
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow do layered double hydroxides evolve? First in situ insights into their synthesis processes†
G. Doungmo ab,
A. F. Moraisc,
D. Mustafac,
T. Kamgaingb,
E. Njanjab,
M. Etterd,
I. K. Tonlé*b and
H. Terraschke*a
ab,
A. F. Moraisc,
D. Mustafac,
T. Kamgaingb,
E. Njanjab,
M. Etterd,
I. K. Tonlé*b and
H. Terraschke*a
aInstitut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max Eyth-Str. 2, 24118 Kiel, Germany. E-mail: hterraschke@ac.uni-kiel.de
bResearch Unit of Noxious Chemistry and Environmental Engineering, Department of Chemistry, Faculty of Science University of Dschang, P.O. Box 67, Dschang, Cameroon. E-mail: ignas.tonle@univ-dschang.org
cInstituto de Física da Universidade de São Paulo, São Paulo 05508-090, Brazil
dDeutsches Elektronen-Synchrotron (DESY), Notkestr. 85, 22607 Hamburg, Germany
First published on 22nd November 2022
Abstract
Despite the importance of layered double hydroxides (LDHs) in catalysis, medicine and water treatment, the crystallisation process of these materials is seldom investigated. In this study, in situ characterisation techniques granted unprecedented experimental access to the formation dynamics of carbonate-intercalated Mg2+/Al3+ LDHs as model system when applying the most relevant co-precipitation approaches by exploring the effects of temperature and concentration of reactants. For this purpose, a combinatorial multi-modal characterisation approach was applied involving in situ measurements of pH, ion conductivity and light scattering, as well as synchrotron-based in situ X-ray diffraction (XRD). Shortly after beginning the addition of basic solutions (i.e., sodium carbonate and sodium hydroxide) to the solutions of magnesium nitrate hexahydrate and aluminium nitrate nonahydrate, a stable pH was reached due to the uptake of hydroxyl ions for nuclei formation. Shortly after, crystal growth phase was detected by an increase in the light scattering signal and confirmed via in situ XRD. Increasing the concentration of reactants accelerated the onset of crystal growth by 70% without significantly changing the crystallite size. On the other hand, increasing the temperature up to 65 °C showed a smaller influence on the reaction kinetics but resulted in a two-fold increase in crystallite size. Adding the solution of metal precursors to the basic solution, saturation was rapidly reached, without an induction period, favouring the formation of very small crystallites of approximately 10 nm.
1. Introduction
Layered double hydroxides (LDHs), also known as anionic clays or hydrotalcite-like compounds, are an important class of ionic lamellar solids.1 Structurally, LDHs consist of positively charged metal hydroxide sheets that are intercalated by anions and water molecules.2 The general chemical formula of these lamellar solids can be expressed as [M1−x2+Mx3+(OH)2]x+[(An−)x/n·yH2O] (abbreviated as M1−x2+Mx3+–A), where M2+/3+ are the divalent/trivalent metal cations within the brucite-like layers and An− are the charge-balancing anions.3 The identities of M2+, M3+ and An− can be varied over a wide range of possibilities, giving rise to a large class of multi-functional materials with potential applications in catalysis,4a drug delivery,4b environmental remediation,4c ion exchange,4d etc.A variety of methods, including co-precipitation,5a,b ion exchange5c and the reconstruction of LDHs from calcined precursors,5d have been reported for the synthesis of LDHs. Co-precipitation is the simplest, least expensive and most utilised technique. Briefly, an aqueous solution with the metal precursor salts and a basic solution with the interlayer anion to be incorporated into the LDH are used. Typically applied M2+/M3+ metals are Mg2+, Fe2+, Co2+, Cu2+, Ni2+, Zn2+, Al3+, Cr3+, Ga3+, In3+, Mn3+ and Fe3+.3a,6 Examples of interlayer anions are F−, Cl−, Br−, CO32−, NO3−, SO42−, MnO4−, V10O286− and Mo7O246−.7
Co-precipitation for LDH formation can be employed under high and low supersaturation conditions. Co-precipitation at low supersaturation conditions involves one of two approaches: (i) the basic solution containing the desired interlayer anion is slowly transferred into a vessel containing the solution with the metal precursors;8 (ii) the solution with the metal precursors and the basic solution are simultaneously transferred into the reaction vessel containing water. In the latter case, the basic solution is also used to regulate and maintain a desired pH.9 Co-precipitation at high supersaturation conditions involves the addition of the solution with the metal precursors into the basic solution.10
In situ characterisation techniques are important for gaining insights on the effects of crucial reaction parameters on the formation of a targeted product; and for obtaining real-time information on the processes involved in the nucleation and crystal growth of solid materials. This allows a better control over the crystallisation process and endowment of the desired structure-related properties of the synthesised materials.11 In situ characterisation techniques have been extensively applied for studying the formation of metal–organic frameworks,12 luminescent complexes,11a,b quantum dots,13 thiostanates,14 etc. Their application in the synthesis of LDHs is limited almost exclusively to their post-synthetic modifications. One example is the application of in situ diffuse reflectance infrared Fourier-transform spectroscopy for investigating the changes of the functional groups in LDH during the intercalation of organic molecules such as (1-[2S]-3-mercapto-2-methylpropionyl)-L-proline and 4-vinylbenzenesulfonate.15 Other important examples are time-resolved in situ energy-dispersive X-ray diffraction (XRD)16 and in situ high-temperature XRD studies for monitoring the post-synthetic structural evolution of LDHs during thermal treatments.17
In the present study, we investigated the formation of Mg0.75Al0.25–CO32− LDH by applying in situ measurements of pH, ion conductivity, light scattering and synchrotron-based XRD. This combinatorial multi-modal approach is important for acquiring fundamental insights into the crystallisation processes of the LDHs at low and high supersaturation regimes as well as understanding the effects of temperature and precursor concentration on LDH nucleation and growth. To the best of our knowledge, this is the first study to report an in situ investigation of the formation processes of LDHs during synthesis.
2. Experimental
2.1. Materials
Magnesium nitrate hexahydrate (Mg(NO3)2·6H2O, 99.999%), aluminium nitrate nonahydrate (Al(NO3)3·9H2O, 99.997%), sodium hydroxide (NaOH, 97%), sodium carbonate (Na2CO3, 99.99%), iron(II) sulphate heptahydrate (FeSO4·7H2O 99.0%) and sulphuric acid (H2SO4, 99.999%) were purchased from Sigma-Aldrich (Darmstadt, Germany). All the chemicals were used without further purification.2.2. Synthesis procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH ratio 1:10) were added to the reaction vessel containing the aqueous solution of Mg(NO3)2·6H2O and Al(NO3)3·9H2O. The concentrations, volumes, dosing rates and temperatures used for each experiment are summarised in Table 1. Following co-precipitation, the reaction solution was aged. This treatment was applied to samples S1X, Ag–S1x–3X and Ag–S40–65°C. Sample S1X was aged at room temperature (RT) for 24 h.18 The abbreviation “Ag” in some sample names indicates an alternative aging step performed at 65 °C for 1 h. The abbreviations “1×”, “2×” and “3×” represent the increases in the concentrations of the reactants, for instance, by a factor of 1, 2 and 3, respectively, while the superscripted values “40 °C” and “65 °C” indicate the additionally applied reaction temperatures.
:NaOH ratio 1:10) were added to the reaction vessel containing the aqueous solution of Mg(NO3)2·6H2O and Al(NO3)3·9H2O. The concentrations, volumes, dosing rates and temperatures used for each experiment are summarised in Table 1. Following co-precipitation, the reaction solution was aged. This treatment was applied to samples S1X, Ag–S1x–3X and Ag–S40–65°C. Sample S1X was aged at room temperature (RT) for 24 h.18 The abbreviation “Ag” in some sample names indicates an alternative aging step performed at 65 °C for 1 h. The abbreviations “1×”, “2×” and “3×” represent the increases in the concentrations of the reactants, for instance, by a factor of 1, 2 and 3, respectively, while the superscripted values “40 °C” and “65 °C” indicate the additionally applied reaction temperatures.
| Sample ID | S1X | Ag–S1X | Ag–S2X | Ag–S3X | Ag–S40°C | Ag–S65°C | SB | SC |
|---|---|---|---|---|---|---|---|---|
| Approach | A | A | A | A | A | A | B | C |
| Measurement set-up | I | II | I | I | I | I | II | I |
| NaOH concentration (mol L−1) | 0.2 | 0.2 | 0.4 | 0.6 | 0.2 | 0.2 | 0.2 | 0.2 |
| Na2CO3 concentration (mol L−1) | 0.02 | 0.02 | 0.04 | 0.06 | 0.02 | 0.02 | 0.02 | 0.02 |
| Volume of basic solution (mL) | 15 | 15 | 15 | 15 | 15 | 15 | 36.3 | 25 |
| Volume of metal precursor solution (mL) | 30 | 30 | 30 | 30 | 30 | 30 | 50 | 25 |
| Mg(NO3)2·6H2O concentration (mol L−1) | 0.06 | 0.06 | 0.12 | 0.18 | 0.06 | 0.06 | 0.06 | 0.06 |
| Al(NO3)3·9H2O concentration (mol L−1) | 0.02 | 0.02 | 0.04 | 0.06 | 0.02 | 0.02 | 0.02 | 0.02 |
| Reaction temperature (°C) | 25 | 25 | 25 | 25 | 40 | 65 | 25 | 25 |
| Addition rate in (mL min−1) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Aging temperature (°C) | RT | 65 | 65 | 65 | 65 | 65 | RT | 65 |
| Aging time (h) | 24 | 1 | 1 | 1 | 1 | 1 | 24 | 1 |
2.3. Characterisation
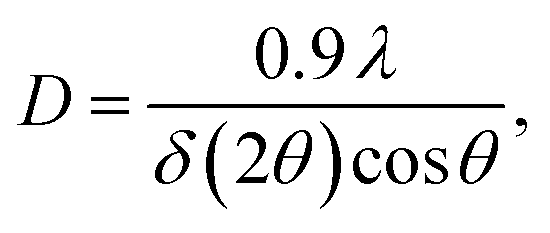 | (1) |
3. Results and discussion
3.1. Co-precipitation under low supersaturation conditions
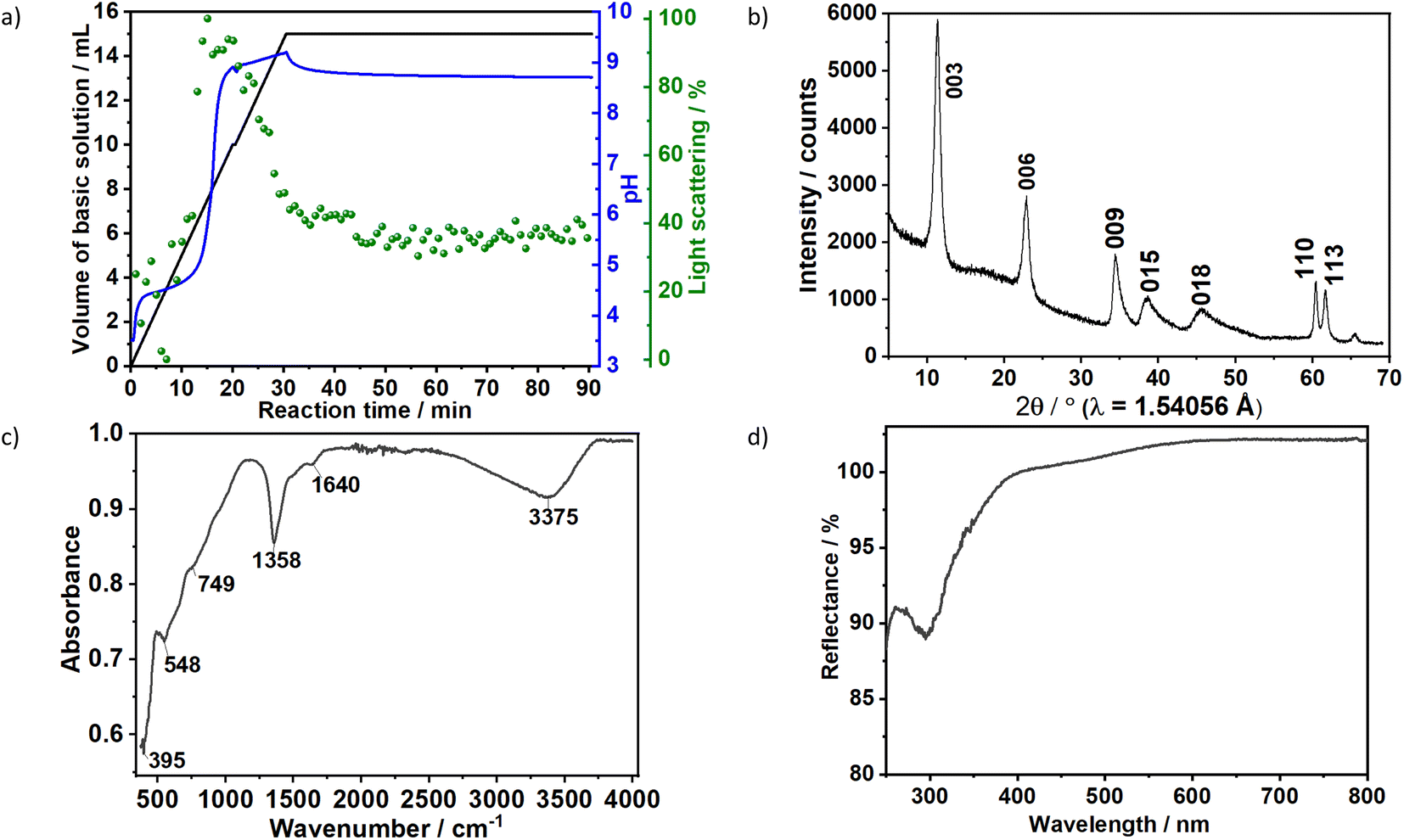 | ||
| Fig. 1 (a) Time dependence of the scattered light intensity at 365 nm (green), pH (blue) and dosed volume of the basic solution (black) during the synthesis of sample S1X. Ex situ (b) XRD pattern, (c) FTIR spectrum and (d) UV/vis reflection spectroscopy. | ||
The formation of carbonate-intercalated Mg0.75Al0.25–CO32− LDHs via Approach A was confirmed using ex situ powder X-ray diffraction (PXRD). Fig. 1b shows the PXRD pattern of sample S1X, which has been indexed based on a 3-layer hexagonal unit cell to facilitate comparison with LDHs in the literature.2a,b,26 At 11.47° 2θ, the Bragg reflection observed is related to the basal spacing c0 of sample S1X. A value of co = d(003) = 7.6 Å, typical of LDHs interspersed by carbonate ions,27 can be calculated using Bragg's law. The Bragg peak around 60.6° 2θ corresponded to the (110) reflection, whose interplanar spacing (d(110) = 1.53 Å) was used to calculate the intermetallic distance (aMM) within the hydroxide layers (aMM = 2d(110) = 3.06 Å).26 The full width at half maximum (FWHM) of the (003) Bragg reflection was δ(2θ) = 0.8°; therefore, the average crystallite size in the stacking direction was estimated to be 15.4 nm, corresponding to the stacking of 20 hydroxide layer slabs in average.
Characteristic vibrations on the IR spectrum of sample S1X (Fig. 1c) further indicated LDH formation. The strong broad absorbance band between 3000 and 3500 cm−1 corresponded to the stretching vibration of hydrogen-bonded hydroxyl groups from the hydroxide layers and interlayer water.27,28 The weak band observed at approximately 1640 cm−1 was attributed to the bending mode of H2O.29 IR absorptions at 749, 1358 and 548 cm−1 reflect the v2, v3 and v4 stretching vibrations of interlayer CO32− anions. In the low energy range, the band located at approximately 395 cm−1 was attributed to the vibrations of O–M2+/3+–O, Mg–OH and Al–OH in the hydroxide layers.30 No additional band was observed between 1390 and 1425 cm−1, indicating that NO3− ions from the metal precursors were successfully substituted by CO32− ions.30 Ultraviolet-Visible reflectance spectroscopy (Fig. 1d) showed that the formed LDH reflects nearly 100% of the light in the visible range. For wavelengths below 400 nm, the reflectance decreased, indicating an increase in optical absorbance in this area, with the highest absorbance observed at approximately 300 nm.
The very thin aspect of the layer slabs was confirmed by transmission electron microscopy (Fig. 2a), which revealed that sample S1X is composed by extremely thin flakes extended between approximately 30 nm and 100 nm. Energy-dispersive X-ray spectroscopy (Fig. 2b) confirmed the S1X composition containing the Mg/Al ratio of 3:1, besides C and O. Even though the sample morphology remained unchanged, the exposition to the electron beam caused the decomposition of the CO32− anions, indicated by the low C concentration as well as the partial conversion of S1X to MgO, shown by the electron diffraction patterns (Fig. 2b, inset).
 | ||
| Fig. 2 (a) TEM image, (b) EDX results, electron diffraction pattern (inset), (c) thermogravimetric and differential thermal analyses of Sample S1X. The asterisks (*) identify the Cu signal from the TEM grid. | ||
Thermal analyses revealed two main endothermic peaks in the intervals 150–250 °C and 250–450 °C, observed during the thermal decomposition of the sample S1X (Fig. 2c). The first peak occurs due to the loss of water molecules contained in the interlayer space. The peak at 400 °C contain a shoulder at 370 °C, attributed in the literature to the loss of hydroxyl groups bound to Al, while the main peak at 400 °C is generated by the loss of hydroxyl groups bound to Mg together with the removal of intercalated carbonate anions.31
To gain additional insight on the time evolution of LDH formation, in situ XRD and light scattering measurements were simultaneously performed during the synthesis of sample Ag–S1X (set-up II at DESY, Fig. 3a), synthesised under similar conditions as those used for obtaining sample S1X. Similar to S1X, the plateau observed for the green curve at t < 10 min suggests the formation of nuclei that are too small to scatter the 365 nm green light,25 which is in-line with the plateau observed during the in situ pH measurements (Fig. 1a). At approximately t = 10 min, 4 mL of the basic solution have been dosed into the metal precursor solution, the synthesis solution became heterogeneous and a steep increase in the scattered light intensity is observed. During the synchrotron-based in situ XRD measurements, the background of the small-angle range (0.15° 2θ) started to increase. This result was assigned to the formation low-crystallinity materials, such as amorphous compounds or nanoparticles,32 in accordance with the increase of light scattering in this reaction period. The low crystallinity of the product was also indicated by the broad Bragg reflections of the synthesised LDHs in the XRD pattern in Fig. 1b. At approximately t = 20–25 min, both a steep increase in the small-angle signal (Fig. 3b) and drastic decrease in the scattered light intensity (Fig. 3a) were observed, indicating the further growth of the LDH crystallites. The decrease in light scattering after approximately t = 20 min can be explained by the increase in turbidity caused by the crystal growth, until reaching saturation at approximately t = 34 min. The increase in temperature to 65 °C at approximately t = 42 min did not significantly affect product formation as suggested by the in situ XRD measurements.
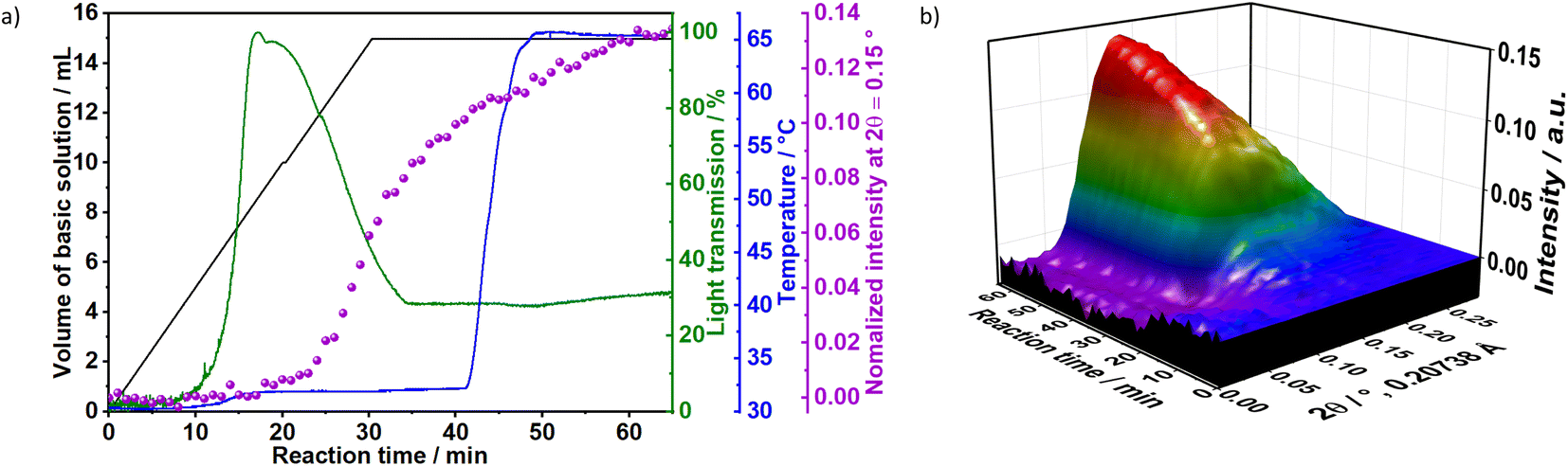 | ||
| Fig. 3 (a) In situ measurements of light scattering at 375 nm (green), temperature (blue), normalised XRD intensity at 2θ = 0.15° (purple) and volume of the basic solution dosed into the reaction vessel (black). (b) In situ XRD during the synthesis of sample Ag–S1X (synchrotron radiation, λ = 0.20738 Å, 60 keV). | ||
3.1.1.1 Influence of the concentrations of reactants. To evaluate the influence of the concentration of reactants on the crystallisation of LDHs, the results for samples Ag–S2X and Ag–S3X were analysed simultaneously (Fig. 4). These samples respectively correspond to a 2-fold and 3-fold increase in the concentration of the basic solution and metal precursor solution in comparison with sample Ag–S1X. The increase in concentration caused the initiation of the nucleation and growth processes earlier, as seen from the scattered light intensity, which started to increase over shorter time intervals for Ag–S2X and Ag–S3X in comparison with S1X and Ag–S1X. This feature is explained by the faster achievement of the critical saturation level when the reactants are dosed at higher concentrations. Moreover, the maximum scattered light intensity occurs at approximately 20, 12 and 6 min for S1X, Ag–S2X and Ag–S3X, respectively, inversely following the 1
:2:3 scaling of the reactant concentrations. Apart from the shorter raising time of the scattered light intensity curves, this inverse scaling is expected from a nucleation process that only starts after a particular critical supersaturation level is achieved.
 | ||
| Fig. 4 Time evolution of in situ light scattering at 365 nm, ion conductivity, pH, temperature (magenta) and dosed volume of the basic solution obtained during the synthesis of the samples Ag–S2X and Ag–S3X. Measurements of ion conductivity during the synthesis of Ag–S2X at t < 12 min and t > 95 min were saturated. | ||
After 90 min of reaction, the temperature was raised from room temperature to 65 °C. For Ag–S2X (Fig. 4), this led to a decrease in the turbidity of the suspension as indicated by the scattered light intensity curve, implying the initiation of Ostwald ripening via the dissolution of small particles in the suspension. In situ measurements of ion conductivity of the solution have confirmed the intense crystal growth at approximately t = 12 min. Before this period, the curve was strongly saturated, while the conductivity sharply decreased afterwards due to the uptake of ions from the solution for further growth of the LDH. In accordance with the in situ pH (Fig. 1 and 3) and in situ XRD (Fig. 3) measurements, the conductivity changes slightly after finishing the dosing process, indicating that the reaction has not reached completion.
Interestingly, samples Ag–S1X, Ag–S2X and Ag–S3X have very similar ex situ XRD patterns (Fig. S5a†), which provides an easy upscaling route via Approach A for which higher amounts of LDHs can be produced without drastically changing the crystalline properties of the product. Indeed, the FWHM of the (003) Bragg reflections of samples Ag–S1X, Ag–S2X and Ag–S3X were 1.67, 1.62 and 1.64° 2θ (λ = 1.54060 Å), corresponding to Scherrer crystallite sizes of 5.0, 5.2 and 5.1 nm, respectively.
3.1.1.2 Influence of reaction temperature. To evaluate the influence of the nucleation temperature (Fig. 5a) on the crystallisation of LDHs, the results for the in situ measurements of light scattering, ion conductivity and pH were plotted for samples S1X, Ag–S40°C and Ag–S65°C, which were synthesised at 25, 40 and 65 °C, respectively. In Fig. 5b, despite the high noise within the nucleation range (marked in grey), in situ light scattering measurements demonstrated in general the influence of the reaction temperature on LDH formation. For example, the decrease in light scattering caused by the strong turbidity resulting from LDH crystal growth started at approximately t = 20 min at 25 °C, t = 14 min at 40 °C and t = 11 min at 65 °C. These results agreed with the ones obtained during the in situ conductivity measurements. For the reaction at 25 °C (S1X), the ion conductivity decreased in two steps: The first step, until approximately t = 13 min, was assigned to the uptake of ions for the formation of LDH nuclei, which did not significantly influence light transmission. The second step (t ≈ 13–15 min) was faster and assigned to crystal growth, which caused the increase of the scattering signal because of the increased dispersion of light by the newly formed particles before causing the decrease on light scattering due to the increase on turbidity, as indicated by in situ XRD results (Fig. 3). Interestingly, Fig. 5c shows that after approximately t = 20 min, the conductivity did not change significantly, suggesting that the crystal growth occurring within this reaction period may take place in parallel with the dissolution of small particles in the suspension. Similar to the light scattering measurements, the temperature rise shortened the time in which the conductivity stopped decreasing from approximately t = 19 min at 25 °C, t = 15 min at 40 °C and t = 13 min for 65 °C. Fig. 5d shows that the pH also increases at different rates after approximately t = 19 min at 25 °C, t = 15 min at 40 °C and t = 13 min for 65 °C, confirming the influence of temperature on LDH formation.
 | ||
| Fig. 5 In situ measurements of (a) temperature, (b) light scattering, (c) ionic conductivity and (d) pH value during synthesis of the samples S1X (green), Ag–S40°C (blue) and Ag–S65°C (grey). The ionic conductivity values at 65 °C reached saturation for t < 12 min. The dosing rate of the basic to metallic solutions applied at this experiment was the same one as shown in Fig. 1–3. | ||
In line with the classical nucleation theory,33 a maximum nucleation rate is reached at a particular temperature (Tmax), above which growth is increasingly favoured over nucleation. This effect can be partially rationalised based on the large critical size of the nucleus and the expected increase in the solubility product of the solid phase with increasing temperature. The latter leads to a decrease in the supersaturation ratio. This drastically decreases the rate of nucleation when compared with the decrease in growth rate, leading to fewer nuclei and larger particles. The effect of this on the turbidity of the solution is immediate, as the scattered intensity is known to scale with the 6th-power of the particle size.34 Thus, the behaviour observed for the scattered light intensities obtained for S1X, Ag–S40°C and Ag–S65°C (Fig. 5b) can be explained by assuming that the reaction temperatures used for these samples were above Tmax. This assumption was further supported by recent observations by Tathod et al.,33 indicating a Tmax value that is lower than or approximately −15 °C.
The effect of an increased reaction temperature can also be seen in the ex situ XRD patterns of samples Ag–S1X, Ag–S40°C and Ag–S65°C (Fig. S6†). With the increase in reaction/aging temperature, sharper Bragg reflections were observed, indicating an increase in crystallite dimensions in the stacking direction and hydroxide plane. Indeed, along the stacking direction, the analysis of the (003) Bragg reflections for samples Ag–S1X, Ag–S40°C and Ag–S65°C gives FWHM values of 1.67, 1.27 and 0.74° 2θ (λ = 1.54060 Å), corresponding to Scherrer crystallite sizes of 5.0, 6.6 and 11.3 nm, respectively. This trend repeats in the hydroxide plane and Gaussian fittings for the (110) Bragg reflections, which gives FWHM values of 1.18, 0.89 and 0.62° 2θ for Ag–S1X, Ag–S40°C and Ag–S65°C, which correspond to crystallite sizes of 8.2, 10.9 and 15.5 nm, respectively.
 | ||
| Fig. 6 Time evolution of the (a) dosed volume of the metal precursor solution (blue) and dosed volume of the basic solution (black) as well as the in situ measurements of (b) pH and (c) light scattering during the synthesis of sample SB, with (d) ex situ XRD results obtained from aliquots removed during the aging step. | ||
3.2. Approach C: dosing the metal precursor solution into a concentrated NaOH solution
Fig. 7 shows the time evolution of pH, the intensity of the light scattered by the synthesis solution, ion conductivity and the dosed volume of the metal precursor solution during the formation and aging step at 65 °C of sample SC as well as the respective FTIR results. At a high starting pH of 14.5, critical supersaturation is reached very rapidly. Indeed, no induction period is observed from the light transmission results, with the reaction solution becoming turbid from the beginning of the dosing process, as indicated by the monotonic decrease of the scattered light intensity at t < 12 min. Approach C produces LDHs with the sharpest (110) Bragg reflections in comparison with those of the other prepared samples. With a FWHM of 0.6°, a crystallite dimension of approximately 16 nm in the hydroxide plane of the LDHs is obtained from the Scherrer equation. Also, a high crystallinity is attained along the stacking direction, with the (003) Bragg reflection (Fig. S8†) having a FWHM of 0.8°, corresponding to a Scherrer crystallite size of approximately 10 nm. The high crystallinity of SC is attributed to the combination of aging and a higher supersaturation rate (when compared with those of the other samples). As known from the classical nucleation theory,33,35 higher supersaturation rates favour nucleation over crystal growth, thereby forming smaller particles by the end of the dosing. However, in the aging step, these small crystals are more likely to be dissolved by the suspension. These redissolved cations can then be recrystallised at high temperatures at a lower supersaturation ratio, which would favour crystal growth.33 Under this mechanism, a suspension with a high fraction of small crystals would be easily converted to a suspension of highly crystalline particles during the aging step. This interpretation is further supported by the very sharp (110) and (113) Bragg reflections of sample SC, indicating high crystallinity in the ab-plane of the LDHs, which is expected to benefit most from Ostwald ripening due to the higher defect density in the edges of the ab-planes.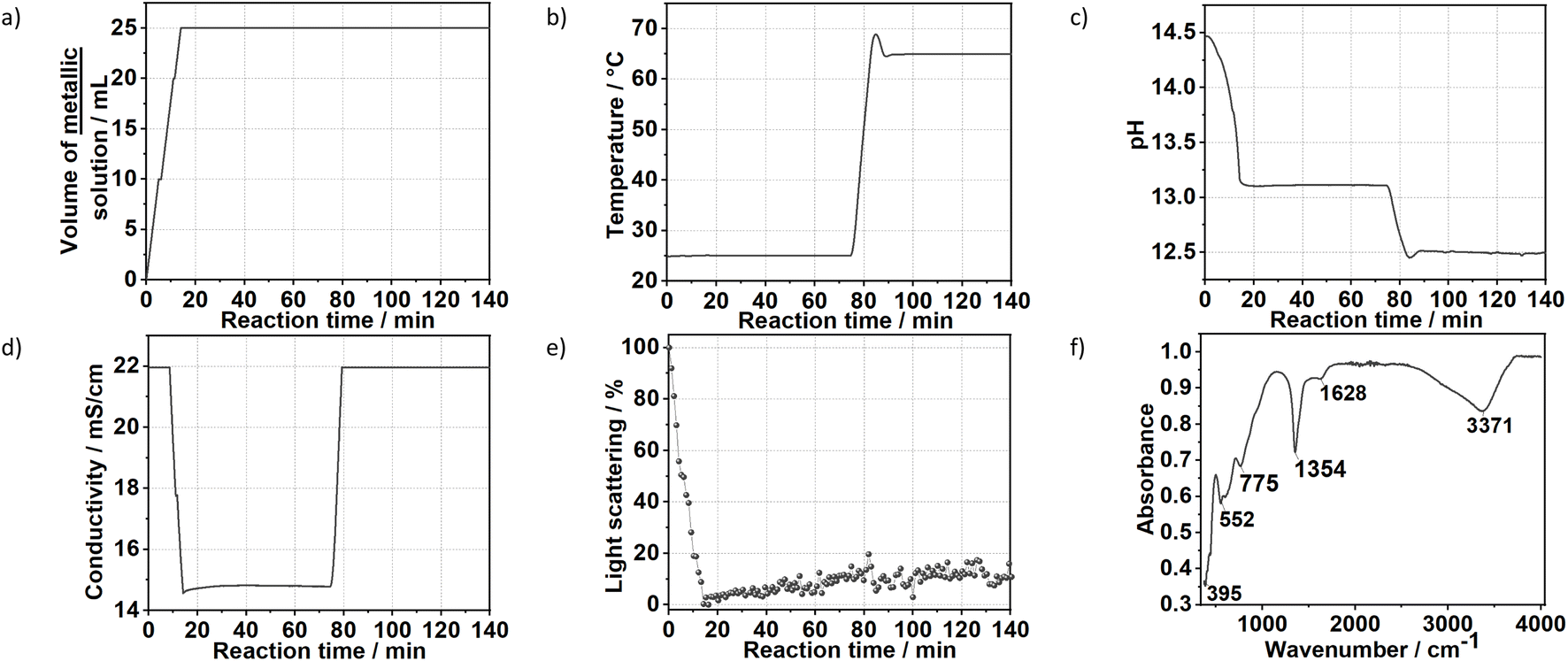 | ||
| Fig. 7 Time evolution of the (a) dosing of the metal precursors solution, (b) temperature, (c) pH value, (d) conductivity, (e) light scattering during the synthesis of the sample SC. (f) FTIR measurement of the final product. The ion conductivity measurements reached saturation at t < 10 min and at t > 85 min. | ||
4. Conclusion
The application of in situ characterisation techniques granted experimental access to the crystallisation dynamics of LDHs. The nucleation process and initiation of crystal growth were both monitored via in situ light scattering measurements. The increase in turbidity could be correlated to the rate of crystal growth in the suspension, and in a more direct way, the appearance of the first LDH particles could be followed using synchrotron-based in situ XRD.Here, three different co-precipitation procedures were investigated. When the basic solution was dosed into the metal precursor solution (approach A), crystal growth was shown to be kinetically favoured over nucleation as the reaction temperature increased, resulting in the formation of large LDH crystallites. This outcome was correlated to the rapid turbidity of the suspension at high temperatures as the scattered intensity scales with the 6th-power of particle size.
Under an increase in the reactant concentration, the initiation of the nucleation and growth processes was brought forward because of the faster achievement of the critical supersaturation level. Under the evaluated conditions of this study, this effect had no impact on the crystalline properties of the final LDH products as observed by ex situ PXRD.
The pH played a vital role in determining the crystallisation kinetics of LDHs. At high pH, here exemplified by approach C (pH between 12.5 and 14.5) in contrast to approach B (pH 10), a higher supersaturation level was achieved for similar dosing rates. Higher supersaturation increasingly favours nucleation over crystal growth, leading to the formation of smaller crystals. By aging at high temperatures, these crystals then underwent Ostwald ripening, being dissolved and recrystallised at lower supersaturation and leading to the formation of LDH particles with high crystallinity.
In summary, in situ characterisation techniques are powerful tools for analysing how parameters such as temperature, reactant concentration and pH influence the nucleation and growth of LDHs. Careful interpretation of the experimental results, considering the classical nucleation theory, can produce a more rational understanding of how to optimise the synthesis parameters of these materials towards the desired physicochemical properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Dr W. Bensch, Prof. Dr Tuczek and Prof. Dr N. Stock for providing the equipment necessary to perform these experiments. We also thank the German Research Foundation's (DFG) priority Program 1415, the German Academic Exchange Service (DAAD), the Alexander von Humboldt Foundation (Bonn, Germany) and the Coordination for the Improvement of Higher Education Personnel “CAPES” for their financial support. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III beamline P02.1 (proposal ID BAG-20170560). Furthermore, we would like to thank S. Senkale, T. Rabe, M. T. Wharmby and N. Heidenreich for their help during the execution of the synchrotron experiments as well as Dr U. Schürmann for the TEM images. D. M. and A. F. M. acknowledge Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants 2015/19210-0, 2018/13837-0 and 2022/01314-8) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, grant 88887.371434/2019-00) for financial support.References
- (a) D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef PubMed; (b) F. Zhang, L. Zhao, H. Chen, S. Xu, D. G. Evans and X. Duan, Angew. Chem., 2008, 120, 2500 CrossRef; (c) P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113 CrossRef CAS.
- (a) A. C. Teixeira, A. F. Morais, I. G. N. Silva, E. Breynaert and D. Mustafa, Crystals, 2019, 153, 1 Search PubMed; (b) A. F. Morais, F. O. Machado, A. C. Teixeira, I. G. N. Silva, E. Breynaert and D. Mustafa, J. Alloys Compd., 2019, 771, 578 CrossRef CAS; (c) C. Mousty, C. Forano, S. Fleutot and J. C. Dupin, Electroanalysis, 2009, 21, 399 CrossRef CAS; (d) B. Ballarin, A. Mignani, E. Scavetta, M. Giorgetti, D. Tonelli, E. Boanini, C. Mousty and V. Prevot, Langmuir, 2012, 28, 15065 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, Adv. Mater., 2017, 29, 1703828 CrossRef; (b) S. Radha, W. Milius, J. Breu and P. V. Kamath, Solid State Chem., 2013, 204, 362 CrossRef CAS; (c) J. Liu, Y. Li, X. Huang, G. Li and Z. Li, Adv. Funct. Mater., 2008, 18, 1448 CrossRef CAS.
- (a) C. G. Silva, Y. Bouizi, V. Fornés and H. García, J. Am. Chem. Soc., 2009, 131, 13833 CrossRef; (b) M. A. Rocha, P. A. D. Petersen, E. Teixeira-Neto, H. M. Petrilli, F. Leroux, C. Taviot-Gueho and V. R. L. Constantino, RSC Adv., 2016, 6, 16419 RSC; (c) A. Khenifi, Z. Derriche, C. Mousty, V. Prévot and C. Forano, Appl. Clay Sci., 2010, 47, 362 CrossRef CAS; (d) M. Everaert, R. Warrinnier, S. Baken, J. P. Gustafsson, D. D. Vos and E. Smolders, ACS Sustainable Chem. Eng., 2016, 4, 4280 CrossRef CAS.
- (a) L. Mohapatra and K. Parida, J. Mater. Chem. A, 2016, 4, 10744 RSC; (b) F. L. Theiss, G. A. Ayoko and R. L. Frost, Appl. Surf. Sci., 2016, 383, 200 CrossRef CAS; (c) Y. Wang, P. Wu, B. Li, N. Zhu and Z. Dang, Appl. Clay Sci., 2011, 53, 615 CrossRef CAS; (d) J. Rocha, D. M. Arco, V. Rives and M. A. Ulibarri, J. Mater. Chem., 1999, 9, 2499 RSC.
- G. Prestopino, G. Arrabito, A. Generosi, A. Mattoccia, B. Paci, G. Perez, G. Verona-Rinati and P. G. Medaglia, Sci. Rep., 2019, 19, 11498 CrossRef.
- (a) Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124 CrossRef CAS; (b) J. Yu, Q. Wang, D. O'Hare and L. Sun, Chem. Soc. Rev., 2017, 46, 5950 RSC.
- (a) H. Yin, L. Cui, S. Ai, H. Fan and L. Zhu, Electrochim. Acta, 2010, 55, 603 CrossRef CAS; (b) B.-K. Kim, G.-H. Gwak, T. Okada and J.-M. Oh, J. Solid State Chem., 2018, 263, 60 CrossRef CAS.
- (a) M. Sun, J. Su, S. Liu, D. Wang, W. Yan, L. Zhang, Y. Xiao and X. Gao, RSC Adv., 2015, 5, 80978 RSC; (b) Y. Xiao, M. Sun, L. Zhang, X. Gao, J. Su and H. Zhu, RSC Adv., 2015, 5, 28369 RSC.
- (a) M. Yang, O. McDermott, J. C. Buffet and D. O'Hare, RSC Adv., 2014, 4, 51676 RSC; (b) C. Nyambo, P. Songtipya, E. Manias, J. M. M. Gasco and C. A. Wilkie, J. Mater. Chem., 2008, 18, 4827 RSC.
- (a) L. Ruiz Arana, P. Lindenberg, H. Said, M. Radke, N. Heidenreich, C. S. Cunha, S. Leubner and H. Terraschke, RSC Adv., 2017, 7, 52794 RSC; (b) P. Polzin, I. V. Eliani, J. Ströh, M. Braun, N. Ruser, N. Heidenreich, P. Rönfeldt, F. Bertram, C. Näther, S. Wöhlbrandt, M. Suta and H. Terraschke, Phys. Chem. Chem. Phys., 2018, 20, 7428 RSC; (c) N. Pienack, P. Lindenberg, G. Doungmo, N. Heidenreich, F. Bertram, M. Etter, M. T. Wharmby and H. Terraschke, Z. Anorg. Allg. Chem., 2018, 644, 1902 CrossRef CAS; (d) P. Lindenberg, L. Ruiz Arana, L. K. Mahnke, P. Rönfeldt, N. Heidenreich, G. Doungmo, N. Guignot, R. Bean, H. N. Chapman, D. Dierksmeyer, J. Knoska, M. Kuhn, J. Garrevoet, V. Mariani, D. Oberthuer, K. Pande, S. Stern, A. Tolstikova, T. A. White, K. R. Beyerlein and H. Terraschke, React. Chem. Eng., 2019, 4, 1757 RSC; (e) H. Terraschke, M. Rothe and P. Lindenberg, Rev. Anal. Chem., 2018, 37, 20170003 CAS.
- (a) P. Rönfeldt, H. Reinsch, M. P. M. Poschmann, H. Terraschke and N. Stock, Cryst. Growth Des., 2020, 20, 4686 CrossRef; (b) P. Rönfeldt, E. S. Grape, A. K. Inge, D. V. Novikov, A. Khadiev, M. Etter, T. Rabe, J. Benecke, H. Terraschke and N. Stock, Inorg. Chem., 2020, 59, 8995 CrossRef PubMed.
- (a) C. Palencia, R. Seher, J. Krohn, F. Thiel, F. Lehmkühler and H. Weller, Nanoscale, 2020, 12, 22928 RSC; (b) H. Huang, M. W. Feil, S. Fuchs, T. Debnath, A. F. Richter, Y. Tong, L. Wu, Y. Wang, M. Döblinger and B. Nickel, Chem. Mater., 2020, 32, 8877 CrossRef CAS.
- (a) N. Pienack, C. Nather and W. Bensch, Eur. J. Inorg. Chem., 2009, 7, 937 CrossRef; (b) B. Seidlhofer, N. Pienack and W. Bensch, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 937 CrossRef CAS; (c) B. Seidlhofer, E. Antonova, J. Wang, D. Schinkel and W. Bensch, Z. Anorg. Allg. Chem., 2012, 638, 2555 CrossRef CAS; (d) N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014 CrossRef CAS PubMed.
- (a) H. Zhang, S. H. Guo, K. Zou and X. Duan, Mater. Res. Bull., 2009, 44, 1062 CrossRef CAS; (b) D. Yan, J. Lu, M. Wei, H. Li, J. Ma, F. Li, D. G. Evans and X. Duan, J. Phys. Chem. A, 2008, 112, 7671 CrossRef CAS.
- (a) F. Millange, R. I. Walton and D. J. O'Hare, Mater. Chem., 2000, 10, 1713 RSC; (b) G. R. Williams and D. O'Hare, J. Mater. Chem., 2006, 16, 3065 RSC.
- L. H. Zhang, F. Li, D. G. Evans and X. Duan, J. Mater. Sci., 2010, 45, 3741 CrossRef CAS.
- P. Vicente, M. Perez-Bernal, R. J. Ruano-Casero, D. Ananias, P. F. A. Almeida, J. Rocha and V. Rives, Microporous Mesoporous Mater., 2016, 226, 209 CrossRef CAS.
- E. Wiberg and A. F. Holleman, Inorganic Chemistry, Walter de Gruyter Press, 2001, 101st edn, ISBN 0-12-352651-5 Search PubMed.
- (a) J. R. Guzman-Sepulveda, J. Deng, J. Y. Fang and A. Dogariu, Soft Matter, 2016, 12, 5986 RSC; (b) Y. Xing, U. O. Koylu and D. E. Rosner, Appl. Opt., 1999, 12, 2686 CrossRef.
- A. C. Dippel, H. P. Liermann, J. T. Delitz, P. Walter, H. S. Schrepping, O. H. Seeck and H. Franz, J. Synchrotron Radiat., 2015, 22, 675 CrossRef CAS.
- (a) J. Ströh, L. Ruiz Arana, P. Polzin, I. V. Eliani, P. Lindenberg, N. Heidenreich, C. S. Cunha, S. Leubner and H. Terraschke, Z. Anorg. Allg. Chem., 2019, 645, 537 CrossRef; (b) H. Terraschke, M. Rothe, A. M. Tsirigoni, P. Lindenberg, A. L. Ruiz, N. Heidenreich, F. Bertram and M. Etter, Inorg. Chem. Front., 2017, 4, 1157 RSC.
- A. E. Platero-Prats, Z. Li, L. C. Gallington, A. W. Peters, J. T. Hupp, O. K. Farha and K. W. Chapman, Faraday Discuss., 2017, 201, 337 RSC.
- L. Ruiz Arana, J. Ströh, J. Amtsfeld, G. Doungmo, D. Novikov, A. Khadiev, M. Etter, M. Wharmby, M. Suta and H. Terraschke, Z. Naturforsch., B: J. Chem. Sci., 2022, 77, 263 CrossRef CAS.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362 CrossRef CAS PubMed.
- X. Duan, D. Evans, J. He, Y. Kang, A. I. Khan, F. Leroux, B. Li, F. Li, D. O'Hare, R. C. T. Slade, C. Taviot-Gueho, M. Wei and G. Williams, Struct. Bonding, 2006, 119, 1 CrossRef.
- M. J. D. Reis, F. Silvério, J. Tronto and J. B. J. Valim, Phys. Chem. Solids, 2004, 65, 487 CrossRef.
- M. R. Othman, N. M. Rasid and W. J. N. Fernando, Chem. Eng. Sci., 2006, 61, 1555 CrossRef CAS.
- M. Bouraada, M. Lafjah, M. S. Ouali and L. C. D. Menorval, J. Hazard. Mater., 2008, 153, 911 CrossRef CAS.
- (a) H. Yin, L. Cui, S. Ai, H. Fan and L. Zhu, Electrochim. Acta, 2010, 55, 603 CrossRef CAS; (b) M. J. H. Moreno, M. A. Ulibarri, J. L. Rendon and C. J. Serna, Phys. Chem. Miner., 1985, 12, 34 Search PubMed.
- C. Forano, T. Hibino, F. Leroux, and C. Taviot-Guého, Handbook of Clay Science, 2006, pp. 1021–1095 Search PubMed.
- M. Wendt, L. K. Mahnke, N. Heidenreich and W. Bensch, Eur. J. Inorg. Chem., 2016, 34, 5393 CrossRef.
- A. P. Tathod and O. M. Gazit, Cryst. Growth Des., 2016, 16, 6709 CrossRef CAS.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409 CrossRef CAS.
- (a) P. Cruz, F. Rocha and A. Ferreira, CrystEngComm, 2016, 18, 9113 RSC; (b) J. MCGinty, N. Yazdanpanah, C. Price, J. H. T. Horst, and J. Sefcik, Handbook of Continuous Crystallization, RSC Publishing, 2020, pp. 1–53, ISBN 978-1-78801-214-0 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental setup, ex situ FITR and XRD results. See DOI: https://doi.org/10.1039/d2ra05269e |
| This journal is © The Royal Society of Chemistry 2022 |