
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn expeditious synthesis of 6,7-dihydrodibenzo[b,j][4,7] phenanthroline derivatives as fluorescent materials†
Kevin George and
Sathananthan Kannadasan*
and
Sathananthan Kannadasan*
Department of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632014, Tamil Nadu, India. E-mail: kannadasan.s@vit.ac.in
First published on 26th September 2022
Abstract
A rapid and efficient method has been developed for the synthesis of 13,14-dimethyl-6,7-dihydrodibenzo[b,j][4,7]phenanthroline derivatives (3a–d) through the Friedländer condensation of 2-aminoarylketone with 1,4-cyclohexanedione under solvent-free conditions using p-toluenesulphonic acid. The synthetic utility of compounds 3a, 3b, and 3c was demonstrated by synthesizing compounds 6a–k via Suzuki coupling, 8 by Buchwald–Hartwig amination, and 9a–b via NBS bromination. Significantly, the emission band corresponding to the π–π* electronic transition of compounds 3a, 6a, 6d, 6f, and 8 showed a redshift with increasing polarity of the solvents. Molar extinction coefficient (ε), Stoke's shift (Δ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) ), and quantum yield (Φf) were calculated for all these compounds.
), and quantum yield (Φf) were calculated for all these compounds.
1. Introduction
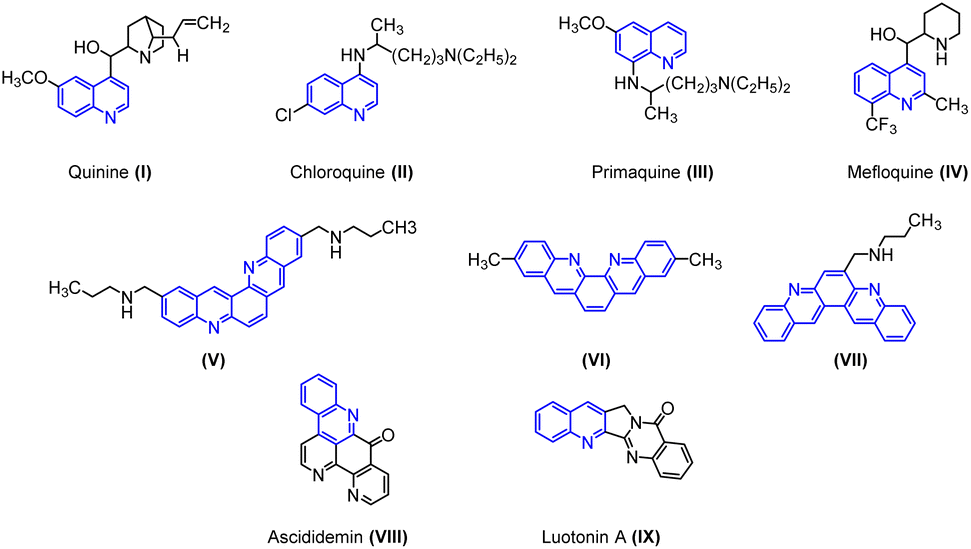
Quinoline cores have been identified as “Privileged Scaffolds” owing to their widespread appearance in natural and synthetic products that exhibit remarkable pharmacological or physical properties.1 Many natural products of quinoline skeleton have been used as medicines or employed as lead molecules for the development of newer and potent molecules. For example, quinine (I) chloroquine (II), primaquine (III), mefloquine (IV) etc.2In particular, halogen-containing quinolines get time-honored attention because the halogen atom plays a pivotal role in the compound's bioactivity. These compounds provide a further avenue for structural elaboration.3 Quinolines are extensively used in industries as dyes, preservatives, ligands, and as fluorescent materials.4
Owing to its immense biological and industrial application, many methods for the synthesis of quinoline have been developed. The conventional approach for the synthesis of quinoline ring involves Skraup, Doebner-von Miller, Friedländer, Pfitzinger, Conrad-Limpach, Combes syntheses.2 Apart from the classical methods, various new methods have been developed for the synthesis of quinoline derivatives by using metallic or organometallic reagents such as CuCN, LiCl,5 ruthenium(III) chloride,6 ytterbium(III) triflate,7 tungsten vinylidene complex,8 boron trifluoride etherate,9,10 benzotriazoleiminium salts, etc.11 Dibenzophenanthroline is an important class of biquinolines found in various natural and synthetic products. They display a broad range of biological activities such as anticancer, antiparasitic, and antibacterial properties. Particularly, the antitumor activity of these heterocyclic compounds has been well explored. For instance, compounds (V), (VI), and (VII) display anticancer activity targeting human telomeric DNA. Ascididemin (VIII) shows significant in vitro and in vivo cytotoxic activities against several tumor cell lines. Luotonin A (IX) is a well-known alkaloid, cytotoxic toward the murine leukemia P-388 cell line (Fig. 1). In addition, certain derivatives of dibenzophenanthroline derivatives are used to synthesize chelating ligands and in the manufacturing of organic semiconductor materials.
 | ||
| Fig. 1 Biologically active polycyclic heteroaromatic compounds. | ||
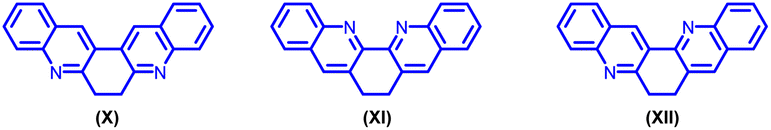
To the best of our knowledge, only three 6,7-dihydro-dibenzo phenanthroline derivatives (X, XI, XII) are documented in the literature.12 (Fig. 2).
 | ||
| Fig. 2 Different types of 6,7 dihydrodibenzo phenanthrolines. | ||
C–C and C–N bond-forming reactions are key steps in synthetic organic chemistry. Aldol reaction is one of the principal tools for the construction of C–C bonds, and amino-carbonyl condensation is a useful reaction for C–N bond formation.12–14 Hence, in this work these two methods were effectively exploited for the synthesis of 6,7-dihydrodibenzo[b,j][4,7]phenanthroline derivatives (3).
Recently, there has been an immense investigation based on conjugated push–pull chromophores of azaheterocyclic fragments. Environmental impulses such as polarity, pH, or the presence of metal cation can be used to tune the photophysical properties of these materials. Pyridine, quinoline, benzodiazines, etc. belong to the six-membered heterocyclic family that is moderate-to-strong electron-withdrawing groups. In the presence of protons, the photophysical properties can be altered due to the interaction with the electron lone pair of the nitrogen atoms in the heterocycle. Such participation of the electron lone pair leads to an increase in the electron-withdrawing character and improves the intramolecular charge transfer (ICT) into the chromophore. This phenomenon has been used to achieve sensors and different optical switches.15
Quinoline-based compounds find applications in various modern analytical platforms because of their tuneable light emission property. These applications take advantage of tagging the objects of interest with fluorescent tracers-compounds that can be readily detected.16
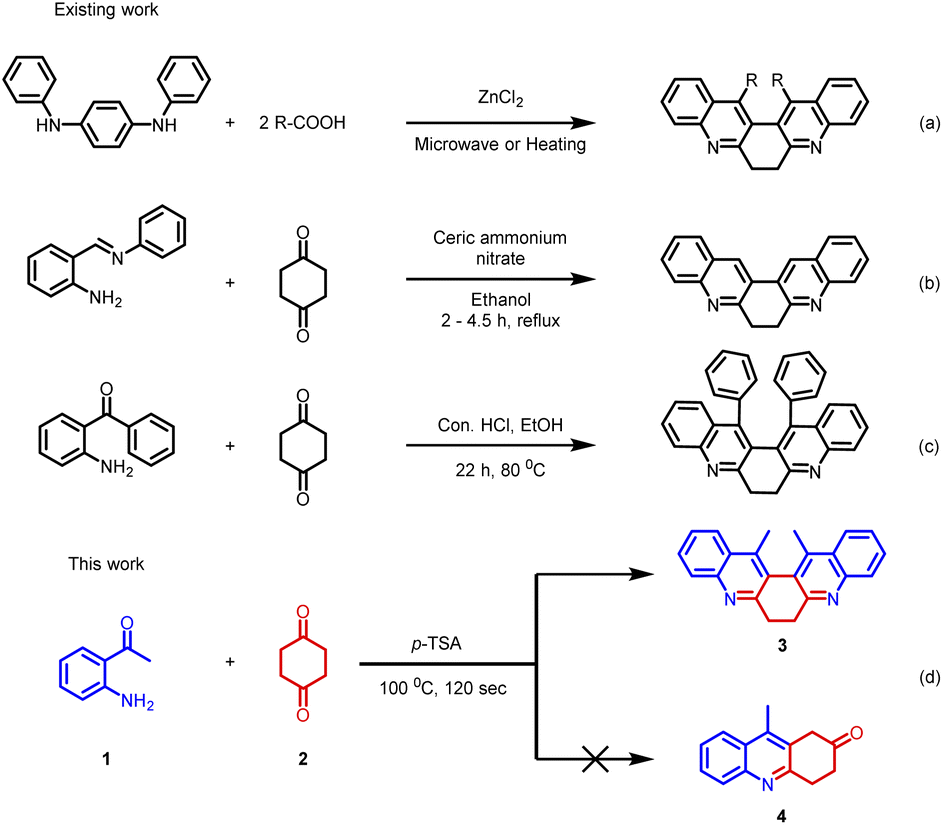
Despite such significant importance of 6,7-dihydrodibenzophenanthroline (Fig. 2) derivatives, the synthesis of these derivatives still remains less explored. In 2004, Masashi Watanabe et al. synthesized 6,7-dihydrodibenzophenanthroline derivatives via a two-step protocol (Scheme 1(a)).17 Later in 2009, J. Carlos Menéndez and co-workers synthesized 6,7-dihydrodibenzo[b,j][4,7]phenanthroline via CAN-catalysed double Friedländer reactions of 2-aminobenzophenones with cyclohexane-1,2-diones (Scheme 1(b)).18 In 2022, Animesh Gosh et al. reported the synthesis of 13,14-diphenyl-6,7-dihydrodibenzo[b,j][4,7]phenanthroline using Con. HCl as the catalyst over a period of 22 hours (Scheme 1(c)).19
 | ||
| Scheme 1 Synthesis of 6,7-dihydrodibenzophenanthroline derivatives. | ||
Herein, we demonstrate a rapid, efficient, and neat method for the synthesis of 6,7-dihydrodibenzo[b,j][4,7]phenanthroline (3) through the Friedländer condensation of 2-aminoarylketone (1) with 1,4-cyclohexanedione (2) in the presence of p-toluenesulphonic acid (p-TSA) as the promoter.
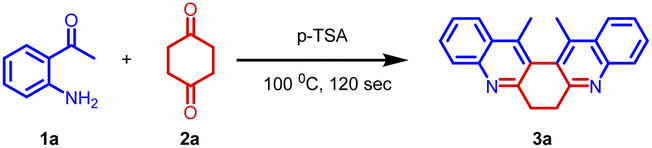
Our initial intention was to synthesize various 9-methyl-3,4-dihydroacridin-2(1H)-one for the synthesis of angularly fused quinoline derivatives. For this, we took 1 equiv. of 2-aminoacetophenone, (1a) 1 equiv. of 1,4-cyclohexanedione (2) and p-TSA in a test tube and heated at 100 °C for 2 minutes. To our surprise, we obtained 13,14-dimethyl-6,7-dihydrodibenzo[b,j][4,7]phenanthroline (3a) precipitated as the sole product in 48% yield along with unreacted starting materials rather than the expected acridinone (4) (Scheme 1(c)). Various concentrations of 2-aminoacetophenone (1a) and 1,4-cyclohexanedione (2) were screened to synthesis 9-methyl-3,4-dihydroacridin-2(1H)-one (4), but 13,14-dimethyl-6,7-dihydrodibenzo [b,j][4,7] phenanthroline (3a) precipitated as the sole product (Table 1, entries 1–5). It was found that the highest yield was obtained when 2.0 equiv of 2-aminoacetophenone (1) and 1.0 equiv. of 1,4-cylohexanedione (2) was used. In order to further evaluate the effect of p-TSA, this reaction was carried out using different amounts of p-TSA, and it has been observed that the maximum yield was obtained when 2 equiv. of p-TSA was used (Table 1, entries 6–8) After determining the optimum quantity of reactants, and p-TSA, various other parameters such as temperature, time, and promoters were screened (Table 1, entries 9–18). The optimum condition for the synthesis of 13,14-dimethyl-6,7-dihydrodibenzo[b,j][4,7] phenanthroline (3a) was found to be 2.0 equiv. of 2-aminoacetophenone, (1a) 1.0 equiv. of 1,4-cyclohexanedione (2) and 2.0 equiv. of p-TSA, at 100 °C for 120 s (Table 1, entry 7).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substra-te ratio 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Promoter (equiv.) | Solvent | Temp. °C | Time (s) | Yield 3aa |
| a Isolated yield. | ||||||
| 1 | 1:1 |
p-TSA (1.0) | Neat | 100 | 120 | 48 |
| 2 | 1.5:1 |
p-TSA (1.0) | Neat | 100 | 120 | 46 |
| 3 | 0.5:1 |
p-TSA (1.0) | Neat | 100 | 120 | 22 |
| 4 | 2:1 |
p-TSA (1.0) | Neat | 100 | 120 | 49 |
| 5 | 2.5:1 |
p-TSA (1.0) | Neat | 100 | 120 | 48 |
| 6 | 2:1 |
p-TSA (1.8) | Neat | 100 | 120 | 90 |
| 7 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1 |
p-TSA (2.0) | Neat | 100 | 120 | 94 |
| 8 | 2:1 |
p-TSA (2.2) | Neat | 100 | 120 | 94 |
| 9 | 2:1 |
Acetic acid (2.0) | Neat | 100 | 120 | 16 |
| 10 | 2:1 |
FeCl3·6H2O (2.0) | Neat | 100 | 120 | 82 |
| 11 | 2:1 |
Con. HCl (2.0) | Neat | 100 | 120 | 26 |
| 12 | 2:1 |
p-TSA (2.0) | DMSO | 100 | 120 | 23 |
| 13 | 2:1 |
p-TSA (2.0) | CH3OH | 100 | 120 | 24 |
| 14 | 2:1 |
p-TSA (2.0) | Toluene | 100 | 120 | 23 |
| 15 | 2:1 |
p-TSA (2.0) | Neat | 90 | 120 | 84 |
| 16 | 2:1 |
p-TSA (2.0) | Neat | 110 | 120 | 93 |
| 17 | 2:1 |
p-TSA (2.0) | Neat | 100 | 110 | 89 |
| 18 | 2:1 |
p-TSA (2.0) | Neat | 100 | 130 | 93 |
Further, the substrate scope of the reaction was explored with various 2-aminoacetophenones, 2-aminobenzophenones and 1,4-diketones. In the cases of 5-nitro-2-aminobenzophenone 1e and acetonylacetone 2b as the starting materials, the desired product was not formed (Fig. 3).
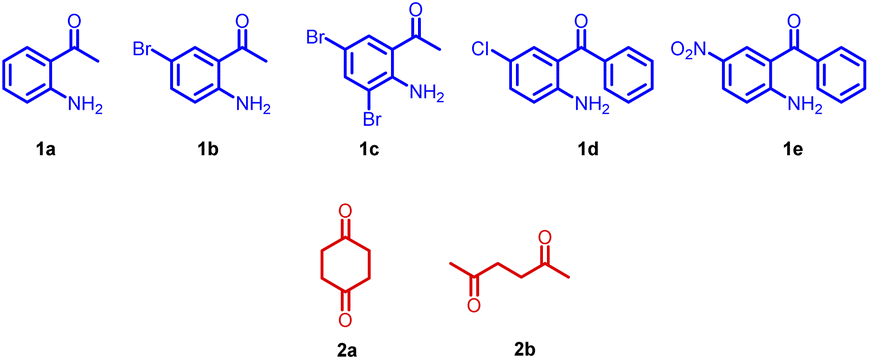 | ||
| Fig. 3 Starting materials screened. | ||
Based on the structure of product 3a, a plausible mechanism is proposed in Scheme 2. Thus, the 2-amino acetophenone 1a undergoes Friedländer condensation with 1,4-cyclohexanedione 2a in the presence of p-TSA to form an unisolable acridinone intermediate, which further undergoes a second Friedländer condensation leading to the formation of stable and isolable compound 3a (Fig. 4).
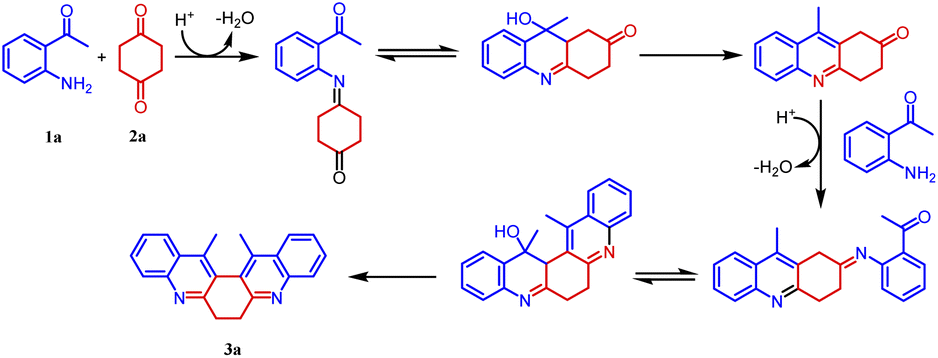 | ||
| Scheme 2 A plausible mechanism for the formation of compound 3. | ||
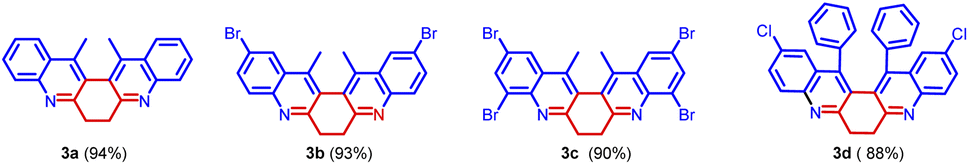 | ||
| Fig. 4 Compounds synthesized. | ||
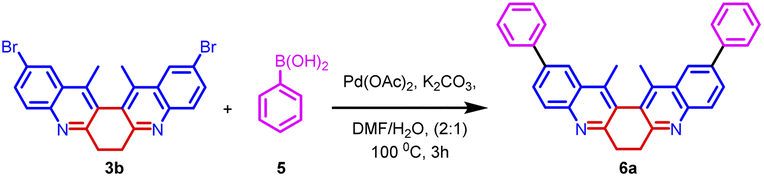
The synthetic utility of biquinoline was explored using Suzuki–Miyaura coupling. Initially, the reaction was performed with 1.0 equiv. of 3b and 2.4 equiv. of benzylboronic acid in the presence of 10 mol% Pd(OAc)2 as catalyst and Na2CO3 as the base in DMF/H2O (2:1) solvent system to give 5a in 80% yield. Further, increase in the loading percentage of the catalyst to 20 mol%, the yield of the product increased to 92%. Also, an increase in the loading percentage of the catalyst did not bring a further increase in the yield. Among the different Pd catalysts screened like Pd(amphos)Cl2, Pd(OAc)2, Pd2(dba)3, and Pd(dba)2 revealed the ascendancy of Pd(OAc)2 in catalyzing the reaction. The introduction of ligands like trimethylolethane could not bring much difference in the yield of the reaction. Different biaryl systems were synthesized using the optimized condition (Table 2, entry 3) (Fig. 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Time (h) | Ligand | Solvent | Product yielda |
| a Isolated yield. | |||||
| 1 | Pd(OAc)2 (20%) | 4.0 | Nil | H2O | 36% |
| 2 | Pd(OAc)2 (10%) | 6.0 | Nil | H2O/DMF | 80% |
| 3 | Pd(OAc)2 (20%) | 3.0 | Nil | H2O/DMF | 92% |
| 4 | Pd(OAc)2 (30%) | 3.0 | Nil | H2O/DMF | 91% |
| 5 | Pd(OAc)2 (20%) | 3.0 | Trimethylolethane | H2O/DMF | 92% |
| 6 | Pd(amphos)Cl2 (20%) | 3.0 | Nil | H2O/DMF | 91% |
| 7 | Pd(dba)2 (20%) | 3.0 | Nil | H2O/DMF | 90% |
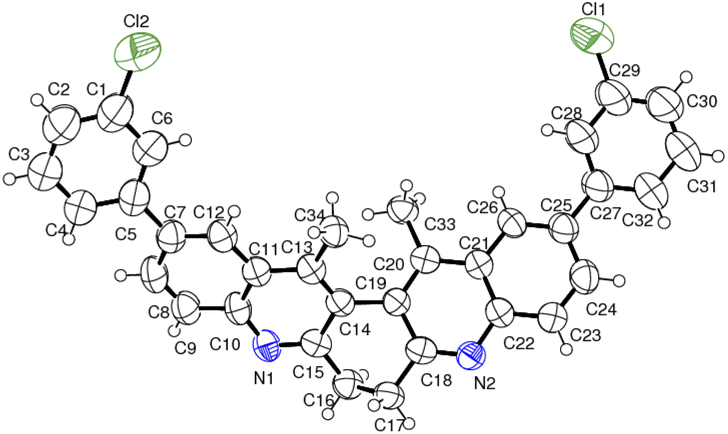 | ||
| Fig. 5 ORTEP diagram of compound 6d. | ||
The structure of the representative compound 6d was confirmed by spectroscopic data analysis (see ESI†) and the structure and relative stereochemistry was assigned based on single-crystal XRD analysis (Scheme 3).20
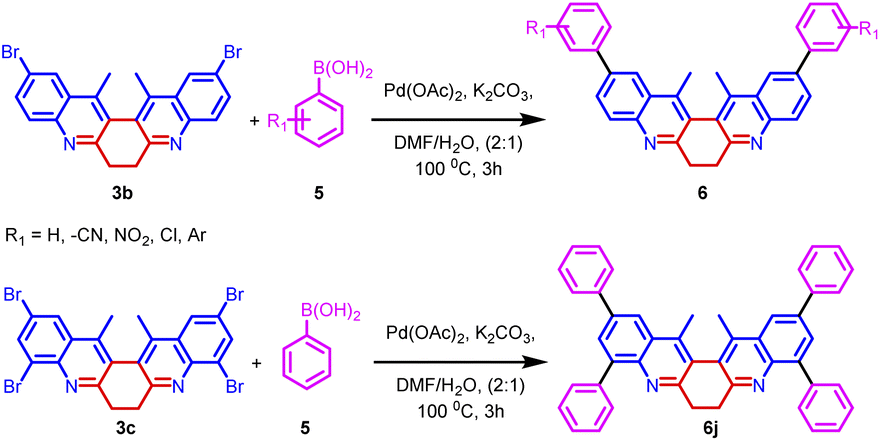 | ||
| Scheme 3 Synthesis of biaryl derivatives 6a–k via Suzuki coupling reaction on derivatives 3a–c. | ||
In order to explore the scope of the palladium-catalysed reaction, compound 3b was subjected to Buchwald–Hartwig amination. In this reaction, 1.0 equiv. of 3b was treated with 2.4 equiv. of 2,3-dimethylaniline 20 mol% of Pd(dppf)Cl2, and 4.0 equiv. of NaOt-Bu. The reaction was facilitated by the ligand SPhos. The reaction yielded product 8 in 89% yield (Scheme 4). The synthesized compound (8) was thoroughly characterized using spectroscopic techniques (Fig. 6).
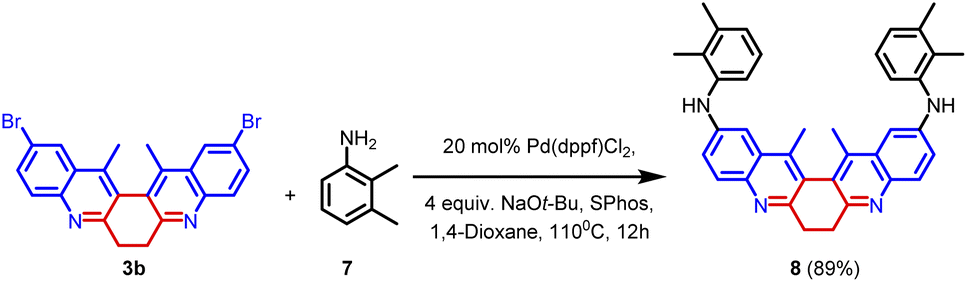 | ||
| Scheme 4 Buchwald–Hartwig reaction. | ||
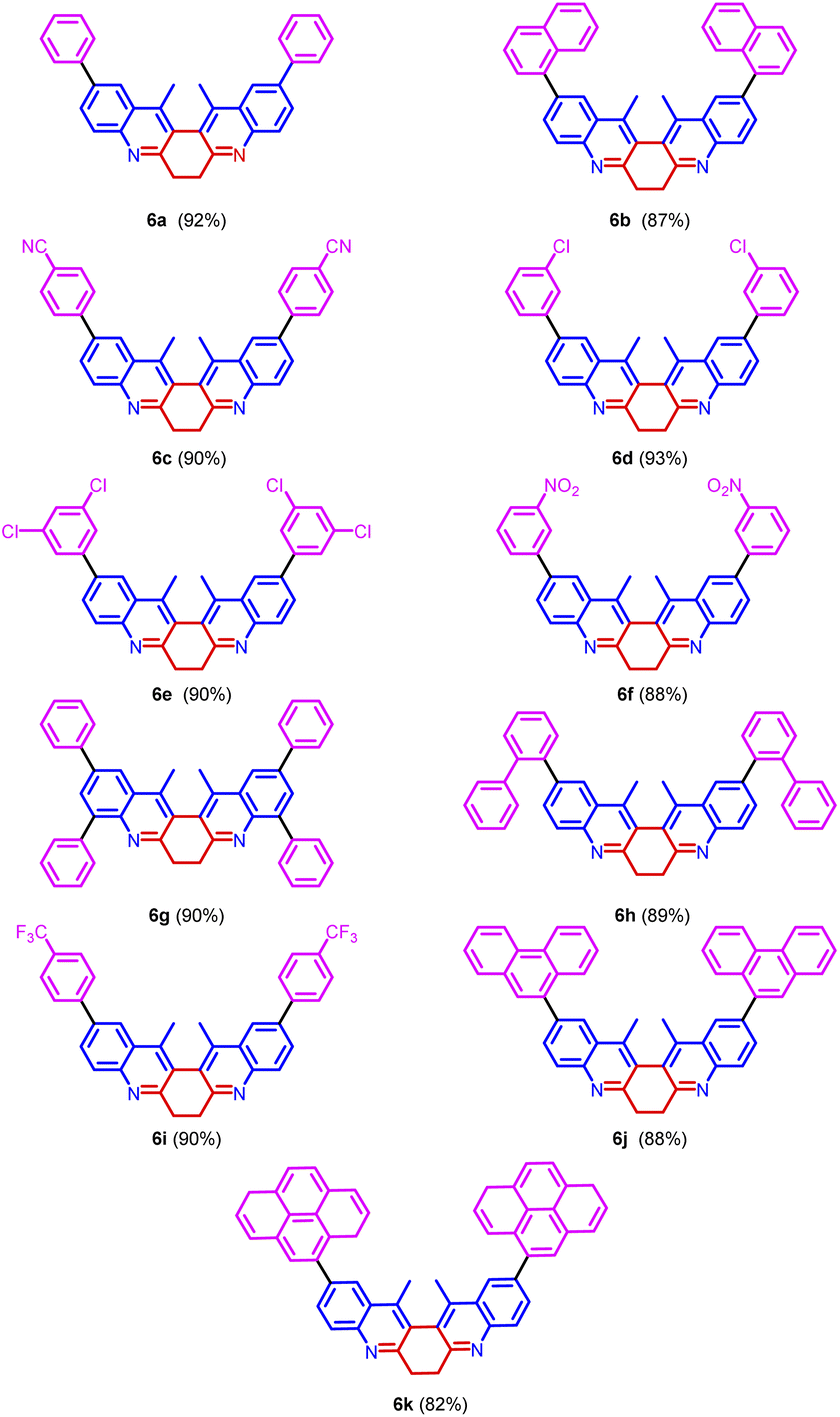 | ||
| Fig. 6 Scope of Suzuki coupling reaction. | ||
Further, compound 3a was treated with 2.0 equiv. of NBS in acetonitrile at 80 °C for 3 hours. We have anticipated the bromination on the cyclic methylene group leading to the formation of 10a and 10b (Scheme 5).12 But we have obtained bromination on the methyl group leading to the formation of 9a and 9b in 68% and 24% yield respectively. Compounds 9a and 9b were thoroughly characterized by spectroscopic methods. The synthetic transformation can provide an avenue for structural elaboration.
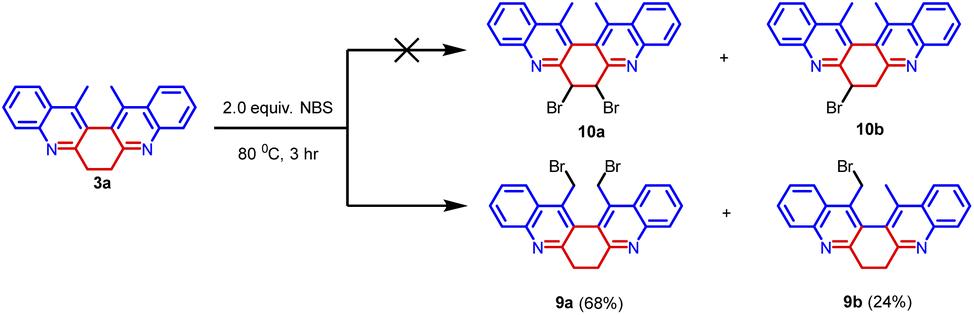 | ||
| Scheme 5 Bromination of compound 3a. | ||
2. Photophysical studies
The structural uniqueness and extended π conjugation induced by the aryl system encouraged us to investigate the photophysical properties of compounds 3a–c and the Suzuki coupled products 4a–k. Thus, absorption and emission spectra were recorded for all the compounds (3a–c, 6a–k) in methanol Fig. 7. In the absorption spectra of compounds (3a–c, 6a–k), a higher energy band in the range of 250 to 300 nm begins with π–π* electronic transition [intramolecular charge transfer (ICT)] and other bands with lower energy n–π* electronic transition in the region 310 to 350 nm were observed. The corresponding emission band was observed in the 350–480 nm region. In the emission spectra, it was observed that with the increase in the size of the aromatic ring connected by the pivotal bond to the biquinoline core, the emission band has been shifted to higher wavelengths.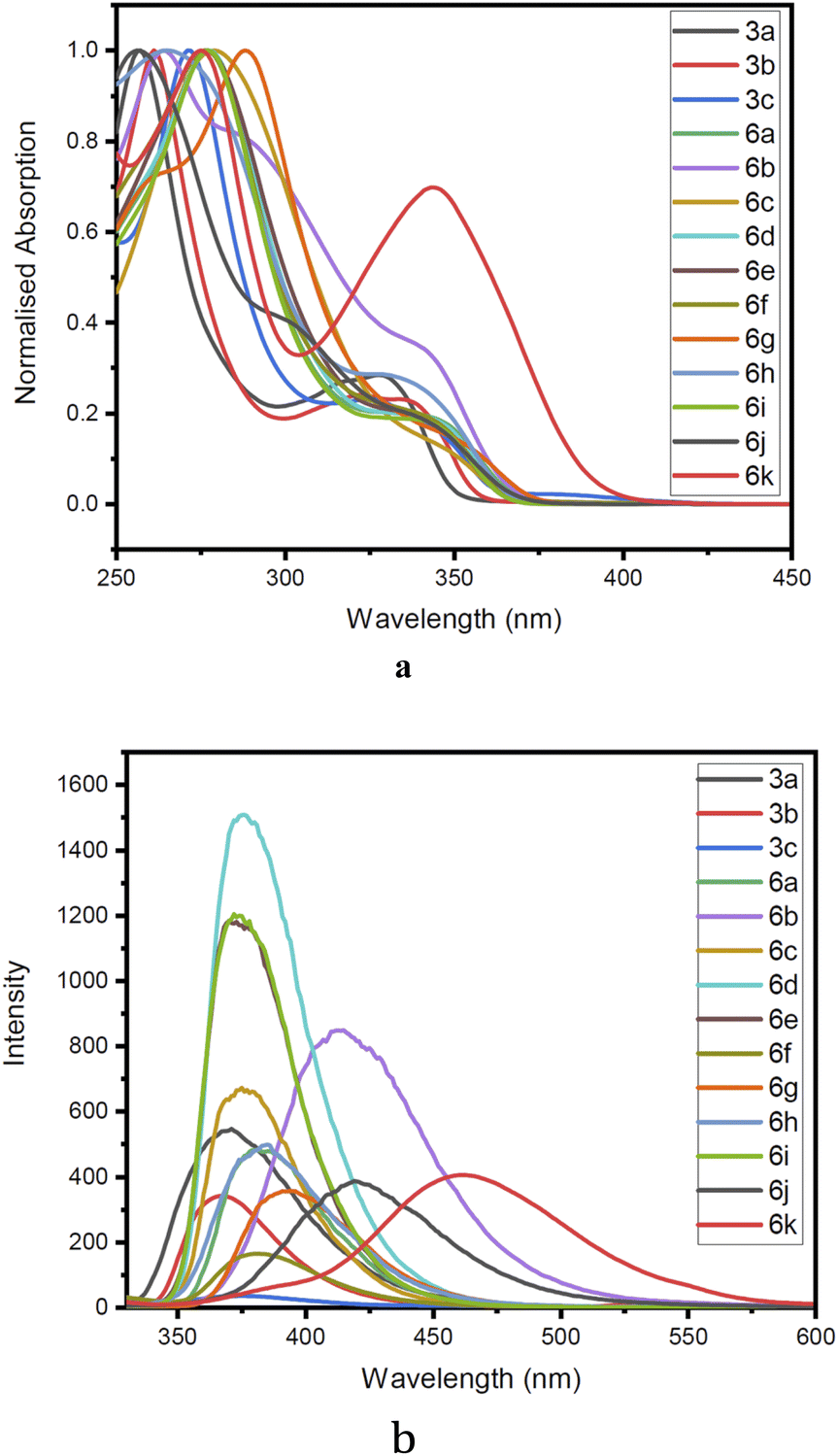 | ||
| Fig. 7 (a) Normalised absorption spectra of compound 3a–c, 6a–k recorded at C 2 × 10−5 M at 298 K in methanol. (b) Normalised emission spectra of compound 3a–c, 6a–k recorded at C 2 × 10−5 M at 298 K in methanol. | ||
Furthermore, quantum yield, Stoke's shift, and molar extinction coefficient were calculated for (3a–c, 6a–k). Quantum yields of compounds were estimated by comparison with the known quantum yields of anthracene in ethanol (Φ = 0.27) at an excitation wavelength of 246 nm using the following equation.
| Φf = ΦR. I/IR. ODR/OD. n2/n2R |
All the derivatives showed good quantum yield (Table 3, entries 1–14). It was observed that the quantum yield of the biquinoline system varied based on the electronic properties of the substituents in the biaryl system. Electron-donating substituents help increased the quantum yield. Whereas, the electron-withdrawing substituents reduced the quantum yield (Table 3, entries 7, and 9). The introduction of higher aromatic systems like naphthalene, phenanthrene, and pyrene to the biquinoline system did not increase the quantum yield of the molecule as expected. But in those derivatives in which the biquinoline system was upended with ring fused aromatic moieties 6b, 6g, 6h, 6j and 6k the emission corresponding to π–π* electronic transition shifted to higher wavelengths (Table 3, entries 5, 10, 11, 13, and 14). This might be due to the highly aromatic system upended to the core dihydrodibenzophenanthroline moiety.
| Entry | Compound | Absorptiona λmax,abs (nm) | Emissiona λmax,emi (nm) | Molar extinction coefficient ×104 (ε) π–π* | Stoke's shiftb Δ ×104 (cm−1) |
Quantum yieldc (Φf) |
|---|---|---|---|---|---|---|
| a Recorded at 298 K.b Stoke's shift = λmax,abs − λmax,emi [cm−1].c Determined with anthracene as a standard Φf = 0.27 at excitation wavelength 246 nm. | ||||||
| 1 | 3a | 257, 327 | 369 | 4.5657 | 1.1810 | 0.1837 |
| 2 | 3b | 261, 322 | 367 | 8.3500 | 1.1066 | 0.0500 |
| 3 | 3c | 271, 325 | 376 | 4.7769 | 0.4537 | 0.0270 |
| 4 | 6a | 277 | 380 | 5.0952 | 0.9785 | 0.1213 |
| 5 | 6b | 264 | 413 | 4.5725 | 1.3665 | 0.3451 |
| 6 | 6c | 279 | 375 | 3.2145 | 0.9175 | 0.2478 |
| 7 | 6d | 277 | 376 | 7.2067 | 0.9505 | 0.2489 |
| 8 | 6e | 276 | 370 | 5.3748 | 0.9204 | 0.2502 |
| 9 | 6f | 276 | 381 | 6.0666 | 0.9985 | 0.0450 |
| 10 | 6g | 288 | 394 | 6.6211 | 0.9341 | 0.0759 |
| 11 | 6h | 265 | 385 | 4.9525 | 1.1761 | 0.1412 |
| 12 | 6i | 277 | 372 | 6.5688 | 0.9219 | 0.2062 |
| 13 | 6j | 256 | 419 | 9.2679 | 1.5196 | 0.0779 |
| 14 | 6k | 236, 275, 344 | 462 | 7.4760 | 1.4718 | 0.1336 |
Also, the molar extinction coefficient (ε) and Stoke's Shift were calculated. The molar extinction coefficient (ε) was calculated using Beer–Lambert's law A = εcl. All the compounds exhibited good molar extinction coefficient (ε). The value of ε varied from 3.2145 × 104 M−1 cm−1 to 9.2679 × 104 M−1 cm−1 (Table 3, entries 1–14). The biquinoline derivatives with the higher aromatic system tethered to it showed the highest molar extinction coefficient (Table 3, entries 13, and 14).
The Stoke's Shift was calculated using the following equation.
| Δ = 107/λmax(Absorption) − 107/λmax(Emission) |
All the compounds exhibited good Stoke's shift values (Table 3, entries 1–14).
To establish solvatochromic property, absorption and emission spectra of the compounds 3a, 6a, 6d and 6f (ESI Fig. 1–4†) were recorded using solvents such as hexane, toluene, dichloromethane, ethyl acetate, methanol, acetonitrile, DMF, and DMSO in the increasing order of polarity. The emission spectra of all the derivatives showed a redshift. But in all the four compounds (3a, 6a, 6d, and 6f) the shift was not prominent. The results revealed that compound 3a, showed an emission maximum of 369 nm in hexane to 377 nm in DMSO giving a redshift of 8 nm. Similarly, compounds 6a, 6d, and 6f showed shifts of 11, 6, and 8 nm respectively.
All the four compounds' 3a, 6a, 6d, and 6f parameters such as quantum yield, molar extinction coefficient, and Stoke's shift were calculated in various solvents (ESI Table 1–4†). All compounds exhibited structureless absorption and emission bands with rather large Stoke's shifts in the 7650–12385 cm−1 range, which is associated with highly polarizable π-conjugated systems due to intramolecular charge transfer (ICT).
Compared to biaryl-appended biquinoline derivatives 6a, those derivatives with electron-donating substituents on the phenyl ring 6d helped to increase the quantum yield. It was also observed that biaryl-appended biquinoline derivatives with electron-withdrawing substitutions 6f reduced the quantum yield in various solvents. Adding nitro groups to aromatic compounds usually quenches their fluorescence via inter-system crossing (ISC) or internal conversion (IC). While strong electronic coupling of the nitro groups with the molecule ensures the benefits from these electron-withdrawing substituents, it also leads to fluorescence quenching.21
None of the above-investigated molecules couldn't give a noticeable shift in the emission band. It has been well known that quinolines processing amino groups have a large redshift. This is due to the stronger electron-withdrawing nature of the quinoline ring and the presence of a strong electron-donating –NH– group,22–24
The absorption and emission of compound 8 in different solvents like hexane, toluene, tetrahydrofuran dioxane, methanol, and acetonitrile were recorded (ESI Fig. 5†) In the absorption spectra of compound 8, a higher energy band in the range 299 to 301 nm begins with π–π* electronic transition [intramolecular charge transfer (ICT)] and other bands with lower energy n–π* electronic transition in the region 379 to 385 nm were observed. The corresponding emission band was observed in the region 421–492 nm (ESI Table 5†). It has also been noted that a large redshift of 71 nm was observed when the solvent polarity was increased. The maximum shift in the emission band was observed when methanol was used as the solvent. This might be due to the stronger electron-withdrawing nature of the quinoline ring and the presence of a strong electron-donating –NH– group in the molecule. Protonation of the compound by the solvent also facilitates the redshift.15
The absorption and emission response of 3a, 6a, 6d, 6f, and 8 in acetonitrile: water media (1:3, 1:1, 3:1) were recorded (†SI Fig. 5–10†). For compound 3a and 6d, the intrinsic fluorescence signals slightly diminishes as a result of intermolecular π–π stacking, which is well-known as the aggregation-caused quenching (ACQ) effect. The irregular intensities observed in the case of compounds 6a, and 6f, might be due to the multiple aromatic rings and/or long conjugated chains, the structural hydrophobicity makes them prone to forming irregular aggregates in aqueous environments. Compound 8 shows enhancement in fluorescence in the aggregated state. This aggregation-induced emission (AIE) might be due to the restriction of intramolecular motions (RIM), including both the restriction of intramolecular rotations (RIR) and the restriction of intramolecular vibrations (RIV).25–27
Furthermore, Stoke's shift, and molar extinction coefficient values were calculated for the compound 8 and showed good Stoke's shift, and molar extinction coefficient values in all the solvents (ESI Table 5†). It was observed that the Stoke's shift and molar extinction coefficient values increased with an increase in the solvent polarity.
These kinds of quinoline-based compounds with tuneable photophysical properties can be used as labels.16 The quinoline cores when exposed to visible or UV light, undergoes absorption and excitation of the molecule. The excited state is unstable and tends to relax emitting light that can be subsequently detected. Detection sensitivity is proportional to the light quanta emitted by the fluorophore, which in turn is a linear function of the intensity of the excitation light. Therefore, for sensitive detection, high-intensity light sources are employed which makes the discrimination between excitation and emission light difficult. This problem can be reduced by using fluorophores with large spectral distances between excitation and emission light (Stoke's shift). Quinoline fluorophores, 3a–c, 6a–k, and 8 represented in the course of the present study, possess the desired property.28
3. Conclusions
In conclusion, a rapid and efficient method was developed for the synthesis of 6,7-dihydrodibenzo[b,j][4,7]phenanthroline derivatives 3 through the Friedländer condensation of 2-aminoarylketone with cyclohexanedione under solvent-free conditions. The reaction is smoothly proceeded using p-TSA as a green promotor. A plausible reaction mechanism is provided and a representative structure of the product 5d was confirmed by single crystal XRD studies. The synthetic utility of compounds 3a, 3b, and 3c was demonstrated by synthesizing compounds 6a–k via Suzuki coupling, 8 by Buchwald–Hartwig coupling, and 9a–b via NBS bromination. Significantly, compounds 3a, 6a, 6d, 6f, and 8 showed a red shift. Molar extinction coefficient (ε), Stoke's shift (Δ), and quantum yield (Φf) were calculated. Also, a larger Stoke's shift was observed which may be used as bio labels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KG thanks VIT, Vellore for the research fellowship. SK thanks to the VIT management for providing infrastructure facilities. The authors thank VIT for providing ‘VIT SEED Grant’ for carrying out this work. The authors thank Dr P. Shanmugam, Chief Scientist, CSIR-CLRI, Chennai and Dr S. K. Ashok Kumar, SAS, VIT-Vellore for giving their valuable suggestions and support.Notes and references
- (a) Y. Morimoto, F. Matsuda and H. Shirahama, Synlett, 1991, 1991, 202–203 CrossRef; (b) J. P. Michael, Nat. Prod. Rep., 1997, 14, 605–618 RSC; (c) J. P. Michael, Nat. Prod. Rep., 2002, 19, 742–760 RSC.
- S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648–1654 CrossRef CAS PubMed.
- (a) B. J. Newhouse, J. Bordner, D. J. Augeri, C. S. Litts and E. F. Kleinman, J. Org. Chem., 1992, 57, 6991–6995 CrossRef CAS; (b) S. Torii, H. Okumoto, L. H. Xu, M. Sadakane, M. V. Shostakovsky, A. B. Ponomaryov and V. N. Kalinin, Tetrahedron, 1993, 49, 6773–6784 CrossRef CAS; (c) Y. S. Kumar, C. Dasaradhan, K. Prabakaran, F. R. N. Khan, E. D. Jeong, E. H. Chung and H. G. Kim, RSC Adv., 2014, 4, 40259–40268 RSC; (d) M. Croisy-Delcey, A. Croisy, D. Carrez, C. Huel, A. Chiaroni, P. Ducrot, E. Bisagni, L. Jin and G. Leclercq, Bioorg. Med. Chem., 2000, 8, 2629–2641 CrossRef CAS PubMed.
- E. S. Sales, J. M. F. M. Schneider, M. J. L. Santos, A. J. Bortoluzzi, D. R. Cardoso, W. G. Santos and A. A. Merlo, J. Braz. Chem. Soc., 2015, 26, 562–571 CAS.
- R. E. Swenson, T. J. Sowin and H. Q. Zhang, J. Org. Chem., 2002, 67, 9182–9185 CrossRef CAS PubMed.
- C. K. Cho, B. Hooh and S. C. Shim, J. Heterocycl. Chem., 1999, 36, 1175–1178 CrossRef CAS.
- N. A. Harry, S. M. Ujwaldev and G. Anilkumar, Org. Biomol. Chem., 2020, 18, 9775–9790 RSC.
- K. Sangu, K. Fuchibe and T. Akiyama, Org. Lett., 2004, 6, 353–355 CrossRef CAS.
- B. Crousse, J.-P. Bégué and D. Bonnet-Delpon, Tetrahedron Lett., 1998, 39, 5765–5768 CrossRef CAS.
- B. Crousse, J.-P. Bégué and D. Bonnet-Delpon, J. Org. Chem., 2000, 65, 5009–5013 CrossRef CAS PubMed.
- A. R. Katrizky and M. A. Arend, J. Org. Chem., 1998, 63, 9989–9991 CrossRef.
- R. R. Rajawinslin, S. S. Ichake, V. Kavala, S. D. Gawande, Y.-H. Huang, C.-W. Kuo and C.-F. Yao, RSC Adv., 2015, 5, 52141–52153 RSC.
- K. George and S. Kannadasan, Curr. Org. Chem., 2021, 25, 1783–1822 CrossRef CAS.
- (a) A. H. Henseler, C. Ayats and M. A. Peric, Adv. Synth. Catal., 2014, 356, 1795–1802 CrossRef CAS; (b) V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593–2595 CrossRef CAS PubMed; (c) S. Luo, J. Li, H. Xu, L. Zhang and J.-P. Cheng, Org. Lett., 2007, 9, 3675–3678 CrossRef CAS PubMed; (d) Z. Tang, Z.-H. Yang, X.-H. Chen, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, J. Am. Chem. Soc., 2005, 127, 9285–9289 CrossRef CAS PubMed; (e) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 958–961 CrossRef CAS; (f) Y. Zhou and Z. Shan, J. Org. Chem., 2006, 71, 9510–9512 CrossRef CAS PubMed.
- Z. I. M. Allaoui, E. le Gall, A. Fihey, R. Plaza-Pedroche, C. Katan, F. R. Guen, L. Rodriguez-Lopez and J. S. Achelle, Chem.–Eur. J., 2020, 26, 8153–8161 CrossRef PubMed.
- S. Pillai, M. Kozlov, S. A. E. Marras, L. N. Krasnoperov and A. Mustaev, J. Fluoresc., 2012, 22, 1021–1032 CrossRef CAS PubMed.
- M. Watanabe, H. Suzuki, Y. Tanaka, T. Ishida, T. Oshikawa and A. Tori-i, J. Org. Chem., 2004, 69, 7794–7801 CrossRef CAS PubMed.
- V. Sridharan, P. Ribelles, M. T. Ramos and J. C. Menéndez, J. Org. Chem., 2009, 74, 5715–5718 CrossRef CAS PubMed.
- A. Ghosh, T. Li, W. Ni, T. Wu, C. Liang, M. Budanovic, S. A. Morris, M. Klein, R. D. Webster, G. G. Gurzadyan and A. C. Grimsdale, Asian J. Org. Chem., 2022, 11, e202100670–e202100684 CAS.
- Compounds 6d (CCDC 2052906).†.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Commun. Chem., 2020, 3, 1–18 CrossRef.
- P. S. Hariharan, E. M. Mothi, D. Moon and S. P. Anthony, ACS Appl. Mater. Interfaces, 2016, 8, 33034–33042 CrossRef CAS.
- Y. Shen, P. Xue, J. Liu, J. Ding, J. Sun and R. Lu, Dyes Pigm., 2019, 163, 71–77 CrossRef CAS.
- K. George, P. Elavarasan, S. Ponnusamy and K. Sathananthan, ACS Omega, 2022, 7, 20605–20618 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Chen and P. R. Selvin, J. Photochem. Photobiol. A: Chem., 2000, 135, 27–32 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2052906. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05198b |
| This journal is © The Royal Society of Chemistry 2022 |