
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompetitive fluorescence immunoassay for the rapid qualitative screening and accurate quantitative analysis of ketamine
Jie Cao *abcd,
Mingjie Li*e and
Xiao-Ying Chenf
*abcd,
Mingjie Li*e and
Xiao-Ying Chenf
aScientific Research and Experiment Center, Fujian Police College, Fuzhou 350007, China. E-mail: caojie@fjpsc.edu.cn
bFuzhou University Postdoctoral Research Station of Chemistry, Fuzhou University, Fuzhou 350108, China
cFujian Police College Judicial Expertise Center, Fuzhou 350007, China
dRegional Counter-Terrorism Research Center, Fujian Police College, Fuzhou 350007, China
eAdam Smith Business School, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: lemmalee@163.com
fCollege of Environment & Safety Engineering, Fuzhou University, Fuzhou 350108, China
First published on 26th October 2022
Abstract
In this paper, a sensitive and specific competitive fluorescence immunoassay (CFIA) method was developed for the qualitative and quantitative analysis of ketamine (KET). A novel competitive model in which ketamine hapten (KET-BSA), coated on microporous plates, competed with ketamine antigen (KET-Ag) in actual samples to bind fluorescein isothiocyanate-labeled antibody (KET-Ab) could be used for rapid and indirect quantitative analysis of KET in human urine, blood, or sewage. In the CFIA method, KET concentration in the sample negatively correlated with the detected fluorescence intensity. The linear correlation coefficient of the competitive quantitative equation was 0.992, the linear range was 0.01–0.5 μg mL−1, and the limit of detection (LOD) was 0.1 pg mL−1. The specificity results showed that the cross-reaction rate of norketamine was less than 10%. Recoveries of spiked samples at low, medium, and high concentrations ranged from 96% to 117%. The CFIA method and classical gas chromatography–tandem mass spectrometry (GC-MS/MS) were used to detect the actual samples simultaneously. The relative deviation of the quantitative results was less than 10%. The LOD value of KET by CFIA was four orders of magnitude lower than that by GC-MS/MS. Additionally, CFIA had great advantages over GC-MS/MS in terms of sample pretreatment and economic investment. In conclusion, this study provided a targeting detection platform for KET, which achieved a rapid, portable, and sensitive analysis of trace KET in various materials.
1. Introduction
Ketamine (KET) is an anesthetic and analgesic agent, achieving its anesthetic effect by inhibiting the N-methyl-D-aspartic acid neurotransmitter receptor and cholinomimetic function transmission in central and peripheral nervous systems.1,2 Recent clinical studies have found the rapid, effective, and lasting antidepressant effects of KET, leading to highlighting of its potential in the field of depression treatment.3However, the biggest safety issue in the clinical application of KET is its unique toxic side effects and the adverse events caused by KET-induced neuropsychiatric disorders. Furthermore, the toxic side effects have been positively correlated with its dosage; the larger the dosage, the stronger the toxic side effects. The abuse of this drug can cause hallucinations and lead to mental disorders. Large doses could lead to spasms and death.4 Approximately 70–90% of KET is metabolized in the liver and excreted in the urine, and its main metabolite in the human body is norketamine. Nevertheless, studies found that KET could not be completely metabolized in the human body, and only part of it was metabolized into norketamine, with KET being excreted in the form of KET itself and norketamine.5–7 Therefore, the rapid and sensitive monitoring of KET concentrations in the blood or urine samples of patients treated with KET as a drug is very necessary.
Drug abuse and drug trafficking are global public hazards of great concern. The illicit use of KET traces back to the late 20th century, and its use for nonmedical purposes has increased since then.8 Southeast Asia is the region with the most serious KET abuse, accounting for 86.2% of the world's KET seizures.9 Over the past decade, KET has also become popular as a drug in China, representing an increasing proportion of the drug market. By 2011, KET was the third most commonly used illicit drug.10 Therefore, it is very important to develop a new method for KET analysis that is more simple, rapid, and accurate. Using the new method, police officers can not only crack down on drug crimes but also monitor people with KET addiction.
Recent studies detected high concentrations of KET in sewages in Taiwan, Hong Kong, and Shenzhen.11–14 Khan et al. also found KET in Beijing's sewage treatment plant but at lower concentrations.11 KET was also widely detected in surface water but in low concentrations, usually in the range of several nanograms to hundreds of nanograms per liter.12,15 Thus, the use of ultrasensitive detection methods to regularly monitor the concentration of KET in sewage is key to assessing and early warning of drug risk.
Due to the universality, importance, and urgency of qualitative and quantitative analyses of KET, almost all known analytical methods have been used for its detection. The classical methods for analyzing KET are chromatography, including gas chromatography (GC);16–19 liquid chromatography (LC);19–21 gas chromatography-mass spectrometry,22,23 and liquid chromatography-mass spectrometry (LC-MS).24–27 Gas chromatography-mass spectrometry was further developed into gas chromatography-single quadrupole mass spectrometry (GC-MS) and gas chromatography-tandem mass spectrometry (GC-MS/MS). Although these techniques offer excellent accuracy and sensitivity, they also display some disadvantages, including lack of portability, long test times, cumbersome and solvent-consuming sample preparation steps, the need for skilled operators, high operation and maintenance costs, and nonsuitability for on-site analyses. Other commonly used methods included capillary electrophoresis,28,29 fluorescence immunoassay (FIA),30,31 electrochemical methods,32,33 surface plasmon resonance (SPR),34 Raman spectroscopy,35–37 and infrared spectroscopy (IR).38–41 Among them, capillary electrophoresis has high separation efficiency but shows the same disadvantages as chromatography. The electrochemical method is simple, has a low cost, and can achieve on-site detections. Nevertheless, it shows poor anti interference abilities, high detection system requirements, complex electrode modifications, and poor stability and reproducibility. The SPR method has the advantages of a fast response, high sensitivity and specificity, and does not require complex sample preparation steps. However, it requires special instruments, shows high requirements for the testing system, and is strongly technical. Raman spectroscopy and IR were simple, fast, sensitive, and nondestructive. However, Raman spectroscopy often had a strong fluorescent background, so surface-enhanced Raman spectroscopy should be made from substrate materials. Moreover, careful and invasive sample preparation is still required, and it is not suitable for routine analysis. IR technology is simple and fast to analyze seized solid synthetic drugs, but the analysis of liquid samples with a complex matrix is complicated by matrix background interference. Among many analytical methods, FIA became a hotspot in the determination of addictive drugs, because of its high sensitivity, simplicity, rapidity, ease of use, and portability.30,31,42–45
According to the literature, Rafał Walczak et al. developed a miniature image-based fluorescence immunochromatography system for the real-time detection of cocaine in human sweat. The LOD of cocaine in sweat was 1 ng mL−1.42 Priya Mishra et al. developed an immunochromatographic dipstick for the rapid screening of heroin metabolites in urine samples. The LOD of the dipstick test was 5 ng mL−1.43 Emine Guler et al. designed a fluorescence sensory device for specific recognition and rapid analysis of cocaine and its metabolite benzoylecgonine (BE). The LOD for cocaine and BE were 0.138 nM and 1.66 μM, respectively.44 Sijia Liang and her colleagues developed a new methamphetamine detection device based on time-resolved FIA methamphetamine test strips. The device could be used to screen methamphetamine abusers through their hair. The LOD of the device was 3.34 × 10−9 mol L−1.45 Qianhua Li et al. introduced a KET electrochemiluminescence (ECL) immunosensor, which can detect KET in plasma samples rapidly and sensitively. The LOD of KET was 0.067 ng mL−1.30 Additionally, Jiuchuan Guo and others had developed a cloud-supported smartphone fluorescence sensor for the quantitative detection of KET in hair samples based on the lateral flow immunoassay, in which UCNPs were introduced as fluorescent labels. The LOD of KET was 1 ng mL−1.31 In summary, although various FIA methods reported in the literature have the advantages of simplicity, rapidity, portability, and suitability for qualitative screening, they also have the disadvantages of high false-positive rates, inaccurate quantitative results, or high LOD for quantitative detection. The problem regarding the quantitative analysis of ultratrace substances in matrix complex samples had remained unsolved to date.
Herein, a new platform for KET specificity analysis was constructed using a CFIA method, using a new idea of competition. The ketamine antibody (KET-Ab) was labeled with fluorescein isothiocyanate (FITC) as the result signal molecule. KET in the sample solution as antigen (KET-Ag) competed with the hapten (KET-BSA) coated on the microporous plates and bound to FITC-labeled KET-Ab. The visual competitive reaction process was shown in schematic diagram (see Fig. 1). This method provided an effective platform for the qualitative and quantitative analysis of ultratrace substances in matrix complex samples and could be used for the analysis of other addictive drugs, alone or mixed.
 | ||
| Fig. 1 Schematic diagram of a competitive reaction process. | ||
2. Experimental
2.1 Statement of human and animal rights
All authors confirm that, first, no human experiments were included in this study. Second, biological samples, including blood and urine, were provided by the Fujian Police College Judicial Expertise Center, and the informed consent of the individuals tested was obtained. Finally, the section of this paper covering antibody acquisition refers to animal experiments and all associated procedures and experiments were approved by the Animal Use and Management Committee of the Fujian Police College.2.2 Instrumentation
Fluorescence measurements on 96-well plates were performed on a Varioskan Flash Multimode microporous plate reader (Thermo Fisher Scientific, USA). By covalently binding KET molecules with bovine serum albumin (BSA), we prepared KET-BSA. The molecular groups of the synthesized KET-BSA were detected via IR spectra (Vertex 70, Bruker Optics, USA), and the exact molecular weight of the synthesized KET-BSA was detected via matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS, autoflex speed, Bruker, USA). KET-Ab was labeled with FITC molecular dye. The concentration of FITC-labeled KET-Ab was measured with a PerkinElmer LS55 fluorescence meter (PerkinElmer Co., Ltd, USA).The pH of the solution was measured using Denver UB-10 (Denver Instruments, USA). All immune experiments were performed in an incubator, and the constant temperature accuracy was 0.1 °C. Ultrapure water was secondary pure water prepared by tap water, successively, through an Aquapro Water Purifier (Chongqing, China) and Milli-Q IQ 7000 Lab Water System (MA, USA).
2.3 Reagents
Reference substances of ketamine hydrochloride (KET·HCl), morphine, heroin, codeine, cocaine, norketamine, and ephedrine were purchased from Shanghai Yuansi Standard Science and Technology Co., Ltd (Shanghai, China). Ovalbumin (OVA), FITC, Tween 20, and BSA were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ethyl acetate, sodium hydroxide (NaOH), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), disodium hydrogen phosphate (Na2HPO4), potassium dihydrogen phosphate (KH2PO4), sodium chloride (NaCl), and potassium chloride (KCl) were all analytically pure and were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The microporous plate (96-well) used in the experiment was from NUNC, Denmark.The buffer solution and its composition involved in the experiment were as follows. Carbonate buffer (CBS), concentration 0.05 mol L−1, pH 9.6, was prepared by weighing 1.59 g sodium carbonate and 2.93 g sodium bicarbonate and adding a constant volume of ultrapure water to 1000 mL. Phosphate buffer (PBS), pH 7.5, was prepared by weighing 8 g sodium chloride, 2.9 g disodium hydrogen phosphate dodecahydrate, 0.2 g potassium dihydrogen phosphate, and 0.2 g potassium chloride and then adding a constant volume of ultrapure water to 1000 mL. PBST buffer with pH 7.2 was prepared by dissolving Tween-20 to obtain 0.05% Tween-20 (v/v) in PBS buffer. KET-BSA was dissolved in CBS buffer to prepare a stock solution. KET-Ab labeled FITC fluorescent probe was prepared and dissolved in PBS solution. The cleaning solution for the 96-well plate was PBST buffer. OVA solution, used as a blocker, was prepared by dissolving OVA to obtain 0.1% OVA (v/v) in PBS buffer. This reagent was used to prevent the nonspecific adsorption of KET-BSA coated on microporous plates.
All experimental data in this paper were the average values of five parallel experiments. The chemical reagents and drugs used in the experiment were analytically pure, and we used ultrapure water.
2.4 KET-BSA preparation
KET-BSA was synthesized as shown in Fig. 2. KET-BSA is a biological macromolecule with immunogenicity formed by coupling the small molecule KET (MW 285) with macromolecular BSA. | ||
| Fig. 2 KET-BSA synthesis process diagram. | ||
Step 1: synthesis of carboxyl ketamine (KET-COOH). KET·HCl was used as raw material. After dissociation, the ester group was introduced by a substitution reaction with ethyl bromoacetate to obtain the ester of KET and then hydrolyzed under alkaline conditions to obtain KET-COOH.
KET·HCl was dissolved in ultrapure water and ammonia was added to adjust the pH value to 10. The organic solvent dichloromethane was used for extraction, and the organic phase extracted three times was collected. The organic phase was then subjected to the removal of water with anhydrous magnesium sulfate, filtration to remove impurities, and reduced pressure evaporation to remove organic solvents. Finally, free KET was obtained, which was dissolved in acetone. Ethyl bromide acetate and potassium carbonate were added, and a reflux reaction was carried out at 70 °C for 16 h. Following cooling to 25 °C and filtration, the filtrate was obtained. KET ester was obtained through vacuum distillation of the filtrate. The obtained KET ester was mixed with tetrahydrofuran and methanol, evenly. A NaOH solution was added and the reaction was carried out for 16 h at 25 °C. The target organic compound KET-COOH was extracted with ethyl acetate after pH was adjusted to 7 by hydrochloric acid. The organic phase was collected and KET-COOH was obtained by vacuum distillation. After purification by high-performance liquid chromatography, the purity of KET-COOH was 99.9%.
Step 2: synthesis of KET-BSA. KET-BSA was prepared by combining KET-COOH with BSA by a carbon diimine method. KET-COOH was mixed evenly with N,N-dimethylformamide, N-hydroxysuccinimide, and cyclohexyl carbamide diimide, and the reaction was performed for 16 h at 25 °C. At the end of the reaction, the solution was centrifuged, and the supernatant was removed as solution A. The solute in solution A is compound 4 (see Fig. 2 for its molecular structure). BSA was added to the PBS buffer and mixed evenly to obtain solution B. Solution A and solution B were mixed and reacted uniformly at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5; that is, compound 4 reacted with BSA at 4 °C for 16 h according to the carbon diimine method, synthesizing KET-BSA. After dialysis with PBS buffer five times, the solution was centrifuged and the supernatant was acquired to obtain KET-BSA.
:5; that is, compound 4 reacted with BSA at 4 °C for 16 h according to the carbon diimine method, synthesizing KET-BSA. After dialysis with PBS buffer five times, the solution was centrifuged and the supernatant was acquired to obtain KET-BSA.
2.5 Verification of KET-BSA
The molecular structure of KET-BSA was identified by IR and MS.First, 1 mg of KET-BSA, BSA, and KET solid powder (purity >95%) were mixed with 100 mg of dried KBr powder (preplaced in the muffle furnace and dried at 200 °C for 10 h) and then placed in the agate mortar for grinding and screening. The screened powder was pressed under the infrared lamp. The KBr tablet pressing method was used to measure it in the wavenumber range of 400–4000 cm−1 by infrared spectrometry, as shown in Fig. 3a.
 | ||
| Fig. 3 (a) IR spectrum of K, K-BSA, and BSA (b) MALDI-TOF-MS spectra of K-BSA and BSA (c) fluorescence spectrum of FITC-labeled KET-Ab. | ||
Second, the conjugation ratio of KET-BSA was determined via MALDI-TOF-MS. MALDI-TOF-MS can be used for the detection of positively charged peptide ions after soft ionization. The soft ionization process is divided into three steps. In the first step, the laser energy is absorbed by the matrix; in the second step, the matrix energy is transferred to the polypeptide; and in the third step, the energy absorbed polypeptide is transformed into charged polypeptide ions by proton transfer. Since the ionization process of the synthesized KET-BSA is similar to that of BSA, it is inferred that the proton transfer associated with the ionization process of KET-BSA is caused by the bound BSA in the molecule. The proteins (0.5 μL BSA or KET-BSA) were mixed with (0.5 μL) sinapinic acid (10 mg mL−1 in 50:50 ACN/H2O with 0.1% TFA), and then pointed it on a polished steel MTP 384 target plate for MALDI-TOF. The protein matrix samples were allowed to crystallize at 25 °C and fed it to an ion source for detection. In the same way, we pointed a BSA standard solution on adjacent target sites for instrument calibration. The raw data was processed using flexAnalysis software. See Fig. 3b.
2.6 KET-Ab preparation
This process was accomplished in four steps. Step 1: BALB/c female mice about 8 weeks old were selected with a body mass of about 20 g. KET-BSA was mixed and emulsified with equal volume of Freund's complete adjuvant, and injected into the armpit, groin, and back of BALB/c mice at multiple points (50 μg per mouse) for basic immunization. At intervals of 2 weeks, the mice were given three booster doses of 50 μg of the above antigens with incomplete Freund's adjuvant. Two weeks after the last booster immunization, serum antibody titers of immunized mice were measured by indirect ELISA. When the antibody titers reached 1:106, the same amount of antigen was injected into the tail vein for additional immunization. Step 2: first, the feeding cells could be obtained by the suspension, centrifugation, and precipitation of mouse celiac cells. Second, mouse myeloma cells (SP2/0) with good growth status and suitable for cell fusion were obtained by subculture. Third, the preparation of the IMDM culture medium, including lymphocytes, was completed by obtaining the immune-treated lymph nodes of mice and then through grinding, centrifugation, and precipitation. Finally, hybridoma cells were obtained by a fusion of lymphocytes, myeloma cells, and feeding cells with polyethylene glycol. Step 3: the hybridoma cells were screened using indirect competitive ELISA with solid phase antigen. KET-BSA was used as the coating antigen. Sera from immunized mice were used as positive control. The supernatant of SP2/0 myeloma cell culture was used as the negative control. The hybridoma cell line with the highest competitive inhibition rate was screened for clonal culture. Clonal culture was carried out via the limited dilution method. Specifically, the clone cells were blown evenly and a microdrop was taken into the culture flask. The number of cells was accurately counted under an inverted microscope and diluted to 70 cells per milliliter. Each 1 mL cell solution was diluted 20 times further. The cell solution was then inoculated into 96 well culture plates for subcloning till the positive rate of monoclonal antibody (McAb) detection in the culture plate reached 100%. Next, the McAb cells were picked and anti-KET McAb was purified after culture expansion. Step 4: after repeated culture, anti-KET McAb with a positive rate of 100% were obtained and refined by affinity column such as the HiTrap PROTEIN G HP. Finally, purified KET-Abs was prepared.
2.7 FITC-labeled KET-Ab
To obtain fluorescent antibodies, KET-Abs was conjugated with FITC. According to the literature,46 the molar concentration ratio of KET-Ab to FITC was 10:1. During labeling, KET-Ab was dispersed in PBS buffer, and the solution concentration was 5 mg mL−1. FITC was dissolved in CBS buffer at a concentration of 0.5 mg mL−1. When mixing, the KET-Ab solution was first added to the reaction bottle. Then, the FITC solution was slowly added and stirred for 2 h at 25 °C in the dark. The FITC-labeled KET-Abs was further purified using a Sephadex G-25 column. The purified 1 mL solution was collected, and the absorbance was measured at 280 nm. The ratio of FITC to protein (F/P) was calculated according to the formula below:47| Molar(F/P) = (2.77 × A495)/(A280 − 0.35 × A495). |
In the above formula, A495 was the absorbance value of the FITC dye. Only the portion of the solution with an F/P coefficient between 1 and 4 was collected to avoid protein over labeling, which would lead to increased nonspecific binding and reduced quantum yield due to the fluorophore self-quenching effect;47–49 see Fig. 3c. The approximate molar ratio of FITC to KET-Ab (nFITC/nKET-Ab) was estimated to be 7.8.
2.8 CFIA method determination process
The entire experiment was performed on a 96-well plate. In the first step, the KET-BSA solution (50 μg mL−1, 100 μL) was slowly dropped into each micropore, coated at 4 °C for 12 h, and then washed with PBST 3 times, for 3 min each time. In the second step, the nonspecific binding sites of KET-BSA were blocked with a 1% OVA solution. We then dropped 350 μL of OVA solution into each well and kept the temperature at 37 °C for 0.5 h, followed by three PBST washes of 3 min each. In the third step, after mixing the FITC-labeled KET-Ab (diluted with PBS, 50 μg mL−1, 100 μL) with the sample solution or standard KET solutions at different concentrations (dissolved in PBS), the mixture was added to the KET-BSA-coated micropores. Then, the incubation was carried out under the optimal temperature and time conditions (37 °C for 2 h). In the fourth step, the optimal excitation and emission wavelengths of the FITC dye were set on the microporous plate reader. The fluorescence intensity of the samples was read by the microporous plate reader. The best excitation wavelength (λex) was 488 nm, and the best emission wavelength (λem) was 525 nm. The fluorescence intensity was negatively correlated with KET concentration, forming a competitive curve. The value of each point on the curve was the average of five parallel experiments. The IC50 value was defined as the concentration of KET-Ag in the corresponding sample at 50% of the maximum fluorescence intensity value. LOD was defined as the KET concentration in which its relative fluorescence intensity was three times the background fluorescence intensity. Maximum fluorescence intensity (Fmax) was defined as the fluorescence intensity at which all the coated KET-BSA on the microporous plate was bound to KET-Ab. In this case, there was no KET-Ag in the sample, and there was no competitive binding of KET-Ag and KET-BSA to the KET-Ab. Fluorescence intensity (F) was defined as the fluorescence value at which partially coated KET-BSA on the microporous plate bound to KET-Ab. In this case, part of the KET-Ag in the sample competed with the KET-BSA for binding KET-Ab. Background fluorescence intensity (F0) was defined as the fluorescence intensity at which the coated KET-BSA on the microporous plate was not bound to KET-Ab at all. In this case, a large number of KET-Ags in the sample was bound directly to KET-Ab, and the KET-BSA could not compete with the KET-Ag for binding the KET-Ab. The relative fluorescence intensity was calculated by F − F0.2.9 GC-MS/MS determination
This method was recognized as an accurate method for the measurement of KET in various fields. This method had lower LOD and more accurate quantitative results than the common GC-MS method. The actual KET sample was measured according to the GC-MS/MS method, which was used to compare and evaluate the qualitative and quantitative results of the new CFIA method. The samples used in this study were provided by the Fujian Police College Judicial Expertise Center. Samples to be tested were stored in sealed glass bottles, and the sample volume was not less than 5 mL. First, urine, serum, or whole blood samples (1 mL) were placed in a centrifuge tube (10 mL) for sample pretreatment. Second, the samples were adjusted to pH 13 with 4 mol per L NaOH and extracted twice with 2 mL ethyl acetate. Third, during extraction, the sample was fully mixed with ethyl acetate and centrifuged at 10000 rpm for 3 min to break the emulsification and promote stratification. The organic layer, extracted twice, was transferred to another glass tube and dried with nitrogen at 40 °C. Finally, methanol (200 μL) was used to dissolve the residue at the bottom of the tube. The above solution (1 μL) was used for GC-MS/MS analysis.
The GC-MS/MS instrument used for chromatographic analysis was Agilent 7890A/7000QQQB. The chromatographic column used was the HP-5 capillary column (30 m × 0.25 mm × 0.25 μm). The oven temperature program was as follows: 120 °C (1 min) to 250 °C (1 min) at a ramp of 20 °C min−1. Helium was used as the carrier gas at a constant flow rate of 1 mL min−1. The split injection mode was used, with a 1:10 split ratio. The injector temperature was 230 °C. For the MS measurement, ionization was executed by electron ionization at 70 eV. The temperatures of the tandem quadrupole and ion source were held at 150 °C and 230 °C, respectively. The MS/MS system was set in multiple response monitoring (MRM) mode and high-purity nitrogen was used as collision gas to break the characteristic precursor ions of the target compound into secondary product ions. Qualitative and quantitative analyses using the MRM mode were carried out by two or three characteristic precursor-product ion pairs of the target compound at optimal collision energies. The ion pairs with the highest peak area were quantitative, and the rest were qualitative. The optimum MS/MS method of KET detection was mainly to determine the parameters of its characteristic precursor-product ion pair and the corresponding collision energy so that the method had high sensitivity and specificity. The best three KET precursor-product ion pairs were 180–116.1, corresponding to CE 25 V (quantitative ion pair); 180–151, corresponding to CE 10 V (qualitative ion pair); and 180–145.1, corresponding to CE 10 V (qualitative ion pair). The retention time of KET was 7.373 min, as shown in Fig. 4.
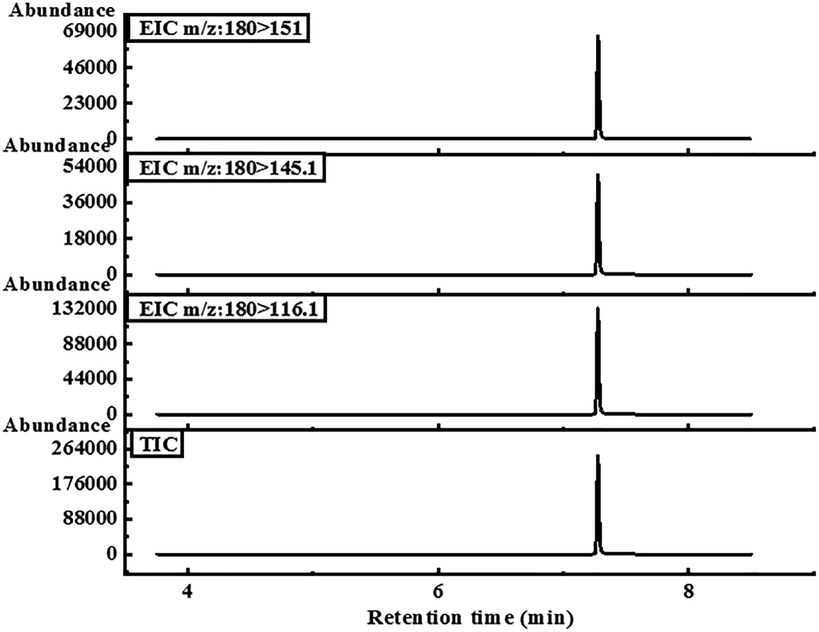 | ||
| Fig. 4 The chromatograms of three characteristic precursor-product ion pairs and total ion flow chromatograms of KET were determined via GC-MS/MS under optimal chromatographic conditions. | ||
3. Results and discussion
3.1 KET-BSA confirmation analysis
The IR spectra of KET-BSA, BSA, and KET were measured by solid KBr tablet, as shown in Fig. 3a. The successful synthesis of KET-BSA was judged by the similarity of absorption peaks of the characteristic groups in the infrared absorption spectra of the three above compounds. First, the infrared spectrum of KET-BSA was compared with that of BSA; the two had similar absorption peaks in the regions around 2800–3400 cm−1 and 1500–1700 cm−1, respectively. These wave number ranges were within the typical absorption regions of amino acids in BSA, which suggested that BSA had been covalently bound in the newly synthesized KET-BSA molecule. Second, the infrared spectra of KET-BSA and KET were compared, and a similar absorption peak was found, which was within the region of 600–1200 cm−1. Since this wave number range is the characteristic absorption region of KET, there was no significant absorption of BSA. Since the characteristic absorption peak of the KET functional group appears in this wave number range, it indicates that KET was covalently bound in the newly synthesized KET-BSA molecule. The abovementioned results confirmed the successful synthesis of KET-BSA from the perspective of infrared absorption by characteristic functional groups (see Fig. 3a).Additionally, MALDI-TOF-MS was used to further verify the successful synthesis of KET-BSA from the perspective of molecular weight. In Fig. 3b, the X-axis was the mass-to-charge ratio, and the Y-axis was the intensity. When the charge was set to 1, the X-axis represented the molecular weight of the substance. KET-BSA was represented by a red line and BSA by a black line. The graphs of the two substances were generally similar; however, the red line shifted significantly to the right compared with the black line. The right shift was in the direction of increased molecular weight, indicating that the molecular weight of the newly synthesized KET-BSA was greater than that of BSA, which reconfirmed the successful synthesis of KET-BSA. The binding ratio of BSA to KET could be calculated from the molecular weight difference between KET-BSA and BSA. As shown in Fig. 3b, the molecular weights of KET-BSA and BSA were 72828.423 and 66399.708, respectively, so the ratio of BSA to KET was around 1:27.
3.2 CFIA method of conditional optimization
Because KET can be widely used in the medical field and illegally abused as an addictive drug, the qualitative and quantitative detection of KET in medical examination, drug abuse identification, and environmental sewage monitoring has become increasingly difficult. The analysis of KET in a simple, fast, portable, sensitive, and accurate manner has become a key problem to be solved urgently by all researchers.Immunoassays have always been employed in biochemical research due to their high specificity and sensitivity. Because KET is a small organic molecule, it must combine with macromolecules to obtain immunogenicity. Thus, an immunoassay must be conducted under a competitive mechanism to obtain accurate detection results. To detect KET qualitatively and quantitatively, KET and BSA proteins were first coupled to form KET-BSA. Next, FITC-labeled KET-Ab was mixed with the sample solution and then added to a microporous plate previously coated with KET-BSA. Finally, the concentration of KET in the sample was calculated by the relative fluorescence intensity of KET. In the new CFIA method, the fluorescence intensity negatively correlated with the concentration of KET in the sample. The optimization of various detection parameters of the CFIA method could improve the sensitivity and reduce the LOD value. Specific parameters included the concentration of coated KET-BSA, the time and temperature of coated KET-BSA, the pH level and ionic force of the buffer solution, the reaction time of KET-Ag/KET-Ab.
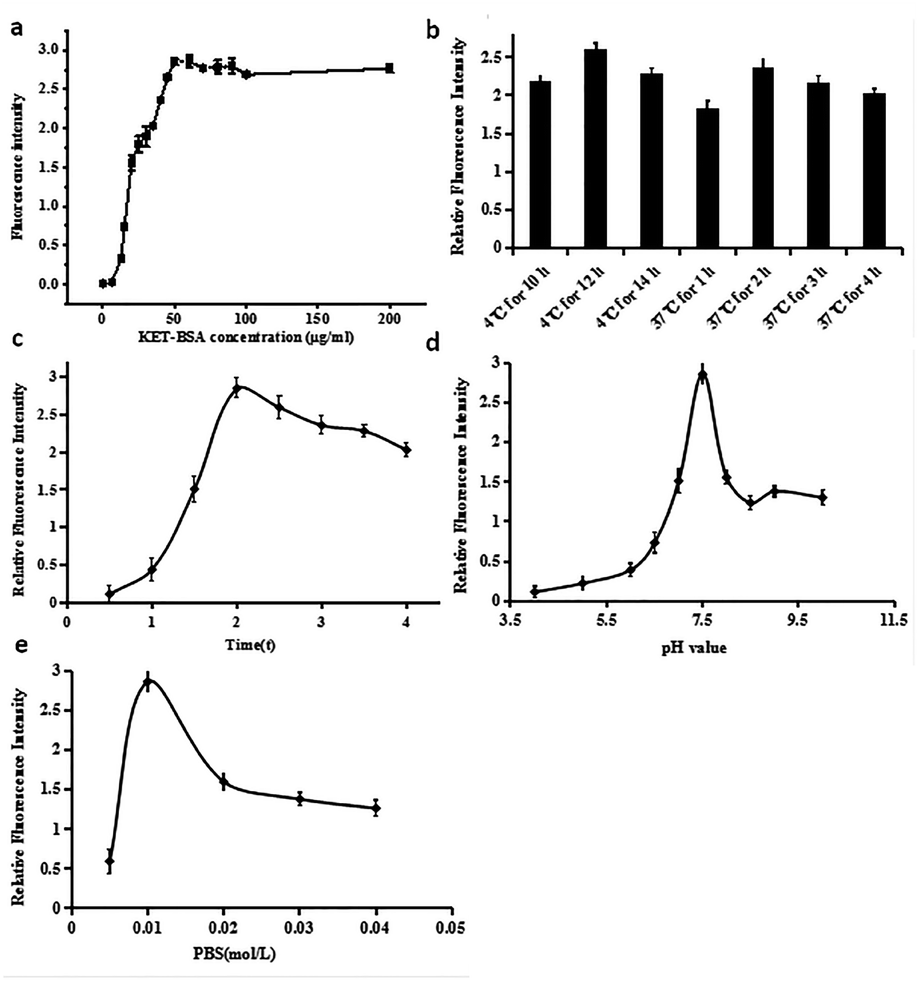 | ||
| Fig. 5 (a) Optimization curve of coated KET-BSA concentration. (b) Schematic diagram showing the immunofluorescence intensity across seven conditions which microporous plates coated with KET-BSA (including coating time and temperature). (c) Diagram showing immunofluorescence intensity against the incubation time of the KET-Ag/KET-Ab reaction. (d) Diagram showing immunofluorescence intensity against PBS buffer pH. (e) Diagram showing immunofluorescence intensity against PBS buffer salt concentration. | ||
3.3 Fluorescence competition curve and quantitative calibration equation
Under optimal conditions, the fluorescence competition curves of different KET concentrations and corresponding fluorescence intensity values were obtained. FITC-labeled KET-Abs mixed with different KET standard solutions (0, 1 × 10−7, 1 × 10−6, 1 × 10−4, 0.01, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.5, and 2.5 μg mL−1) were added dropwise onto a microporous plate of coated KET-BSA (50 μg mL−1). The fluorescence intensity and fluorescence competition curves of the KET standard solution in the range of 0–2.5 μg mL−1 are shown in Fig. 6. In this figure, the X-axis and Y-axis represent the concentration and fluorescence intensity of KET, respectively. The value of each point on the competition curve was the average of five parallel experiments. According to the competition curve, the IC50 value of 1 pg mL−1 (the concentration of chemicals used to reduce to 50% the Fmax) was determined.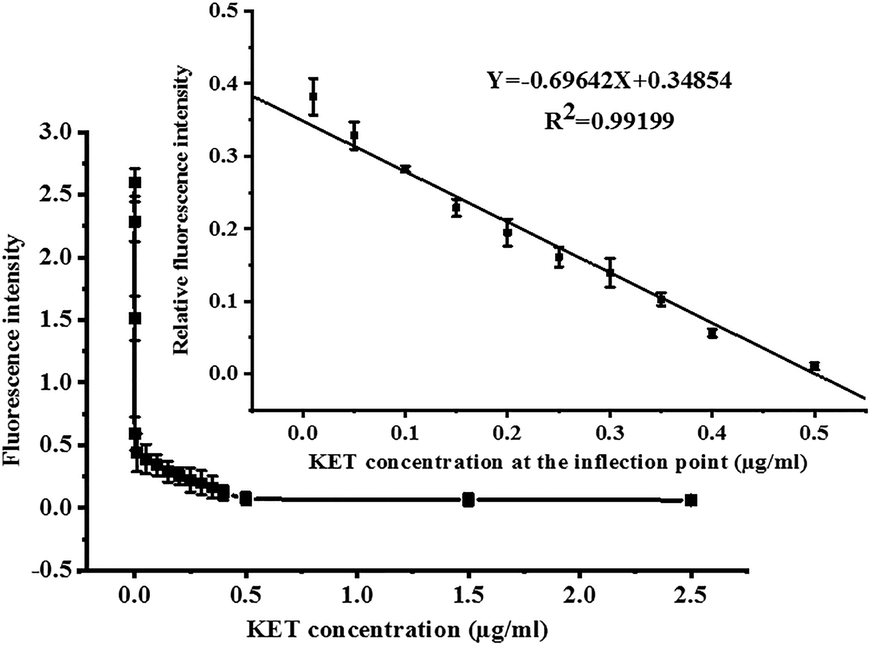 | ||
| Fig. 6 Competition curve of KET and quantitative linear equation of KET (illustration). | ||
The inflection point of the competition curve was analyzed, and a KET quantitative calibration linear equation was drawn according to KET concentration and relative fluorescence intensity at the inflection point. The quantitative equation and linear range were Y = −0.69642X + 0.34854 (where X was KET concentration and Y was relative fluorescence intensity) and 10–500 ng mL−1, respectively. The linear correlation coefficient (R2) was 0.992. The LOD for qualitative analysis was 0.1 pg mL−1 (Fig. 6 illustration). The parameters of the two KET analysis methods (GC-MS/MS method and the new CFIA method) are listed in Table 1, such as linear range, linear equation, R2, and LOD. Table 2 shows the standard addition and recovery results of KET in human urine samples.
| Method | CFIA | GC-MS/MS |
|---|---|---|
| Linear range (μg mL−1) | 0.01–0.5 | 2.0–7.5 |
| Linear regression equation | Y = −0.69642X + 0.34854 | Y = 5.7390X − 12022.8302 |
| R2 | 0.992 | 0.998 |
| LOD (ng mL−1) | 1 × 10−4 | 1.4 |
| Detection method | Drug added (μg mL−1) | Recovery (%) | RSD (%) |
|---|---|---|---|
| CFIA | 0.01 | 96.03 | 2.74 |
| 0.07 | 103.30 | 9.15 | |
| 0.5 | 101.40 | 9.49 | |
| GC-MS/MS | 2.0 | 116.20 | 7.37 |
| 5.0 | 91.56 | 3.97 | |
| 7.5 | 100.42 | 6.17 |
KET analyses of blood, urine, and sewage samples were mainly based on the GC-MS technique. Nevertheless, when traditional GC-MS was used for the analysis of matrix complex samples containing trace KET, the sensitivity and selectivity of the method was often low and the target KET could not be detected.19,20 GC-MS/MS is a reliable technology which has developed rapidly in recent years. The results of its qualitative and quantitative analyses are accurate and have high sensitivity and selectivity. Thus, the technique is effective for the targeted analysis of trace substances in matrix complex samples. According to Table 1, which compares GC-MS/MS to CFIA, the CFIA method had a wider quantitative range and a lower LOD. The R2 values of the quantitative equations of the two methods were greater than 0.99. When the R2 of the quantitative equation is greater than 0.99, the linearity of the quantitative curve is high; hence, the quantitative result is reliable. The LOD value of KET by the CFIA method was 1 × 104 times lower than that of the GC-MS/MS method. As shown in Table 2, the concentration of the standard substance added to the urine matrix was selected from a linear range of low, medium, and high of the quantitative equation. The recoveries of both the CFIA and GC-MS/MS methods were less than 10%, showing that the quantitative results of the two methods were accurate and reliable for each concentration. Nevertheless, in the actual sample detection, a complex and time-consuming sample preparation process was required for the GC-MS/MS method, whereas the new CFIA method did not need sample pretreatment. Hence, the new CFIA method was more advantageous and prospective for practical applications than the GC-MS/MS method.
3.4 CFIA method specificity
The specificity of the immunoassay is very important for the accuracy of quantitative results. Thus, the potential cross-reactivity (CR) of drug analogs with a similar molecular structure to the target KET, such as morphine, heroin, codeine, cocaine, norketamine, and ephedrine, was evaluated. These drugs are also popular among drug abusers, so multiple drugs and their metabolites could be present in the samples of urine, blood, and sewage at the same time. The solution to these new problems depended on the preparation of highly specific antibodies and the development of a highly specific immunoassay system. The CR ratio is calculated according to the following formula.
In this formula, IC50 values of all drugs were determined according to the steps described in section 2.8, under the optimized conditions. When the fluorescence intensity value was reduced to 50% of the original intensity, the concentration of the added drug was the IC50 value of the drug.
Table 3 shows the CR ratio of morphine, heroin, codeine, cocaine, norketamine, ephedrine, and KET. The immunoassay was highly specific for KET. The molecular recognition of antigens by antibodies was based on their unique structural characteristics. Among the coexisting drugs, norketamine was the metabolite of KET and exhibited the most similar structure. Thus, the CR value of norketamine was the highest (9.6%, see Table 3) and its interference with antibodies was the strongest. Additionally, the CR values of the other coexisting drugs were almost undetectable.
| Cross-reactant | CR ratio (%) |
|---|---|
| KET | 100 |
| Norketamine | 9.6 |
| Morphine | <0.05 |
| Heroin | <0.05 |
| Codeine | <0.05 |
| Cocaine | <0.05 |
| Ephedrine | <0.05 |
To summarize, we found that the CFIA method had the several advantages. First, the LOD of CFIA was significantly low, which had obvious advantages in the analysis of KET in matrix complex samples. Second, the specificity of the CFIA method for KET was high and the interference of various impurities in the sample matrix was reduced. Moreover, it had significantly enhanced qualitative and quantitative accuracy.
3.5 Actual sample analysis
Blood, urine, and sewage samples were obtained from Fujian Police College Judicial Expertise Center. Different types of samples were used for research, and the CFIA method was used for analysis under the best conditions. KET concentration in human blood and urine decreased with the increase of taking time. The actual concentration of KET-positive samples in the blood and urine of KET abusers was generally 1–6 μg mL−1. According to the quantitative range of the CFIA method, the actual sample should be diluted 10–100 times before analysis. Additionally, various samples were analyzed via GC-MS/MS. If the actual sample concentration was below the LOD of the instrument, enrichment steps could be added to the pretreatment. In Table 4, the results of various samples (including blood, urine, and sewage samples) tested by the CFIA and GC-MS/MS methods are listed. By comparison, it was found that the relative deviation of the same sample is less than 10%, regardless of the sample type and detection method used. The results show that the quantitative results for KET in the different sample types were accurate when using the new CFIA method, which was more suitable for the real-time, in situ, and rapid quantitative analysis of various samples and could be performed without sample pretreatment steps.| Sample code | Sample types | Quantitative results (μg mL−1) | Relative deviation (%) | |
|---|---|---|---|---|
| CFIA | GC-MS/MS | |||
| 1 | Urine | 2.65 | 2.83 | 6.57 |
| 2 | Urine | 1.28 | 1.4 | 8.96 |
| 3 | Blood | 4.75 | 4.58 | 3.64 |
| 4 | Blood | 1.43 | 1.39 | 2.84 |
| 5 | Sewage | 0.083 | 0.091 | 9.20 |
| 6 | Sewage | 0.045 | 0.049 | 8.51 |
4. Conclusions
To conclude, a simple, efficient, and sensitive KET analysis method was successfully established, which was the CFIA method. This new method could be used for accurate qualitative and quantitative analyses of KET in complex matrix samples such as blood, urine, and sewage samples. It could obtain the highest sensitivity, the lowest detection limit, and the best specificity by optimizing various parameters. The optimized parameters included coated KET-BSA concentration, coating time and temperature of KET-BSA; the incubation time of KET-Ag/KET-Ab reaction; and pH value and phosphate ion strength of the PBS buffer. Under optimal parameters, the fluorescence competition experiment was conducted, and the LOD of KET was obtained as 0.1 pg mL−1. The working range of the quantification curve was 10–500 ng mL−1. The LOD value of KET by CFIA was 1 × 104 times lower than that by GC-MS/MS. Using the CFIA method, KET could be recognized with high specificity. Among the six drug analogs, norketamine had the highest CR value (9.6%), because its molecular structure was most similar to that of KET. The CR values of the other five drug analogs were all less than 0.05%, indicating that they did not affect the accuracy of KET analysis results in actual samples. The classical GC-MS/MS and CFIA methods were used to analyze spiked samples and actual samples simultaneously, and the relative deviations of the quantitative results were less than 10%. The quantitative results of the new method were verified to be accurate. Altogether, the new method was simple and rapid and did not require sample pretreatment. It could be used for the qualitative and quantitative determination of KET in blood, urine, and sewage samples. Additionally, the fluorescence analysis method adopted in this study could easily integrate multitarget detection into a microarray platform, providing a new approach for the simultaneous detection of multiple addictive drugs.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was supported by the Young and middle-aged teacher foundation of Education Department of Fujian Province [grant number JAT200377], the start-up funds for new teachers in Fujian Police College, the Natural Science Foundation of Fujian Province [grant number 2022J01516], the Application innovation Program of The Ministry of Public Security of P.R.C. [grant number 2021YY21] and Special scientific research projects of Fujian Provincial Department of Finance for the directly affiliated institutions in 2020.References
- C. J. A. Morgan, H. V. Curran and Independent Scientific Committee on Drugs, Addiction, 2012, 107(1), 27–38 CrossRef PubMed.
- S. Marland, J. Ellerton, G. Andolfatto, G. Strapazzon, O. Thomassen, B. Brandner, A. Weatherall and P. Paal, CNS Neurosci. Ther., 2013, 19(6), 381–389 CrossRef CAS PubMed.
- J. Xu and H. Lei, CNS Neurosci. Ther., 2014, 20(12), 1015–1020 CrossRef CAS PubMed.
- R. Quibell, E. E. Prommer, M. Mihalyo, R. Twycross and A. Wilcock, J. Pain Symptom Manage., 2011, 41(3), 640–649 CrossRef CAS PubMed.
- K. A. Moore, J. Sklerov, B. Levine and A. J. Jacobs, J. Anal. Toxicol., 2001, 25(7), 583–588 CrossRef CAS PubMed.
- C.-Y. Chen, M.-R. Lee, F.-C. Cheng and G.-J. Wu, Talanta, 2007, 72(3), 1217–1222 CrossRef CAS PubMed.
- M. C. Parkin, S. C. Turfus, N. W. Smith, J. M. Halket, R. A. Braithwaite, S. P. Elliott, M. D. Osselton, D. A. Cowan and A. T. Kicman, J. Chromatogr. B, 2008, 876, 137–142 CrossRef CAS.
- W. V. Bobo and S. C. Miller, Am. J. Addict., 2002, 11(4), 332–334 CrossRef.
- D. K. Lim, Singapore Med. J., 2003, 44(1), 31–34 CAS.
- G. Bokor and P. D. Anderson, J. Pharm. Pract., 2014, 27(6), 582–586 CrossRef PubMed.
- U. Khan, A. L. N. van Nuijs, J. Li, W. Maho, P. Du, K. Y. Li, L. L. Hou, J. Y. Zhang, X. Z. Meng, X. Q. Li and A. Covaci, Sci. Total Environ., 2014, 487, 710–721 CrossRef CAS PubMed.
- A. Y. C. Lin, W. N. Lee and X. H. Wang, Water Res., 2014, 53, 351–360 CrossRef CAS PubMed.
- F. Y. Lai, R. Bruno, H. W. Leung, P. K. Thai, C. Ort, S. Carter, K. Thompson, P. K. S. Lam and J. F. Mueller, Forensic Sci. Int., 2013, 233(1–3), 126–132 CrossRef CAS PubMed.
- J. F. Gao, J. O'Brien, F. Y. Lai, A. L. N. van Nuijs, J. He, J. F. Mueller, J. S. Xu and P. K. Thai, J. Environ. Sci., 2015, 27, 70–79 CrossRef PubMed.
- A. L. N. van Nuijs, S. Castiglioni, I. Tarcomnicu, C. Postigo, M. L. Lopez de Alda, H. Neels, E. Zuccato, D. Barcelo and A. Covaci, Sci. Total Environ., 2011, 409(19), 3564–3577 CrossRef CAS PubMed.
- N. Raikos, G. Theodoridis, E. Alexiadou, H. Gika, H. Argiriadou, H. Parlapani and H. Tsoukali, J. Sep. Sci., 2009, 32(7), 1018–1026 CrossRef CAS PubMed.
- B. L. Zhu, L. Meng and K. F. Zheng, Forensic Sci. Int., 2014, 242, e44–e47 CrossRef CAS PubMed.
- T. C. VandenBoer, S. A. Grummett and J. H. Watterson, J. Forensic Sci., 2008, 53(6), 1474–1482 CAS.
- C. Dogaroiu and R. Sugden, Rom. J. Leg. Med., 2008, 16(2), 95–102 Search PubMed.
- H. Y. Aboul-Enein and M. M. Hefnawy, Talanta, 2005, 65(1), 67–73 CAS.
- A. Turcant, M. Deguigne, S. Ferec, C. Bruneau, I. Leborgne, B. Lelievre, C. Gegu, F. Jegou, C. Abbara, G. Le Roux and D. Boels, Toxicol. Anal. Clin., 2017, 29(1), 18–33 Search PubMed.
- Y. S. X. Liu, Y. L. Fan, Z. P. Huang, H. J. Liu, L. L. Wang, Z. L. Shen and I. Watanabe, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1153, 122275 CrossRef CAS.
- Z. B. Lin, J. L. Li, X. Y. Zhang, M. H. Qiu, Z. B. Huang and Y. L. Rao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 177–184 CrossRef CAS PubMed.
- H. B. Jin, D. Yang, Y. B. Hao, J. Y. Zhang, P. F. Wu, W. P. Liu and M. R. Zhao, Sci. Total Environ., 2021, 778, 146370 CrossRef CAS.
- L. Schenkel, I. Vogel Kahmann and C. Steuer, Hosp. Pharm., 2022, 57(2), 246–252 CrossRef CAS PubMed.
- S. Yuan, X. Wang, R. J. Wang, R. X. Luo, Y. Shi, B. H. Shen, W. Liu, Z. G. Yu and P. Xiang, Water Sci. Technol., 2020, 82(9), 1771–1780 CrossRef PubMed.
- L. Kurzweil, L. Danyeli, Z. D. Şen, A. Fejtova, M. Walter and S. Gensberger-Reigl, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1152, 122214 CrossRef CAS.
- M. Świądro, P. Stelmaszczyk, I. Lenart and R. Wietecha-Posłuszny, Molecules, 2021, 26(4), 813–822 CrossRef PubMed.
- N. Porpiglia, G. Musile, F. Bortolotti, E. F. De Palo and F. Tagliaro, Forensic Sci. Int., 2016, 266, 304–310 CrossRef CAS PubMed.
- Q. H. Li, W. J. Tang, Y. Wang, J. W. Di, J. P. Yang and Y. Wu, J. Solid State Electrochem., 2015, 19(10), 2973–2980 CrossRef CAS.
- J. C. Guo, S. L. Tian, K. Liu and J. H. Guo, IEEE Trans. Nanobiosci., 2021, 20(1), 2–8 Search PubMed.
- H. S. M. Abd-Rabboh, A. El-G. E. Amr, E. A. Elsayed, A. Y. A. Sayed and A. H. Kamel, RSC Adv., 2021, 11(20), 12227–12234 RSC.
- J. Schram, M. Parrilla, N. Sleegers, N. Samyn, S. M. Bijvoets, M. W. J. Heerschop, A. L. N. van Nuijs and K. De Wael, Anal. Chem., 2020, 92(19), 13485–13492 CrossRef CAS PubMed.
- L. J. Huang, T. T. Li, Y. J. Zhang, X. H. Sun, Y. A. Wang and Z. Y. Nie, J. Pharm. Biomed. Anal., 2020, 186, 113174 CrossRef CAS PubMed.
- K. Mao, Z. G. Yang, H. Zhang, X. Q. Li and J. M. Cooper, Water Res., 2021, 189, 116559 CrossRef CAS PubMed.
- C. M. Liu, H. Y. He, L. Xu and Z. D. Hua, Drug Test. Anal., 2021, 13(3), 720–728 CrossRef CAS PubMed.
- S. J. Sun, M. Guan, C. Guo, L. Ma, H. Zhou, X. M. Wang, F. Mi and J. T. Li, RSC Adv., 2020, 10(60), 36609–36616 RSC.
- E. Deconinck, C. Aït-Kaci, A. Raes, M. Canfyn, J. L. Bothy, C. Duchateau, C. Mees, K. De Braekeleer, L. Gremeaux and P. Blanckaert, Drug Test. Anal., 2021, 13(3), 679–693 CrossRef CAS PubMed.
- C. M. Liu, Y. Han, S. G. Min, W. Jia, X. Meng and P. P. Liu, Forensic Sci. Int., 2018, 290, 162–168 CrossRef CAS PubMed.
- C. M. Liu, Y. Han and S. G. Min, Spectrosc. Spectrom. Anal., 2019, 39(7), 2136–2141 CAS.
- R. M. Correia, E. Domingos, F. Tosato, N. A. dos Santos, J. d. A. Leite, M. da Silva, M. C. A. Marcelo, R. S. Ortiz, P. R. Filgueiras and W. Romão, Anal. Methods, 2018, 10(6), 593–603 RSC.
- R. Walczak, J. Krüger and S. Moynihan, Meas. Sci. Technol., 2015, 26(8), 085401 CrossRef.
- P. Mishra, I. Banga, R. Tyagi, T. Munjal, A. Goel, N. Capalash, P. Sharma, C. R. Suri and S. Gandhi, RSC Adv., 2018, 8(41), 23163–23170 RSC.
- E. Guler, G. Bozokalfa, B. Demir, Z. P. Gumus, B. Guler, E. Aldemir, S. Timur and H. Coskunol, Drug Test. Anal., 2017, 9(4), 578–587 CrossRef CAS PubMed.
- S. J. Liang, Q. Y. Wu, J. K. Mao, C. Gong, D. D. Yu and J. G. Zhou, Mater. Express, 2020, 10(10), 1638–1645 CrossRef CAS.
- H. Vázquez-Becerra, E. Pérez-Cárdenas, S. Muñiz-Hernández, V. Izquierdo-Sánchez and L. A. Medina, J. Liposome Res., 2017, 27(4), 274–282 CrossRef PubMed.
- T. H. The and T. E. W. Feltkamp, Immunology, 1970, 18(6), 865–873 CAS.
- T. H. The and T. E. W. Feltkamp, Immunology, 1970, 18(6), 875–881 CAS.
- A. F. Wells, C. E. Miller and M. K. Nadel, Appl. Microbiol., 1966, 14(2), 271–275 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |