
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into nanocrystal synthesis: from precursor decomposition to combustion
Buzuayehu Abebe
 *,
Dereje Tsegaye
and
H. C. Ananda Murthy
*
*,
Dereje Tsegaye
and
H. C. Ananda Murthy
*
Adama Science and Technology University, Department of Applied Chemistry, 1888, Adama, Ethiopia. E-mail: buzea8@gmail.com; anandkps350@gmail.com
First published on 30th August 2022
Abstract
Nanotechnology-based synthesis of nanoscale materials has appealed to the attention of scientists in the modern scientific community. In the bottom-up approach, atoms start to aggregate/agglomerate and form nuclei within the minimum and maximum supersaturation range. Once nuclei are generated above the critical-free energy/radius, the growth is initiated by obeying the LaMar model with a slight extra simple growth by diffusion advancement. The in situ real-time liquid phase analysis using STEM, AFM, and XAS techniques is used to control precursor decomposition to the nanocrystal formation process and should be a non-stoppable technique. Solution combustion synthesis (SCS) is a time-/energy-efficient self-sustained process that produces mass-/ion transport active porous materials. SCS also permits the synthesis of evenly distributed-doped and hybrid-nanomaterials, which are beneficial in tuning crucial properties of the materials. The growth and development of nanocrystals, dehydrating the sol in the presence of a surfactant or/and fuel results in combustion once it arrives at the ignition temperature. Besides, the kinetic and thermodynamics controlled architecture-directing agent-assisted SCS offers colloidal nanocrystal framework formation, which is currently highly applicable for energy devices. This short review provides insightful information that adds to the existing nanocrystal synthesis process and solution combustion synthesis and recommends future directions in the field.
 Buzuayehu Abebe | Dr. Buzuayehu Abebe is currently doing his research work on nanomaterials, specifically, nanocomposite materials (heterojunction/doping) for photocatalysis and antimicrobial activity. He published more than 27 research and review articles in the SCI/Scopus/PubMed-indexed journals. He also presented many papers at national and international conferences. Buzuayehu is working as an academic editor, review editor, and reviewer in the world's most prominent and highest-cited publishers. He has successfully completed a 1 projects certified by Adama science and Technology University, Ethiopia. He is currently supervising 2 M.Sc. students. |
 Dereje Tsegaye | Dr. Dereje Tsegaye has served in industry, academics and research for the last 30 years. He is currently working as Assistant Professor, Department of Applied Chemistry, Adama Science and Technology University, Adama, Ethiopia, East Africa. Dr Dereje has published 5 research articles in national and international journals and presented some papers in national and international conferences. He has taught various chemistry courses to UG, PG, and PhD students in various Ethiopian universities and supervised more than 20 M.Sc. and 1 PhD students. He is currently supervising 4 M.Sc. and 2 PhD students. His research is mainly focused on the synthesis and application of environment friendly nanomaterials. |
 H. C. Ananda Murthy | Dr. H. C. Ananda Murthy has been a sincere, committed and dedicated faculty member at various prestigious universities in India, Tanzania and Ethiopia for the last 24 years. He is currently working as Associate Professor, Department of Applied Chemistry, Adama Science and Technology University, Adama, Ethiopia, East Africa. Prof. Ananda has authored number of books, compendia, book chapters and published more than 100 research articles in the journals of international repute and presented many papers in national as well as international conferences. He has been a guest editor for journal of nanomaterials and journal of renewable materials. He is also a review editor of Frontiers in Catalysis journal and Editorial board member of Annals of Applied Science journal. He has 4 patents to his credit. He has delivered many invited talks at various platforms. He supervised 7 M.Sc. and 1 PhD students. He is currently guiding 2 M.Sc. and 7 PhD students (1 awarded). He has successfully completed a 2 projects sanctioned by Adama science and Technology University, Ethiopia and National Innovation Foundation of India (NIFI). He is currently associated with research projects related to green synthesis of metal and metal oxide nanoparticles/nanocomposites for multifunctional applications. |
1. Introduction
Nanotechnology-based nanomaterials (NMs) synthesis has attracted the attention of researchers in the modern scientific world compared to that of bulk materials.1 Improvement in the synthesis approach, along with its crucial outlooks, is the current worldwide consideration, ordinarily, for industrially-scalable applications. Understanding the process from precursor decomposition to nanocrystal formation using advanced instruments allows for controlling the materials' size, morphology, and crystallinity. After passing the sequence of precursor decomposition-reduction, nucleation due to supersaturation, and growth by an atomic diffusion process, the nanocrystal formation still follows the LaMer model,2 with further support for the interpretation of the characteristics of the growth process by Reiss3 and Matijevic.4The classical (CNT) and non-classical nucleation theory (NCT) gives the overall nucleation process. As described by the CNT theory, the precursors decomposed non-spontaneously to give a supersaturated saturation. Within the minimum and maximum supersaturation points, nucleation starts either homogenously, in which nucleation occurs due to self-aggregation of reduced atoms, or heterogeneously in which nucleation occurs on some surface.5–7 The total free energy (ΔG) of nucleation and growth processes is the sum of surface- and bulk-free energy. Nucleation is a non-spontaneous aggregation process, and growth is a spontaneous process. At the critical free energy (ΔG*)/critical radius (Rc) point, the nuclei formation ceases, and the growth starts either via diffusion of atoms towards the nuclei (classical growth theory) or via an oriented attachment (non-classical growth theory).8 Besides, the nucleation and growth process is also affected by different factors such as temperature, surface heterogeneity, and wettability (affinities of the nuclei towards the surface).9 Nowadays, based on the in situ real-time liquid phase analysis, the process of precursor decomposition to the nanocrystal formation can be controlled by advanced instruments such as scanning transmission electron microscopy (STEM) with its direct electron detectors,10–12 atomic force microscopy (AFM),13–15 and X-ray absorption spectroscopy (XAS).16,17
Solution combustion synthesis (SCS) is an effortless, time-/energy-efficient method and creates regularly ordered porous materials18 that have significance in the ion-/mass-transport phenomenon.19 The stoichiometry balance between the oxidizer/precursor and reducer/fuel is calculated based on the propellant chemistry.20 Colloidal solution (sol) formations from precursors decomposition, dehydration of the sol to form a gel by heating, combustion at the ignition temperature, and quenching of the process through the evolution of gasses are the crucial steps that occur in the SCS.21 The self-generated energy/temperature, once the ignition temperature is reached, may go up to 4000 °C.22 Herein, metal oxide reduction to metal may also occur through interaction with reducing gases formed during combustion in the presence of oxygen.23 The SCS approach favors a low-cost instrument, time-/energy-efficient, and creates a well-defined porosity during gas evolution.24 The porosity assists in the interfacial mass-/ion transport, which is crucial for several applications such as energy storage, sensing, and catalysis.25,26 It also reduces the time of purification and the post-synthesis treatment process.27,28 Of course, different factors such as solution pH18,29 that has great use in complex formation, precursor and stabilizing agent nature,30,31 and temperature32 has beneficial effects on the final products. Besides, the approach also comprises a decent dopant–host distribution or heterojunction via Pearson's hard/soft acids/bases (HSAB) theory. The doping/heterojunction can tune the materials' optical, magnetic, and electrical properties, thus, boosting materials application through charge transfer or/and synergistic roles.33,34
Nanocrystal has higher surface energy than the bulk equivalent that supports its aggregation. Surfactants can be used as NPs stabilizers and structure-directing agents.10,11 Since the nanocrystal size is small, it has a high surface free energy; during capping/stabilization, the surface density increases and prevents aggregation. Besides, different facets have different interactions/reactivity with different surfactants, which assists in tuning the geometry of the materials.35,36 The architecture-directing agents (ADAs) assisted SCS can also produce ordered colloidal nanocrystal frameworks (CNFs), which are functional in mass-/ion-transport in energy-converting devices.37,38 This paper gives detailed information and recommends future outlooks on the fundamental concepts of precursor decomposition to nanocrystal formation, instruments to follow crystal growth, solution combustion synthesis in the presence of surfactants, and ADA-assisted SCS to produce ordered colloidal nanocrystal frameworks.
2. Fundamentals in the bottom-up synthesis approach
Owing to the crucial properties of the nanoscale size materials compared to the bulk counterparts, it has drawn interest in several fields. By understanding the precursor decomposition, nucleation, and growth process, under critical exterior and interior conditions, the synthesis of nanocrystals based on the bottom-up approach with precise morphology, size, and structure is the current progress. Before forming the nanocrystals, controlling the nucleation and growth developments processes are the basic and significant processes. LaMer's group theory2 is the first model that gave a clue for precursor decomposition and reduction, nucleation, development into seeds, and finally growth into nanocrystals in solution. The nanocrystals' overall shape and size is dependent on the growth of the seeds and surfactant reactivity with different facets of the nanocrystals. Depending on the different conditions, the surfactants such as poly(vinyl alcohol) (PVA) and poly(vinyl pyrrolidone) (PVP) are usually used as reducing agents.40,41According to LaMer's model (Fig. 1), first, the precursor decomposes and elevates the concentration of its ions, which are then reduced into atoms in the solution up to a supersaturation point (step I). During the precursor decomposition, first, it is converted into either monomeric or/and larger units by forming complexes with anions, solvents, and surfactant molecules.41,42 The nucleation, tiny clusters formation, starts either via random self-aggregation of a few atoms in solution to relieve the supersaturation (homogenous nucleation) or on-site/surface of impurities (heterogeneous nucleation) (step II).43 The source for the impurities is mostly from the precursor used. The homogenous nucleation occurs only between the minimum and maximum supersaturation ranges due to the stochastic instabilities of monomer connection.44 Once the burst nucleation starts, the supersaturation decreases due to the rapid formation of nuclei compared to the precursor decomposition. Below the minimum supersaturation points, the self-aggregation/nuclei creation process is reduced, and growth starts via atomic addition (by diffusion) (step III). The higher the supersaturation is, the lower the free energy (energy required to stabilize nuclei) and the higher the chance of forming smaller nuclei.45
 | ||
| Fig. 1 The LaMer model of nucleation and growth: the diagram show the generation of atoms, nucleation, and subsequent growth of colloidal systems. Reproduced/Adapted from ref. 39 with permission from The Royal Society of Chemistry. | ||
Beneath the minimum supersaturation point, the birth of the seeds (such as single, twinned, or multiply twinned structures) starts. These twinned or multiply twined types of seeds then converted to a single crystal based on oxidative etching, which means the re-oxidation of reduced atoms into ions in the presence of oxygen and a capping agent. The twinned, stacking faults, and boundary defect accompanied structures have a greater oxidative surface environment, which is highly affected by the oxidative etching than the single crystal. The kinetic and thermodynamic factors are the critical conditions for controlling seed formation. In the case of thermodynamically controlled reaction, the total interfacial free energy, the energy needed for producing a unit area of a “new” surface, is reduced in the given volume, and a more stable product is formed. By controlling various thermodynamic and kinetics parameters, developing nanocrystals into specific shapes is possible.46 Temperature is the most common parameter for synthesizing materials either thermodynamically or kinetically, in which thermodynamic reaction occurs by increasing the temperature and kinetics reaction occurs by decreasing the temperature. According to the Arrhenius equation, the reaction temperature has a direct relationship with the activation energy barrier; the higher the temperature, the lower the activation energy barrier for the reaction to pass and form a product. A thermodynamic reaction occurs at a minimum Gibbs free energy and cares only about the final state, while a kinetically controlled reaction occurs on all other free energy values except that on the global minimum value and cares all the process. The thermodynamically controlled synthesized material is the most stable than the kinetically controlled reaction. Because in a thermodynamically controlled reaction, the atoms reach their final destination due to the availability of enough time to move and attain minimum energy. During the precursor's decomposition process, the atoms diffuse and need to settle in the site where lower free energy position. However, the reduced atoms can be adsorbed in different thermodynamically less stable locations, in which the reaction leads to under kinetics control.47 To indicate, the {111} facet of the fcc structure (that has either tetrahedral or octahedral structure) highly reduces the total energy of the system and has a greater surface area compared to the {100} and {110} facets. However, if the decomposition or/and reduction speed is slow, the kinetics controlled reaction results, so as the domination of the twinned, or multiply twined and stacking faults structure in contrary to the thermodynamically controlled paths.48
A combination of classical nucleation theory (CNT) that describes the nucleus formation quantitatively and non-classical theory (NCNT) that gives the intermediate progression can fully explain the nucleation process (Fig. 2(a) and (b)). According to the CNT, which assumes the nucleus as a sphere, the total free energy (ΔG) of nucleation is equal to the sum of surface- and bulk-free energy. At the point of maximum free energy, also known as the point of nucleation energy barrier (ΔG*), the nuclei start to grow and form nanocrystal. Even though the CNT gives information about the critical radius/critical free energy and nucleation rate, it delivers no facts about the developments of random metastable intermediate phase type nucleation processes (atoms/molecules nucleation occurs in other ways than the proposed nucleation processes by the classical nucleation theory). Besides, the CNT only gives information about the aggregate, which contains approximately greater than hundred molecules.49 Thus, the NCNT theory can be used as an alternative way to show the multiple intermediate metastable prenucleation stages both computationally (such as modelling using molecular dynamics) and experimentally (such as titration) before crystal nuclei formation.5–7 The seed to nanocrystal growth is under the control of both surface free energy (ΔG) (free energy present between large-size particles and the solute in solution, which is positive) and bulk-free energy (free energy present between surface particle and the bulk particle, which is negative). The decrease in surface-free energy and bulk-free energy favours the cluster growth process. A cluster with a smaller radius R than critical radius Rc has less thermodynamic stability and favours dissolution, and clusters with greater thermodynamic stability (greater R values than Rc value) can persevere in solution and grow to nanocrystal. Thus, at the point of critical radius (Rc) and binding free energy (maximum free energy) (ΔGc), stable nuclei formed, which promotes to the nanocrystal growth (Fig. 2(c)).9,43 The nucleation can also be affected by other factors such as surface free energy, temperature, and substrate wettability, in which nucleation is facilitated more with increasing the temperature and supersaturation and with lower surface-free energy of materials.9
 | ||
| Fig. 2 Classical nucleation theory. (a) Energy landscapes for classical and non-classical nucleation. (b) Classical and non-classical nucleation pathways.5 (c) Nucleation barrier; Nuclei have to reach a critical size before they become thermodynamically stable. (d) Heterogeneous nucleation on a flat substrate; nucleation preferentially occurs on substrates due to the reduced nucleation barrier. (e) Heterogeneous nucleation on a curved substrate. Reproduced/Adapted from ref. 50 with permission from The Royal Society of Chemistry. | ||
In the case of the wettability of the substrate, the angle of contact (θ) between the embryo/nuclei and substrate is considered. The higher the affinities of the nuclei towards the substrate (θ < 180°), the lower the interface free energy and the higher probabilities of the nucleation formation process (see Fig. 2(d)). While, if θ > 180°, the affinities of the nuclei towards the substrate decrease (that increases the free energy) and results in the dissolution of the nuclei (see Fig. 2(e)). If θ = 180°, the binding free energy for heterogeneous and homogenous nucleation becomes equivalent.
Thanks to technological advancements, the in situ STEM analysis shows all the processes from the precursor's decomposition to the nanocrystal formation. Recently, the in situ liquid cell STEM nucleation and growth analysis of Pt nanocrystal by Dachraoui et al.8 showed an increase in the number and size of particles and then a sudden decrease in the number of particles as the size increases (at t ∼180 s). In this in situ liquid cell STEM analysis, the reduction of platinum precursor (Na2PtCl4·2H2O), aggregation of the atoms to cluster (at 18 s), diffusion of atoms to the preformed cluster to form amorphous seed crystal (up to 33 s), and finally, 1 nm crystalline Pt with (200) facet is captured from the movie of in situ real-time liquid phase reaction process of STEM instrument. Then, the nanocrystal that has comparable sizes start to come close to each other (at 55 s) and grow through an oriented attachment (OA), and finally totally merge to form one single nanocrystal (at 85 s) as shown in Fig. 3(a). Fig. 3(b) is the false colour supported OA process.
 | ||
| Fig. 3 Oriented attachment via nanobridge-induced contact. (a) Atomic resolution ADF-STEM time series show the OA process of two small Pt nanocrystals, indicating the approach of nanocrystals and pre-alignment by rotation, followed by nano bridge formation (marked by the black arrow in the inset at 85 s) and subsequent fusion of the nanocrystals. (b) Schematic illustration of the nano bridge-induced OA process. Images in the insets are shown in false colour. Reproduced/Adapted from ref. 8 with permission from The Springer Nature. | ||
During the reaction process, particles rotate randomly when the separation distance is greater than 1.5 nm, and then the rotation is aligned to form {111} facet by an OA at a lesser distance.8 Similar citrate stabilized OA nanocrystal growth process between gold nanocrystals (from HAuCl4 precursor) was studied by Zhu et al. using the in situ liquid cell TEM investigation. Herein, the rotational alignment of the nanocrystals was guided by the citrate ligand to share a common {111} facet orientation. The citrate ligand acts the opposite activity, facilitating the aggregation/coalescence instated of stabilizing it. These opposite activities indicate that surfactants' activities for different facets are different.51 In addition to the OA, a monomer (single atom) attachment on the nanocrystals with ≥1 nm size known as jump-to-coalescence/monomer attachment also occurs, as shown in Fig. 4(a). According to the first-principles of molecular dynamics models, the precursor's compound (with higher reduction potential) may also form nuclei and grow to the nanocrystals without reduction into atoms.9,50 The jump-to-coalescence process of nanoparticles occurs mainly due to the reduction of surface free energy. It assists the formation of larger particle sizes within a short time compared to the atom-by-atom growth process, which decreases the active sites of the surface.52 The jump-to-coalescence process of gold nanoparticle growth had been examined by Neng et al. based on the in situ AC-HRTEM analysis. In this study, they also proposed new crystal growth mechanisms based on the atoms present within the nanoparticles acting as a catalyst at some distance.52
 | ||
| Fig. 4 Nanocrystal growth by monomer attachment and Ostwald ripening. (a) ADF-STEM time-series showed a particle growing by monomer attachment, where visible attachment of free Pt atoms (white arrows) forms new atomic layers on top of the old ones (red arrows). (b) ADF-STEM time-series showed a particle growing by Ostwald ripening, where single atoms and clusters migrate through a liquid from the smaller particle (top) to the larger particle (bottom). At the bottom is a schematic illustration of the Ostwald ripening process. The yellow arrows highlight the single atoms and clusters detached from the smaller particle. Images are shown in false colour. Reproduced/Adapted from ref. 8 with permission from The Springer Nature. | ||
Fig. 4(b) shows the ADF-STEM time-series controlled particle growth by Ostwald ripening (thermodynamically-driven spontaneous deposition of tiny crystals onto more energetically favored larger crystals), and at the bottom, the process is shown in false color.
Ou et al. also used the liquid-phase TEM analysis to control the transition of a gold prism to a hexagonal lattice and its interaction processes to form gold nanoparticles from HAuCl4·3H2O precursor. They identified the presence of an amorphous intermediate, superlattice ordering, and line tension with the help of Monte Carlo simulation.54 Monte Carlo simulation is commonly used to control the reasonable aggregate structure and aggregation behavior of nanoparticles with less computational time.55 Recently, Gao et al. synthesized high-entropy alloy NPs by facile fast-moving bed pyrolysis (FMBP) approach. Herein, the FMBP formation follows the high supersaturation, smaller nuclei development, and mixed alloys (MnCoNiCuRhPdSnIrPtAu) loaded graphene oxide substrate without phase separation process, as seen in the Scheme 1, which is not possible by conventional fixed bed pyrolysis (FBP) approach. The binding free energy for FMBP is much lower (because it needs a fewer energy of nucleation) than the FBP, since the FMBP works at high temperatures than FBM.53
 | ||
| Scheme 1 Schematic diagrams show synthesizing homogeneous and phase-separated HEA-NPs by FMBP and FBP strategies, respectively. Reproduced/Adapted from ref. 53 with permission from The Springer Nature. | ||
Finally, nanocrystal formation occurs through the diffusion of atoms towards the pre-formed nuclei/seed surface until equilibration between atoms in solution and atoms on the crystal is reached. The monomeric diffusion towards the pre-existing seeds is also explained by the classical growth (CGT) and non-classical growth (NCGT) theories. As explained by CGT, a spiral, mass transfer, and two-dimensional types of growth may occur depending on different factors such as the activity and solubility properties of ions, temperature, kink shape, and growth mechanisms. According to the NCGT, the growth may also occur via oriented attachments of the crystals following the self-assembly, reorganization through rotation, and aggregation/agglomeration processes. The two main driving forces for aggregation/agglomeration (of the same or different-sized crystals) are the interfacial water structuring and macroscopic interparticle interactions (van der Waals (VdW) and electrostatic interaction, respectively). Besides, additional factors such as substrate facets and defects (such as faults, steps, and point defects), surface heterogeneity, ions and water at interfaces, confinement, and electronic structure also affect the nucleation and growth process of nanocrystals. Different facets have different free energy of nucleation and growth rate, the rate needed to grow and enable better stability.
The decomposition of the precursor is continuous, and atoms diffuse around the seeds and are attached once a free and favourable site is obtained. The LaMer model is still working with a small constraint on interpreting the growth characteristics. The declaration by LaMer on growth (which says, “all particles in an assembly grow the squares of their radii at equal rates”) did not confirm. Besides, the LaMer declaration was based on the Langmuir equation (which undertakes a steady state), which is invalid for the growth of most of the sols. Besides, the Langmuir equation says nothing about the relative rates of particle growth. One year ahead of LaMer's theory, Reiss developed a simplified “growth by diffusion” model that states the growth rate is only dependent on the monomer flux without considering other factors such as Ostwald ripening and aggregation.3 Then, Sugimoto continued Reiss's growth by diffusion model, which considers the Ostwald ripening factor by applying the Gibbs–Thomson equation.4 On the contrary, the nucleation and growth process may deviate, specifically for metal oxides, from the valid LaMer's model. This deviation may depend on the reactivity of precursors and surfactants used and different conditions. This type of deviation was confirmed by Chang et al. during iron oxide NPs growth (from iron-oleate precursor) in the presence of carboxylate moieties chelating agent. Here in Chang et al. study, the analysis of matrix-assisted-laser-desorption ionization time-of-flight mass spectrometry confirmed the formation of a tri-iron-oxo-carboxylate complex [Fe3O(C17H33COO)6]+ nuclei, instead of monomer aggregation as usual.56 Inconstant to the normal LaMer's nucleation and growth model, Baumgartner et al. also studied and confirmed the rapid aggregation of primary particles (from iron chloride precursor) to form Fe3O4 nanocrystal without forming an amorphous phase.57
According to the (Verwey and Overbeek) and (Derjaguin and Landau) (DLVO) theory, the particle growth (colloidal stabilization) in solution is controlled by the two forces, the VdW attraction and electrical double layer (EDL) repulsion forces (Fig. 5(a)). The aggregation of NPs is highly dependent on their size. The smaller the size, the lower the aggregation barrier, thus increases in the aggregation processes. However, after a particular particle size, the aggregation barrier becomes self-subsistent, which means the particles attain their stability, and the thermal energy becomes insufficient to overcome the increase in aggregation barrier (Fig. 5(c)). Fig. 5(d) shows the potential energy versus particle distance, indicating the dependency of the aggregation barrier on the thermal energy after some point, in which particles can aggregate within section-I and no aggregation within section-II. Fig. 5(e) shows the reliance of the critical radius RkT in a different system. EDL combines the inner compact and outer diffused layer between two colloids existing in the solution. The interaction between the nuclei and nanocrystals may also lead to the process of agglomeration or/and aggregation that diminishes the properties and application of the nanoscale materials. The irreversible aggregation and reversible agglomeration of nanoparticles occur due to the presence of VdW forces, instead of the electrostatic repulsion of NPs enclosed by the solvated ions. The potential increase is due to the VdW attraction, decreasing due to the EDL repulsion. Of course, these interactions are also dependent on different conditions such as size (the larger the particle, the higher the stability), surface potential, and type and concentration of the ion. The larger the counterion concentration, the lower the EDL size (Fig. 5(b)). As the distance increase from the surface, the counterions distribution decreases; this means high distribution in the Stern layer, medium in the diffused layer, and low within the Debye region. Water creates an electrostatic interaction at the interface by breaking its hydrogen bond, which aligns ions and solvents differently to generate EDL. Thus, the generated EDL also alters the nucleation and growth rate of the nanocrystal.43
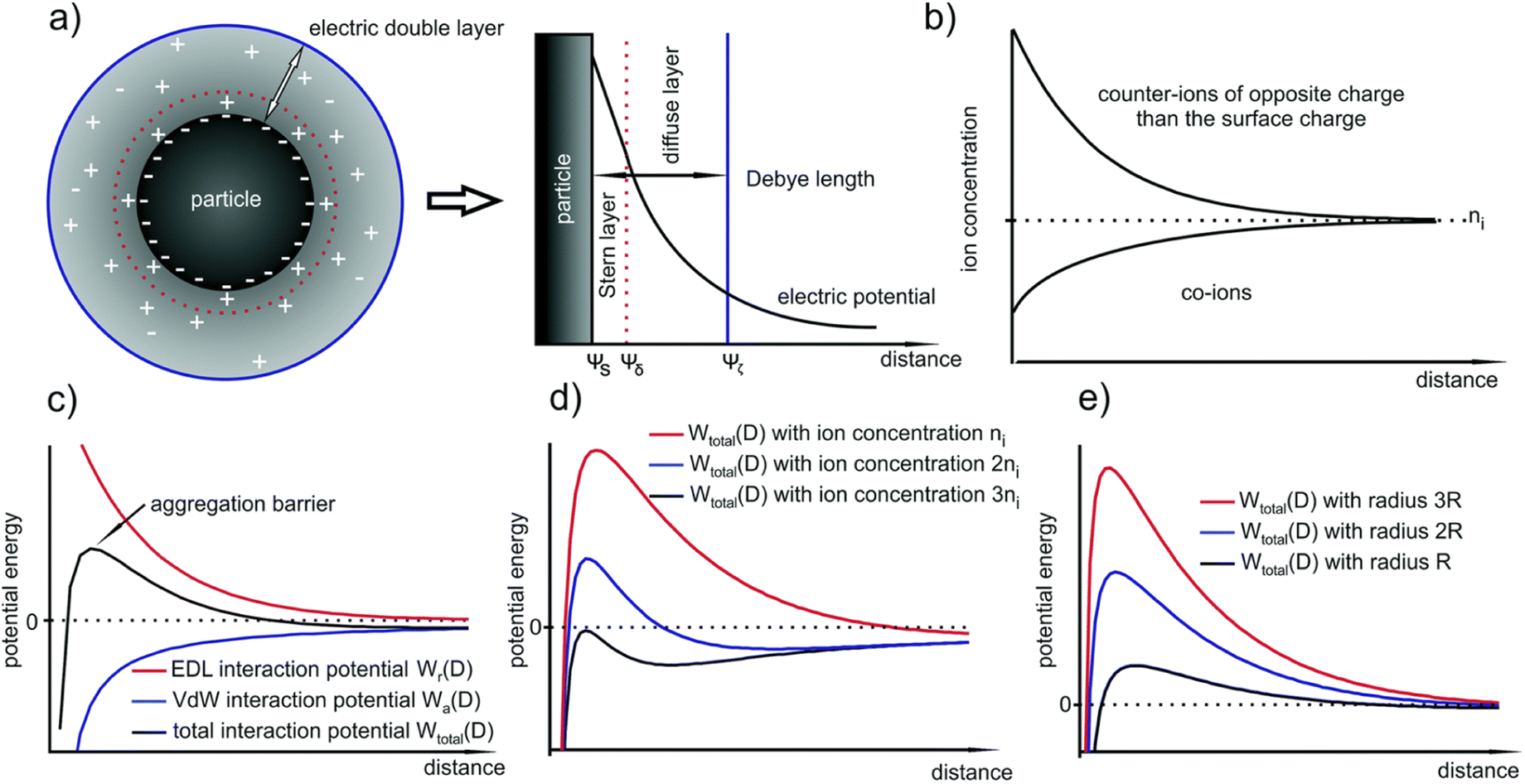 | ||
| Fig. 5 (a) Formed EDL around a NP due to the Gouy–Chapman model which consists of the inner Stern layer and the outer diffuse layer (b) corresponding decrease in the counter- and co-ion concentrations with respect to the distance from the particle surface; (c) schematic of the EDL, van der Waals and total interaction potentials of two NPs; (d) and (e) influence of the ion concentration and the particle size on the TIP. Reproduced/Adapted from ref. 43 with permission from The Royal Society of Chemistry. | ||
2.1. Stabilizing and geometry controlling agents
Polymers are used as both stabilization and reducing agent in nanomaterials synthesis. Besides, they also act as a structural directing agent that helps to tune the structure of materials towards a specific shape.59 Polyvinylpyridine (PVP) is a polymer commonly used as stabilizing and reducing agent. PVP is a non-toxic and water-soluble polymer at room temperature with a large amount of carbonyl group, which has adhesive forces with NPs. The networked type of cross-linking between the oxygen group of PVP and gold NPs is depicted in Fig. 6. Thus, once the NPs are capped by the polymer, the aggregation due to surface energy decreases.58 The stabilization of surfactants occurs through adsorption on the surface of the nanocrystal. The smaller the distance between the nanocrystal, the higher the density of the surfactants, which decreases the entropy (increases the Gibbs free energy) to prevent aggregation/agglomeration. The shape and internal structure of the face-centred cubic (fcc) nanocrystals determine their total free energy, in which crystal that has global minimum free energy is under thermodynamic control and kinetics control for all forms that deviate from global minimum free energy. For instance, the {110} facets of fcc nanocrystal have more dangling bonds (has closely packed facet) that decrease the stability, as compared to the {111} and {100} facets. Once the surfactants are adsorbed on the surface, they interact to decrease the crystal's free energy. These surfactants are adsorbed, change the order of surface free energies of crystal planes thermodynamically and tune crystal geometry based on the difference in the chemical interactions/binding affinities of different crystal facets.61 The selection of a specific capping agent for specific facets that leads to altering the order of surface free energy was reported by Xia et al., as shown in Fig. 7(a). Herein, selecting a specific capping agent for one facet (example {100}) results in increasing the free energy of the other facets ({111}), thereby rising growth in the {111} direction.46 | ||
| Fig. 6 Adsorption and cobridging of (a) PVP molecules in (b) a surface layer via Au-atoms on a gold particle. Reproduced/Adapted from ref. 58 with permission from The American Chemical Society. | ||
 | ||
| Fig. 7 (a) Schematic illustration of the role of a capping agent in directing the growth of a single-crystal seed into nanocrystals with different shapes Reproduced/Adapted from ref. 46 with permission from The American Chemical Society. (b) Overgrowth process of Ag nanocrystals, in which Ag atoms are continuously deposited onto the {100} facets of Ag nanocube to eventually result in an octahedron enclosed by {111} facets.60 | ||
To indicate, the PVP preferentially binds to the {100} facet of the Ag crystal and hinders its growth; this results in the elongation of the {110} and {111} facets to decrease their surface free energy, as show in Fig. 7(b).35,60 In contrast to the PVP, the citrate ions have greater binding affinities for {111} facets and result in elongation towards the {100} and {110} facets.62 However, thermodynamically controlling the growth rate based on the surface free energy works only if the crystallite size of the crystal is greater than its critical particle size. Xie et al. synthesized Pd–Rh nanocrystals by capping the Pd{100} facets (by Br− or I− ions) of Pd polyhedral seeds. Herein, after decomposition of Na3RhCl6 precursor at 140 °C, the nucleation and growth, in the island growth mode, start in the Pd{111} facets of preformed Pd seeds. The island model of growth, growth with the unoccupied free surface of Pd{111} facets, obtained at 140 °C is due to the domination of Rh–Rh interaction (greater surface ΔG). In their study, the possibilities of tuning the island growth to the layered growth model, which means growth with fully covering the free Pd{111} facets surface, were confirmed by increasing the temperature to 160 °C and extending deposition time.36
2.2. Advanced characterization techniques
Nowadays, the nucleation, growth, and nanocrystals formation are understood with rapidly developed experimental instruments, including the optical absorption and emission spectroscopy (in situ), mass spectroscopy (ex situ), electrospray photoelectron spectroscopy (EPS), and electron microscopic (specifically, transmission electron microscopy (both in situ and ex situ)). The advancements in the TEM detector, capturing the in situ real-time liquid-phase processes, are now possible in the atomic level mapping to control the processes of atomic diffusion, nucleation, growth, and nanocrystals formation under different conditions.10,11,63 For example, the high-resolution ADF-STEM analysis controlled the nanocrystal's early-stage growth from dissolved precursors.8 Loh et al. reported spinodal gold precursor decomposition, amorphous nanoclusters formation, and crystallization (multistep nucleation process) using the in situ real-time electron microscopy (Fig. 8). Au3+(aq), the decomposed precursors, have been reduced to gold atoms by the solvated electrons (electrons from the TEM source). The measured crystallinity score of sensitive view, fcc {111} plane, showed 27% crystallinity. The other 73% are either amorphous or not detected due to instrumental detection limits.12 | ||
| Fig. 8 A proposed three-step pathway for gold nucleation in solution. (a) TEM images show the intermediate steps in nucleating gold nanocrystals from a supersaturated aqueous Au0 solution. From 3.0 to 9.2 s, the supersaturated Au0 solution spontaneously demixes into gold-poor and gold-rich liquid phases (lighter and darker regions, respectively) via spinodal decomposition. Amorphous gold nanoclusters emerge (11.3 s) from the gold-rich phases that crystallize (15.4 s). Insets show Fourier transforms of cropped square regions (orange) with the Au(111) fcc reciprocal lattice spacing circled in red. (b) Schematic of the proposed steps in nucleation (gold as orange spheres, with surrounding water as blue bent lines). Reproduced/Adapted from ref. 12 with permission from The Springer Nature. | ||
The in situ AFM also gives detailed information about the two-dimensional nucleation critical step lengths, growth, and interaction between nanocrystals.13–15 Luchkin et al. confirmed in situ AFM measurements to control the solid-electrolyte interphase nucleation and growth formation in Li-ion batteries.64 The X-ray scattering investigates the nanocrystals' arrangement, size, aggregation/agglomeration, and shape.65 The in situ X-ray absorption spectroscopy (XAS) also used to understand the nucleation and growth process within few seconds, which is impossible with most of the above discussed analytical techniques.
From XAS, the X-ray absorption near-edge structure (XANES) regimes of XAS give information about local chemical environment and the extended X-ray absorption fine structure (EXAFS) about the chemical state and elements local coordination. Just et al. studied the mechanistic path of copper thiolate intermediate formation, its decomposition to copper sulfide, and the formation of Cu2ZnSnS4 nanorods product upon incorporating Sn and Zn from their respective acetate precursors within a few seconds.17 Using the small-angle X-ray scattering technique, Naik et al. also showed the “LaMer type” nucleation, growth, and spherical Ag NPs synthesis process in the presence of the Nafion-117 membrane. Herein, the nucleation and growth of the reaction process are controlled by tuning the concentration of silver citrate precursor (controlling the supersaturation by limiting the precursor concentration) and the reaction temperature. Increasing the temperature increased the nucleation and growth rates due to the raising of the precursor decomposition, fast supersaturation, and boosting the atomic diffusion toward the nuclei.16
3. Solution combustion synthesis
The LaMer model gives insight into the overall process of the nanocrystal formation starting from the precursor decomposition. However, in addition to the reduction, growth, and nanocrystal formation process, the development of complexes between the precursor's anions and fuels/surfactants/solvents, which result in the combustion process once the ignition temperature is reached, should also be critical within this document. The nanocrystal probably contains complexes formed between the precursor's anions and surfactants/fuels. Further dehydration and heating to ignition temperature results in combustion, commonly known as solution combustion synthesis (SCS). SCS is a complex self-sustained exothermic reaction process, initiated between the surfactants or/and organic fuel and metal nitrates. The SCS follows (i) a molecular level mixing to form the colloidal NPs (sol), (ii) evaporating the water molecules to form a gel, (iii) creating a high locally initiated reaction temperature, which is activated by low preheating temperature,66 (iv) the combustion front spreads along the rest of the media, and (v) finally, diminishing of the temperature of the overall system due to the quenching effect through the evolution of gases. The occurrence of gas evolution results in decreasing the combustion front velocity and temperature by blocking the crystal growth. This in turn increases the final product's total porosity/textural properties.21Fig. 9(a) shows the mechanism of microstructures synthesis (combustion wave capturing) under the controlled infrared (IR) measurement. The in situ high-speed IR camera uses to control the combustion wave and measure the width of the combustion wave.67 Herein, two cases may exist; the first case is a reaction of the gaseous products during the nitrate decomposition, in which combustion starts from many points and results in the formation of spongy/foam-like structure of metal oxides (Fig. 9(b)). In the second case, the reaction may start at one spot/point, through which the bright reaction front starts to propagate in one direction and form a long wire metal oxide68 (Fig. 9(c)). Finally, the primarily developed metal oxide may react with the reducing gases/atmosphere (H2 and NH3, produced during gas-phase combustion) or/and with oxygen in the atmosphere resulting in reduction of metal oxides to zerovalent metal.23 Following the SCS procedures, the combusted materials may be further calcined at high temperatures to decompose the un-burnt impurities and improve the crystallinity of the materials.69 Of course, increasing the furnace temperature may facilitate the detonation time and crystallinity, although it increases the crystallite sizes of the material. The increasing crystallite size increase/agglomeration process with increasing temperature is due to the melting of the particles and fusing into a larger aggregate, commonly known as melt-induced aggregation. The aggregation mechanism is by diffusion of surface matter (such as ash, which melts and facilitates the adhesion process) around the particles resulting in the assembly.70
 | ||
| Fig. 9 Scheme representation of the self-sustained combustion process: (a) infrared images for reaction front propagation (b) SEM images of quenched front in a nickel nitrate–glycine gel32 and (c) iron nitrate–glycine. Reproduced/Adapted from ref. 32 with permission from The American Chemical Society. | ||
SCS creates a pure metal oxide material with very high stability and a good surface area within a short reaction time.71 This approach-based synthesis of semiconductor metal oxide materials is time-/energy-efficient and creates a well-defined porosity due to the evolution of gases.24 The porosity in the materials assists in the interfacial mass-/ion transport, especially in sorption, energy storage, catalysis, and sensing applications.25,26 Besides, the technique is also simple, needs a low-cost instrument, and yields highly pure products. It neglects the time consumed by the purification or the post-synthesis treatment procedures compared to the other established approaches.27,28 In this approach, once the mixture reaches the ignition temperature, self-generated energy raises the reaction temperature to 4000 °C,22 and within seconds per minutes, the precursor is converted into the corresponding stable metal/metal oxides.
According to propellant chemistry,20 the total oxidizing valences (OV) and reducing valences (RV) serve as a stoichiometric balance for the fuels and oxidizers, respectively during solution combustion synthesis. Based on the approach developed, the value of the fuel/oxidizer ratio φe was calculated using eqn (1). Where the RV/OV ratio is equal to Σ (coefficients of the reducing elements vs. valencies)/Σ(coefficients of the oxidant elements vs. valences),18 n is the number of moles of reducer/mole of the oxidant.72 During calculation, nitrogen and oxygen oxidizer elements should have 0 and −2 valances, respectively. Carbon and hydrogen reducing elements should have +1 and +4 valances and water is not incorporated in this calculation. The valence of all the metal ions is equal to the respective metal valances. All the reducing and oxidizing elements are treated similarly irrespective of whether they exist in the oxidizer or fuel parts.20 When φe = 1, it indicates that the reducing and oxidizing species are completely balanced, in which total decomposition took place to give metal oxide and gaseous molecules. φe > 1 indicates the fuel-rich mixture, and φe < 1 is the indication of the fuel-poor mixture.
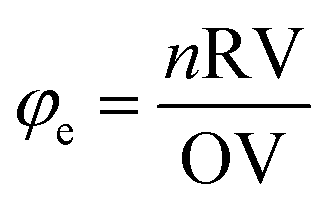 | (1) |
Besides, based on the φe ratio, the combustion reaction can be classified into extremely fuel-excess, fuel excess, stoichiometric, fuel deficient, and extremely fuel-deficient. The extremely fuel-deficient and fuel-deficient combustion reaction types need external heat for the reaction completion and have a less exothermic nature. The latter shows a good surface area and particle dispersion compared to the former. Besides, the latter produces a more toxic gaseous by-product, NOx. A highly exothermic reaction occurs in the stoichiometric and fuel excess combustion type, although the agglomerations increase due to this inner sintering process. However, an extremely fuel-excess reaction results in the decomposition of the gel without completing the self-sustained reaction.18
As an example, Gao and his group calculated the RV/OV ratio for the mixture of nickel(II) nitrate hexahydrate, aluminium nitrate nonahydrate, and CO(NH2)2 reagents. In this study, a Ni–Al2O3 catalyst is synthesized (using a microwave-assisted SCS approach) by slowly adding the urea to the nickel and aluminium nitrate solution mixture with continuous stirring at 60 °C. Microwave-assisted heating of the mixture on the evaporating dish results in dehydration (gel formation) and gas evolution (see Fig. 10). The un-burnt urea and nitrates are further decomposed by heating in a furnace at 400 °C. In their calculation, for optimum stoichiometry (RV/OV = 1), 1 mol of Ni–Al2O3 involves 13.07 mol of urea (CO(NH2)2) (so n = 13.07). Therefore, the value of RV/OV is determined as: [n × (4 × 1 + (−2 × 1) + 0 × 2 + 1 × 4)]/[1(2 × 1 + 0 × 2 + (−2 × 6)) + 4.56(3 × 1 + 0 × 3 + (−2 × 9))].69 The chemical formula of common organic fuels and their reducing valances are illustrated in Table 1.73
 | ||
| Fig. 10 Schematic diagram of the synthesis process of catalysts by solution combustion. Reproduced/Adapted from ref. 69 with permission from The Royal Society of Chemistry. | ||
| aFuel CF | bRV | Fuel CFa | bRV |
|---|---|---|---|
| a Fuel chemical formula.b Reducing valence. | |||
| C11H12N2O2 | −52 | C6H8O7 | −18 |
| C12H22O11 | −48 | C2H8N2 | −16 |
| C9H11NO2 | −43 | C3H7NO2 | −15 |
| C6H12N4 | −36 | C3H5NO | −15 |
| C6H14N4O2 | −34 | C4H6O6 | −10 |
| C6H15NO3 | −33 | C2H6O2 | −10 |
| C5H11NO2 | −27 | C4H7NO4 | −15 |
| C4H11N | −27 | C2H5NO2 | −9 |
| C6H14O6 | −26 | CH6N4O | −8 |
| C6H12O6 | −24 | H4N2O | −6 |
| C5H9NO4 | −21 | N2H4 | −4 |
| C6H8O6 | −20 | C2H2O4 | −2 |
Based on the applied temperature, the SCS is categorized as controlled and uncontrolled reaction. Uncontrolled SCS reaction, which took place under the higher redox mixture ignition temperature, can give nanosized metal/metal oxides, although it is explosive. Whereas controlled SCS reaction, a reaction that took place under near redox mixture temperature, used for synthesizing the well-defined catalysts, herein it controls the occurrence of explosive reaction.74 The gas evolution and combustion enthalpy volume are calculated by assuming that the reaction are complete (adiabatic conditions, in which heat loss through conduction and radiation are assumed to be zero). This crucial parameter in SCS, which means the reaction temperature/adiabatic flame (eqn (4)) is predicted by solving eqn (3).66,75
 | (2) |
| Tc = To + (ΔHor − ΔHop)/Cp | (3) |
The SCS is also classified into gas, liquid and solid-phase combustion processes. The gas-phase synthesis process is a reaction process in which some parts of the synthesis involve gas. The liquid phase combustion type (also known as SCS) is a common and manageable approach used to synthesize a wide range of materials by taking a stoichiometric proportion of the oxidizer and fuel. This combustion process can be divided into sol–gel combustion, gel-combustion, and emulsion combustion.76 The gel formation parameters (gelation time, solution pH, temperature, and ageing) are the necessary conditions in the SCS, as it maintains chelating processes between fuel and oxidant, which is crucial for the combustion process. The sol–gel process in the SCS is similar to the process that occurred in the sol–gel methods. The solution pH also has a vital function in the fuel/oxidant gelation/chelation process. It is also possible to control the gel formation by measuring pH during solvent evaporation. As the solvent evaporates, hydrolyzed precursors release H+ ions, which results in a decreased solution pH.18,29 Temperature is an endothermic process; an increase in the temperature of the mixture solution results in solvent evaporation and therefore increases gelation.32 Ageing time also facilitates network formation between the mixtures, although it increases the pre-ignition temperature.77
Sadabadi and his co-workers synthesized LaCoO3 perovskite by the sol–gel auto-combustion approach to reduce nitrogen oxide (NOx) gases. A Perovskite oxide contains rare-/alkaline-earth metal, transition metal, and octahedral structured with six oxygen atoms. Herein, the lanthanum-/cobalt-nitrate as an oxidizer, glycine- and urea-fuel as a reducer, and PVP surfactant as shape and size distribution controlling agent reactants were used. Besides, the fuel type and fuel ratio were also studied, in which the LaCoO3 synthesized by glycine fuel showed good surface area.78 Using the same sol–gel auto-combustion approach, Dara and his groups synthesized double perovskite Tb2ZnMnO6 NPs to degrade methyl violet and methyl orange dyes. Hydrated terbium, manganese, and zinc nitrate salts as an oxidizer, sugars (lactose, fructose, and maltose) and liquorice powder fuels as reducers have been used as reactants. Among these fuels, the maltose showed good dispersion and surface area for the synthesized NPs. Besides, taking the optimum maltose fuel and two different fuel/terbium ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 3:2), the fuel-lean ratio (1:2) showed a good surface area and morphology.79
:2 and 3:2), the fuel-lean ratio (1:2) showed a good surface area and morphology.79
The salt oxidizers used in the SCS can be reducers, neutrals/chloride-based, or oxidants/nitrate-based. For the SCS based nanomaterials synthesis using the reducer type precursors, additional oxidants such as NH4NO3 and HNO3 are required. Also, it is possible to use other oxidizing agents in the case of neutral precursors. However, the neutral/chlorine-based ones that release HCl result in the contamination of the final products. The oxidants, precursors containing nitrates, are the best oxidizer due to their crucial properties such as (i) adequate oxidizing potential (NO−3, a negative charge), and (ii) stable decomposition temperature starting from the low temperature, which results in high evolution of more gaseous byproducts, consequently enhancing the purity and porosity of the final product30 and (iii) noble solubility compared to the other salts.31 Of course, the properties mentioned above are also related to the metal-cation charge density, exchange kinetics, and cations' standard molar Gibbs free energies of hydration.80,81
Solution pH has a critical function in the metal ions and nitrates chelation. Combustion of nitrates during the drying and pre-igniting stage results in a decrease in the maximum combustion temperature. Thus, the optimum chelation solution pH point is selected to protect the oxidant (nitrates) from degradation before the combustion reaction starts and to control the final product morphology.82 Junliang and his groups studied the solution pH and citric acid to metal ions (CA/M) ratio on the maximum combustion temperature and auto-igniting time. Herein, the barium hexaferrite powders were synthesized by the sol–gel auto-combustion approach from barium(II) nitrate/iron(III) nitrate nonahydrate oxidant and HOC(COOH)(CH2COOH)2·H2O reducer. In this work, increasing the pH and CA/M ratio results in an increase in the maximum combustion temperature and auto-ignition time. In their conclusion, the pH and CA/M ratio increase the chelation properties between the cation (Fe3+/Ba2+) and CA.83
Pourgolmohammad and his group have also synthesized cobalt ferrite NPs from the hydrated ferric and cobalt nitrates as an oxidant and glycine as fuel to confirm the effect of solution pH on the shape of the by-product. As concluded, solution pH has a critical role in the auto-combustion reaction rate and distribution of cations. This intern affects the specific surface area and morphology as confirmed by the adsorption–desorption isotherms and SEM/TEM image analysis. Here, an increase in the solution pH results in the shifting of the morphology from porous and good particle distribution to agglomerated/bulk morphology.84
In addition to the aforementioned parameters, the effects of injecting O2, N2, and CO2 gases in line with microwave-assisted SCS procedures for synthesizing ternary CuO/ZnO/Al2O3 composites were also reported. This study used ethylene glycol fuel and copper-, zinc-, and aluminium-nitrate oxidants. Compared to the other gases, a significant influence on the CuO dispersion, morphology, surface area change, and good interaction between CuO and ZnO is confirmed for N2 gas injection. In their work, the optimization of fuel/oxidants and microwave irradiation resulted in the surface area and better dispersion of NPs. Besides, the creation of porous by-product resulting from the combustion of gases has been of great importance in the availability of the reactant molecules to the inner active sites.24
3.1. The effect of parameters on SCS
In SCS, the reducers should be soluble in water and complexant to the metal cations in an aqueous solution. Depending on the nature of oxidizer/reducers and several parameters, the melting/ignition temperature lies between 150–500 °C and the combustion temperature lies in the range of 500–4000 °C.23 The combustion process will not start till all the adsorbed water and water of crystallization are evaporated. Thus, depending on the chemical reaction, it may be needed to heat to 500 °C in the furnace to convert the sol into gel.85The complexation property of the reducers as a result of their chelating agents facilitates the homogeneous dispersion of metal cations. Consequently, it prevents premature precipitation by forming strong coordinate bonds (gel network after solvent evaporation) and diminishes the aggregation/agglomeration process during water evaporation. A structural scheme developed based on the carbon-13 nuclear magnetic resonance (CP/MAS 13C-NMR) and Fourier-transform infrared spectroscopy (FTIR) shows urea is located as a second- and nitrate as a third/outer-coordination sphere (Fig. 11).31 Herein, the formation of oxygen-to-metal coordination is understood from the FTIR spectrum. The presence of carbon atoms in urea and the octahedral AlO6 coordinate bond was confirmed from the CP/MAS 13C-NMR spectra.
 | ||
| Fig. 11 Expected structure of Fe (Al)–urea–nitrate complex Reproduced/Adapted from ref. 31 with permission from The American Chemical Society. | ||
In addition to activating the combustion process, fuels also act as architecture directing templates and complexing agents. Besides, based on the type of bond formed between the reactive groups to the hydrocarbon chain, fuels can be classified as carboxylic (–COOH), hydroxyl (–OH), amino (–NH2), and other multifunctional fuels.68 As confirmed in several studies, the amino > hydroxy > carboxyl is an order of organic fuels complexation/combustion reactivity.
Xanthopoulou et al. synthesized glycine reducer-based flowerlike Ni–NiO microstructure nanocomposites by SCS from Ni(NO3)2·6H2O and glycine. Here in this study, an increase in φe results in a decrease in a combustion reaction. For a lower φe-value, a combustion regime wave occurred ‘‘in the volume,” and increasing the φe amount shifted the combustion wave regime to the self-propagating range.67 Combustion regime shift from volumetric to self-propagating mode, with an increase in the φe-value, is also seen in Wang and his group's studies indicating that the ignition starts at a point/spot. Besides, increasing the value of φe results in a morphology shift from nanoneedles to porous nanosheets and finally to aggregated NPs. This work synthesized iron oxides from ferric nitrate and glycine using the SCS approach with improved electrochemical properties.86
Recently, Novitskaya et al. summarized the effects of different SCS parameters.66 In this review work, longer molecular chain fuels (higher molecular weight) such as triethanolamine, citric acid, and sorbitol were reported to have greater combustion behaviours and more sites for complex formation. The greater combustion behaviour of organic fuels results in the evolution of large amounts of gases, creating assembled pores and active sites. In addition to the molecular weight, the heat of the reaction66 and the type of the functional groups are also the other essential parameters that control the property of the material. As concluded based on the several assessments, the heat of reaction is dependent on the saturation and unsaturation, in which unsaturated fuels have greater heat of reaction, so highly reactive and saturated fuels have less reactivity.33
The occurrence of a series of endothermic and exothermic events in the combustion reaction was studied by Kanakala and his co-workers using the differential scanning calorimetry (DSC) technique. An endothermic peak detected at a temperature of 76 °C was attributed to the melting of the precursor's nitrates. The peak at 170 °C was attributed to the melting of carbohydrazide and forming a complex between metal ions and carbohydrazide. Finally, the combustion and crystallization process took place to form a powder. The first exothermic peak at ∼290 °C is due to the combustion initiation, and the final two exothermic peaks at 339 and 379 °C are due to the total combustion and crystallization for the formation of nanoscale size powder. Besides, the heating rate and sequence in the addition of precursors, fuels, and other reagents should also be considered for complete reaction and prevent covering the nitrates with other reagents or powder.85
Reducing-gases-assisted sol–gel auto-combustion-based reduction of the preformed metal oxides/complex multicomponent oxides was reported in Niu and his group's study. This study synthesized CoCrCuNiAl high-entropy alloys from the respective hydrated salt oxidizer and citric acid fuel. Based on the thermogravimetric-differential scanning calorimetry-mass spectrometry (TGA–DSC–MS) analysis, first, the (Cu, Mg, Ni, Co, Zn)O formed, and then the oxides are reduced to metal alloys with the existence of reducing gases that vaporized during the self-sustained combustion reaction (Fig. 12(a–c)). The synthesized materials' crystal structure and type of phase formation are studied using the XRD pattern by taking different fuel/oxidant ratios (Fig. 12(d)). Fig. 12(e) shows the XRD patterns of metal alloys synthesized by mechanical alloying and combustion for comparison, in which both methods show the same phases. A schematic illustration of high-entropy alloys is depicted in Fig. 12(f) inset. Herein, the complete reduction of the oxides through the ejection of gasses to pure alloys takes place at a fuel/oxidant ratio of 1:1. The role of the fuel/oxidant ratio is also studied in their work, with the 1:1 ratio obtained to be optimal. The synthesized alloy also exhibited characteristic magnetic properties.87 The role of the reducing gas (NH3) on the reduction of MoO3 to MoO2 was also reported in the Gu et al. work. The hexaammonium molybdate [(NH4)6Mo7O24·4H2O], ammonium nitrate, and glycine reactants were used to synthesize foam-like MoO2 material as an organic dye degradation agent. Herein, among five different fuel/oxidant ratios, the 0.5 (fuel-lean) ratio was reported to give foam-like MoO2 structure, stable, and excellent potential in photocatalytic activity.88
 | ||
| Fig. 12 (a–c) TG–DSC–MA results are obtained for the dried gel prepared from the sol with a fuel-oxidant ratio of 1:1; (d) XRD patterns revealing the crystal structures of the combustion products of the dried gels for different fuel-oxidant ratios; (e) comparison of XRD patterns of CoCrCuNiAl high-entropy alloys synthesized by sol–gel auto combustion and mechanical alloying; (f) XRD patterns of the combustion product of the gels with a fuel–oxidant ratio of 0.8:1 and the schematic illustration of high-entropy alloys Reproduced/Adapted from ref. 87 with permission from The Springer Nature. | ||
In the Manukyan et al. study, a spongy/foam-like microstructure product was synthesized using nickel nitrate hydrate oxidizer and glycine fuel with the self-propagation reaction mode. Here in this study, the occurrence of a ∼10 μm thickness quenching front due to gas evolution is confirmed with the help of energy dispersive spectroscopy map analysis. A combustion front propagation occurred due to the N2O and NH3 reactions. As they understood from the TGA–DSC–MS glycine fuel decomposition analysis, three decomposed gases, H2O, NH3, and CO2, were detected in the temperature range of 240–280 °C. The produced NH3 reduced nickel oxide to the nickel-metal in the reaction front. Besides, the TGA–DSC analysis showed the occurrence of three oxidizer decomposition processes as a result of adsorbed water (at 70 °C), the water of crystallization (at 160 °C), and nitrate decomposition (at 250 °C). Based on the TGA–DSC and MS analysis, the overall combustion reaction mechanism of nickel nitrate–glycine gels was summarized in Fig. 13. The figure shows a time–temperature profile of the combustion process in the case of the self-propagation reaction mode. In which the locally initiated combustion is completed within two seconds (2 s) (elevating the temperature from ∼150–1100 °C), and finally, the cooling stage takes place. Within these 2 s (i) the sequential evaporation of water up to 150 °C from the hydrated nickel nitrate, (ii) up to 250 °C, further decomposition of the dehydrated zinc nitrate to NiO and Ni2O with the release of gaseous products, and (iii) finally, the reaction of ammonia gas (produced from glycine) with zinc oxide, which results in the reduction of metal oxide to the nickel metal with the release of nitrogen gas took place.32
 | ||
| Fig. 13 A schematic representation of the mechanism for Ni formation during self-sustained reactions of nickel nitrate–glycine gels Reproduced/Adapted from ref. 32 with permission from The American Chemical Society. | ||
3.2. Beyond solution combustion synthesis
Nowadays, synthesizing a metal oxide with well-defined porosity and stability using time-/energy efficient (economical) and industrial-scalable SCS routes got great attention.18 However, large bandgap metal oxides are inactive in reaping the abundant visible light. Hence, proper modifications such as creating surface vacancies, foreign impurities (doping/co-doping) inclusion, and heterojunction have been discovered for visible light activity improvements. The created surface oxygen vacancies assist as an electron capture site that prevents the recombination process in pristine metal oxide materials. An oxygen vacancy also has bandgap narrowing properties by creating band energy below the CB of the large bandgap materials.89 In addition to forming metal oxide materials, the combustion process allows the processes such as homogenous in situ metal/metal ions doping, vacancies, and construction of a heterojunction.33,90Doping or co-doping of metal/metal ions can fine-tune the host materials' optical, magnetic, and optical properties. Besides, a proper heterojunction or doping inhibits the photo-induced electron–hole recombination through the charge transfer process91 or by acting as an electron reservoir,92 which has great importance in several applications like photocatalysis. The synergistic and charger transfer improvement role through doping and heterojunction/hybrid has also been reported in recent works. The HSAB theory is a crucial clue for selecting appropriate dopant–host materials. The dopant should have less hardness for successful exchange and diffuse to the host lattice in a hard base solvent like water.93,94 However, if the hardness of the dopant is greater than that of the host, surface adsorption occurs and, therefore, formation of hybrid/heterojunction up on the oxidation.
A hard carbon template-assisted-porous CeO2–MnOx synthesized by the SCS technique showed improved catalytic activity towards the oxidation of CO. This improved activity is a result of the synergistic CeO2 and MnOx components. Herein this study, the time-/energy-efficient and green properties of the SCS are confirmed without using any organic solvents and surfactants.95 Delfani et al. synthesized a mesoporous Co3O4–Sb2O3–SnO2 heterojunction using the SCS for capacitive deionization. The materials were synthesized using the cobaltous nitrate hexahydrate, tin(II) chloride dihydrate, antimony trichloride, HNO3 oxidizer, and CO(NH2)2 fuel. Herein, the HNO3 was utilized as a combustion aid since the chloride salt (SnCl2·2H2O) was used as a precursor.27 The synthesized ternary hybrid electrode capacitive deionization system exhibited better ion removal potential than single Co3O4. In addition to the synergistic effect of ion removal efficiency, incorporating Sb2O3 and SnO2 improved material conductivity and cycling stability.
Ni-based hybrid metal oxides (NixCo1−xOy) were also synthesized by using the SCS as improved electrocatalytic activity. The material was synthesized using the respective nitrate oxidizers salt precursors and glycine as a fuel. This improvement is also attributed to the synergism effects resulting from heterojunction, although attributed to the electronic structure improvement of Ni (oxidation of Ni from Ni2+ to Ni3+).28 Besides, Wrzesińska et al. synthesized a rare-earth (RE) doped BiFeO3 (Bi0.9RE0.1FeO3) material using the SCS method. This work confirmed the promising properties of SCS for the synthesis of a neat and doped material with enhanced application potential. In addition to doping, the creation of oxygen vacancies due to the gas evolution also enhanced the applications of materials. The doped Bi0.9RE0.1FeO3 material has significantly improved its magnetic and ferroelectric properties.96
However, the SCS may also have drawbacks, such as powder agglomeration during evaporation of water and pore shrinkage at high temperature, lack of shape control, and organic and carbonaceous byproduct impurities remaining due to incomplete combustion.33,97,98 The shape and properties of the products depend on the solution pH, nature of the fuel, heating rate, fuel/oxidizer ratio φe, and gel characteristics. The essential factors are the reaction temperature that enables agglomeration and the amount of gas product that inhibits the agglomeration/aggregation processes.23
3.3. Porous crystalline framework
Still, synthesizing ordered porous nanomaterial is challenging using the SCS because of the rapid and high-temperature nature of the process. Tuning the material structure to the secondary structure, ordered porous crystalline framework, improves the mass-/ion-transport functions of energy-converting devices.99 Thus, selecting an appropriate solvent, ADA, and temperature is vital.93,100 Subsequently, ageing/condensation, dehydration, and thermal/chemical treatments are followed to form assembled frameworks.101 The ADA templates can be soft or hard, which can be removed by either physical or chemical processes to obtain stable assembled ordered nanocrystal frameworks. The soft/co-polymeric ADAs can be small colloidal templates used to assemble mesoporous materials or large colloidal templates used to synthesize macroporous materials.38Voskanyan et al. synthesized an ordered mesoporous CeO2 NPs by combining the time-/energy efficient SCS approach and SiO2 as a hard template, architecture directing agents, which assists as a morphology monitoring agent. This work utilized the colloidal SCS in the place of the SCS procedure, which can give an ordered structure unlike that of disordered morphology obtained in the SCS. The materials are synthesized by directly adding the SiO2 template in cerium nitrate oxidizer and glycine fuel. Upon heating, evaporation of water and gel formation between SiO2 colloidal resulted. Here in this work, the initial ignition starts at 150 °C. Formerly, locally initiated rapid combustion and cooling took place by releasing a gas product resulting in the SeO2–SiO2 composite formation, as shown in Fig. 14. Finally, the SeO2 NPs obtained by removing the SiO2 with the help of an alkaline etching technique.82
 | ||
| Fig. 14 Schematic of the CSCS method for synthesizing crystalline mesoporous CeO2 with tailored porosity: (a) evaporation of water (b) combustion process (c) cooling (d) etching. Reproduced/Adapted from ref. 82 with permission from The American Chemical Society. | ||
Recently, Wang et al. have also reported the synthesis of SiO2 assisted active and stable optimum Ni loaded CeO2 for CO2 methanation. The Ce(NO3)2·6H2O and Ni(NO3)2·6H2O oxidizer and glycine fuel were used as a reactant and SiO2 as a template. The presence of SiO2 here also assists in the formation of ordered mesoporous morphology.102
Taking the reproducibility, cost-effectiveness, and reliability of the self-sustained SCS, Zarezadeh et al. also proved the time-/energy-efficient salient features of the SCS. The carbonaceous by-product residual left due to the incomplete combustion is the main drawback of the SCS. Here in this work, the ammonium nitrate-rich in hydrogen, oxygen, and nitrogen was used as a fuel to reduce the carbonaceous by-product. The FESEM and TEM bright-field images confirmed the formation of porous NPs that have a crystallite size of 20 nm.103
The salient features of the SCS technique as compared to the conventional impregnation and co-precipitation methods are confirmed in the study conducted by Wanga and Liu. Herein, a high dopant dispersion and close dopant contact with the host matrix, which is challenging in convectional synthesis techniques, was accompanied by dispersing Co impurities in the CeO2 matrix.104 Kang et al. also synthesized CeO2 powders by varying the oxidizer (cerium ammonium nitrate and cerium nitrate hexahydrate) and fuel (glycine and hydrous hydrazine) type. In their study, the combustion mechanism, ignition temperature, and gas evolution properties of the self-sustained reaction are dependent on the fuel and oxidizer type. The SeO2 synthesized using hydrous hydrazine fuel with a φe ratio of 2, and an ammonium nitrate/metal nitrate ratio of 4 showed a high surface area.105
4. Conclusions
In conclusion, the LaMar model is still vital to visualize the process of the nanocrystal formation from the precursor decomposition with the next advancement that assists the growth characteristics. Thanks to technological advancements, capturing the in situ real-time decompositions to the crystal formation process is the current progress and should be the unstoppable outlook. In addition, stabilizing the nanoparticles and tuning their geometry based on the facets' surfactant reactivity is critical for crystal designing for a specific application. The solution combustion synthesis (SCS) approach is a noble methodology with efficient management of time and energy aspects resulting in highly stable and porous nanomaterials, which can be applied easily for industrially scalable purposes. Tuning materials to the secondary structure, ordered porous colloidal frameworks, improves the mass-/ion-transport functions of energy-converting devices. The architecture-directing agents assisted SCS can produce ordered colloidal nanocrystal frameworks. Thus, under the optimized conditions of crucial parameters and templates, the SCS approach becomes noble as future outlooks.Author contributions
Writing – original draft and Investigation by Buzuayehu Abebe. The writing – review & editing by Dereje Tsegaye and H. C Ananda Murthy. All authors read in details and approved the final manuscript.Abbreviations
| SCS | Solution combustion synthesis |
| HSAB | Pearson's hard/soft acids/bases |
| NMs | Nanomaterials |
| ADA | Architecture-directing agent |
| CNFs | Colloidal nanocrystal frameworks |
| φe | Fuel/oxidizer ratio |
| CP/MAS 13C-NMR | Carbon-13 nuclear magnetic resonance |
| FTIR | Fourier-transform infrared spectroscopy |
| DSC | Differential scanning calorimetry |
| TGA–DSC–MS | Thermogravimetric analysis–differential scanning calorimetry–mass spectrometry |
| EDS | Energy dispersive spectroscopy |
| FESEM | Field emission scanning electron microscope |
| TEM | Transmission electron microscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Adama Science and Technology University.Notes and references
- A. Khort, J. Hedberg, N. Mei, V. Romanovski, E. Blomberg and I. Odnevall, Sci. Rep., 2021, 11, 7860 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- H. Reiss, J. Chem. Phys., 1951, 19, 482–487 CrossRef CAS.
- E. Matijevic, Chem. Mater., 1993, 5, 412–426 CrossRef CAS.
- Q. Li and Y.-S. Jun, Commun. Chem., 2018, 1, 56 CrossRef.
- D. Erdemir, A. Y. Lee and A. S. Myerson, in Handbook of Industrial Crystallization, Cambridge University Press, 2019, pp. 76–114 Search PubMed.
- J. Lee, J. Yang, S. G. Kwon and T. Hyeon, Nat. Rev. Mater., 2016, 1, 16034 CrossRef CAS.
- W. Dachraoui, T. R. Henninen, D. Keller and R. Erni, Sci. Rep., 2021, 11, 23965 CrossRef CAS PubMed.
- M. Abbas, L. Zeng, F. Guo, M. Rauf, X.-C. Yuan and B. Cai, Materials, 2020, 13, 4851 CrossRef CAS PubMed.
- Y. Xu, K. C. H. Tijssen, P. H. H. Bomans, A. Akiva, H. Friedrich, A. P. M. Kentgens and N. A. J. M. Sommerdijk, Nat. Commun., 2018, 9, 2582 CrossRef PubMed.
- Q. Chen, J. M. Yuk, M. R. Hauwiller, J. Park, K. S. Dae, J. S. Kim and A. P. Alivisatos, MRS Bull., 2020, 45, 713–726 CrossRef.
- N. D. Loh, S. Sen, M. Bosman, S. F. Tan, J. Zhong, C. A. Nijhuis, P. Král, P. Matsudaira and U. Mirsaidov, Nat. Chem., 2017, 9, 77–82 CrossRef CAS PubMed.
- X. Zhang, Z. Shen, J. Liu, S. N. Kerisit, M. E. Bowden, M. L. Sushko, J. J. De Yoreo and K. M. Rosso, Nat. Commun., 2017, 8, 835 CrossRef CAS PubMed.
- Y. Cao, M. Li, M. Cheng, J. Song and Z. Hu, J. Cryst. Growth, 2014, 388, 22–28 CrossRef CAS.
- B. Calderón-Jiménez, A. R. Montoro Bustos, R. Pereira Reyes, S. A. Paniagua and J. R. Vega-Baudrit, Sci. Rep., 2022, 12, 882 CrossRef PubMed.
- A. N. Naik, S. Patra, D. Sen and A. Goswami, Phys. Chem. Chem. Phys., 2019, 21, 4193–4199 RSC.
- J. Just, C. Coughlan, S. Singh, H. Ren, O. Müller, P. Becker, T. Unold and K. M. Ryan, ACS Nano, 2021, 15, 6439–6447 CrossRef CAS PubMed.
- F. Deganello and A. K. Tyagi, Prog. Cryst. Growth Charact. Mater., 2018, 64, 23–61 CrossRef CAS.
- R. Buonsanti, T. E. Pick, N. Krins, T. J. Richardson, B. A. Helms and D. J. Milliron, Nano Lett., 2012, 12, 3872–3877 CrossRef CAS PubMed.
- S. R. Jain, K. C. Adiga and V. R. Pai Verneker, Combust. Flame, 1981, 40, 71–79 CrossRef CAS.
- S. L. González-Cortés and F. E. Imbert, Appl. Catal., A, 2013, 452, 117–131 CrossRef.
- K. Rajeshwar and N. R. de Tacconi, Chem. Soc. Rev., 2009, 38, 1984 RSC.
- H. H. Nersisyan, J. H. Lee, J.-R. Ding, K.-S. Kim, K. V. Manukyan and A. S. Mukasyan, Prog. Energy Combust. Sci., 2017, 63, 79–118 CrossRef.
- H. Ajamein and M. Haghighi, Ceram. Int., 2016, 42, 17978–17989 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909 RSC.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- E. Delfani, A. Khodabakhshi, S. Habibzadeh, L. Naji and M. R. Ganjali, RSC Adv., 2022, 12, 907–920 RSC.
- A. S. Abu Hatab, Y. H. Ahmad, M. B. Abdul Rahman and S. Y. Al-Qaradawi, RSC Adv., 2022, 12, 1694–1703 RSC.
- D.-M. Liu, T. Troczynski and W. J. Tseng, Biomaterials, 2002, 23, 1227–1236 CrossRef CAS PubMed.
- S. J. Kim, S. Yoon and H. J. Kim, Jpn. J. Appl. Phys., 2014, 53, 02BA02 CrossRef CAS.
- J. Zhang, Q. Guo, Y. Liu and Y. Cheng, Ind. Eng. Chem. Res., 2012, 51, 12773–12781 CrossRef CAS.
- K. V. Manukyan, A. Cross, S. Roslyakov, S. Rouvimov, A. S. Rogachev, E. E. Wolf and A. S. Mukasyan, J. Phys. Chem. C, 2013, 117, 24417–24427 CrossRef CAS.
- F. Li, J. Ran, M. Jaroniec and S. Z. Qiao, Nanoscale, 2015, 7, 17590–17610 RSC.
- B. Abebe and H. C. A. Murthy, RSC Adv., 2022, 12, 5816–5833 RSC.
- Y. Sun, B. Mayers, T. Herricks and Y. Xia, Nano Lett., 2003, 3, 955–960 CrossRef CAS.
- S. Xie, H.-C. Peng, N. Lu, J. Wang, M. J. Kim, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2013, 135, 16658–16667 CrossRef CAS PubMed.
- T. E. Williams, D. Ushizima, C. Zhu, A. Anders, D. J. Milliron and B. A. Helms, Chem. Commun., 2017, 53, 4853–4856 RSC.
- B. A. Helms, T. E. Williams, R. Buonsanti and D. J. Milliron, Adv. Mater., 2015, 27, 5820–5829 CrossRef CAS PubMed.
- P. W. Dunne, A. S. Munn, C. L. Starkey, T. A. Huddle and E. H. Lester, Philos. Trans. R. Soc., A, 2015, 373, 20150015 CrossRef PubMed.
- Y. Wu, D. Wang and Y. Li, Sci. China Mater., 2016, 59, 938–996 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- A. Kirakosyan, J. Kim, S. W. Lee, I. Swathi, S.-G. Yoon and J. Choi, Cryst. Growth Des., 2017, 17, 794–799 CrossRef CAS.
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC.
- D. Qin, Z. He, P. Li and S. Zhang, Front. Chem., 2022, 10, 1–11 Search PubMed.
- P. W. Dunne, A. S. Munn, C. L. Starkey, T. A. Huddle and E. H. Lester, Philos. Trans. R. Soc., A, 2015, 373, 20150015 CrossRef PubMed.
- Y. Xia, X. Xia and H.-C. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022–2051 CrossRef CAS PubMed.
- Y.-S. Jun, Y. Zhu, Y. Wang, D. Ghim, X. Wu, D. Kim and H. Jung, Annu. Rev. Phys. Chem., 2022, 73, 1–25 CrossRef PubMed.
- B. Jin, Z. Liu and R. Tang, CrystEngComm, 2020, 22, 4057–4073 RSC.
- T. H. Zhang and X. Y. Liu, Chem. Soc. Rev., 2014, 43, 2324–2347 RSC.
- C. Zhu, S. Liang, E. Song, Y. Zhou, W. Wang, F. Shan, Y. Shi, C. Hao, K. Yin, T. Zhang, J. Liu, H. Zheng and L. Sun, Nat. Commun., 2018, 9, 421 CrossRef PubMed.
- W. Neng, L. Shuang-ying, X. Jun and M. Martini, Nanotechnology, 2016, 27, 205605 CrossRef PubMed.
- S. Gao, S. Hao, Z. Huang, Y. Yuan, S. Han, L. Lei, X. Zhang, R. Shahbazian-Yassar and J. Lu, Nat. Commun., 2020, 11, 2016 CrossRef CAS PubMed.
- Z. Ou, Z. Wang, B. Luo, E. Luijten and Q. Chen, Nat. Mater., 2020, 19, 450–455 CrossRef CAS PubMed.
- X. Deng, Z. Huang, W. Wang and R. N. Davé, Adv. Powder Technol., 2016, 27, 1971–1979 CrossRef CAS.
- H. Chang, B. H. Kim, H. Y. Jeong, J. H. Moon, M. Park, K. Shin, S. I. Chae, J. Lee, T. Kang, B. K. Choi, J. Yang, M. S. Bootharaju, H. Song, S. H. An, K. M. Park, J. Y. Oh, H. Lee, M. S. Kim, J. Park and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 7037–7045 CrossRef CAS PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- S. Ram and H.-J. Fecht, J. Phys. Chem. C, 2011, 115, 7817–7828 CrossRef CAS.
- A.-Q. Zhang, L.-J. Cai, L. Sui, D.-J. Qian and M. Chen, Polym. Rev., 2013, 53, 240–276 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 4597–4601 CrossRef CAS PubMed.
- T. H. Yang, Y. Shi, A. Janssen and Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 15378–15401 CrossRef CAS PubMed.
- Y. Xiong, J. M. McLellan, Y. Yin and Y. Xia, Angew. Chem., Int. Ed., 2007, 46, 790–794 CrossRef CAS PubMed.
- J. M. Yuk, J. Park, P. Ercius, K. Kim, D. J. Hellebusch, M. F. Crommie, J. Y. Lee, A. Zettl and A. P. Alivisatos, Science, 2012, 336, 61–64 CrossRef CAS PubMed.
- S. Y. Luchkin, S. A. Lipovskikh, N. S. Katorova, A. A. Savina, A. M. Abakumov and K. J. Stevenson, Sci. Rep., 2020, 10, 8550 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- E. Novitskaya, J. P. Kelly, S. Bhaduri and O. A. Graeve, Int. Mater. Rev., 2021, 66, 188–214 CrossRef CAS.
- G. Xanthopoulou, O. Thoda, S. Roslyakov, A. Steinman, D. Kovalev, E. Levashov, G. Vekinis, A. Sytschev and A. Chroneos, J. Catal., 2018, 364, 112–124 CrossRef CAS.
- E. Carlos, R. Martins, E. Fortunato and R. Branquinho, Chem.–Eur. J., 2020, 26, 9099–9125 CrossRef CAS PubMed.
- Y. Gao, F. Meng, X. Li, J. Z. Wen and Z. Li, Catal. Sci. Technol., 2016, 6, 7800–7811 RSC.
- J. D. Morris, S. S. Daood, S. Chilton and W. Nimmo, Fuel, 2018, 230, 452–473 CrossRef CAS.
- B. Cai, H. Liu and W. Han, Catalysts, 2020, 10, 1027 CrossRef CAS.
- R. Branquinho, A. Santa, E. Carlos, D. Salgueiro, P. Barquinha, R. Martins and E. Fortunato, in Developments in Combustion Technology, InTech, 2016 Search PubMed.
- L. E. Shea, J. McKittrick, O. A. Lopez and E. Sluzky, J. Am. Ceram. Soc., 1996, 79, 3257–3265 CrossRef CAS.
- G. Avgouropoulos, J. Papavasiliou and T. Ioannides, Chem. Eng. J., 2009, 154, 274–280 CrossRef CAS.
- J. C. Toniolo, A. S. Takimi and C. P. Bergmann, Mater. Res. Bull., 2010, 45, 672–676 CrossRef CAS.
- S. Challagulla and S. Roy, J. Mater. Res., 2017, 32, 2764–2772 CrossRef CAS.
- J. Wang and L. L. Shaw, J. Mater. Sci.: Mater. Med., 2009, 20, 1223–1227 CrossRef CAS PubMed.
- H. Sadabadi, S. R. Allahkaram, A. Kordijazi, O. Akbarzadeh and P. K. Rohatgi, Eng. Rep., 2021, 3, 1–7 Search PubMed.
- M. Dara, M. Hassanpour, H. A. Alshamsi, M. Baladi and M. Salavati-Niasari, RSC Adv., 2021, 11, 8228–8238 RSC.
- E. A. Cochran, K. N. Woods, D. W. Johnson, C. J. Page and S. W. Boettcher, J. Mater. Chem. A, 2019, 7, 24124–24149 RSC.
- S. Yuvaraj, L. Fan-Yuan, C. Tsong-Huei and Y. Chuin-Tih, J. Phys. Chem. B, 2003, 107, 1044–1047 CrossRef CAS.
- A. A. Voskanyan, K.-Y. Chan and C.-Y. V. Li, Chem. Mater., 2016, 28, 2768–2775 CrossRef CAS.
- L. Junliang, Z. Wei, G. Cuijing and Z. Yanwei, J. Alloys Compd., 2009, 479, 863–869 CrossRef.
- B. Pourgolmohammad, S. M. Masoudpanah and M. R. Aboutalebi, J. Magn. Magn. Mater., 2017, 424, 352–358 CrossRef CAS.
- R. Kanakala, G. Rojas-George and O. A. Graeve, J. Am. Ceram. Soc., 2010, 93, 3136–3141 CrossRef CAS.
- X. Wang, M. Qin, F. Fang, B. Jia, H. Wu, X. Qu and A. A. Volinsky, Ceram. Int., 2018, 44, 4237–4247 CrossRef CAS.
- B. Niu, F. Zhang, H. Ping, N. Li, J. Zhou, L. Lei, J. Xie, J. Zhang, W. Wang and Z. Fu, Sci. Rep., 2017, 7, 3421 CrossRef PubMed.
- S. Gu, M. Qin, H. Zhang, J. Ma, H. Wu and X. Qu, CrystEngComm, 2017, 19, 6516–6526 RSC.
- H. Li, S. Yin, Y. Wang, T. Sekino, S. W. Lee and T. Sato, J. Catal., 2013, 297, 65–69 CrossRef CAS.
- B. Abebe, E. A. Zereffa, H. C. A. Murthy and C. R. Ravikumar, J. Mater. Sci.: Mater. Electron., 2021, 32, 7778–7790 CrossRef CAS.
- H. Lachheb, F. Ajala, A. Hamrouni, A. Houas, F. Parrino and L. Palmisano, Catal. Sci. Technol., 2017, 7, 4041–4047 RSC.
- V. P. Dinesh, P. Biji, A. Ashok, S. K. Dhara, M. Kamruddin, A. K. Tyagi and B. Raj, RSC Adv., 2014, 4, 58930–58940 RSC.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed.
- J. Gui, M. Ji, J. Liu, M. Xu, J. Zhang and H. Zhu, Angew. Chem., 2015, 127, 3754–3758 CrossRef.
- G. Chen, G. Song, W. Zhao, D. Gao, Y. Wei and C. Li, Chem. Eng. J., 2018, 352, 64–70 CrossRef CAS.
- A. Wrzesińska, A. Khort, M. Witkowski, J. Szczytko, J. Ryl, J. Gurgul, D. S. Kharitonov, K. Łątka, T. Szumiata and A. Wypych-Puszkarz, Sci. Rep., 2021, 11, 22746 CrossRef PubMed.
- O. Thoda, G. Xanthopoulou, G. Vekinis and A. Chroneos, Adv. Eng. Mater., 2018, 20, 1800047 CrossRef.
- A. Kumar, E. E. Wolf and A. S. Mukasyan, AIChE J., 2011, 57, 2207–2214 CrossRef CAS.
- B. Abebe, Mater. Res. Express, 2021, 8, 125005 CrossRef CAS.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- Z. Lu and Y. Yin, Chem. Soc. Rev., 2012, 41, 6874 RSC.
- L. Wang, J. Hu, H. Liu, Q. Wei, D. Gong, L. Mo, H. Tao and C. Zhang, Catalysts, 2020, 10, 523 CrossRef CAS.
- M. Zarezadeh Mehrizi, S. Ahmadi, R. Beygi and M. Asadi, Russ. J. Non-Ferrous Metals, 2018, 59, 111–116 CrossRef.
- L. Wang and H. Liu, Catal. Today, 2018, 316, 155–161 CrossRef CAS.
- W. Kang, D. O. Ozgur and A. Varma, ACS Appl. Nano Mater., 2018, 1, 675–685 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |