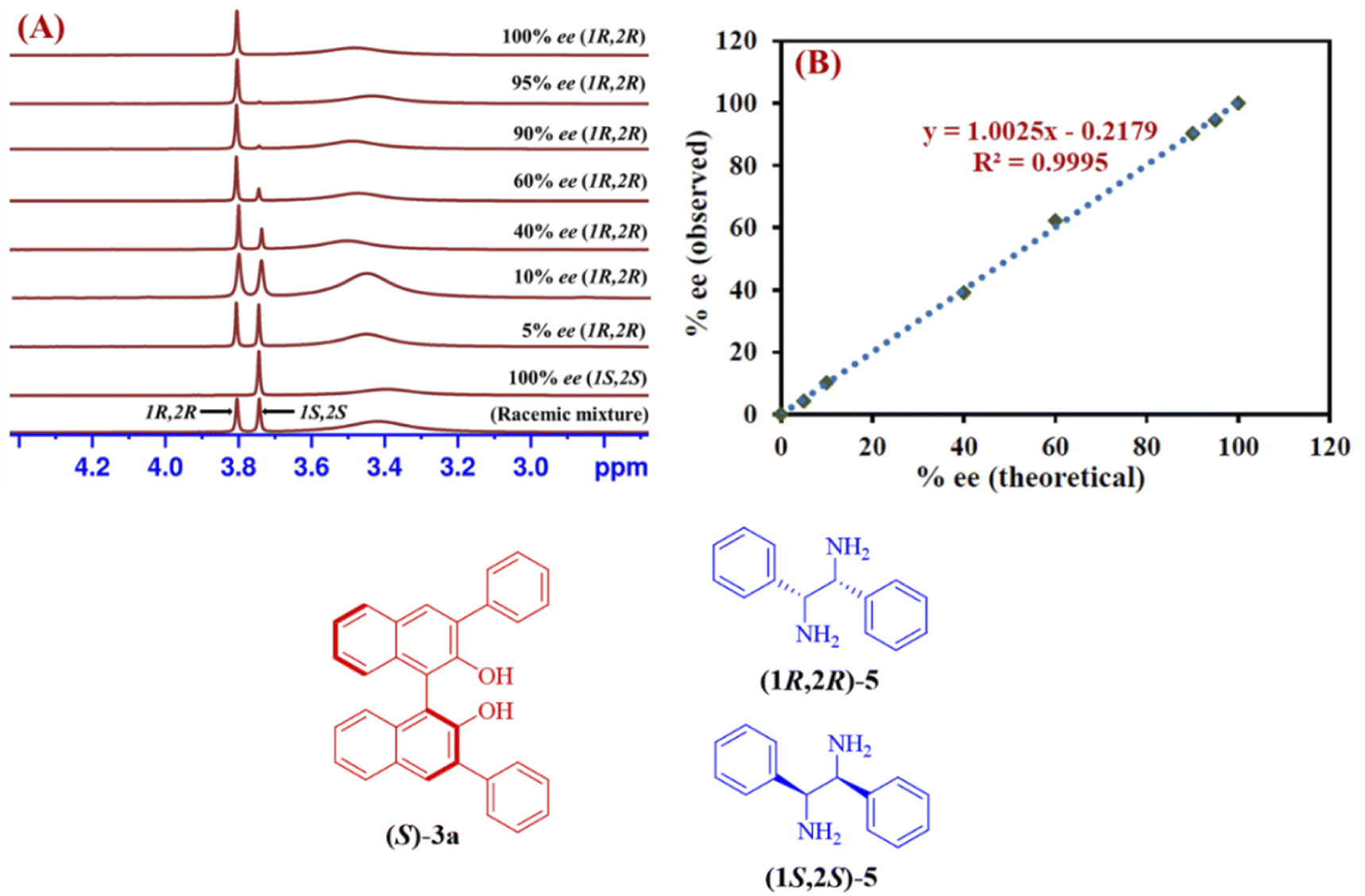 | ||
| Fig. 4 (A) Partial stacked plot of 1H-NMR spectrum (400 MHz, CDCl3) for the C resonance of 5 for six scalemic mixtures of enantiopure (1R,2R/1S,2S)-1,2-diphenylethylenediamine (5) with (S)-3a in 0.6 mL chloroform-d at 25 °C. (B) Linear relationship between theoretically determined ee values and the ee values determined by 1H-NMR spectra. | ||
UV-visible spectroscopy is one of the best technique to study the extent of binding between both the enantiomers of analyte with CSA due to its easy handling and high sensitivity. Herein, we determine the binding constant for both the enantiomers of analyte 5 with CSA (S)-3a. Initially, the absorbance was measured for free CSA (S)-3a and then optically pure form of both the enantiomers of analyte 5 was added separately to get a two sets of readings. With the increase in equivalent of enantiomerically pure analyte 5, the absorption intensity was also increased, which indicates the formation of diastereomeric host guest complex (see ESI, Fig. 3 and 4†). The change in the absorption intensity for both the enantiomers of analyte 5, indicate their different extent of binding with CSA (S)-3a. A graph was plotted between the change in absorbance (ΔA) and the analyte concentration (C). The plot of 1/[ΔA] versus 1/[C] gives a straight line with correlation coefficients (R2) 0.99925 and 0.99743 for R and S enantiomers respectively (see ESI, Fig. 5†). The binding constants (KR and KS) for analyte 5 were calculated from the slope of the plot using modified Benesi–Hildebrand equation given below:25
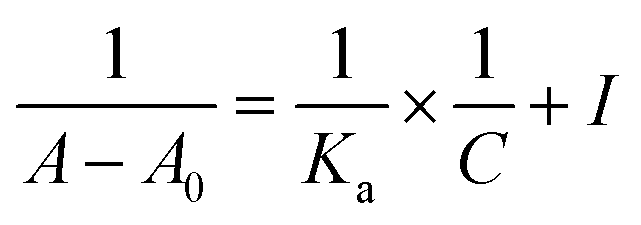
The Gibbs free energy (ΔG) for diastereomeric complexes formed between CSA (S)-3a and both the enantiomer of analyte 5 were calculated from the binding constant (KR/KS) using following equation:
ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK lnK |
The calculated binding constant and ΔG values for diastereomeric complexes formed between both the enantiomers of analyte 5 with CSA (S)-3a are summarized in Table 4.
| Substrate | K (L mol−1) | ΔG (kJ mol−1) |
|---|---|---|
| a Calculated from equation ΔG = −RTlnK, where R = 0.008314 kJ mol−1 K−1, T = 298.15 K. |
||
| (R)-5 | 617.28 | −15.91 |
| (S)-5 | 900.90 | −16.85 |
The results in Table 4 shows that the KS value is more compared to KR value for analyte 5, which conclude that CSA (S)-3a form stronger diastereomeric complex with (S)-5 compared to (R)-5 enantiomer.
The NMR enantiodifferentiation of rac-2-amino-3-phenylpropan-1-ol (6) (0.025 mmol) with 0.1 mmol of CSAs 1, and 3a–d was evaluated in 0.6 mL of chloroform-d at 25 °C. The multiplet signal that appeared at 3.11 ppm for C resonance of rac-6 was separated into two distinct resonance peaks with a ΔδR/S value of 0.09 ppm in the presence of (S)-BINOL (1) (Table 5 and see ESI, Fig. 6†). The 1H-nuclei nearby to the amine group shows significant shielding due to the hydrogen bond formation between NH2 and OH group of analyte 6 and two OH group of (S)-BINOL (1) and the diastereomeric host guest complexes were formed between (S)-BINOL (1) and two enantiomers of analyte 6. No clear baseline separation was observed for the C signal of analyte 6, if 3,3-disubstituted (S)-BINOLs (3a–d) were used as chiral solvating agents. This may be due to the steric repulsion exhibited by bulky substituents attached at 3,3′-position of (S)-BINOL to form the hydrogen bond between analyte 6 and CSAs. Therefore, (S)-BINOL (1) was found to be the effective chiral solvating agent for the NMR enantiodifferentiation of analyte 6.
| Entry | Chiral solvating agents (CSAs) | (ΔδR/S)b (ppm) |
|---|---|---|
| a In NMR tube, the CSAs (0.1 mmol) and rac-6 (0.025 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at 25 °C.b Calculated from the 1H-NMR spectrum. | ||
| 1 | (S)-1 | 0.09 |
| 2 | (S)-3a | 0.06 |
| 3 | (S)-3b | 0.05 |
| 4 | (S)-3c | 0.05 |
| 5 | (S)-3d | No resol. |
The effects of change in molar ratios of (S)-BINOL (1) and analyte 6, were also studied for enantiodifferentiation of analyte 6. With a fixed amount (0.1 mmol) of (S)-BINOL (1), the NMR experiments were performed by varying the amount of analyte 6 from 0.0125 mmol to 0.125 mmol (Table 6, entries 1–6, and see ESI, Fig. 7†). With 0.0125 mmol of analyte 6, the maximum enantiodifferentiation (ΔδR/S = 0.09 ppm) was observed but the integration of resolved peaks for C protons was found to be unequal. After then the amount of analyte 6 was increased from 0.0125 mmol to 0.125 mmol and best baseline separation and integration of resolved peaks for C resonance was observed at 0.025 mmol of analyte 6. Hence, 0.1 mmol of (S)-BINOL (1) and 0.025 mmol of analyte 6 in 0.6 mL of chloroform-d were found to be the optimum condition to get the clear baseline separation for C resonance of analyte 6.
| Entry | Amount of rac-6 (mmol) | (ΔδR/S)b (ppm) |
|---|---|---|
| a In NMR tube, the (S)-BINOL (1) (0.1 mmol) and rac-6 (0.0125–0.125 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at 25 °C.b Calculated from the 1H-NMR spectrum. | ||
| 1 | 0.0125 | 0.09 |
| 2 | 0.025 | 0.09 |
| 3 | 0.05 | 0.08 |
| 4 | 0.075 | 0.07 |
| 5 | 0.1 | 0.07 |
| 6 | 0.125 | 0.06 |
We have also screened the (S)-BINOL derived chiral solvating agents to determine the enantiodifferentiation of secondary amines (7–11) and none of the BINOL derivatives showed separation of C proton signals for secondary amines (see ESI, Fig. 8†). The (R)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (4) undergoes a neutralization reaction with chiral amines and forms an ion-pair in chloroform-d, resulted in the separation of proton signals of analyte 7–11.19,54 Initially, we optimized the molar ratio of CSA 4 and analyte 8 to get a good enantiodifferentiation of secondary amines. With the fixed amount of CSA 4 (0.1 mmol), the amounts of analyte 8 were varied from 0.0125 mmol to 0.2 mmol (Table 7, entries 1–5, and see ESI, Fig. 9†) and results show that 0.1 mmol of CSA 4 and 0.1 mmol of analyte 8 was found to be optimal amount to get the clear baseline separation in analyte 8. At this molar ratio, the maximum baseline separation for the C3 (ΔδR/S = 0.09 ppm), aromatic C3 (ΔδR/S = 0.13 ppm) and C (ΔδR/S = 0.16 ppm) resonance of analyte 8 were observed (Table 7, entry 4). With a decrease in the amount of rac-8, the overlapping of C and C2 resonances were observed and also the ΔδR/S values of C3 and aromatic C3 resonances were lowered. If we increased the amount of rac-8 from 0.1 mmol to 0.2 mmol, the decreased in ΔδR/S value was observed. Hence, it is essential to find the optimum amounts of CSAs and analytes to accomplish a good splitting of signals and to avoid overlapping of signals.
| Entry | Amount of rac-8 (mmol) | (ΔδR/S)b (ppm) | ||
|---|---|---|---|---|
| C |
Ar-C3 |
C3 |
||
| a In NMR tube, the CSA (R)-4 (0.1 mmol) and rac-8 (0.0125–0.2 mmol) were dissolved in chloroform-d (0.6 mL) and after shaking the NMR tube for 30 seconds, 1H-NMR spectrum was recorded on a 400 MHz NMR spectrometer at 25 °C.b Calculated from the 1H-NMR spectrum. | ||||
| 1 | 0.0125 | No resol. | 0.04 | 0.04 |
| 2 | 0.025 | No resol. | 0.05 | 0.05 |
| 3 | 0.05 | No resol. | 0.05 | 0.06 |
| 4 | 0.1 | 0.16 | 0.13 | 0.09 |
| 5 | 0.2 | 0.09 | 0.06 | 0.05 |
To know the generality of this protocol, the enantiodifferentiation of different secondary amines (7–11) (0.1 mmol) was investigated using (R)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (4) (0.1 mmol) as a chiral solvating agent in 0.6 mL of chloroform-d at 25 °C. 1H-, 13C-, and 19F-NMR spectra recorded for secondary amines are given in ESI (see ESI, Fig. 10–21†). The ΔδR/S values of clear baseline resolved proton, carbon, and fluorine signals of secondary amines (7–11) are given in Scheme 2. As shown in 1H-NMR spectrum (see ESI, Fig. 10†) of analyte 7, the overlapping of C and C2 resonances create inaccuracy in integrating the two distinct resonance peaks obtained for C and C2 protons which led to inaccurate determination of enantiopurity of amines via 1H-NMR spectroscopy.
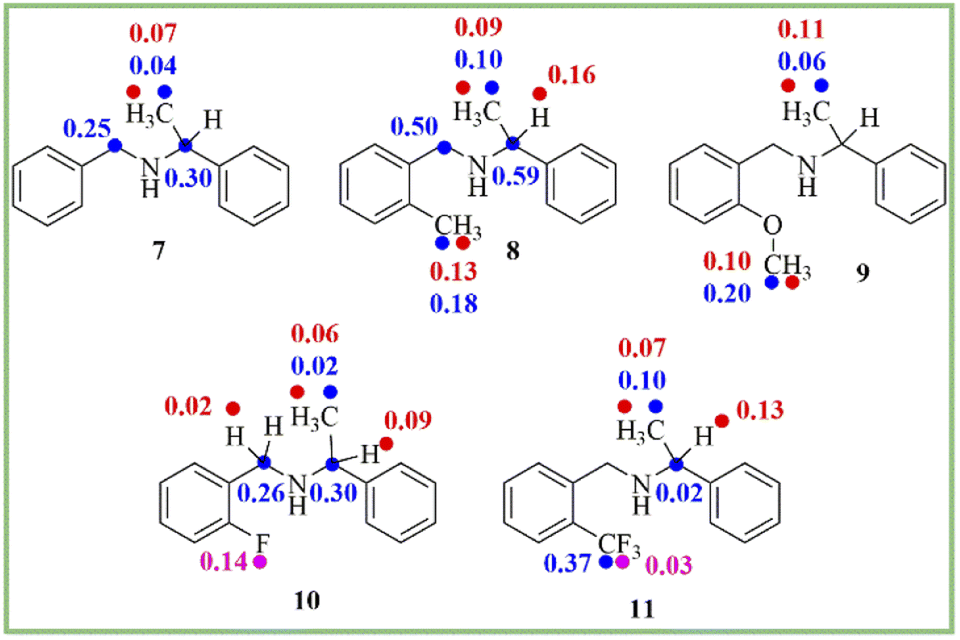 | ||
| Scheme 2 The chemical shift difference (ΔδR/S) values of clearly baseline resolved proton (red), carbon (blue), and fluorine (pink) signals of secondary amines (7–11). | ||
In that case, the 13C- and 19F-NMR spectroscopy plays a crucial role to determine the enantiopurity of amines. The 13C- and 19F-NMR spectra were also recorded for secondary amines at 25 °C. The 19F-NMR spectroscopy is also beneficial due to its 100% natural abundance and 83% signal sensitivity relative to 1H-NMR spectroscopy and better separation of the signal due to less number of nuclei present in the analyte.55 In this study, two examples of fluorine containing analytes (10 and 11) are included. The analyte 10 (ΔδR/S = 0.14 ppm) shows more splitting of a signal as compared to analyte 11, which is having CF3 group (ΔδR/S = 0.03 ppm) (see ESI, Fig. 18 and 21†).
Experimental
General procedure for NMR sample preparation
The analyte (0.0125–0.2 mmol) and chiral solvating agent (0.0125–0.1 mmol) were directly mixed in an NMR tube and dissolved in chloroform-d (0.6 mL). After shaking the NMR tube for 30 seconds, 1H-, 13C-, and 19F-NMR spectra were recorded on a 400 MHz NMR spectrometer at 25 °C and well-resolved resonance peaks were observed for both the enantiomers present in an analyte. The enantiopurity was calculated by integrating the resonance peaks observed for each of the enantiomers of an analyte from their respective 1H, 13C-, and 19F-NMR spectra.Conclusions
In summary, a simple and fast protocol for enantiopurity determination of different types of amines (analytes) is described here via 1H- and 19F-NMR spectroscopy (for fluorinated analytes) in chloroform-d at 25 °C. (S)-3a, having phenyl group at 3,3′ positions act as a better CSA for NMR enantiodifferentiation of 1,2-diphenylethylenediamine while simple (S)-BINOL (1) shows better enantiodifferentiation for rac-2-amino-3-phenylpropan-1-ol. (R)-1,1′-Binaphthyl-2,2′-diyl hydrogenphosphate (4) acts as an effective chiral solvating agent for NMR enantiodifferentiation of different secondary amines. The enantiopurity of analyte 5 was determined and linear relationship with coefficient of R2 = 0.9995 was observed. Using UV-visible spectroscopy, the binding constant and associated ΔG values were also calculated for diastereomeric complexes formed between both the enantiomers of analyte 5 with CSA (S)-3a. This protocol is very efficient and less time-consuming as compared to the derivatization protocol.Author contributions
Pooja Chaudhary and Dr Geeta Devi Yadav were involved in the investigation, validation and data writing of this manuscript. Prof. Surendra Singh is involved conceptualization, funding acquisition, supervision of the research activity and writing of original draft of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SS acknowledges the CSIR grant no. 02(317)/17/EMR-II. PC is thankful to CSIR for the Senior Research Fellowship (SRF). We are thankful to USIC, University of Delhi, Delhi, India for the instrumentation facility. We are also thankful to Synthesis with Catalyst Pvt. Ltd, India for gifting the (1R,2R)-1,2-diphenylethylenediamine and (1S,2S)-1,2-diphenylethylenediamine.References
- B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC.
- K. C. Nicolaou, J. S. Chen and S. M. Dalby, Bioorg. Med. Chem., 2009, 17, 2290–2303 CrossRef CAS.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- R. Wohlgemuth, Curr. Opin. Microbiol., 2010, 13, 283–292 CrossRef CAS PubMed.
- J. A. Tao, G.-Q. Lin and A. Liese, Biocatalysis for the pharmaceutical industry: discovery, development and manufacturing, John Wiley & Sons, 2009 Search PubMed.
- T. C. Nugent, Chiral amine synthesis: methods, developments and applications, John Wiley & Sons, 2010 Search PubMed.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS.
- P. Metola, E. V. Anslyn, T. D. James and S. D. Bull, Chem. Sci., 2012, 3, 156–161 RSC.
- M. O. Okuom, R. Burks, C. Naylor and A. E. Holmes, J. Anal. Methods Chem., 2015, 865605 Search PubMed.
- P. J. Walsh, D. K. Smith and C. Castello, J. Chem. Educ., 1998, 75, 1459–1462 CrossRef CAS.
- A. Kaddoumi, M. N. Nakashima and K. Nakashima, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 763, 79–90 CrossRef CAS.
- I. Ilisz, A. Aranyi, Z. Pataj and A. Peter, J. Pharm. Biomed. Anal., 2012, 69, 28–41 CrossRef CAS.
- V. Schurig and H.-P. Nowotny, Angew. Chem., Int. Ed., 1990, 29, 939–1076 CrossRef.
- M. A. Shishovska and M. T. Stefova, J. Chromatogr. Sci., 2012, 50, 43–50 CAS.
- F. Chen, B. Fang and S. Wang, J. Anal. Methods Chem., 2021, 8821126 CAS.
- G. Roskam, B. Velde, A. Gargano and I. Kohler, LCGC Europe, 2022, 353, pp. 83–92 Search PubMed.
- L. Liu, C. Ma, Q. He, Y. Huang and W. Duan, Org. Chem. Front., 2021, 8, 4144–4152 RSC.
- D. Prasad, S. Mogurampelly and S. R. Chaudhari, RSC Adv., 2020, 10, 2303–2312 RSC.
- B. Benedict, C. E. Lietz and T. J. Wenzel, Tetrahedron, 2018, 74, 4846–4856 CrossRef CAS.
- S. H. Jung, K. Y. Kim, A. Ahn, S. S. Lee, M. Y. Choi, J. Jaworski and J. H. Jung, New J. Chem., 2016, 40, 7917–7922 RSC.
- C.-X. Liu, L. Zheng, L. Zhu, H.-P. Xiao, X. Li and J. Jiang, Org. Biomol. Chem., 2017, 15, 4314–4319 RSC.
- N. Jain, R. B. Patel and A. V. Bedekar, RSC Adv., 2015, 5, 45943–45955 RSC.
- H. B. Raval and A. V. Bedekar, ChemistrySelect, 2020, 5, 6927–6932 CrossRef CAS.
- R. Gupta, R. G. Gonnade and A. V. Bedekar, J. Org. Chem., 2016, 81, 7384–7392 CrossRef CAS PubMed.
- R. Gupta, R. G. Gonnade and A. V. Bedekar, ChemistrySelect, 2020, 5, 13183–13190 CrossRef CAS.
- J. C. Merino, A. F. Keppler and M. S. Silva, J. Braz. Chem. Soc., 2018, 29, 1638–1644 CAS.
- P. Borowiecki, Tetrahedron: Asymmetry, 2015, 26, 16–23 CrossRef CAS.
- F. Yuste, R. Sánchez-Obregón, E. Díaz and M. A. García-Carrillo, Tetrahedron: Asymmetry, 2014, 25, 224–228 CrossRef CAS.
- J. S. Salsbury and P. K. Isbester, Magn. Reson. Chem., 2005, 43, 910–917 CrossRef CAS.
- F. Toda, K. Mori, J. Okada, M. Node, A. Itoh, K. Oomine and K. Fuji, Chem. Lett., 1988, 17, 131–134 CrossRef.
- S. K. Mishra and N. Suryaprakash, Tetrahedron: Asymmetry, 2017, 28, 1220–1232 CrossRef CAS.
- L.-P. Li and B.-H. Ye, Inorg. Chem., 2017, 56, 10717–10723 CrossRef CAS PubMed.
- N. V. Orlov and V. P. Ananikov, Green Chem., 2011, 13, 1735–1744 RSC.
- Y. Pérez-Fuertes, A. M. Kelly, J. S. Fossey, M. E. Powell, S. D. Bull and T. D. James, Nat. Protoc., 2008, 3, 210–214 CrossRef PubMed.
- A. M. Kelly, S. D. Bull and T. D. James, Tetrahedron: Asymmetry, 2008, 19, 489–494 CrossRef CAS.
- Y. Pérez-Fuertes, A. M. Kelly, A. L. Johnson, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 609–612 CrossRef PubMed.
- H. J. C. Yeh, S. K. Balani, H. Yagi, R. M. E. Greene, N. D. Sharma, D. R. Boyd and D. M. Jerina, J. Org. Chem., 1986, 51, 5439–5443 CrossRef CAS.
- K. Omata, S. Aoyagi and K. Kabuto, Tetrahedron: Asymmetry, 2004, 15, 2351–2356 CrossRef CAS.
- I. Ghosh, H. Zeng and Y. Kishi, Org. Lett., 2004, 6, 4715–4718 CrossRef CAS PubMed.
- J. Yi, G. Du, Y. Yang, Y. Li, Y. Li and F. Guo, Tetrahedron: Asymmetry, 2016, 27, 1153–1159 CrossRef CAS.
- M. Ardej-Jakubisiak and R. Kawecki, Tetrahedron: Asymmetry, 2008, 19, 2645–2647 CrossRef CAS.
- J. Redondo, A. Capdevila and I. Latorre, Chirality, 2010, 22, 472–478 CAS.
- G. Du, Y. Li, S. Ma, R. Wang, B. Li, F. Guo, W. Zhu and Y. Li, J. Nat. Prod., 2015, 78, 2968–2974 CrossRef CAS PubMed.
- W. H. Pirkle and D. J. Hoover, Top. Stereochem., 1982, 13, 263–331 CAS.
- T. R. Wu, L. Shen and J. M. Chong, Org. Lett., 2004, 6, 2701–2704 CrossRef CAS PubMed.
- C. Recsei and C. S. P. McErlean, Tetrahedron, 2012, 68, 464–480 CrossRef CAS.
- W. Hu, J. Zhou, X. Xu, W. Liu and L. Gong, Org. Synth., 2011, 88, 406–417 CrossRef CAS.
- Y. Liu, S. Zhang, Q. Miao, L. Zheng, L. Zong and Y. Cheng, Macromolecules, 2007, 40, 4839–4847 CrossRef CAS.
- L.-J. Yan, X. Liu, P.-A. Wang, H.-F. Nie and S.-Y. Zhang, Org. Prep. Proced. Int., 2013, 45, 473–482 CrossRef CAS.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS.
- P. Chaudhary, G. D. Yadav, K. K. Damodaran and S. Singh, New J. Chem., 2022, 46, 1308–1318 RSC.
- J. H. Shim, M.-J. Kim, J. Y. Lee, K. H. Kim and D.-C. Ha, Tetrahedron Lett., 2020, 61, 152295 CrossRef CAS.
- M. Ratajczak-Sitarz, A. Katrusiak, K. Gawrońska and J. Gawroński, Tetrahedron: Asymmetry, 2007, 18, 765–773 CrossRef CAS.
- J.-H. Tay and P. Nagorny, Synlett, 2016, 27, 551–554 CAS.
- S. Jang and H. Kim, Org. Lett., 2020, 22, 7804–7808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05291a |
| This journal is © The Royal Society of Chemistry 2022 |