
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDearomative gem-diprenylation of hydroxynaphthalenes by an engineered fungal prenyltransferase†
Yuanyuan Xu‡
ac,
Dan Li‡ac,
Wenxuan Wangac,
Kangping Xuac,
Guishan Tanab,
Jing Lib,
Shu-Ming Li d and
Xia Yu*ac
d and
Xia Yu*ac
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, People's Republic of China. E-mail: xyu226@csu.edu.cn
bXiangya Hospital of Central South University, Central South University, Changsha, Hunan 410008, People's Republic of China
cHunan Key Laboratory of Diagnostic and Therapeutic Drug Research for Chronic Diseases, Central South University, Changsha 410013, P. R. China
dInstitut für Pharmazeutische Biologie und Biotechnologie, Philipps-Universität Marburg, Robert-Koch Straße 4, 35037 Marburg, Germany
First published on 27th September 2022
Abstract
Prenylation usually improves structural diversity and bioactivity in natural products. Unlike the discovered enzymatic gem-diprenylation of mono- and tri-cyclic aromatic systems, the enzymatic approach for gem-diprenylation of bi-cyclic hydroxynaphthalenes is new to science. Here we report an enzymatic example for dearomative C4 gem-diprenylation of α-hydroxynaphthalenes, by the F253G mutant of a fungal prenyltransferase CdpC3PT. Experimental evidence suggests a sequential electrophilic substitution mechanism. We also explained the alteration of catalytic properties on CdpC3PT after mutation on F253 by modeling. This study provides a valuable addition to the synthetic toolkit for compound prenylation and it also contributes to the mechanistic study of prenylating enzymes.
Introduction
Prenylation usually plays an important role in increasing the structural diversity and biological activities of natural products.1–4 Gem-diprenylation on aromatic substrates is a noteworthy derivatization observed in plant natural products such as β-bitter acids lupulone and colupulone from Humulus lupulus, patulone from Hypericum patulum, and ferruginin A from vismia species (Fig. 1).5–8 These compounds were reported with various pharmacological activities including sedative effects, anti-bacterial activities, cytotoxic activities, etc.5–8 Development of new methods to prepare more compounds with a gem-diprenylation feature is valuable for drug discovery purposes. The gem-dialkylation on aromatics such as naphthol disrupts the aromaticity of the substrate and chemical processes to perform this type of modification usually involve catalysts such as transition metal complexes.9,10 In nature, specific prenyltransferases from corresponding biosynthetic pathways were reported to catalyze the formation of the gem-diprenyl moiety. For example, membrane-bound aromatic prenyltransferases HlPT1L and HlPT2 from hops (Humulus lupulus) were identified to be responsible for the sequential prenylation steps on the acylphloroglucinol substrate in the biosynthesis of β-bitter acid.11 Hs/HcPT8px and Hs/HcPTpat from Hypericum sp. were demonstrated to catalyze a stepwise prenylation of 1,3,6,7-tetrahydroxyxanthone (1367THX) with DMAPP to produce gem-diprenylated patulone using recombinant protein expressed in heterologous hosts such as Saccharomyces cerevisiae and Nicotiana benthamiana.12 Likewise, fungal prenyltransferase AtaPT provided examples for gem-diprenylation of acylphloroglucinols.13 | ||
| Fig. 1 Examples of gem-diprenylated compounds in nature. | ||
Hydroxynaphthalenes, especially 1,3,6,8-tetrahydroxynaphthalene (THN), are cores of many natural products. Prenylated hydroxynaphthalenes were reported to display intriguing biological and pharmacological activities.14,15 Unlike the known enzymatic gem-diprenylation of mono- and tri-cyclic aromatic systems, the enzyme for gem-diprenylation of bi-cyclic hydroxynaphthalenes is new to science. The fungal prenyltransferase CdpC3PT from Neosartorya fischeri showed mono-prenylation activities towards several types of substrates such as hydroxynaphthalenes and biflavonoids,16,17 while its mutation on F253 altered donor selectivity towards biflavonoids.17 In this study, we discovered novel gem-diprenylation activities catalyzed by the CdpC3PT_F253G toward α-hydroxynaphthalenes, which expands enzymatic tools for gem-diprenylation of aromatic substrates. Here we also confirmed the substitution process and explained the alteration of catalytic property after the mutation.
Results and discussion
To exam its prenylation activities towards the hydroxynaphthalenes 1–10 (Fig. 2), 50 μg purified CdpC3PT_F253G were incubated with 1 mM substrates at 37 °C for 12 h in the presence of 1 mM DMAPP (Fig. 3). In comparison, control assays were carried out by incubation of 1–10 with wild-type CdpC3PT under the same conditions. The assays with inactivated CdpC3PT_F253G or inactivated CdpC3PT were used as negative controls. All the assays were subjected to HPLC analysis.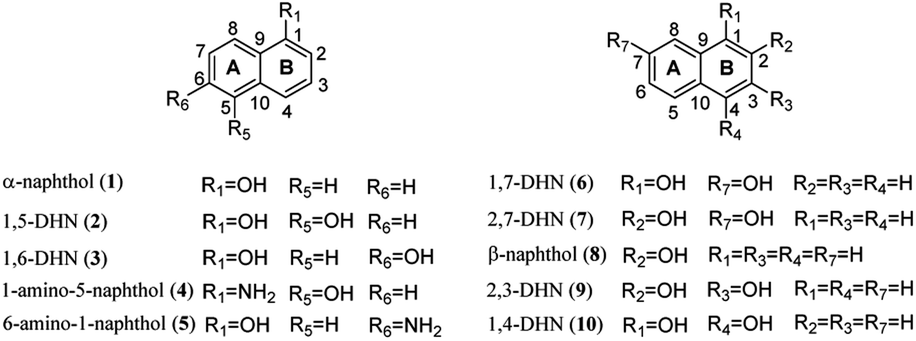 | ||
| Fig. 2 Substrates tested in this study. | ||
 | ||
| Fig. 3 (A)–(E) HPLC analysis of the reaction mixtures of 1–5 with CdpC3PT wild-type and CdpC3PT_F253G in the presence of DMAPP. Red lines (top): analysis for enzyme reaction of respective substrate with CdpC3PT and DMAPP. Blue lines (below): analysis for enzyme reaction of respective substrate with CdpC3PT_F253G and DMAPP. (4PN: 4-prenylated-α-naphthol.) | ||
In a first round of assays, substrates 1–5 were incubated. A noteworthy phenomenon was observed in the prenylation assays with CdpC3PT_F253G and DMAPP. In comparison to the controls with the wild-type CdpC3PT, a major product 1D1 was formed with a yield of 52.5 ± 1.0% in the assay for 1, differing from the wildtype product 4-prenylated-α-naphthol16 in retention time and UV absorption (Fig. 3 and S1, ESI†). Similarly, 3D2 was found as the major product in the assay of 3 with a yield of 33.1 ± 1.8% and differed clearly from the CdpC3PT product 3D1 in UV and elution profiles. Two series of peaks were detected in the assay of CdpC3PT_F253G with 2, 4 or 5, the more polar products 2D1, 4D1, and 5D1 and the less polar products 2D2, 4D2, and 5D2. Total yields were calculated to be 46.6 ± 2.4%, 33.2 ± 2.7%, and 31.7 ± 1.0% for 2, 4, and 5, respectively.
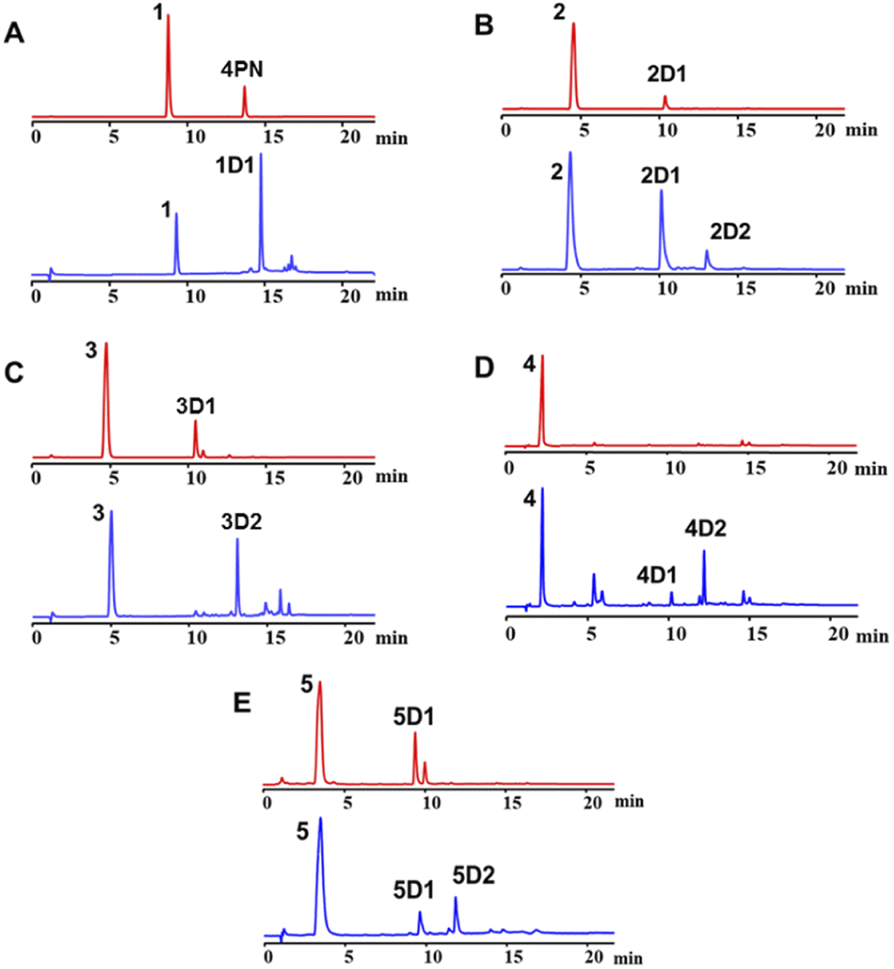
For structure elucidation, 1D1, 2D2, 3D2, 4D2, and 5D2 were purified by semi-preparation HPLC from the assays with CdpC3PT_F253G and subjected to MS and NMR analyses (Fig. 4 and Tables S1, S2, ESI†). HR-ESI-MS data supported a diprenylation by detection of a 136 Da increment in molecular weight from the substrate 1. In the 1H NMR spectrum of 1D1, signals at δH 1.45 (6H, s, H-4′/H-4′′), 1.48 (6H, s, H-5′/H-5′′), 2.80 (2H, dd, H-1′ and H-1′′), and 2.63 (2H, dd, H-1′ and H-1′′) further indicated the presence of two identical prenyl groups in the structure. In comparison to substrate 1, the disappearance of protons for the three coupling aromatic protons in ring B and appearance of two coupling, i.e. δH 6.45 (1H, d, 10.2 Hz, H-2) and 7.04 (1H, d, 10.2 Hz, H-3), suggested the attachment of the prenyl moieties at ring B. In the 13C NMR spectrum of 1D1, a characteristic downfield chemical shift of δC 188.0 (C-1) indicated the formation of a ketone. The correlations in the HMBC spectrum from 7.04 (1H, d, 10.2 Hz, H-3) to the ketone δC 188.0 revealed the α,β-unsaturated ketone fragment in 1D1. HMBC correlations from δH 2.80 (2H, dd, H-1′ and H-1′′) or 2.63 (2H, dd, H-1′ and H-1′′) to δC 158.8 (C-3)/149.3 (C-10)/49.1 (C-4) confirmed the two prenyl moieties in 1D1 at C-4. Like 1D1, the HR-ESI-MS and 1H NMR data of 2D2, 3D2, 4D2, and 5D2 suggest the presence of two identical regular C-attached prenyl groups. In addition, an α,β-unsaturated ketone group in each of the four products was identified by appearance of two coupling aromatic protons at ring B, i.e. δH 6.29–6.40 (1H, d, 10.1–10.2 Hz, H-2) and 6.81–6.97 (1H, d, 10.1–10.2 Hz, H-3), and clear HMBC correlations from δH 6.81–6.97 (1H, d, 10.1–10.2 Hz, H-3) to the ketone carbon C1 (δC 187.2–189.1). The attachment of two prenyl moieties at C-4 was unambiguously proven by the HMBC correlations from δH 2.42–2.48/3.26–3.52 (each 2H, dd, H-1′ and H-1′′) to δC 161.5–161.6 (C-3)/49.3–49.4 (C-4)/131.6–134.6 (C-10) for 2D2 and 4D2, or from δH 2.56–2.70 (4H, dd, H-1′ and H-1′′) to δC 156.9–157.8 (C-3)/48.3–48.6 (C-4)/152.0–152.4 (C-10) for 3D2 and 5D2.
 | ||
| Fig. 4 The structures of enzyme products of CdpC3PT_F253G identified in this study. | ||
The products 2D1, 4D1, and 5D1 were isolated from the assays of CdpC3PT_F253G with 2, 4, and 5 in the presence of DMAPP, respectively. The HR-ESI-MS data revealed the presence of one prenyl moiety in 2D1, 4D1, and 5D1, by showing 68 Da larger molecular ions than those of the respect substrates. The proton signals at δH 3.52–3.96 (2H, d, H-1′) indicated the presence of one regular prenyl moiety attached to a C-atom.18 NOESY correlations between δH 3.52 (2H, d, H-1′) with 6.95 (1H, d, H-3) and 7.11 (1H, d, H-5) unambiguously proved the attachment of the prenyl moiety at C-4 in 5D1. The disappearance of one triplet in the 1H NMR spectra of 2D1 and 4D1 in comparison with those of the respective substrates, indicated the prenylation at position 2, 4, 6 or 8 of 2D1 and 4D1. In the HMBC spectra, connectivities from δH 6.66 (1H, d, 7.7 Hz, H-2), 6.94–6.95 (1H, d, 7.7–7.8 Hz, H-3), and 7.67–7.68 (1H, dd, 8.4, 1.2–1.3 Hz, H-8) to δC 152.3–153.3 (C-1) excluded the prenylation at C-2 and C-8. Correlations between δH 3.87–3.96 (2H, d, H-1′) and 6.94–6.95 (1H, d, 7.7–7.8 Hz, H-3) in the NOESY spectrum confirmed the prenylation at C-4 rather than C-6. The product 3D1 was also purified from the assay of CdpC3PT with 3 and DMAPP. In a previous study, minor product formation was observed when assays were performed with lower concentration of CdpC3PT.16 Here we identify the structure of 3D1 to be 4-prenylated 1,6-DHN (Table S2 and Fig. S1, ESI†), of which the 1H NMR data correspond well to those reported previously.16
In the assay of CdpC3PT_F253G towards 6 with DMAPP, HPLC analysis showed the presence of a mono-prenylated product 6D1 as major peak and a gem-diprenylated product with very low yield, which wasn't isolated in this study. The structure of 6D1 was identified to be 4-prenylated 1,7-DHN, corresponding well to the 1H NMR data reported before.16 No obvious product was detected in assays of CdpC3PT_F253G and DMAPP towards 7–9 with hydroxyl groups at β positions (position 2), indicating that a hydroxyl group at α position (position 1) is essential for the formation of gem-diprenylated products for hydroxynaphthalenes. When the para-position of α-OH is substituted with hydroxyl group, i.e. 1,4-DHN (10), no noticeable product was found.
Kinetic parameters including Michaelis constant (KM) and turnover numbers (kcat) were determined by nonlinear regression. Assays contained different concentrations of hydroxynaphthalenes, 2 mM DMAPP and lower enzyme amounts were used (details were described in Material and methods). For reactions with DMAPP as donor, the KM values for substrates 1–5 were in the range of 0.41–1.14 mM, while the catalytic efficiencies (kcat/KM) were of values 46.1–297.8 s−1 M−1 (Table S3, ESI†). In comparison, lower or similar KM values were found for Hs/HcPT8px and Hs/HcPTpat towards 1367THX or AtaPT towards acylphloroglucinols.12,13 The kcat/KM values of AtaPT towards PIVP and PBZP were in a similar range as those for CdpC3PT_F253G towards 1–5, while AtaPT showed a much higher catalytic efficiency towards PIBP.13
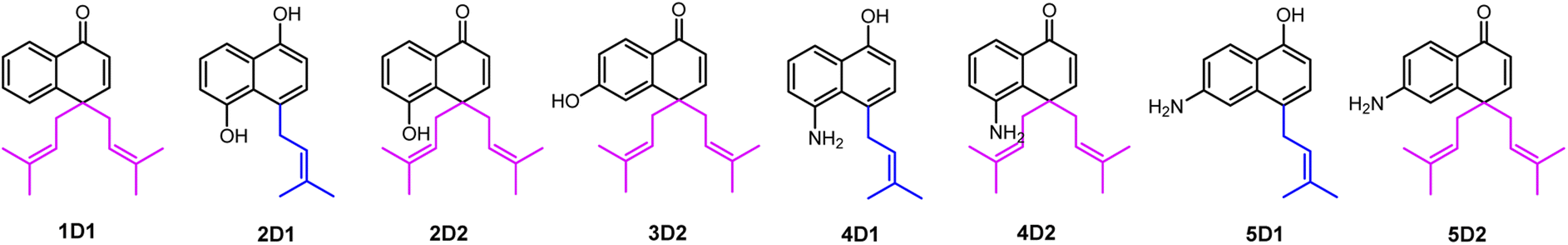
As mentioned above, besides C4-gem-diprenylated products, C4-monoprenylated derivatives were also detected in the assays of 2, 4, and 5 with CdpC3PT_F253G in the presence of DMAPP. This interesting phenomenon encouraged us to investigate the formation of 2D2, 4D2, and 5D2 by using 2D1, 4D1, and 5D1 as substrates. Incubation of 2D1, 4D1, and 5D1 with CdpC3PT_F253G in the presence of DMAPP, gem-diprenylated products 2D2, 4D2, and 5D2 were detected as predominant products (Fig. 5), proving that the monoprenylated products sever as substrates for diprenylation. The catalytic process of the dearomative gem-diprenylation of the hydroxynaphthalenes is confirmed to be an electrophilic substitution on the electron-rich aromatics. The reaction involves the leaving of the diphosphate group (PPi) from the donor DMAPP, which is catalysed through the positively charged lysine and/or arginine residues,19 followed by deprotonation of the hydroxynaphthalenes, employing a general base on the enzyme. Accordingly, a graphical mechanism of hydroxynaphthalene dearomative gem-diprenylation is given in Scheme 1. For substrate 1 and 3, the second prenylation was kinetically faster than the first, which led to the dominant gem-diprenylated products.
 | ||
| Fig. 5 (A)–(C) HPLC analysis of the enzyme assays of 2D1, 4D1, and 5D1 with CdpC3PT_F253G in the presence of DMAPP. Traces on top row indicate substrate-only control assays and the corresponding traces below are assays with CdpC3PT_F253G. | ||
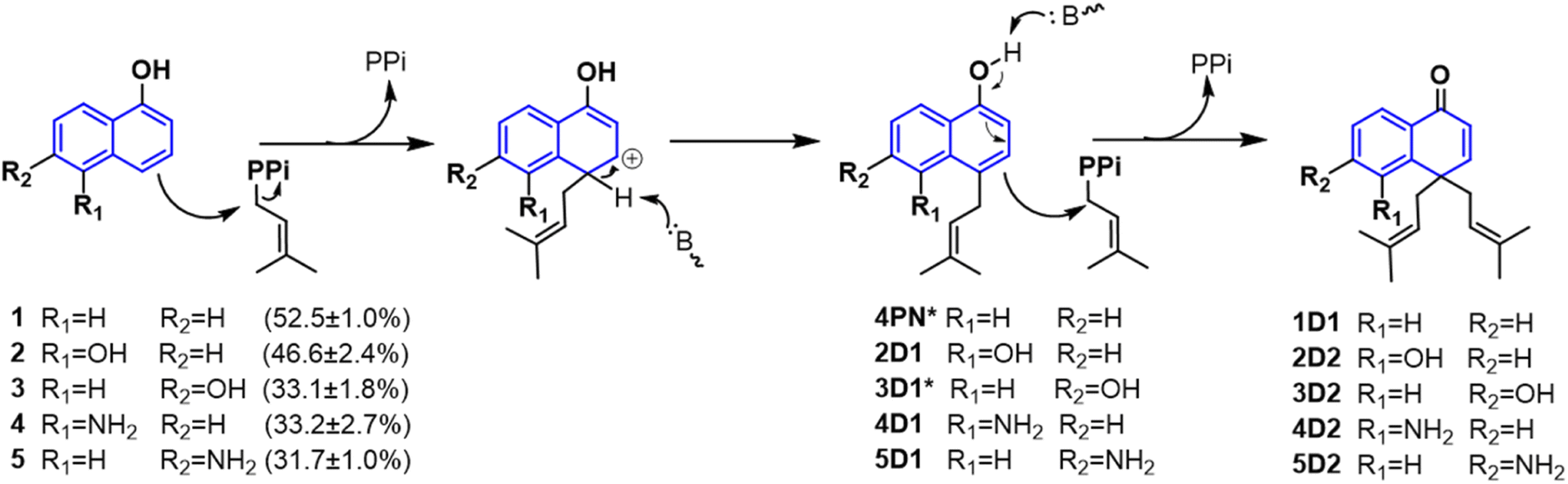 | ||
| Scheme 1 Mechanism of hydroxynaphthalene dearomative gem-diprenylation catalyzed by CdpC3PT_F253G in this study. (* Not isolated from the assays catalyzed by CdpC3PT_F253G.) | ||
To explain the alteration of catalytic property on CdpC3PT after the mutation on F253, we performed homology modelling and molecular docking by using Molecular Operating Environment.20 The crystal structure of the prenyltransferase CdpNPT (4E0U), with highest sequence identity of 54% to CdpC3PT, was chosen as template for modelling. 2D1 was docked into the active site of the CdpC3PT_F253G mutant using the induced fit protocol. After that, we aligned and overlapped this docking model with the donor-containing crystal structure of FgaPT2 (PDB entry: 3I4X) (Fig. S6, ESI†). The prenyl donor analogue DMSPP in 3I4X is observed at a position next to the docked-in substrate 2D1. Interestingly, it revealed that the prenyl moiety of the substrate 2D1 is directing to the F253G residue. The bulky phenylalanine in the wild-type CdpC3PT at this position results in a close contact with the substrate, as shown in Fig. 6. The smaller glycine leaves more space for accommodation of the second prenyl group in the gem-diprenylated products, which allows the second prenylation step for the generation of the gem-diprenyl moiety.
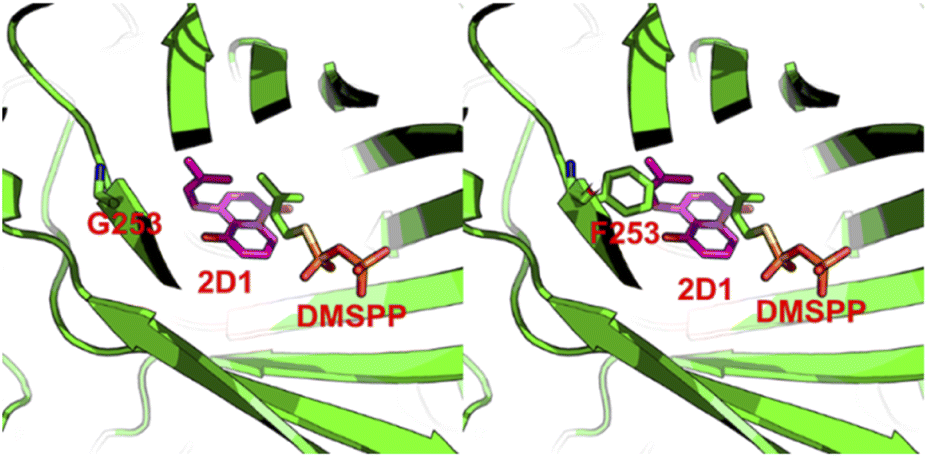 | ||
| Fig. 6 Docking model of 2D1 with CdpC3PT_F253G (left), replacing the glycine back to phenylalanine (right). The carbon atoms in substrate 2D1 were colored as magenta and those in protein or DMSPP were colored as green. | ||
Conclusions
In summary, we have discovered the first enzymatic example for dearomative gem-diprenylation on α-hydroxynaphthalenes by an engineered enzyme CdpC3PT_F253G and confirmed the electrophilic substitution processes for the dearomative reactions. Several enzymatic products were synthesized by these biocatalytic processes and their structures were characterized, which indicates the great potential in structure diversity by the enzyme CdpC3PT_F253G, and expands the chemical diversity of prenylated hydroxynaphthalenes.Experimental
Chemicals
DMAPP was synthesized according to the method reported by Woodside and co-workers.21 Substrates used in this work were purchased from TCI, Aladdin and Yuanye Bio-Technology Co. Ltd and the purity of these substrates is greater than 97%.Enzyme assays
CdpC3PT and CdpC3PT_F253G were successfully created, overproduced and purified as described in our previous study.17 The purity of protein was checked on SDS-PAGE analysis. To test enzyme activities towards hydroxynaphthalenes, the assays (50 μL) containing 50 mM Tris–HCl (pH = 7.5), 1 mM DMAPP, 5 mM Ca2+, 5% DMSO, and 50 μg of purified recombinant protein CdpC3PT_F253G or CdpC3PT wild type were incubated with 1 mM of each substrate. Negative controls were performed with inactivated CdpC3PT_F253G or CdpC3PT by boiling for 20 min. The reaction mixtures were incubated at 37 °C for 12 h and terminated by addition of 100 μL methanol. For precipitation removing, the mixtures were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min, and 100 μL supernatant was subjected to HPLC analysis. The amount of enzyme product was quantified by the absorption coefficient relative to the substrate, which was calculated from the UV intensity of the product and the consumption of substrate in three assays with an initial substrate concentration of 0.1 mM.
000 rpm for 15 min, and 100 μL supernatant was subjected to HPLC analysis. The amount of enzyme product was quantified by the absorption coefficient relative to the substrate, which was calculated from the UV intensity of the product and the consumption of substrate in three assays with an initial substrate concentration of 0.1 mM.
Enzymatic kinetic analysis
To determine the kinetic parameters of CdpC3PT_F253G towards hydroxynaphthalenes, 50 μL assays each contained 50 mM Tris–HCl (pH 7.5), 10 mM CaCl2, 2 mM DMAPP, 5% (v/v) DMSO, 0.02, 0.05, 0.1, 0.2, 0.5, 1, 2, or 5 mM substrate and purified enzyme. For assays with DMAPP as prenyl donor, 25 μg of the purified CdpC3PT_F253G were used for 1, 3–5 and 12.5 μg proteins were used for 2. All reaction mixtures were incubated at 37 °C for 30 min. Kinetic parameters were determined using the “Michaelis–Menten” nonlinear regression tools by GraphPad Prism software.HPLC analysis for enzyme reactions
HPLC analysis was performed on an Agilent 1200 series (Agilent Technologies) or Acquity Arc 1718093S (Waters) with Venusil MP C18 column (4.6 × 100 mm, 5 μm, Agela Technologies). Water with 0.5% acetic acid (solvent A) and methanol (solvent B) with 0.5% acetic acid were used as elution solvents. A liner gradient from 40% to 100% of solvent B was used for 15 min, then the column was washed with 100% of solvent B for 5 min and equilibrated with 40% of solvent B for 5 min at a rate of 1 mL min−1. Detection was carried out on a photodiode array detector and illustrated for absorption at 296 nm.Isolation of enzyme products
For product isolation, large scales of enzyme reactions were carried out at 40 mL. The reaction mixtures were extracted with double volume of ethyl acetate for three times. The organic phase was then evaporated on rotary evaporator at 30 °C and the residue was resolved in 1 mL methanol for isolation on a HPLC system (Agilent 1200 series). A semipreparative column YMC Innoval ODS-2 (10 × 250 mm, 5 μm) was used for isolation of the enzyme products with the same solvents mentioned above at a flow rate of 2.5 mL min−1. A liner gradient from 80%–95% of solvent B for 10 min was eluted, then the column was washed with 95% of solvent B for 10 min, and equilibrated with 80% of solvent B for 5 min.NMR and mass spectra analysis
All the isolated products were dissolved in CD3OD and subjected to NMR analysis. The data were recorded at room temperature on a Bruker AV400 or AV500 spectrometer. The pulse program hsqcetgp (1024/2048 and 256 points for F2 and F1, respectively) was used to acquire the HSQC data. The pulse program hmbcgpnd (4096 and 128 points for F2 and F1, respectively) was used to acquire the HMBC data. The pulse program noesyphsw (2048 and 256 points for F2 and F1, respectively) was used to acquire the NOESY data. Acquisition times in 1H NMR, 13C NMR, HSQC, HMBC, NOESY experiments were 2.7–4.1, 0.9–1.4, 0.05–0.10, 0.2–0.4 s, respectively. Chemical shifts were referenced to the solvent signal at δH 3.31 ppm and δC 49.01 ppm for CD3OD and the NMR spectra were processed with MestReNova software.High resolution electrospray ionization mass spectrometry was carried out on Agilent 6540 Q-TOF. The ionization method was electrospray ionization (ESI), and the Agilent standard tuning solution ESI-L Low Concentration Tuning Mix (G1969-85000) was used to calibrate the accurate mass before sample injection and analysis. The detection range of primary mass spectrometry scanning was m/z 100–1700. Nitrogen was used as solvent removal drying gas, with the temperature at 325–350 °C and flow rate at 6.8–11 L min−1. The sheath gas temperature was 350 °C. The capillary voltage was 4.0 kV, and the fragment voltage was 150–180 V. All the spectra were acquired in positive ion mode with scan rates of 1–4 spectra per s and mass resolutions of >11000.
Homology modeling and docking
Homology modeling and molecular docking were performed using MOE 2014 (Molecular Operating Environment) software.20 Molecular docking is carried out through the homology model constructed in the previous stage.17 2D1 was constructed using molecule builder module and energy minimized. The top 15 conformations of 2D1 were docked into the active site of CdpC3PT_F253G or CdpC3PT using an induced fit protocol. Pymol was used to overlay models, which is aligned with the backbone atoms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support by the National Natural Science Foundation of China (No. 21877129) and the “Huxiang Young Talents Plan” Support Project of Hunan Province (No. 2020RC3007). We thank Dr Guogen Liu (Central South University) and Dr Hongping Long (Hunan University of Chinese Medicine) for taking NMR and HR-ESI-MS spectra.Notes and references
- K. Yazaki, K. Sasaki and Y. Tsurumaru, Phytochemistry, 2009, 70, 1739–1745 CrossRef CAS.
- B. Botta, A. Vitali, P. Menendez, D. Misiti and G. Delle Monache, Curr. Med. Chem., 2005, 12, 713–739 CrossRef CAS.
- X. Chen, E. Mukwaya, M.-S. Wong and Y. Zhang, Pharm. Biol., 2014, 52, 655–660 CrossRef CAS PubMed.
- O. Wesolowska, J. Gasiorowska, J. Petrus, B. Czarnik-Matusewicz and K. Michalak, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 173–184 CrossRef CAS PubMed.
- A. A. Hussein, B. Bozzi, M. Correa, T. L. Capson, T. A. Kursar, P. D. Coley, P. N. Solis and M. P. Gupta, J. Nat. Prod., 2003, 66, 858–860 CrossRef CAS.
- H. Oku, Y. Ueda, M. Iinuma and K. Ishiguro, Planta Med., 2005, 71, 90–92 CrossRef CAS PubMed.
- M. Saugspier, C. Dorn, B. Czech, M. Gehrig, J. Heilmann and C. Hellerbrand, Oncol. Rep., 2012, 28, 1423–1428 CrossRef CAS.
- M. Van Cleemput, K. Cattoor, K. De Bosscher, G. Haegeman, D. De Keukeleire and A. Heyerick, J. Nat. Prod., 2009, 72, 1220–1230 CrossRef CAS PubMed.
- J. Hu, S. Pan, S. Zhu, P. Yu, R. Xu, G. Zhong and X. Zeng, J. Org. Chem., 2020, 85, 7896–7904 CrossRef CAS PubMed.
- X. Fang, Y. Zeng, Q. Li, Z. Wu, H. Yao and A. Lin, Org. Lett., 2018, 20, 2530–2533 CrossRef CAS PubMed.
- H. Li, Z. Ban, H. Qin, L. Ma, A. J. King and G. Wang, Plant Physiol., 2015, 167, 650–659 CrossRef CAS PubMed.
- M. Nagia, M. Gaid, E. Biedermann, T. Fiesel, I. El-Awaad, R. Haensch, U. Wittstock and L. Beerhues, New Phytol., 2019, 222, 318–334 CrossRef CAS.
- K. Zhou, C. Wunsch, J. Dai and S.-M. Li, Org. Lett., 2017, 19, 388–391 CrossRef CAS PubMed.
- A. A. Hussein, I. Barberena, T. L. Capson, T. A. Kursar, P. D. Coley, P. N. Solis and M. P. Gupta, J. Nat. Prod., 2004, 67, 451–453 CrossRef CAS PubMed.
- M. Ishibashi, S. Funayama, Y. Anraku, K. Komiyama and S. Omura, J. Antibiot., 1991, 44, 390–395 CrossRef CAS PubMed.
- X. Yu, X. Xie and S.-M. Li, Appl. Microbiol. Biotechnol., 2011, 92, 737–748 CrossRef CAS.
- Y. Xu, D. Li, G. Tan, Y. Zhang, Z. Li, K. Xu, S.-M. Li and X. Yu, Org. Lett., 2021, 23, 497–502 CrossRef CAS PubMed.
- K. Xu, C. Yang, Y. Xu, D. Li, S. Bao, Z. Zou, F. Kang, G. Tan, S.-M. Li and X. Yu, Org. Biomol. Chem., 2020, 18, 28–31 RSC.
- U. Metzger, C. Schall, G. Zocher, I. Unsöld, E. Stec, S.-M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS PubMed.
- Chemical Computing Group ULC, Molecular Operating Environment (MOE), 2019.01, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite # 910, Montreal, QC, Canada, H3A 2R7, 2021 Search PubMed.
- A. B. Woodside, Z. Huang and C. D. Poulter, Org. Synth., 1988, 66, 211–215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinetic parameters, F253G_2D1 docking model, CdpC3PT model, HR-ESI-MS data, and NMR data. See https://doi.org/10.1039/d2ra04837j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |