
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChronoampermetric detection of enzymatic glucose sensor based on doped polyindole/MWCNT composites modified onto screen-printed carbon electrode as portable sensing device for diabetes†
Katesara Phasuksom* and
Anuvat Sirivat *
*
Conductive and Electroactive Polymers Research Unit, Petroleum and Petrochemical College, Chulalongkorn University, 254 Chula 12 Phayathai Rd. Pathumwan, Bangkok, 10330, Thailand. E-mail: pkatesara.p@outlook.com; anuvat.s@chula.ac.th
First published on 6th October 2022
Abstract
Doped-polyindole (dPIn) mixed with multi-walled carbon nanotubes (MWCNTs) were coated on a screen-printed electrode to improve the electroactive surface area and current response of the chronoamperometric enzymatic glucose sensor. Glucose oxidase mixed with chitosan (CHI-GOx) was immobilized on the electrode. (3-Aminopropyl) triethoxysilane (APTES) was used as a linker between the CHI-GOx and the dPIn. The current response of the glucose sensor increased with increasing glucose concentration according to a power law relation. The sensitivity of the CHI-GOx/APTES/dPIn was 55.7 μA mM−1 cm−2 with an LOD (limit of detection) of 0.01 mM, where the detectable glucose concentration range was 0.01–50 mM. The sensitivity of the CHI-GOx/APTES/1.5%MWCNT-dPIn was 182.9 μA mM−1 cm−2 with an LOD of 0.01 mM, where the detectable glucose concentration range was 0.01–100 mM. The detectable concentration ranges of glucose well cover the glucose concentrations in urine and blood. The fabricated enzymatic glucose sensors showed high stability during a storage period of four weeks and high selectivity relative to other interferences. Moreover, the sensor was successfully demonstrated as a continuous or step-wise glucose monitoring device. The preparation method employed here was facile and suitable for large quantity production. The glucose sensor fabricated here, consisting of the three-electrode cell of SPCE, were simple to use for glucose detection. Thus, it is promising to use as a prototype for real glucose monitoring for diabetic patients in the future.
1. Introduction
Glucose monitoring is widely applied in clinics and hospitals for the symptoms assessment of diabetes mellitus. Glucose level is essential for diabetic patients as an excess glucose level may induce many comorbidities such as vision loss, renal failure, retinopathy, and strokes.1 Thus, the monitoring becomes an important part of the patients life. Glucose level can be detected in various biological fluids such as blood, interstitial fluid (ISF), sweat, saliva, tears, and urine. However, glucose concentrations in those biological fluids are vastly different, yet they are closely related. Glucose concentrations typically found in diabetic patients are 2–40 mM in blood, 0.01–1 mM in sweat, 0.55–1.77 mM in saliva, and >5.55 mM in urine.2 Presently, the glucose monitoring in blood, sweat, saliva, and urine has become common and increasingly essential to be easily accessible through various new methods or techniques.Electrochemical measurements based on converting the analyte concentration into an electrochemical signal have been widely used for the glucose monitoring because of high sensitivity, good selectivity, and simple operation.3 In particular, the chronoamperometry is an effective electrochemical technique for glucose sensors offering several advantages such as ease of use, short time for analysis, high sensitivity, low limit of detection, and low cost.4,5 A constant voltage is applied between the working and reference electrodes for a period of time generating a current between the working and counter electrodes.6 Recently, the glucose sensors fabricated from screen printed electrodes (SPEs) have received numerous interests as they are of low cost and easy to use for the on-site analysis as the electrochemical biosensors fabricated from various materials with surface modifications requiring only small volumes of sample.7 Moreover, SPEs can provide rapid responses with high sensitivity and selectivity.8 Consequently, SPEs are available as the portable9–11 and wearable sensors12,13 for clinical diagnostics and self-healthcare monitoring. SPEs can be prepared in various types of substrates such as paper sheets, plastic films, and ceramics. They generally consist of three electrodes on the same substrate namely the working, counter, and reference electrodes, the last one typically made of Ag or Ag/AgCl, which are highly accurate and can be adaptable to the electrochemical analysis.14
Enzymatic glucose sensors which are currently available in market are highly selective towards glucose molecules providing precise glucose concentration and are able to operate under a neutral electrolyte condition such as in a phosphate buffer solution (PBS) (pH 7–7.4). Although, non-enzymatic glucose sensors using noble metals and metal oxides have been extensively reported recently, the direct electro-oxidation and electro-reduction mostly occur in the harsh electrolyte conditions such as in an acidic solution (pH <2) or alkaline solution (pH >11).15,16 Glucose-oxidase (GOx) is the key enzyme commonly used for medical electrochemical glucose sensors;17,18 it is highly selective and responsive to glucose molecules and it is the most stable enzyme compared to other enzymes.1 Thus, one of the critical factor of a enzymatic glucose sensor is the immobilization of enzyme layer on the electrode as the firmly adhered enzyme on the electrode leads to a reliable sensor performance in a long-lasting operation.19 Generally, the immobilization can be classified into 2 methods: physical and chemical immobilizations possessing different advantages and disadvantages.20 Physical immobilization is a reversible method with the weak interactions between the enzyme and the support material such as hydrogen bonds, hydrophobic interactions, van der Waals forces, affinity binding, ionic binding causing high enzyme activity but providing enzyme leakage and low stability.20,21 Chemical immobilization is an irreversible and stable method with the strong interaction of covalent bonds through functional groups or crosslinkers such as glutaraldehyde or EDC/NHS between the enzyme and the support material, providing high stability but low enzyme activity as an enzyme can be denatured by toxic chemical reagents.20,21 Although, the chemical immobilization of enzyme provides the high sensor stability, the enzyme is denatured and lost its activity because toxic chemicals are often used. Herein, chitosan was used as a support material for the enzyme immobilization; chitosan possesses a variety of functional groups such as polyamine, amino and hydroxyl, facilitating efficient binding with enzyme. Moreover, it is biocompatible, biodegradable, and non-toxic.22
To improve biosensor performances, various materials have been used namely metal, metal oxide, carbon-based materials (reduced-graphene oxide, graphene oxide, graphene, MWCNT, and SWCNT), metal organic frameworks (MOFs), conductive polymers, and their hybrid/composite materials. These materials can increase the surface for the immobilization of enzyme, electrocatalytic activity, and electron transfer ability.23–25 The glucose sensors fabricated from metals and metal oxides, provide the high electron transfer capability, large surface area, and high electrocatalytic activity. However, these properties are ideally possible when they are of nanosized.26 Moreover, the main disadvantage of noble metals such as gold, and platinum is the high cost for commercial use, and the metal oxides are of poor conductivity with high oxidation potential.27
Recently, the conductive polymers on modified electrodes become one of the favourable materials because of the direct and easy deposition on the electrode by the electrochemistry or drop coating. They provide a stable and porous matrix for the immobilization of the biocomponents and act as a mediator effectively transporting electrons from enzyme active sites to the electrode with adjustable electrical conductivity.28 Moreover, the conducting polymers are compatible with biological molecules in neutral aqueous solutions. Glucose sensors prepared by conducting polymers as a support material are expected to provide a fast response time with a high storage and operational stability.28 Polyindole (PIn) is a non-toxic conductive polymer with excellent thermal stability, high redox activity, and high stability.29 However, only few publications on biosensors based on PIn have been reported namely the cholesterol sensor,30 dopamine sensor31,32 and glucose sensors.33,34 A publication of an enzymatic electrochemical glucose sensor based on polyindole was reported in 1988, in which PIn was deposited on the platinum foil by electro-polymerization and used as a working electrode, and the glucose sensor showed a fast response time and long-term stability.33
Carbon nanotubes are a material possessing a high surface area-to-volume ratio, high conductivity; they play many material roles in biosensor electrode modification namely enhancing the rate of electron transfer, sensitivity, and electrocatalytic activity towards glucose oxidation during electrochemical analysis.35 Compared with single-walled carbon nanotubes (SWCNTs), multi-walled carbon nanotubes (MWCNTs) provide a higher yield of production and lower cost per unit, and higher thermochemical stability. Electrode modification with MWCNTs composites yields long-term stability and selectivity for biosensors as a result of its potential synergistic effects.36 Most biosensors based on conductive polymers were fabricated by electrochemical polymerization as it provides a better control of polymer deposition.37 However, this technique requires time for preparation and it is difficult to prepare in large quantities. Herein, the MWCNT/doped-polyindole solutions were easily prepared and directly drop coated on to SPCE.
The motivation of the present work is to utilize a conductive polymer as a component of glucose sensing materials to replace the typical metals and metals oxides as it provides a stable immobilization of biocomponents and can act as a mediator, effectively transporting electrons from enzyme active sites to the electrode with adjustable electrical conductivity. Moreover, the conducting polymers are compatible with biological molecules in neutral aqueous solutions. The objective of this work is to present the feasibility of using dPIn and MWCNT/dPIn composites as potential materials for a glucose sensor, consisting of a modified screen-printed electrode from the facile drop coating method. The fabricated glucose sensor can be expected to be utilized conveniently for the on-site analysis as a portable electrochemical biosensor. This work is evident that the utilizations of dPIn and MWCNT in combination with glucose oxidase and chitosan as a support material provide an effective portable glucose sensor with high sensitivity, selectivity, repeatability, and stability, suitable for glucose level monitoring in blood or urine for diabetes. The fabricated enzymatic glucose sensor can be easily connected to the PalmSens4 portable device. Moreover, it is promising to use as a prototype for real use applications in the future.
2. Materials and methods
2.1 Materials
Indole monomer for the synthesis and ferric chloride (FeCl3) were purchased from Merck, and Sigma-Aldrich, respectively. 37% v/v Hydrochloric acid (HCl) and ethanol were obtained from RCI Labscan. 70% v/v Perchloric acid (HClO4) for the doping was purchased from Panreac. Triton X-100 as a dispersant for MWCNT was obtained from Sigma-Aldrich. MWCNT (length ∼20 μm, purity >95%) was obtained from AlphaNano Technology Co., Ltd. Screen-printed carbon electrodes (SPCE, CI1703OR) with 12.5 mm in width and 30 mm in length were purchased from Quasense Co., Ltd., Thailand. Sodium hydroxide pellet (NaOH) was obtained from Sigma-Aldrich. Ethylene glycol was obtained from Merck. (3-Aminopropyl) triethoxysilane (APTES) was received from Aldrich. Chitosan (Mw 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–310000 Da, 75–85% deacetylated) and acetic acid were acquired from Sigma-Aldrich and ACI Labscan, respectively. D (+) Glucose anhydrous for analysis was purchased from Carlo Erba. Glucose Oxidase (GOx) from Aspergillus Niger (G2133-10KU, Type VII, 248878 U g−1, without added oxygen) was purchased from Sigma. Sodium phosphate monobasic (NaH2PO4) and sodium phosphate dibasic (Na2HPO4) purchased from Sigma-Aldrich were used to prepare the phosphate buffer solution (PBS) (2.34 g of NaH2PO4 and 15.13 g of Na2HPO4 were dissolved in deionized water to obtain 1000 mL of PBS at 0.1 M, pH 7.4). Potassium hexacyanoferrate (II) and (III), K3[Fe(CN6)]/K4[Fe(CN6)] with purity >99% were purchased from Sigma-Aldrich. All chemicals are of analytical reagent grades.
000–310000 Da, 75–85% deacetylated) and acetic acid were acquired from Sigma-Aldrich and ACI Labscan, respectively. D (+) Glucose anhydrous for analysis was purchased from Carlo Erba. Glucose Oxidase (GOx) from Aspergillus Niger (G2133-10KU, Type VII, 248878 U g−1, without added oxygen) was purchased from Sigma. Sodium phosphate monobasic (NaH2PO4) and sodium phosphate dibasic (Na2HPO4) purchased from Sigma-Aldrich were used to prepare the phosphate buffer solution (PBS) (2.34 g of NaH2PO4 and 15.13 g of Na2HPO4 were dissolved in deionized water to obtain 1000 mL of PBS at 0.1 M, pH 7.4). Potassium hexacyanoferrate (II) and (III), K3[Fe(CN6)]/K4[Fe(CN6)] with purity >99% were purchased from Sigma-Aldrich. All chemicals are of analytical reagent grades.
2.2 Polyindole synthesis and doping
PIn was synthesized by chemical oxidative polymerization in 0.1 M HCl at room temperature. FeCl3 was used as an oxidizing agent. The synthesis procedure was previously reported in a previous work.29 3 g of indole monomer in 10 mL ethanol was slowly dropped into the FeCl3 oxidant solution (10.67 g in 180 mL of 0.1 M HCl). The monomer: oxidant mole ratio was fixed at 1:2.57, and the mixture solution was continuously stirred for 24 h. Then, 1 M of HCl (200 mL) was poured over the mixture solution, and collected the precipitate by filtration, washed by distillated water several times and dried in hot air oven at 70 °C overnight to obtain a PIn powder. PIn powder was dedoped with 5M NaOH at the 10:1 of NaOH:indole mole ratio, and then filtered and rinsed by DI water before drying at 70 °C for 24 h. The dedoped PIn powder was subsequently doped by 2.5 M HClO4 at the 10:1 of HClO4: indole mole ratio. Then, the doped PIn (dPIn) was filtered by a cellulose acetate filter paper without water rinsing and dried at 70 °C for 24 h. The dPIn was slightly grind with a mortar to obtain the dPIn powder with the particle size of 325 ± 79 nm. The electrical conductivity was 1.26 ± 0.08 S cm−1.29
2.3 Preparation of dPIn and MWCNT-dPIn solutions
20 mg of the dPIn powder was dissolved in 1 mL of ethylene glycol and then continuously stirred for 24 h at room temperature to obtain the dPIn solution. The MWCNT solutions at various concentrations (0.5–2.0% w/v) were prepared by mixing 10 mL of 1 %w/v Triton X-100 (TX-100) in deionized water and sonicating for 1 h. To prepare the MWCNT-dPIn solution, the dPIn solution (20 mg mL−1) was mixed with each MWCNT solution at the 1:1 volume ratio. Next, the MWCNT-dPIn solutions were sonicated for 30 min and continuously stirred at room temperature for 2 h. Herein, the MWCNT-dPIn solutions (with the 0.5, 1.0, 1.5, 2.0 %w/v MWCNT solutions) are now called 0.5%MWCNT-dPIn, 1.0%MWCNT-dPIn, 1.5%MWCNT-dPIn, and 2.0%MWCNT-dPIn, respectively.
2.4 Electrode modification
The working electrode of SPCE (Ø = 3 mm, Area = 7.065 mm2) was pre-treated with 10 μL of 5M NaOH solution for 1 h to create the hydroxyl groups herein called b-SPCE. Next, the electrode was rinsed with DI water several times. The b-SPCE was dried at room temperature before drop-coated with 4.5 μL of the dPIn solution or the MWCNT-dPIn solution. Then, the modified electrode was dried at 70 °C for 2 h. Finally, the modified electrode was coated with 5 μL of 2.5% v/v APTES at the 3:1 volume ratio of water: ethanol, and then dried at room temperature, and then rinsed with deionized water. APTES has been used extensively for the functionalization of bioanalytical platforms.38
2.5 Immobilization of GOx
1% w/v Chitosan (CHI) in 2% acetic acid was mixed with 2.5 mg mL−1 GOx in PBS (0.1 M, pH 7.4) at the 1:2 volume ratio. The CHI-GOx solution was stirred 30 min and stored at 4 °C prior to use. 5 μL of CHI-GOx was immobilized on APTES/dPIn and APTES/MWCNT-dPIn by drop-coating and dried at room temperature. The immobilized electrode was stored at 4 °C prior to use. The modification steps of the working electrode are shown in Scheme 1(a).
 | ||
| Scheme 1 (a) Working electrode modification steps to prepare the CHI-GOx/APTES/1.5%MWCNT-dPIn; and (b) hydrolysis of APTES. | ||
2.6 Electrochemical analysis
Glucose solution was prepared in 0.1 M PBS at pH 7.4 and kept overnight at room temperature to reach the equilibrium mutarotation of glucose.1 Chronoamperometry (CA) was the electrochemical technique to evaluate the glucose sensor performance. The electrode was incubated with the glucose solution for 3 min before applying a constant potential. The screen-printed electrodes, consisting of the modified carbon electrode as the working electrode, Ag/AgCl as a reference electrode, and carbon as a counter electrode, were used. Cyclic voltammetry (CV) was also used to investigate the electroactivity and electron transfer of the modified electrode. For the determination of electroactive surface area and electroactivity of the modified electrodes, they were tested in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl. The potential range was between −1 to 1 V with a scan rate between 10–50 mV s−1 in 5 cycles. For the glucose mechanism, the electrodes were tested in 1 mM glucose in PBS by CV. The potential range was −0.8 to 0.8 V with a scan rate between 10–50 mV s−1 in 5 cycles. All electrochemical methods were analyzed at room temperature by the electrochemical analyzer (PalmSens BV, PalmSens4, PSTrace software). Electrochemical impedance spectroscopy (EIS) was carried out in 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl within 100000 to 0.1 Hz frequency range with an amplitude of 10 mV.
2.7 Characterizations
The functional groups of the modified electrodes were characterized by a Fourier transform infrared spectrometer (Thermo Scientific™, Nicolet™ iS™5), using the ATR-Diamond for the attenuated total reflection mode with the iD7 ATR Accessory in which air was used as a reference. The wavenumber range was between 4000–650 cm−1 with a resolution of 64 cm−1 and a scan number of 64. Element and chemical bonding analysis were analyzed and carried out by XPS (Kratos Analytical Shimadzu Group Company, Axis Ultra DLD), operated by a monochromated Al Kα X-ray radiation source. XPS survey spectra and high-resolution spectra were obtained at a pass energy of 160 eV and 40 eV, respectively. The Casa-XPS software was used to evaluate XPS spectra, C 1s was a reference for the calibration of binding energy (binding energy of C 1s is 284.8 eV). The morphological properties of the modified electrode were investigated by FE-SEM (HITACHI, S-4800), with an accelerating voltage of 5 kV and an emission current of 10 mA. All samples were coated with pure platinum before testing.3. Results and discussion
3.1 Structural and element analysis
The modifications of working electrode as shown in Scheme 1(a) were confirmed by ATR-FTIR as shown in Fig. 1. The evidence of SPCE treated with NaOH is not evident by ATR-FTIR. Therefore, it was further verified by XPS. The dPIn coated on b-SPCE shows the FTIR characteristic peaks at 3223 cm−1, 1570 cm−1, and 738 cm−1 which can be attributed to the N–H stretching, N–H deformation, and out of plane deformation of benzene ring, respectively.29 Peaks at 1056 and 1031 cm−1 are due to the ClO4− vibration, owing to the doping with HClO4.29 The dPIn shows a small peak at 1701 cm−1 (carbonyl groups) implying the overoxidation of the dPIn.29 The ATR-FTIR spectrum of the 1.5%MWCNT-dPIn shows the N–H stretching peak at 3266 cm−1; the peak is shifted to a higher wavenumber relative to the dPIn peak at 3223 cm−1, owing to the hydrogen bonding interaction between the N–H group of the dPIn and the hydroxyl group on the MWCNT structure. Moreover, the 1.5%MWCNT-dPIn spectrum clearly shows the asymmetric and symmetric vibration peaks of CH2 at 2947 and 2871 cm−1 belonging to MWCNT indicating the incorporation of MWCNT with the dPIn, in which MWCNT is covered with the dPIn as shown in SEM image (Fig. S1†). Another interaction between the conductive polymer based aromatic structure and MWCNT is the π–π interaction (aromatic π systems binding face to face with one another).35 It can be suggested that the interaction of the dPIn and MWCNT is the π–π interaction, occurring at the aromatic rings of the dPIn and the MWCNT. With the 1.5%MWCNT-dPIn modified with APTES, the spectrum shows the stretching peak of Si–OH at 3568–3552 cm−1, the asymmetric and symmetric vibration peaks of CH2 at ∼2932 and ∼2888 cm−1, and the two stretching peaks of NH2 at ∼1567 and ∼1486 cm−1 which can be distinguished owing to the incorporation of the hydrolyzed APTES as shown in Scheme 1(b).39 After the APTES coating, the stretching of N–H group shifts from 3222 to 3211 cm−1 for the dPIn and from 3266 to 3173 cm−1 for the 1.5%MWCNT-dPIn, indicating the hydrogen bonding between the Si–OH or NH2 groups of APTES with the N–H group of the dPIn. With CHI-GOx immobilized on APTES/dPIn and APTES/1.5%MWCNT-dPIn, two distinct FTIR spectrum peaks at ∼1651 and ∼1538 cm−1 can be observed, belonging to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the amide bond and the N–H bending vibration of the secondary amide of GOx, respectively.37. Moreover, the broad O–H stretching peaks for both modified electrodes resulted from the intermolecular hydrogen bonds between chitosan and the GOx molecules.40 Thus, the FTIR results confirm the functional groups of the dPIn, 1.5%MWCNT-dPIn, before and after the APTES and CHI-GOx modifications, and the interaction of these materials in each modifying step.
O stretching of the amide bond and the N–H bending vibration of the secondary amide of GOx, respectively.37. Moreover, the broad O–H stretching peaks for both modified electrodes resulted from the intermolecular hydrogen bonds between chitosan and the GOx molecules.40 Thus, the FTIR results confirm the functional groups of the dPIn, 1.5%MWCNT-dPIn, before and after the APTES and CHI-GOx modifications, and the interaction of these materials in each modifying step.
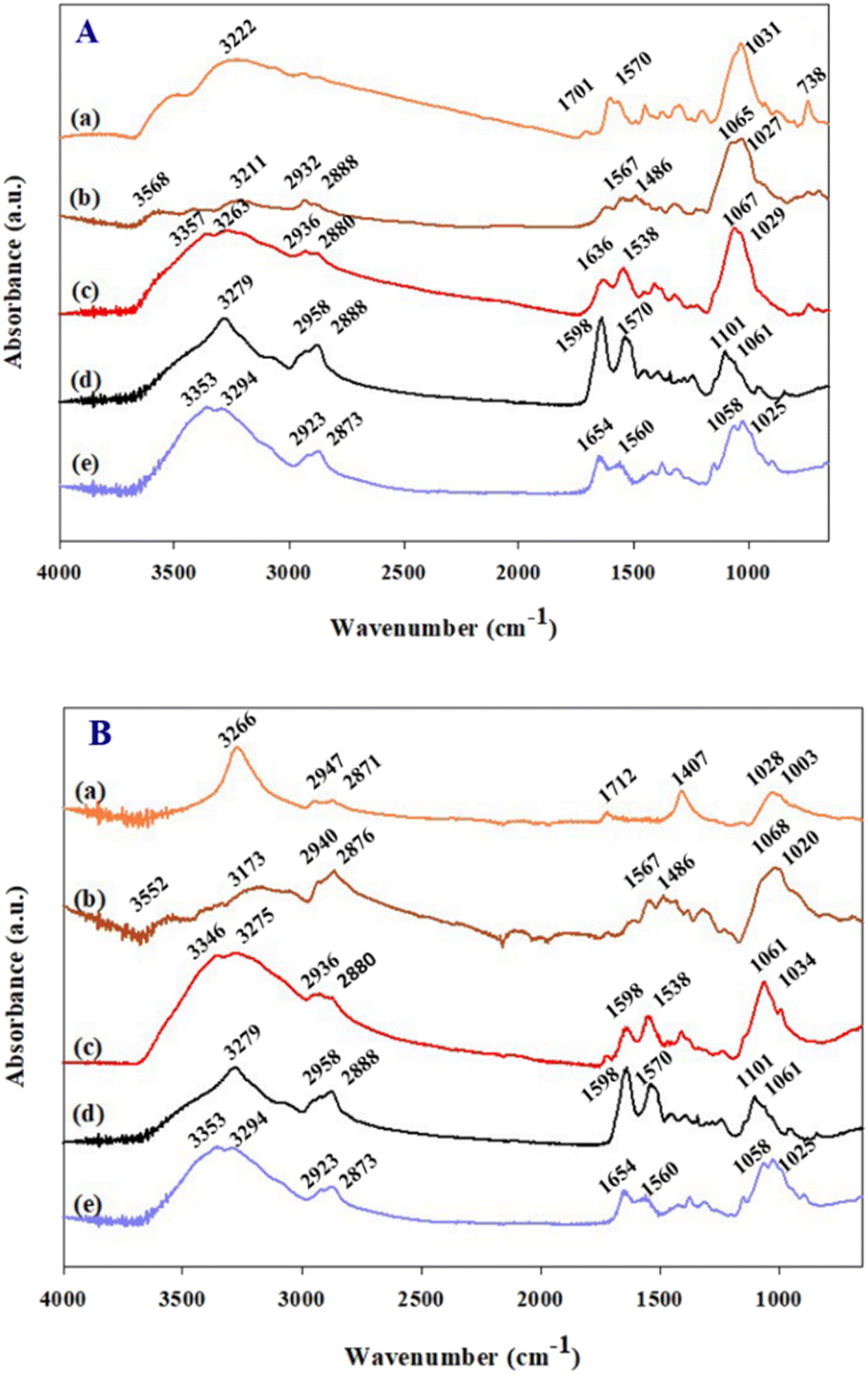 | ||
| Fig. 1 ATR-FTIR analysis of (A-a) dPIn; (A-b) APTES/dPIn; (A-c) CHI-GOx/APTES/dPIn; (B-a) 1.5%MWCNT-dPIn; (B-b) APTES/1.5%MWCNT-dPIn; (B-c) CHI-GOx/APTES/1.5%MWCNT-dPIn; (A-d) or (B-d) GOx; and (A-e) or (B-e) CHI. | ||
The element contents and the XPS spectra of the modified electrode were verified by XPS as shown in Table 1 and Fig. S2.† The amount of oxygen increases when treated with 5 M NaOH as confirmed by the increase in O 1s from 11.29 to 12.84% atom due to the increase of hydroxyl groups on the working electrode surface. The presence of 7.45% atom N 1s of the dPIn indicates the existence of the dPIn on the working electrode surface and 3.21% atom N 1s of the 1.5%MWCNT-dPIn implies the incorporation of the dPIn in MWCNT. After coating with APTES, the amounts of O 1s and N 1s and Si 2p increase, demonstrating the incorporation of the hydrolyzed APTES molecules into the dPIn and the 1.5%MWCNT-dPIn. After the immobilization of CHI-GOx, there are increments in the amounts of O 1s and N 1s implying that the CHI-GOx was embedded on the modified electrode as oxygen and nitrogen are the main elements of the CHI and GOx structures. The presence of a small amount of Na atoms is owed to the fact that PBS was used as a solvent for dissolving glucose oxidase.
| Electrodes | % Atomic composition | |||||
|---|---|---|---|---|---|---|
| O 1s | C 1s | N 1s | Cl 2p | Si 2p | Na1s | |
| a The presence of Si in the modified electrode without APTES is a contaminant. The presence of Cl in SPCE is a part of component in carbon ink. | ||||||
| SPCE | 11.29 | 84.59 | — | 2.68 | 1.44 | — |
| b-SPCE | 12.84 | 83.30 | — | 2.45 | 1.40 | — |
| dPIn | 27.68 | 56.19 | 7.45 | 3.01 | 3.67 | — |
| APTES/dPIn | 28.74 | 52.11 | 8.05 | 2.60 | 8.50 | — |
| CHI-GOx/APTES/dPIn | 30.09 | 54.53 | 8.46 | — | 5.60 | 1.32 |
| 1.5%MWCNT-dPIn | 14.53 | 80.78 | 3.21 | 1.48 | — | — |
| APTES/1.5%MWCNT-dPIn | 17.08 | 73.59 | 5.13 | 0.32 | 3.88 | — |
| CHI-GOx/APTES/1.5%MWCNT-dPIn | 31.44 | 55.85 | 8.16 | — | 3.62 | 0.94 |
The deconvolution of N 1s at each modification step is shown in Fig. S3.† SPCE and b-SPCE do not possess the N 1s peak. The N 1s peak was observed after the dPIn and 1.5%MWCNT-dPIn coating on the b-SPCE. The existing 4 peaks, with the binding energies of 398.5–398.7, 399.2–399.6, 400.7–401.2, and 401.6–402.1 eV, can be attributed to = N– (imine), N–H, –N·+H– (polaron), and = N+H– (bi-polaron), respectively.41 After the APTES coating, the 2 peaks with the binding energies of 399.6–399.8 eV for NH2 belonging to free amine, and 401.4–401.6 eV for the hydrogen bonding or the protonated amine (–NH2/NH3+).42 The hydrogen bond can occur between the dPIn and the protonated amine is arisen from the APTES molecules bound by the silane group.43 After the immobilization of CHI-GOx, the 4 peaks, with the binding energies at 398.9 eV, 399.5, 400.7, and 401.5 eV, can be attributed to NH2 (amine) arisen from chitosan and GOx, C–N of peptide bonds within protein of GOx, OC–NH- (amide linkage of chitosan), and NH3+ (protonated amine of chitosan), respectively.44,45
3.2 Morphological characterization
Surface morphologies of the working electrodes before and after modifications were characterized by FE-SEM as shown in Fig. 2. The SPCE and b-SPCE morphologies are not different, the morphologies appear as porous structures of carbon particles mixed with graphite flakes as can be seen in Fig. 2(a) and (b), respectively. The morphology change can be observed after coating with the dPIn as shown in Fig. 2(c); the dPIn is completely distributed on the b-SPCE without a porous structure, indicating the successful coating of dPIn. When coated with APTES and immobilized with CHI-GOx on the dPIn, the CHI-GOx/APTES/dPIn has less roughness than the dPIn alone because CHI-GOx was additionally coated on the dPIn surface as shown in Fig. 2(d). The morphology of the 1.5%MWCNT-dPIn coated on the b-SPCE is illustrated in Fig. 2(e); it shows long fibers of MWCNT covered with the dPIn, as evidenced by the different MWCNT morphology in Fig. S1.† When coated with APTES and immobilized with CHI-GOx on the 1.5%MWCNT-dPIn, CHI-GOx was completely deposited on the 1.5%MWCNT-dPIn surface leading to a smoother surface as shown in Fig. 2(f). Thus, these results suggest the successful immobilization of the CHI-GOx on the dPIn and the 1.5%MWCNT-dPIn surfaces. | ||
| Fig. 2 Surface morphologies at 30000× of magnification of various modified SPCEs: (a) SPCE; (b) b-SPCE; and the b-SPCE modified with various chemicals; (c) dPIn; (d) CHI-GOx/APTES/dPIn; (e) 1.5%MWCNT-dPIn; (f) CHI-GOx/APTES/1.5%MWCNT-dPIn. | ||
3.3 Electroactivity and electroactive surface area
Electrochemical activity of the modified electrodes was analyzed by cyclic voltammetry in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl as illustrated in Fig. 3(a). The current responses of the pristine SPCE and the b-SPCE are similar. However, after coating with dPIn and the 1.5%MWCNT-dPIn showed the higher current response as compared to the pristine SPCE and b-SPCE. Thus, coating with the dPIn and the 1.5%MWCNT-dPIn improved the current response of the electrode because the electroactive surface area of the electrode increased. The peak-to-peak separations (ΔEp) of anodic and cathodic peaks of the SPCE (0.220 V), the b-SPCE (0.180 V), the dPIn (0.340 V), the 1.5%MWCNT (0.360 V and 0.919 V) are higher than 0.059 V (the ΔEp theoretical value of an ideal electron transfer), implying the quasi-reversible electron transfer process.46 | ||
| Fig. 3 Electrochemical analysis: (a) cyclic voltammograms; and (b) Nyquist plots of various modified electrodes with the inset. The anodic peaks current vs. the square root of scan rates (ν0.5) and insets of cyclic voltammograms with different scan rates of: (c) CHI-GOx/APTES/dPIn; and (d) CHI-GOx/APTES/1.5%MWCNT-dPIn. The electrodes were tested in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl. | ||
The quasi-reversible electron transfer is further confirmed by the potential shift of anodic and cathodic peaks with increasing scan rate;46 the anodic peak shifts to a more positive potential and the cathodic peak shifts to a negative potential. The 1.5%MWCNT-dPIn shows the higher potential shift than the pristine dPIn and the SPCE, respectively as shown in Fig. S4.† The ipa/ipc values of the SPCE, the b-SPCE, the dPIn, and the 1.5%MWCNT-dPIn are 1.06, 1.11, 1.68, and 2.78, respectively. The ratios of ipa/ipc of the SPCE and b-SPCE are close to 1 revealing a fast electron transfer rate of [Fe(CN)6]3−/4 (redox probe), whereas those of the dPIn and the 1.5%MWCNT-dPIn are larger than 1 indicating the slower electron transfer rate of [Fe(CN)6]3−/4.46 After immobilization with CHI-GOx on the dPIn, the current response of the CHI-GOx/APTES/dPIn decreases because CHI-GOx acting as an electrostatic barrier blocks the electron transfer of [Fe(CN)6]3−/4 and obstructs the diffusion ability to the inner layer.47 Surprisingly, CHI-GOx immobilized on APTES/1.5%MWCNT leads to a higher current response. The charge transfer resistance (Rct) of the modified electrodes was measured by the electrochemical impedance spectroscopy (EIS) as shown in Fig. 3(b) whereas the Nyquist plots are shown in Fig. S5.† Generally, the charge transfer resistance of the electrode surface is equal to the semicircle diameter of the Nyquist diagram.48,49 The larger semicircle portion can be referred to the higher Rct related to the low electron transfer of [Fe(CN)6]3−/4.50 The larger semicircle portion can be referred to the higher Rct related to the low electron transfer of [Fe(CN)6]3−/4.50 The large semicircle portion can be seen for the SPCE (∼9 kΩ) and it becomes larger for the b-SPCE (∼18.5 kΩ) due to the creation of hydroxyl groups after the NaOH treatment leading the higher Rct. The small semicircles of the Nyquist plots can be observed when the b-SPCE was modified with the dPIn (∼0.0175 kΩ) and the 1.5%MWCNT-dPIn (∼0.055 kΩ) indicating the high electron transfer capability of [Fe(CN)6]3−/4 to the electrode surface. After immobilization, the semicircle portions became larger for CHI-GOx/APTES/dPIn (∼0.05 kΩ) and CHI-GOx/APTES/1.5%MWCNT-dPIn (∼0.29 kΩ) due to the addition of CHI-GOx.
The electroactive surface areas of the dPIn and the 1.5%MWCNT were determined by cyclic voltammetry at various scan rates as shown in Fig. 3(c) and (d), respectively. The inset figure is the plot between anodic peak current vs. the square root of scan rate. The electroactive surface areas as calculated by the Randles–Sevcik eqn (1)51,52 were 31.5 mm2 for the dPIn and 50.3 mm2 for the 1.5%MWCNT-dPIn; both are higher than the electroactive surface area of the pristine SPCE (8.81 mm2). This clearly implies that the 1.5%MWCNT provides a higher electroactive surface area than the dPIn because its surface contains the 1D structure of MWCNT fibres providing the larger surface area.
The anodic current can be represented by eqn (1)51,52 as:
 | (1) |
485C mol−1), R is the gas constant (8.314 J mol−1 K), T is the absolute temperature (298 K), A is the electroactive surface area (cm2), n is the number of electron involved in the redox reaction (n = 1 for [Fe(CN)6]3−/[Fe(CN)6]4−), D is the diffusion coefficient of the [Fe(CN)6]3− = 7.6 × 10−6 cm2 s−1,53 C0 is the [Fe(CN)6]3−/[Fe(CN)6]4− concentration (5 mM), and ν is the scan rate (V s−1).
3.4 Effect of MWCNT concentrations
MWCNT solutions of various concentrations (0.5, 1.0, 1.5, 2.0 %w/v) were mixed with the dPIn solution before coating onto the b-SPCE. The electrochemical behaviour of various MWCNT solutions coated on the b-SPCE was determined by cyclic voltammetry in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl as shown in Fig. 4(a). The 0.5%MWCNT-dPIn and 1.0%MWCNT-dPIn demonstrate a distinct oxidation peak and a reduction peak at 0.659 V and 0.639 V, respectively. The cyclic voltammograms of the 1.5%MWCNT-dPIn and 2.0%MWCNT-dPIn show two apparent oxidation peaks at 0.339, 0.619 V and two reduction peaks at 0.399, 0.679 V, corresponding to the oxidation and reduction of [Fe(CN)6]3−/4 with the bare MWCNT and the dPIn, respectively. The clear redox peaks with the enhanced current response of the 1.5%MWCNT-dPIn and 2.0%MWCNT-dPIn can be related to the increase in electrical conductivity of the composites and fast electron transfer.54,55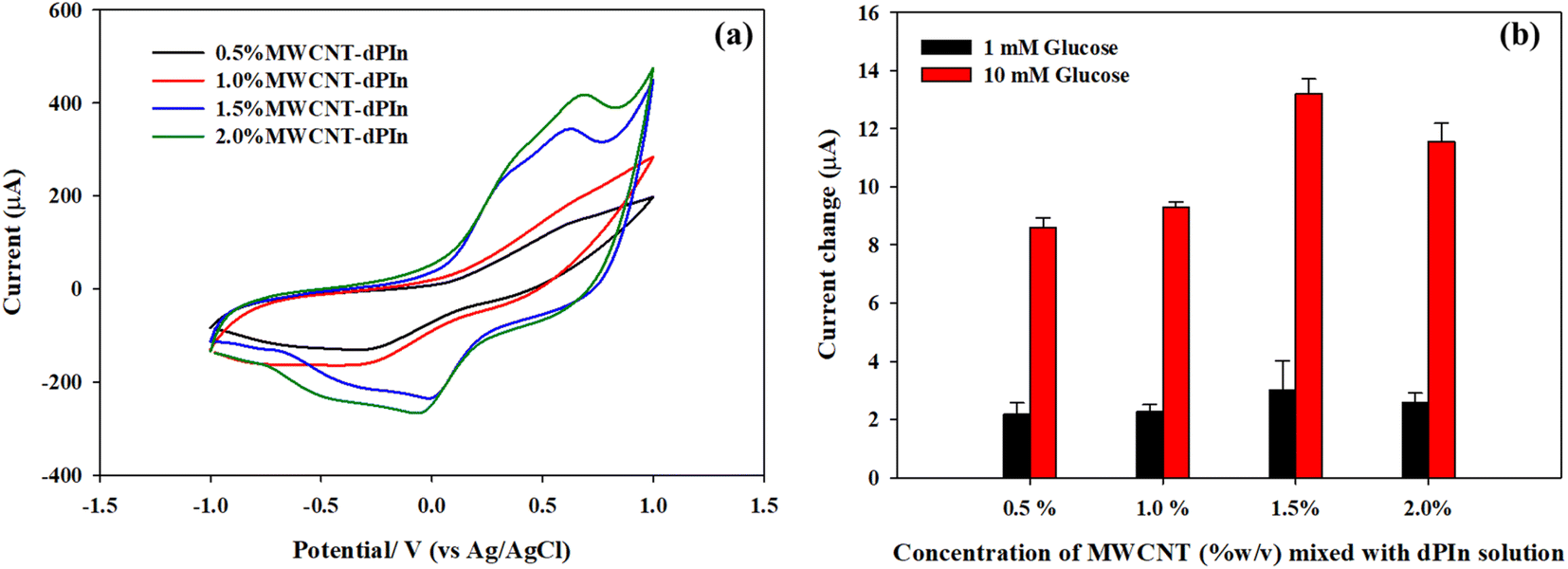 | ||
| Fig. 4 (a) Cyclic voltammograms in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl; and (b) current changes (ΔI = Iglc-IPBS) at +0.6 V in 1 mM and 10 mM glucose solutions of the dPIn mixed with various concentrations of MWCNT solutions. | ||
The cyclic voltammograms of CHI-GOx immobilized on various APTES modified on MWCNTs-dPIn electrodes are shown in Fig. S6.† The current responses are related to the amounts of MWCNT presence; 0.5%MWCNT-dPIn < 1.0%MWCNT-dPIn < 1.5%MWCNT-dPIn < 2.0%MWCNT-dPIn.
The electroactive surface area of the CHI-GOx immobilized on various electrodes modified with MWCNT-dPIn solution was analyzed by cyclic voltammetry in a solution of 5 mM of K3[Fe(CN6)]/K4[Fe(CN6)] containing 0.1 M PBS solution (pH 7.4) and 0.1 M KCl. The electroactive surface areas of the CHI-GOx/APTES/0.5%MWCNT-dPIn, the CHI-GOx/APTES/1.0%MWCNT-dPIn, the CHI-GOx/APTES/1.5%MWCNT-dPIn, and the CHI-GOx/APTES/2.0%MWCNT-dPIn are 33.9, 36.2, 53.8, 39.9 mm2, respectively. This demonstrates that the appropriate concentration of MWCNT-dPIn solution is 1.5%MWCNT-dPIn providing the largest electroactive surface area leading to the higher available sites for enzyme immobilization. At 2.0%MWCNT-dPIn, the electroactive surface area decreases possibly because of the agglomeration of MWCNT. The current changes in 1 mM and 10 mM glucose solutions of the modified electrodes with various MWCNT-dPIn solutions are illustrated in Fig. 4(b). The highest current change was obtained with the 1.5%MWCNT-dPIn, corresponding to the largest electroactive surface area.
3.5 Chronoamperometric detection of glucose solution
Glucose detection was analyzed by chronoamperometry. Each electrode was used at each glucose concentration, in which 200 μL of a glucose solution was dropped to cover the three-electrode cell as shown in Scheme 2(a). The experimental setup is shown in Fig. S7.† The glucose solution was incubated 3 min prior to applying a constant oxidation potential to electrochemical system of +0.6 V vs. Ag/AgCl. The glucose concentration range was from 0.01 mM to 100 mM. The current responses vs. time are shown in Fig. 5(a) and (b) for the CHI-GOx/APTES/dPIn and CHI-GOx/APTES/1.5%MWCNT-dPIn, respectively. It is apparent that the current response increases with increasing glucose concentration which can be related to the glucose oxidation by glucose oxidase with the FAD cofactor, generating H2O2. Finally, the H2O2 was electrochemically oxidized under an applied constant voltage at the electrode surface releasing electron leading to the increase in the current response.56 | ||
| Scheme 2 (a) Components of SPCE; and (b) the possible glucose mechanism of the fabricated glucose sensor. | ||
 | ||
| Fig. 5 Chronoamperometric current responses vs. time at +0.6 V vs. Ag/AgCl and the current change (ΔI = Iglc-IPBS) vs. glucose concentration (C, mM) of: (a), (c) CHI-GOx/APTES/dPIn; and (b), (d) CHI-GOx/APTES/1.5%MWCNT-dPIn. Inset in figures (c) and (d) are logarithmic scale of ΔI vs. C. | ||
Fig. 5(c) and (d) demonstrate the increases in the current changes (ΔI = IGlc-IPBS) with increasing glucose concentration with a power law relation (non-linear relation); the data were plotted on the logarithmic scales (linear relation) as shown in the inset figures. The power law equation of the CHI-GOx/APTES/dPIn is y = 0.8312 × 0.5251, r2 = 0.9805 in glucose concentration range between 0.01 and 50 mM. The power law equation of the CHI-GOx/APTES/1.5%MWCNT-dPIn is y = 3.0722 × 0.5667, r2 = 0.9927 in glucose concentration range between 0.01–100 mM. The linear relation between current and concentration of the glucose sensors can be obtained in logarithmic scale as shown in inset of Fig. 5c and d. Sensitivity is defined as dy/dx divided by geometric surface area (AG), where y is the current change (ΔI) and x is the glucose concentration. Herein, the sensitivity of the CHI-GOx/APTES/dPIn is 55.7 μA mM−1 cm−2 with the LOD of 0.01 mM or 10 μM, and the detectable glucose concentration range is 0.01–50 mM. The sensitivity of the CHI-GOx/APTES/1.5%MWCNT-dPIn is 182.9 μA mM−1 cm−2 with the LOD of 0.01 mM or 10 μM, and the detectable glucose concentration range is 0.01–100 mM. The calculations of sensitivity are shown in the ESI.† It can be seen that the composite of 1.5%MWCNT-dPIn provides a higher sensitivity than the dPIn because MWCNT effectively facilitates the electron transfer rate of the working electrode and enhances electroactive surface area of electrode.57
The apparent LOD is defined as the lowest glucose concentration detectable by the fabricated glucose sensor as observed from the logarithmic scale of ΔI vs. C in inset Fig. 5(c) and (d). The apparent LOD value of the CHI-GOx/APTES/dPIn is 0.01 mM or 10 μM and the apparent LOD value of the CHI-GOx/APTES/1.5%MWCNT-dPIn is 0.01 mM or 10 μM. Herein, the LOD and the detectable glucose concentration ranges of the fabricated glucose sensors well cover the glucose detections in urine (2.78–5.55 mM for healthy patients and more than 5.55 mM for diabetic patients) and blood (4.9–6.9 mM for healthy patients and 2–40 mM for diabetic patients).2,58
The response times of the glucose sensors are longer than 150 s for the CHI-GOx/APTES/dPIn and 200 s for the CHI-GOx/APTES/1.5%MWCNT-dPIn when analyzed by chronoamperometry. This long response times might be related to a possible cause in which the electron takes a long time to move from the active sites of GOx to the surface of electrode.
The comparison of current responses in 1 mM and 10 mM of glucose solution of enzymatic and non-enzymatic glucose sensors is reported in Fig. S8 in the ESI.† The present enzymatic glucose sensors show that the current change increases with increasing glucose concentration because of the oxidation of glucose leading to the H2O2 production, releasing electron through the electrode surface, and leading to the increase in current response. On the other hand, the non-enzymatic glucose sensors (the dPIn and the 1.5%MWCNT-dPIn) demonstrate the decreases in the current change with increasing glucose concentration because the glucose oxidation does not occur on the electrode surface and the glucose molecules are acting as a non-electrolyte blocking the oxidation of ions in the glucose solution leading to the decrease in current response.
The step-wise glucose detection of the glucose sensors based on the CHI-GOx/APTES/1.5%MWCNT-dPIn and the CHI-GOx/APTES/dPIn was investigated by chronoamperometry at +0.6 V vs. Ag/AgCl. First, 200 μL of 0.1 M PBS solution was dropped on the three-electrode cell to obtain the current response. Then, 50 μL of a glucose solution (0.01−100 mM) was added into the PBS solution covering the modified electrode surface at every 50 s.
The current responses are illustrated in Fig. 6(a) and (b). The current response increases with increasing glucose concentration, and tends to decrease with further additions of the PBS solutions to reduce the glucose concentration. Thus, the CHI-GOx/APTES/dPIn can be used up to the glucose concentration of 50 mM, because the current response decreased when exposed to the 100 mM of glucose concentration. The CHI-GOx/APTES/1.5%MWCNT-dPIn can be used up to the 100 mM glucose concentration. The CHI-GOx/APTES/1.5%MWCNT-dPIn shows the higher current response than the CHI-GOx/APTES/dPIn due to its higher electrocatalytic efficiency as well as the synergistic effect of MWCNTs and the dPIn.55
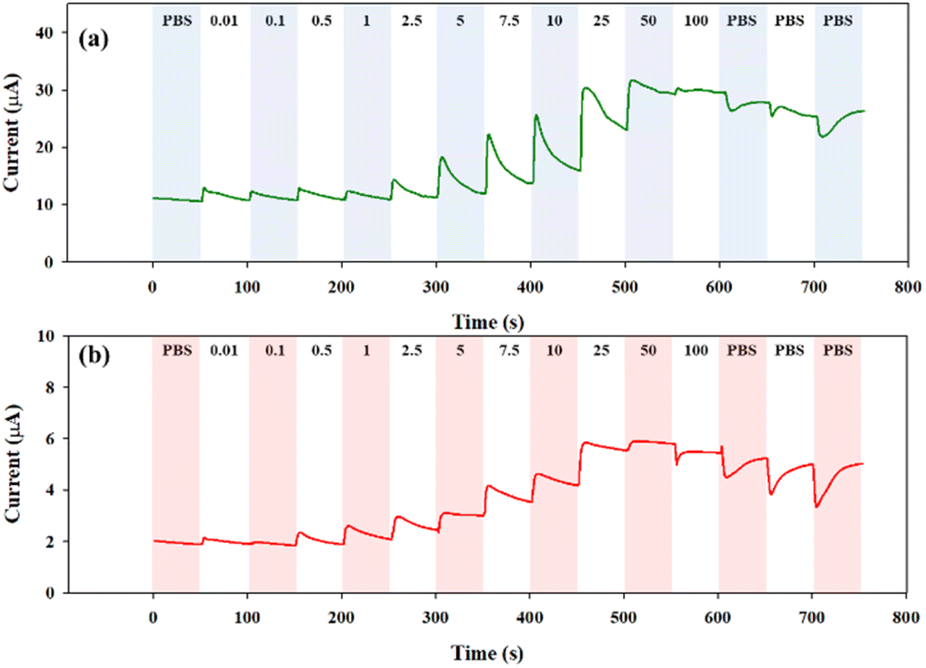 | ||
| Fig. 6 Continuous current response vs. time at +0.6 V vs. Ag/AgCl in various glucose concentrations of: (a) CHI-GOx/APTES/1.5%MWCNT-dPIn; and (b) CHI-GOx/APTES/dPIn. | ||
3.6 Glucose mechanism
The glucose mechanism of the previously fabricated sensors is shown in Scheme 2(b), in which glucose is catalyzed by GOx(FAD) to gluconolactone (reaction 2), and the GOx(FAD) is converted to its reduced form GOx (FADH2). Gluconolactone is hydrolyzed to gluconic acid by water in the solution (reaction 3). The GOx(FADH2) is re-oxidized by oxygen in the solution to reproduce GOx(FAD), and H2O2 is released (reaction 4). The generated H2O2 is oxidized at the electrode surface and produces the electron (reaction 5). The mechanisms of the reactions (2) to (5) can be referred to as in the previous reports.59 The oxidation of H2O2 is proportional to the glucose concentration. In the present work, the MWCNT-dPIn2+ (bipolaron) is suggested to act as an electron acceptor and transfer electron to electrode (reaction 6). The glucose sensor mechanism was confirmed by the reaction with o-dianisidine used as a chromogenic oxygen acceptor in the presence of enzyme peroxidase. The result confirms that H2O2 was generated by the interaction of glucose and GOx as observed by the solution color suddenly changed from colorless to brown as shown in Fig. S9 in the ESI.†59,60| Glucose + GOx (FAD) → Gluconolactone + GOx (FADH2) | (2) |
| Gluconolactone → Gluconic acid | (3) |
| GOx (FADH2) + O2 + 2e− + 2H+ → H2O2 + GOx (FAD) | (4) |
| H2O2 → O2 + 2e− + 2H+ | (5) |
| MWCNT-dPIn 2+(bipolaron) + 2e− → MWCNT-dPIn | (6) |
The cyclic voltammograms and the plots of anodic and cathodic peak current at various scan rates (10–50 mV s−1) in 1 mM glucose solution for CHI-GOx/APTES/dPIn are illustrated in Fig. 7(a) and (b), and for CHI-GOx/APTES/1.5%MWCNT-dPIn in Fig. 7(c) and (d). The anodic peak current linearly increases with increasing scan rate and the cathodic peak linearly decreases with increasing scan rate, with the correlation coefficients, r2, are close to 1. This result suggests the surface controlled redox reaction occurred.61,62
 | ||
| Fig. 7 Electrochemical characterization by in 1 mM glucose solution: cyclic voltammograms and the anodic and cathodic currents vs. scan rate of (a) and (b) for CHI-GOx/APTES/dPIn; and (c) and (d) for CHI-GOx/APTES/1.5%MWCNT-dPIn. | ||
3.7 Reproducibility, repeatability, stability, and selectivity of glucose sensor
The glucose sensor reproducibility, repeatability, and stability were analyzed by chronoamperometry in 1 mM glucose solution at + 0.6 V. The CHI-GOx/APTES/dPIn and the CHI-GOx/APTES/1.5%MWCNT-dPIn have the reproducibility (n = 5) with the 6.2 %RSD and 7.5 %RSD, respectively, as shown in Fig. 8(a). Thus, the fabricated glucose sensors show good reproducibility owing to the low percentages of relative standard deviation (%RSD). | ||
| Fig. 8 (a) Electrode reproducibility; (b) electrode repeatability; (c) electrode stability at different storage times at 4 °C, the electrodes were tested in 1 mM glucose solution; and (d) selectivity in 1 mM of interference chemicals. | ||
The repeatability test is shown in Fig. 8(b). The data clearly show that the glucose sensors are not suitable for reuse because the current responses decrease to 69.6% and 42.3% for the second time use and 57% and 37.8% for the third time use for the CHI-GOx/APTES/dPIn and the CHI-GOx/APTES/1.5%MWCNT-dPIn, respectively. The reason is the leakage of enzyme, a common drawback derived from the immobilization via physical adsorption in which enzymes were adsorbed to the support matrix by the weak interactions such as hydrogen bonding, van der Waals forces, or hydrophobic interaction.63 Hence, these glucose sensors are more suitable for a disposable use.
For long term stability, the sensor was stored in refrigerator at 4 °C for various time periods before testing in the 1 mM glucose solution. The glucose sensors are stable up to 28 days as observed from the current responses to be at 76.70% and 81.09% relative to the fresh CHI-GOx/APTES/dPIn and the fresh CHI-GOx/APTES/1.5%MWCNT-dPIn, respectively, as illustrated in Fig. 8(c). The results here indicate that these two glucose sensors have high storage stability during a period of at least 28 days.
Most interferences of glucose found in blood are such as Na+, K+, ascorbic acid (AA), lactic acid (LA), uric acid (UA). Generally, the interferences can be found at very low concentrations in blood relative to the glucose concentration.64 Herein, the selectivity was each tested with the 1 mM of interference as compared to the 10 mM glucose solution in 0.1 M PBS. In Fig. 8(d), the sensors show high selectivity to glucose relative to other interference. The current change ratios [ΔI(Glucose)/ΔI(interference)] of the CHI-GOx/APTES/dPIn are 15.70, 8.93, 9.69, −6.57, −63.86 for the AA, LA, UA, K+, Na+, respectively. The current change ratios of CHI-GOx/APTES/1.5%MWCNT-dPIn are 5.25, 7.56, 64.45, −12.97, −10.62, for the AA, LA, UA, K+, Na+, respectively. The NaCl and KCl show negative responses; the current in KCl is lower than in 0.1 M PBS, owing to the electron repulsion with the dPIn or 1.5%MWCNT-dPIn on the surface of SPCE65
3.8 Comparison of glucose sensors fabricated from conductive polymers
The enzymatic glucose sensors based on the dPIn and the 1.5%MWCNT-dPIn are compared to the glucose sensors based on conductive polymers from previous reports as listed in Table 2. Although the fabricated glucose sensors in this work have lower sensitivity values than some in the previous works, it is higher than the minimum requirement for a blood glucose sensor (1 μA mM−1 cm−2).66 Moreover, the present glucose sensors can detect glucose concentrations in a wider range that well covers the glucose detection in urine and blood of healthy person and diabetes patients.2,58 The preparation method of the glucose sensors by drop coating on SPCE in our work is facile and easy to prepare in large quantities when compared to the other works with electrochemistry. The advantages of the fabricated glucose sensor based on SPCE with three-electrodes cell is its low cost when manufactured in large volume, and a wider detectable range of glucose concentration when compared to other fabricated material electrodes such as gold/platinum electrode, glassy carbon electrode, indium tin oxide (ITO) and etc. as reported and compared in Table 2. The sensors are ready to be used with any glucose sample solutions as they were prepared on the screen-printed electrode consisting of the three-electrodes cell coupled with the portable PalmSen4 unit.| Glucose sensor | E (v) | Sensitivity (μA mM−1 cm−2) | Dynamic range (mM) | LOD | Ref. |
|---|---|---|---|---|---|
| a Spin coating on SiO2.b Electrodeposition on gold electrode.c Screen printed paper.d Electrodeposition on platinum electrode.e Electrodeposition on graphite rod.f Electrodeposition on platinum electrode.g Electrodeposition on ITO.h Electrodeposition of SPCE.i Electrodeposition on carbon fiber microelectrode (CF).j Drop coating on SPCE. | |||||
| GOx/PEDOT:PSSa | −1.50 | 1.65 | 1.1–16.5 | — | 67 |
| CS–GOD/3DOM SPAN/PBb | 0 | 99.4 | 0.002–1.6 | 0.4 μM | 68 |
| GOx (ePAD)/PB/Paperc | −0.10 | 0.58 | 0–33.1 | — | 69 |
| GOx/Nafion/CNT/PPyd | + 0.52 | 54.2 | 1–4.1 | 5.0 μM | 35 |
| GOx/GP/PB/PPye | + 0.05 | 1.9 | 0.1–20 | 0.1 mM | 70 |
| GOx/PEDOT-PAAf | + 0.60 | 0.0274 | 0.98–30 | 0.29 μM | 59 |
| GOx/poly(Th-co-TCA)/ITOg | −0.175 | 38.75 | 0.001–27.0 | 0.1 μM | 71 |
| GOx/Pt/rGO/P3ABAh | +0.50 | 22.01 | 0.26–6.0 | 44.3 μM | 49 |
| GOx/PCBe | +0.35 | 14 | 1.0–4.9 | 0.14 mM | 51 |
| GOx/PEDOT/CFi | −0.65 | 8.5 | 0.5–15.0 | — | 72 |
| CHI-GOx/APTES/dPInj | +0.60 | 55.7 | 0.01–50 | 10 μM | This work |
| CHI-GOx/APTES/1.5%MWCNT-dPInj | +0.60 | 182.9 | 0.01–100 | 10 μM | |
4. Conclusion
The chronoamperometric enzymatic glucose sensors were easily fabricated by drop coating on the SPCE. The SPCE was modified by the NaOH treatment to modify hydrophilic surface property by the creation of hydroxyl groups to obtain b-SPCE. The dPIn and its composites with MWCNT at various MWCNT concentrations were drop coated onto the b-SPCE.The most suitable MWCNT concentration was 1.5 %w/v providing the highest current response toward glucose. The SPCEs modified with the dPIn and the 1.5%MWCNT-dPIn showed the higher increases in the electroactive surface area and current response when compared to the SPCE as confirmed by cyclic voltammetry.
The chitosan-glucose oxidase (CHI-GOx) was immobilized on the modified electrode by using APTES as a linker by the hydrogen bonding between the enzyme and the dPIn. The enzymatic glucose sensor involved in H2O2 as evidenced by the o-dianisidine-glucose solution color change from colorless to brown in the presence of enzyme peroxidase. The sensitivity of the fabricated enzymatic glucose sensor based on the 1.5%MWCNT-dPIn showed the higher sensitivity than the dPIn because of the larger electroactive surface area and higher electrical conductivity properties which increase the glucose oxidation and induce the higher electron transfer through the electrode surface. The fabricated enzymatic glucose sensors demonstrated the increases in current response with increasing glucose concentration at the applied potential at +0.6 V vs. Ag/AgCl confirming the successful chronoamperometric enzymatic glucose sensor. The current response increased with increasing glucose concentration with the power law relation. The sensitivity of the CHI-GOx/APTES/dPIn was 55.7 μA mM−1 cm−2 at LOD of 0.01 mM or 10 μM and the detectable glucose concentration range was 0.01–50 mM. The sensitivity of the CHI-GOx/APTES/1.5%MWCNT-dPIn was 182.9 μA mM−1 cm−2 at LOD of 0.01 mM or 10 μM and the detectable glucose concentration range was 0.01–100 mM. The fabricated glucose sensor is thus promising to detect glucose in urine and blood. The herein glucose sensors were suitable as the disposal glucose sensors, and they can also be used for the continuous or step-wise glucose monitoring.
Author contributions
KP: Conceptualization, methodology, formal analysis, writing–original draft. AS: Conceptualization, writing–review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors particularly grateful for the assistance and financial supports from the Conductive and Electroactive Polymers Research Unit of Chulalongkorn University; the Thailand Science Research and Innovation Fund of Chulalongkorn University (TSRI-CU) (HEA666300110), the National Research Council of Thailand (NRCT), and the Ratchadapisek Somphot Fund for Postdoctoral Fellowship, Chulalongkorn University.References
- D.-W. Hwang, S. Lee, M. Seo and T. D. Chung, Anal. Chim. Acta, 2018, 1033, 1–34 Search PubMed.
- D. Bruen, C. Delaney, L. Florea and D. Diamond, Sensors, 2017, 17, 1866 Search PubMed.
- J. Kim, A. S. Campbell and J. Wang, Talanta, 2018, 177, 163–170 Search PubMed.
- T. Sridara, J. Upan, G. Saianand, A. Tuantranont, C. Karuwan and J. Jakmunee, Sensors, 2020, 20, 808 Search PubMed.
- A. Safavi and F. Farjami, Biosens. Bioelectron., 2011, 26, 2547–2552 Search PubMed.
- S. Damiati and B. Schuster, Sensors, 2020, 20, 1721 Search PubMed.
- R. A. S. Couto, J. L. F. C. Lima and M. B. Quinaz, Talanta, 2016, 146, 801–814 Search PubMed.
- M. Metto, S. Eramias, B. Gelagay and A. P. Washe, Int. J. Electrochem., 2019, 2019, 6318515 Search PubMed.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865–891 Search PubMed.
- D. Ji, L. Liu, S. Li, C. Chen, Y. Lu, J. Wu and Q. Liu, Biosens. Bioelectron., 2017, 98, 449–456 Search PubMed.
- M. Bauer, L. Wunderlich, F. Weinzierl, Y. Lei, A. Duerkop, H. N. Alshareef and A. J. Baeumner, Anal. Bioanal. Chem., 2021, 413, 763–777 Search PubMed.
- A. Martín, J. Kim, J. F. Kurniawan, J. R. Sempionatto, J. R. Moreto, G. Tang, A. S. Campbell, A. Shin, M. Y. Lee, X. Liu and J. Wang, ACS Sens., 2017, 2, 1860–1868 Search PubMed.
- S. Emaminejad, W. Gao, E. Wu, Z. A. Davies, H. Yin Yin Nyein, S. Challa, S. P. Ryan, H. M. Fahad, K. Chen, Z. Shahpar, S. Talebi, C. Milla, A. Javey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4625 Search PubMed.
- X. Li, M. Zhang, Y. Hu, J. Xu, D. Sun, T. Hu and Z. Ni, Biomed. Microdevices, 2020, 22, 17 Search PubMed.
- M. Adeel, M. M. Rahman, I. Caligiuri, V. Canzonieri, F. Rizzolio and S. Daniele, Biosens. Bioelectron., 2020, 165, 112331 Search PubMed.
- M. H. Hassan, C. Vyas, B. Grieve and P. Bartolo, Sensors, 2021, 21, 4672 Search PubMed.
- J. R. Sempionatto, T. Nakagawa, A. Pavinatto, S. T. Mensah, S. Imani, P. Mercier and J. Wang, Lab Chip, 2017, 17, 1834–1842 Search PubMed.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927–938 Search PubMed.
- H. Lee, Y. J. Hong, S. Baik, T. Hyeon and D.-H. Kim, Adv. Healthcare Mater., 2018, 7, 1701150 Search PubMed.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 Search PubMed.
- A. Taheri-Kafrani, S. Kharazmi, M. Nasrollahzadeh, A. Soozanipour, F. Ejeian, P. Etedali, H.-A. Mansouri-Tehrani, A. Razmjou, S. M.-G. Yek and R. S. Varma, Crit. Rev. Food Sci. Nutr., 2021, 3160–3196 Search PubMed.
- M. L. Verma, S. Kumar, A. Das, J. S. Randhawa and M. Chamundeeswari, Environ. Chem. Lett., 2020, 18, 315–323 Search PubMed.
- D. B. Gorle, S. Ponnada, M. S. Kiai, K. K. Nair, A. Nowduri, H. C. Swart, E. H. Ang and K. K. Nanda, J. Mater. Chem. B, 2021, 9, 7927–7954 Search PubMed.
- S. Ponnada, D. B. Gorle, M. S. Kiai, C. V. Raju, M. Faraji, R. K. Sharma and A. Nowduri, Anal. Methods, 2022, 14, 560–573 Search PubMed.
- S. Ponnada, D. B. Gorle, M. S. Kiai, S. Rajagopal, R. K. Sharma and A. Nowduri, Adv. Mater., 2021, 2, 5986–5996 Search PubMed.
- M. A. Anees A. Ansari, M. S. Alsalhi and A. S. Aldwayyan, Nanostructured Metal Oxides Based Enzymatic Electrochemical Biosensors, Intechopen, 2010 Search PubMed.
- E. Sehit and Z. Altintas, Biosens. Bioelectron., 2020, 159, 112165 Search PubMed.
- J. Branimir, G. Branimir, A. Mirjana, K.-J. Zorica, S. Jasmina and G. Milica, Int. J. Electrochem. Sci., 2016, 11, 1152–1161 Search PubMed.
- K. Phasuksom and A. Sirivat, Synth. Met., 2016, 219, 142–153 Search PubMed.
- S. Sharma, P. Joshi, S. Mehtab, M. G. H. Zaidi, K. Singhal and T. I. Siddiqi, J. Anal. Test., 2020, 4, 13–22 Search PubMed.
- M. Ghita and D. W. M. Arrigan, Electrochim. Acta, 2004, 49, 4743–4751 Search PubMed.
- P. C. Pandey, D. S. Chauhan and V. Singh, Mater. Sci. Eng. C, 2012, 32, 1–11 Search PubMed.
- P. C. Pandey, J. Chem. Soc., Faraday Trans., 1988, 84(7), 2259–2265 Search PubMed.
- L. Kumar, R. Gupta, D. Thakar, V. Vibhu and S. Annapoorni, Plasmonics, 2013, 8, 487–494 Search PubMed.
- B. K. Shrestha, R. Ahmad, S. Shrestha, C. H. Park and C. S. Kim, Sci. Rep., 2017, 7, 16191 Search PubMed.
- I. Anshori, L. Nuraviana Rizalputri, R. Rona Althof, S. Sean Surjadi, S. Harimurti, G. Gumilar, B. Yuliarto and M. Handayani, Nanocomposites, 2021, 7, 97–108 Search PubMed.
- B. Lakard, Appl. Sci., 2020, 10, 6614 Search PubMed.
- S. K. Vashist, E. Lam, S. Hrapovic, K. B. Male and J. H. T. Luong, Chem. Rev., 2014, 114, 11083–11130 Search PubMed.
- N. Majoul, S. Aouida and B. Bessaïs, Appl. Surf. Sci., 2015, 331, 388–391 Search PubMed.
- L. F. Ang, L. Y. Por and M. F. Yam, PLoS One, 2013, 8, e70597 Search PubMed.
- M.-Y. Hua, C.-J. Chen, H.-C. Chen, R.-Y. Tsai, W. Cheng, C.-L. Cheng and Y.-C. Liu, Sensors, 2011, 11, 5873–5885 Search PubMed.
- H. Min, P.-L. Girard-Lauriault, T. Gross, A. Lippitz, P. Dietrich and W. E. S. Unger, Anal. Bioanal. Chem., 2012, 403, 613–623 Search PubMed.
- T. Serodre, N. Oliveira, D. Miquita, M. Ferreira, A. Santos, V. Resende and C. Furtado, J. Braz. Chem. Soc., 2019, 30, 2488–2499 Search PubMed.
- V. T. K. P. Fidal, S. Inguva, S. Krishnamurthy, E. Marsili, J.-P. Mosnier and T. S. Chandra, Enzyme Microb. Technol., 2017, 96, 67–74 Search PubMed.
- B. Zhang, R. Hu, D. Sun, T. Wu and Y. Li, Sci. Rep., 2018, 8, 15397 Search PubMed.
- W. T. Wahyuni, R. Heryanto, E. Rohaeti, A. Fauzi and B. R. Putra, J. Kim. Sains Apl., 2021, 2021(24), 8 Search PubMed.
- E. Djaalab, M. E. Samar, S. Zougar and R. Kherrat, Catalysts, 2018, 8, 233 Search PubMed.
- N. Huang, M. Liu, H. Li, Y. Zhang and S. Yao, Anal. Chim. Acta, 2015, 853, 249–257 Search PubMed.
- S. Phetsang, J. Jakmunee, P. Mungkornasawakul, R. Laocharoensuk and K. Ounnunkad, Bioelectrochemistry, 2019, 127, 125–135 Search PubMed.
- S. Chaiyo, E. Mehmeti, W. Siangproh, T. L. Hoang, H. P. Nguyen, O. Chailapakul and K. Kalcher, Biosens. Bioelectron., 2018, 102, 113–120 Search PubMed.
- G. Bagdžiūnas and D. Palinauskas, Biosensors, 2020, 10, 104 Search PubMed.
- H. Zare, G. D. Najafpour, M. Jahanshahi, M. Rahimnejad and M. Rezvani, Rom. Biotechnol. Lett., 2017, 22, 12611–12619 Search PubMed.
- C.-H. Hsu, A. K. Gupta, A. Purwidyantri, B. A. Prabowo, C.-H. Chen, C.-C. Chuang, Y.-C. Tian, Y.-J. Lu and C.-S. Lai, Chemosensors, 2022, 10, 94 Search PubMed.
- N. I. Ibrahim and A. S. Wasfi, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 757, 012036 Search PubMed.
- V. Kathiresan, D. Thirumalai, T. Rajarathinam, M. Yeom, J. Lee, S. Kim, J.-H. Yoon and S.-C. Chang, J. Anal. Sci. Technol., 2021, 12, 5 Search PubMed.
- R. Tipnis, F. Vaddiraju S Fau - Jain, D. J. Jain F Fau - Burgess, F. Burgess Dj Fau - Papadimitrakopoulos and F. Papadimitrakopoulos, J. Diabetes Sci. Technol., 2007, 1, 193–200 Search PubMed.
- S. Madhurantakam, J. B. Karnam, J. B. B. Rayappan and U. M. Krishnan, Appl. Nanosci., 2017, 7, 773–780 Search PubMed.
- A. Müsse, F. La Malfa, V. Brunetti, F. Rizzi and M. De Vittorio, Micromachines, 2021, 12, 805 Search PubMed.
- P. Krzyczmonik, E. Socha and S. Skrzypek, Electrocatalysis, 2018, 9, 380–387 Search PubMed.
- M. Pohanka, Int. J. Electrochem. Sci., 2021, 16, 210719 Search PubMed.
- R. S. Alruwais, W. A. Adeosun, H. M. Marwani, M. Jawaid, A. M. Asiri and A. Khan, Polymers, 2021, 13, 2562 Search PubMed.
- M. Zidan, T. W. Tee, A. H. Abdullah, Z. Zainal and G. Joo Kheng, Int. J. Electrochem. Sci., 2011, 6, 289–300 Search PubMed.
- H. H. Nguyen and M. Kim, Appl. Sci. Converg. Technol., 2017, 26, 157–163 Search PubMed.
- M. Dervisevic, M. Alba, B. Prieto-Simon and N. H. Voelcker, Nano Today, 2020, 30, 100828 Search PubMed.
- W.-Y. Jeon, H.-H. Kim and Y.-B. Choi, Membranes, 2021, 11, 384 Search PubMed.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246–1301 Search PubMed.
- J. Liu, M. Agarwal and K. Varahramyan, Sens. Actuators, B, 2008, 135, 195–199 Search PubMed.
- X. Chen, Z. Chen, R. Tian, W. Yan and C. Yao, Anal. Chim. Acta, 2012, 723, 94–100 Search PubMed.
- J. Noiphung, T. Songjaroen, W. Dungchai, C. S. Henry, O. Chailapakul and W. Laiwattanapaisal, Anal. Chim. Acta, 2013, 788, 39–45 Search PubMed.
- A. Ramanavicius, A. I. Rekertaitė, R. Valiūnas and A. Valiūnienė, Sens. Actuators, B, 2017, 240, 220–223 Search PubMed.
- S.-Y. Kim, H.-J. Jo and S.-H. Choi, J. Korean Wood Sci. Technol., 2019, 36, 269–278 Search PubMed.
- J. Chen, X. Zheng, Y. Li, H. Zheng, Y. Liu and S.-i. Suye, J. Electrochem. Soc., 2020, 167, 067502 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04947c |
| This journal is © The Royal Society of Chemistry 2022 |