
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ESIPT-ICT steered naphthylthioic-based ionic probe with dual emissive channels exhibiting CHEF and CHEQ effects†
Martha N. Amputu,
Johannes Naimhwaka and
Veikko Uahengo *
*
Department of Physics, Chemistry and Material Science, University of Namibia, 340 Mandume Ndemufayo Avenue, Windhoek, 9000, Namibia. E-mail: vuahengo@unam.na; vuahengo@gmail.com; Tel: +264 61 206 3465
First published on 21st September 2022
Abstract
A naphthylthioic-based emissive probe (M) bearing a hydroxyl and amine group was designed and synthesized via a one-step Schiff base reaction process. The probe was characterized spectroscopically using 1H NMR, UV-Vis and fluorescence spectrophotometers. The probe turned out to be spectroscopically and colorimetrically selective and sensitive to multiple cations and anions. Interestingly, the probe displayed characteristics of excited-state intramolecular proton transfer (ESIPT)-driven dual emissive channels; experiencing fluorescence enhancement upon the molar additions of Al3+ as well as the anions used, events presumably ascribed to chelation fluorescence enhancement (CHEF), hydrogen bonding and deprotonation effects. Moreover, the fluorometric titration with Hg2+ resulted in ratiometric spectral behaviors of M, with the disappearance of the peak at 450 nm, concomitant with the appearance of a new peak at 520 nm, distinguished by a clear isosbestic point, the same behaviors exhibited by Sn2+ and Ag+ analytes towards M. The introduction of all other cations used, resulted in fluorescence quenching, attributable to chelation enhanced fluorescence quenching (CHEQ), thereby inhibiting the ESIPT process. The experiments were all carried out in the aqueous environment medium of DMSO–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at ambient temperature. Theoretical density functional theory calculations were carried out to gain insight into the interaction of M with cations and anions, and their influence on the HOMO–LUMO energy gaps.
:1) at ambient temperature. Theoretical density functional theory calculations were carried out to gain insight into the interaction of M with cations and anions, and their influence on the HOMO–LUMO energy gaps.
1. Introduction
Chemosensing techniques have been at the forefront of analytical chemistry research in recent times, due to their simplicity in application and cost-effectiveness, in preparation and easy access. The quest to develop simpler and more convenient analytical probes for the detection of heavy metal cations and biological anions, has seen chemosensing techniques emerging as substantive preferences to replace the costly traditional techniques such as ICP and AAS.1–4 The demands for more efficient and easy-to-use sensing probes in the detection of heavy metals such as mercury (Hg2+), nickel (Ni2+), iron (Fe3+ or Fe2+), zinc (Zn2+), copper (Cu2+) and aluminum (Al3+) have been high on the priority list.5–15 The ascendance of chemosensing techniques is largely intensified by the upsurge of industrial effluents, increasingly being discharged in natural streams on a daily basis, resulting in an unprecedented rise in toxic wastes, causing environmental hazards and health problems.16,17Recent developments have on record that, heavy metal contamination has become the epicenters of environmental pollution, especially in developing countries, by discharging industrial wastes into underground aquifers. Among the common industrial pollutants is organic mercury (CH3Hg2+) which leads to neurological diseases, termed as a “Minamata Disease” normally caused by excess Hg2+, which is highly lethal to the physiological system. Ionic and elemental mercury, usually soluble in water, enters the environment through metal mining activities, industrial wastes, agricultural pesticides and volcanic activities. Ionic mercury once consumed does accumulate in physiological organs through food chain, which results in severe damage to the nervous system, brain, kidney and endocrine system.18 Generally, the accumulation of any heavy metal concentrations above their thresholds, in physiological systems, has severe damages to the body.19–25
Exclusively, Al3+ is the third most abundant metal in the periodic table, highly versatile with distinctively excellent chemistry. It has the ability to form amphoteric compounds, especially in its oxidic forms, exhibiting both basicity and acidity properties, depending on the chemical environment. Thus, its versatility nature can have adverse effects to environmental and physiological systems in living organisms, should its concentration left unmonitored. Numerous chemosensing modulus sensitive and selective to Al3+, have been developed and reported in literature, particularly the fluorometric probes capable of discriminating Al3+ in environmental samples.26,27 However, this is such a tenacious field in contemporary space, the pursuit of fluorogenic probes for heavy metals and other toxic ionic species including Al3+ still remain high on the priority list.28
Correspondingly, anions too have been attracting concerted interests in many research fraternities, owing to their various roles in environmental, clinical and biological applications. In particular, cyanide has been used traditionally in several applications such metallurgical processes in mining, fibers, resin industries, and many others. Consequently, cyanide concentration in environmental spaces has been accumulating, due to voluminous usage in industries, yielding about 140 k tons per year of cyanide.29–32 Cyanide is viewed as highly toxic, that its presence induces the susceptibility of coordinating to iron ion in biological systems, thus blocking the electron transfer chain in mitochondria.10,33–36 In addition, other anions too, have similar adverse effects when present in excess or uncontrolled quantities. The effects seen with fluoride (fluorosis and thyroid activities),32,37,38 acetate (as acetyl coenzymes and food preservation),39–42 cyanate (chronic kidney diseases, CKD),43,44 hydroxide (corrosive effect at high concentration)35,45,46 and inorganic dihydrogen phosphate (effect on signal transduction and energy storage in biological systems).47–49
Furthermore, excited-state intramolecular proton transfer (ESIPT), single or dual emitters which are important proton transfer reactions with efficient fluorophores, have been receiving a fair share of attention lately, due to their attractive optoelectronic properties, with remarkable large Stokes shifts, strong solid-state luminescence efficiency and environment-sensitive emission bands.50–55 The applications of fluorophores guided by ESIPT mechanisms are widely applied in many fields of scientific research, ranging from metabolic process of living systems, biochemistry, analytical chemistry, laser dyes, molecular memory storage devices, electrophonic modulation and fluorescent probes.56–62 In combination with intramolecular charge transfer (ICT) mechanism, the two (ESIPT and ICT) mechanisms have seen applications extended to optoelectronic materials, solar cells, ion detectors, chemosensors, and thin film transistors.63–65 The robustness of the two mechanisms (ESIPT and ICT) coupled in a single molecule have been investigated in organic molecules over the past years. Recently, there has been reports on comparative studies and their effects on ESIPT and ICT mechanisms. For instance, studies on ICT versus ESIPT in pyridine imidazole and in cyclodextrin complexes have been reported, which included fluorescent sensing, based on ESIPT and ICT. In other accounts, benzothiazoles have been reported, describing pH effect on ESIPT and ICT, harnessing ESIPT and ICT processes in imines derivatives, triple-signaling mechanisms in Schiff bases and a dual mechanism strategy in imidazole.66–70
In this work, we are reporting a fluorescent dual molecular probe (M) bearing a hydroxyl and an amine group, thus exhibiting the ESIPT mechanism. The probe was designed to exploit the ESIPT mechanism known to exhibit excellent molecular recognition properties towards cations and anions, capable of forming tautomeric isomers. The probe was synthesized through a one-step Schiff base reaction mechanism at room temperature. The reaction mechanism of M is shown in Scheme 1.
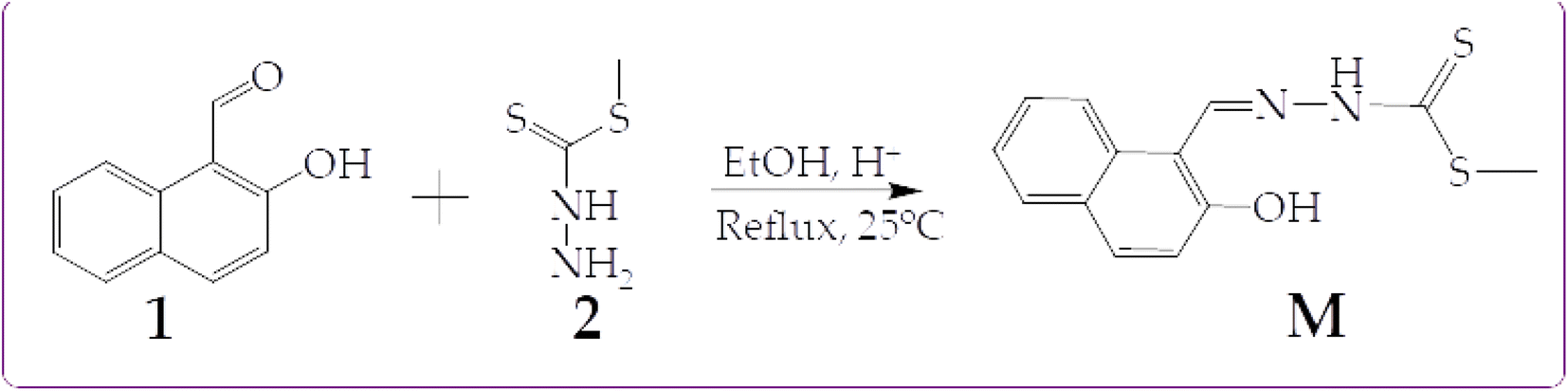 | ||
| Scheme 1 Synthetic method of M. | ||
2. Experimental
2.1 General
All reagents were of analytical grade, and were used as received from the commercial sources, unless mentioned. UV-Vis spectroscopy was performed on PerkinElmer Lambda 35 spectrophotometer in a standard 3.0 ml quartz cuvette with 1 cm path length. 1H NMR spectra were recorded on a Varian Mercury VX-300 MHz spectrometer in DMSO-d6, elemental analyses (CHN) were carried out on a PerkinElmer 240C analytical instrument. Fluorescence studies were carried out on a steady state excitation and emission spectra on the Molecular Device SpectraMax M2, Plate Reader. All the measurements were carried out at ambient temperature. The density functional theory (DFT) calculations at B3LYP/6-31G** were performed using a Spartan ’14 package software.292.2 Synthesis procedures
The synthesis of M was as follows. The ethanolic solution (15 ml) of 2-hydroxy-1-naphthaldehyde (1) (1.00 g, 5.81 mmol) was mixed drop-wisely with a solution (15 ml) of methyl hydrazinecarbodithioic acid (2) (0.71 g 5.81 mmol) while magnetically stirred, and catalyzed with a few drops of acetic acid. The solution mixture was stirred for up to one hour at room temperature, before an orange precipitate appeared in the solution. The orange precipitate was filtered off and washed several times with hot ethanol. The product was dried in vacuum at room temperature and recrystallized from ethanol (the recrystallized product was further subjected to column chromatography packed with alumina stationary phase, yielding the pure product). Yield 1.2811 g, 79.6%. 1H-NMR (300 MHz, DMSO-d6) δH: 13.37 (s, 1H, –OH), 11.06 (s, 1H, –NH–), 9.21 (s, 1H, H7) 8.76 (d, J = 8.58 Hz, 1H, H1), 7.94 (d, J = 8.94 Hz, 1H, H5), 7.87 (d, J = 7.62 Hz, 1H, H4), 7.59 (ddd, J = 1.32 Hz, J = 6.93 Hz, J = 8.46 Hz, 1H, H3), 7.40 (m, 1H, H2) and 7.23 (d, J = 8.94, 1H, H6) (Fig. S7†). Elemental analysis (C13H12N2OS2), calcd. (%) for C, 56.50; H, 4.38; N, 10.14; O, 5.74; S, 23.20; found: C, 55.32; H, 4.25; N, 9.07.3. Results and discussions
3.1 Photophysical property studies of M
The physical appearance of M was defined by a light yellow colour, with good stability at room temperature (Fig. 1a). Spectroscopically, the identity of M in dimethyl sulfoxide–water (DMSO–H2O), was characterized by a few spectral humps with two prominent absorption peaks; a high-energy band at 267 nm, as well as the intense band at 384 nm (Fig. 1b). Generally, absorption bands in the shorter wavelength region are attributed to localized π–π* transitions, while the moderate band towards the longer wavelength is ascribed to intramolecular charge transfer, occasionally mixed with a more delocalized π–π* transitions. In addition to the two peaks, M was further characterized by a low energy band in the region of 490 nm, attributed to the visible light-induced charge transfer. The existence of a visible-light-induced charge transfer band is a good indication of a potential dye sensitizers for solar cell application, whereby through judicious tuning of optoelectronic properties, M can be a potential organic dye sensitizer. The spectral fingerprint was influenced and characterized by possible photoinduced tautomeric forms, enol and keto (ESIPT), as a result of sample exposure to light, during analysis. Concurrently, the optoelectronic properties of a dye sensitizer are normally closely related to the chemosensing properties, since the same charge transfer mechanisms are liable for both enhanced functions in a molecule. | ||
| Fig. 1 (a) Physical appearance of M, and (b) UV-Vis absorption spectrum of free sensor M (1 × 10−5 M) in DMSO–H2O (9:1). | ||
3.2 Chemosensing property studies
 | ||
| Fig. 2 Observable colorimetric changes of cations (1 mL drop) upon interacting with M (1 × 10−5 M) in DMSO–H2O, at room temperature. | ||
Spectrally, the molar titration of M with cations, both in DMSO–H2O resulted in distinctive shifts in spectra based on the identity of cations (Fig. 3). The molar addition of Cu2+ to M saw the emergence of a band in the region of 300 nm simultaneous with the disappearance of the initially intense peak at 384 nm, as well as the appearance of another peak at 430 nm (Fig. 3a). The spectral shifts were concomitantly evolving with colorimetric changes observed (from yellow to olive green) upon adding copper ions. Spectral changes were attributed to the charge transfer induced by coordination between M and Cu2+ in a 1:1 interaction ratio. The formation of complexed state of M–Cu pedant was further complemented by two isosbestic points at 355 nm and 412 nm, showing more than one species in the system at equilibrium. The ratiometric patterns could be seen via the titration profile as displayed (Fig. 3a inset). Similarly, the titrations with other metal cations (Fe2+, Ni2+, Zn2+) could induce spectral changes upon interacting with M (Fig. 3b–d), with titrations profiles inserted, respectively. The other cations that displayed responsive actions were also investigated, and provided as addendum in the ESI, as shown (Fig. S1†). A significant number of cations used did not display any significant spectral changes (Fig. S2†), as observed in colorimetric studies.
 | ||
| Fig. 3 The absorption titration spectra and titration profiles (insets) of M (1 × 10−5 M) in DMSO–H2O, with 3 equiv. of (a) Cu2+, (b) Fe2+, (c) Ni2+ and (d) Zn2+, at room temperature. | ||
 | ||
| Fig. 4 Visually observable color changes M (1 × 10−5 M) upon interacting with anions (1 mL drop) in DMSO–H2O, at room temperature. | ||
Furthermore, the naked eye observable colour changes were complemented by spectroscopic analysis, via UV-Vis titration of M with anions. As expected, the molar addition of anions, displayed distinctive spectral shifts, confirming the colour changes initially observed. The probe has displayed the ability to discriminate a few anions (CN−, F−, AcO−, OCN−, OH−, N3− and H2PO4−) all together in a similar fashion, after each was added to the sensor. Generally, when each of the anion was added to M, a prominent high-energy peak at 384 nm gradually decreased to disappearance, with slightly bathochromic shift characteristics (Fig. 5). The disappearance of the peak at 384 nm happened concomitantly with the gradual formation of a new broader absorption peak in the region of 420–500 nm (Fig. 5), with vibronic characteristics. Specifically, the molar addition of 3 equiv. of H2PO4− and CN− yielded similar spectral behaviors, where a peak at 384 nm disappeared completely (Fig. 5a & b), while the same amount of F− and AcO− displayed similar patterns (Fig. 5c & d). In both occasions, the isosbestic points were observed, indicating the transformation from M to the complexed state (M–A). Spectra similarity was an indication that the nature of interaction between M and these anions had resemblance. Evidently, the interaction between M and the anions was through hydrogen bonding mechanism, either through the –OH or –NH group in the structure of M. Other anions which have displayed spectral changes such as N3−, OH− and OCN− are displayed (Fig. S3†), while those with no significant changes, either colorimetrically or spectrally have also been shown (Fig. S4†).
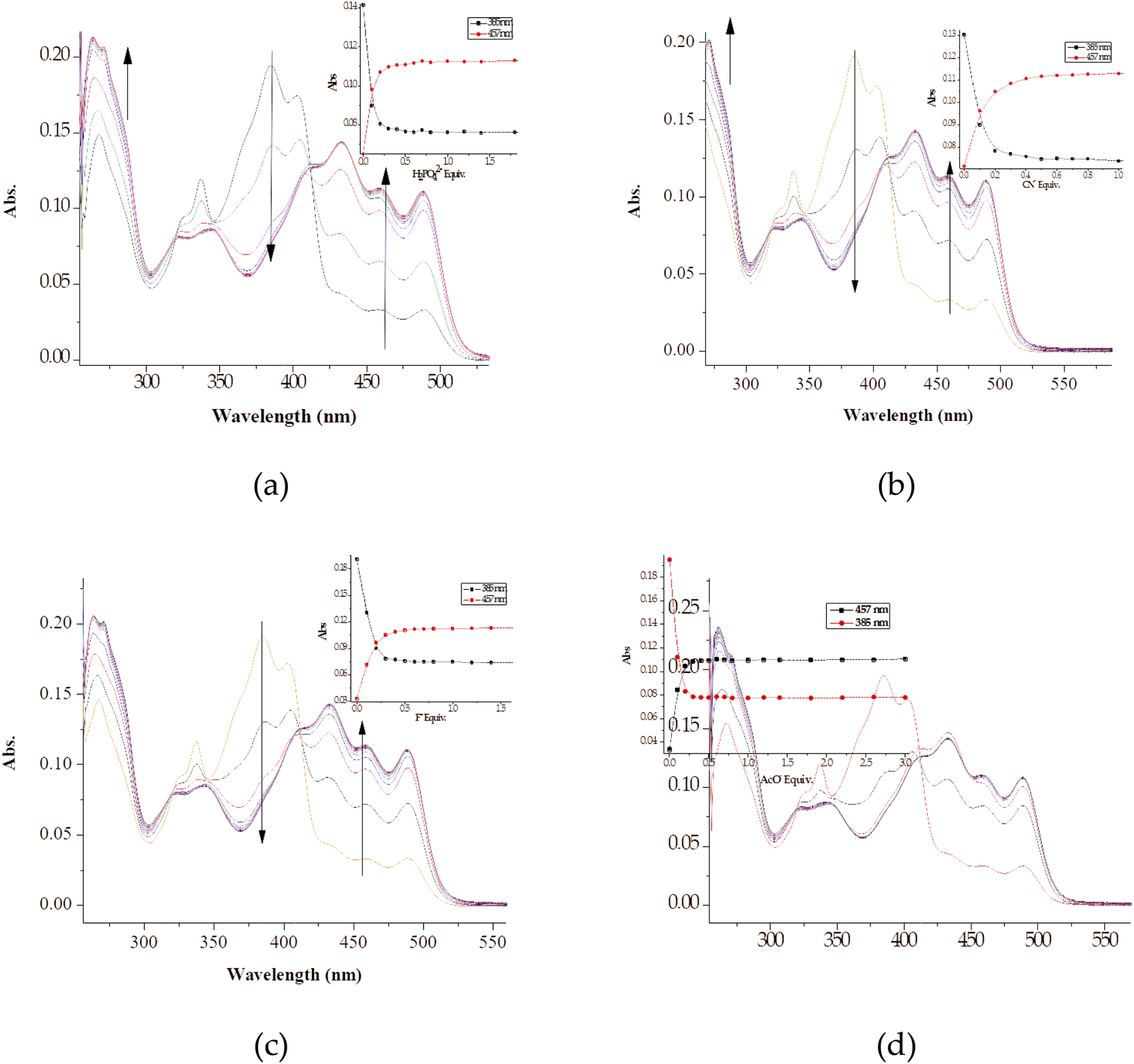 | ||
| Fig. 5 The absorption titration spectra and titration profiles (insets) of M (1 × 10−5 M) in DMSO–H2O, with up to 3 equiv. of (a) H2PO4−, (b) CN−, (c) F− and (d) AcO− at room temperature. | ||
Intrinsically, the Job plot was conducted to determine the interaction ratios between the host and the guest, experimentally. Thus, upon carrying out the Job plot experiments for selected anions (AcO−, CN−, F− and OH−), resulting spectra were quantitatively used to determine the interaction ratio. The spectral data from the Job's plot showed that the interaction ratio between the host (M) and the guests (anions) had distinctive similarity (Fig. 6). Accordingly, the interaction ratio between M and the selected anions was hinted to be a 1:1 (Fig. 6). The structural framework of M displayed strong characteristics of an anion sensor, with the presence of –OH and –NH groups, which possess acidic protons, normally attracting negatively charged (basicity) anions.
 | ||
| Fig. 6 The Job plots of (a) [M–CN], (b) [M–AcO], (c) [M–F] and (d) [M–OH] at 1 × 10−4 M in DMSO–H2O. | ||
In addition, the Job plots have presented a glimpse in the nature of interaction between M and the selected anions. The proposed interaction mode and mechanism for the anions were indicated accordingly (Scheme 2), all by way of hydrogen bonding/deprotonation (M–F, M–CN, M–AcO, M–OH). The interaction modes (1:1) with all anions were by hydrogen bonding (Scheme 2a–d), which led to deprotonation, when excess of the analytes were added. The mechanism for the complexes (M–F, M–CN, M–AcO, M–OH) were commonly through hydrogen bonding via the two electrophilic protons (–OH and –NH) in the structure.
 | ||
| Scheme 2 The proposed binding models of (a) M–F, (b) M–CN, (c) M–AcO and (d) M–OH. | ||
Similarly, to study the interaction nature and the bonding modes of M with guest cations, Job plot method was used again (Fig. 7). Accordingly, the spectra obtained revealed that the interaction ratio between M and the selected cations (Cu2+, Ni2+, Zn2+ and Fe2+) is mixed, with a 1:1 and 1:2 strongly exhibited. The spectra suggest that the interaction for M–Cu and M–Zn, with an absorption maxima centered around 7.0, translating into a 2:1 (2M:Cu and 2M:Zn) as displayed (Fig. 7a & c). Contrastingly, Job plot spectra for the other two complexes (M:Ni and M:Fe) exhibited strong characterizes of a 1:1 interaction ratio. The absorption maximum of the two plots are centered at 5.0, which translates into a 1:1 interaction ratio (Fig. 7b & d).
 | ||
| Fig. 7 The Job plots of (a) [M–Cu], (b) [M–Ni], (c) [M–Zn] and (d) [M–Fe] at 1 × 10−4 M in DMSO–H2O. | ||
Accordingly, the nature of the probe or ligand (M) can be classified as a tridentate, possessing two donor/soft atoms (N, S & O), which are proper recipes for coordination induced charge transfer mechanism. Thus, resulting from the Job plot investigation, the proposed interacting modes have been suggested, based on a 1:1 and 1:2 ratio (Scheme 3), accordingly. Clearly, the interaction modes of M with cations was subsequently based on coordination through the oxygen, sulfur and nitrogen atoms in the structure, in which M–Fe and M–Ni (Scheme 3a & c), exhibit a 1:1 binding mode, predicted to form a 6-coordinate-bonded pedants, high likely to be of tetrahedral and square planar geometries, respectively.
 | ||
| Scheme 3 The proposed binding models of M with (a) Fe2+, (b) Zn2+, (c) Ni2+ and (d) Cu2+. | ||
3.3 Fluorescence studies of M
All fluorescence titrations were performed by drawing 3 ml of M (1 × 10−5 M) of the solvent mixture (DMSO–H2O) and measure the fluorescence intensity at ambient temperature, where the excitation wavelength was 390 nm. To this solution (3 ml of M, 1 × 10−5 M), the molar additions of different ions (cations or anions) were incrementally added, each one separately, in 0.2 equivalents intervals, till no spectral changes were observed, as a result the upper titration limit was set at varying equivalents, based on individual ions, once no changes were observed upon the addition of certain quantities.Generally, fluorophore probes are often designed using a strategy where the structure bears oxygen (from hydroxyl groups) and/nitrogen donor atoms, with the functions of coordinating to metal ions. Thus, with appropriate functional groups incorporated in the structure, for instance a rich in π-electron naphthyl moiety, such is highly likely to exhibit the excited-state intramolecular charge transfer (ESIPT) mechanism. Moreover, upon interacting with cations or anions, the fluorescence properties of the probe are highly likely to be affected, attributed to chelation-enhanced (or quenching) fluorescence mechanism (CHEF and CHEQ). The CHEF mechanism normally results in the inhibition of the ESIPT process and the corresponding C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N isomerization, which at times increases the rigidity of the structure. Basically, the emissions of ESPIT-driven systems are characterized by dual emissions, indicating the coexistence of two tautomeric forms (an enol and keto) throughout the excitation process.71–73 The ESIPT and restriction in rotation of CN bond phenomena in M are normally responsible for the observed optical outputs changes in the presence of cations and anions, as displayed (Scheme 4).
N isomerization, which at times increases the rigidity of the structure. Basically, the emissions of ESPIT-driven systems are characterized by dual emissions, indicating the coexistence of two tautomeric forms (an enol and keto) throughout the excitation process.71–73 The ESIPT and restriction in rotation of CN bond phenomena in M are normally responsible for the observed optical outputs changes in the presence of cations and anions, as displayed (Scheme 4).
 | ||
| Scheme 4 Proton transfer model of M detailing the ESIPT process. | ||
The excited state fluorescence emission model for M (Scheme 4) initially starts on the ground state (E), before excited into a phototautomer, due to ESIPT process (E*). Sequentially, the excited state form (E*) then decays by emitting fluorescence, normally towards the longer wavelength (K*). Subsequently, the proton transfer process in the structure is central to driving the ESIPT, ascribed to the redistribution of the excited state electron density, which results in the –OH group getting more acidic. Theoretically, the structural features of M made it suspect to interaction with ionic species, due to the presence of soft donor atoms such as O, S & N capable of interacting coordinatively with cations, while the hydroxyl (–OH) and amine (–NH–) group are fitting for anion interactions. These interactions have a significant impact on the charge transfer mechanisms (ICT and ESIPT), thus affecting the emission properties of M, upon the introduction of cations or anions.
N group, which normally results in fluorescence quenching during the excited state. The other factor was the ESIPT process which M was highly likely to go through in the excited state, which led to non-radiative decay of the excited state, and thus leading to fluorescence quenching.70,74
 | ||
| Fig. 8 The fluorescence emission spectra of (a) M (1 × 10−5 M) in DMSO–H2O, excited at 390 nm, (b) a combined normalized absorption and fluorescence spectrum of M, with a Stokes shift of 3700 cm−1, (c) under UV-light at 254 nm, (d) under UV-light at 365 nm, and (e) under day-light conditions. | ||
It is generally known that ESIPT single or dual emitters exhibit fluorophore properties due to their smart photosensitive and optical properties, which are normally defined by sizeable Stokes shifts. Thus, M was no exception in this case, showing a Stokes shift of 3700 cm−1 (63 nm), obtained from the normalized absorption and fluorescence data (Fig. 8b). The hydroxyl-based group integrated into the naphthyl moiety coupled with the hydrazone group (–CHN–NH–) were duly responsible for M exhibiting the ESIPT mechanism in the probe. The ESIPT emitters underwent certain molecular vibrations and rotations (centered at the –CN–) in solution leading to the manifestation of non-radiative deactivation pathways, by increasing the rigidity of the molecular cluster, which in turn affected the emission quantum yield.
The fluorescence properties of M were further investigated by exposing the solid sample to daylight and UV-light. Despite M exhibiting weak emission properties due to the ESIPT process (Fig. 8a), the powder could still display glowing behaviors under UV-light (Fig. 8c, at 254 nm and Fig. 8d, at 365 nm), showing a bright intense yellow colour, while under daylight, the colour of the probe was plain yellow (Fig. 8e).
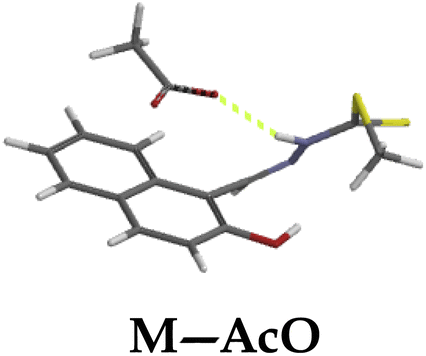
The molar addition of anions (AcO−, CN−, F−, OCN−, H2PO4−, N3− and OH−) to M in a DMSO–H2O medium was generally characterized by fluorescence emission enhancement, at the excitation wavelength of 390 nm. In addition, the prominent peak of M at 445 nm, a new peak appeared in the region of 520 nm. Precisely, the molar titration of M with AcO− led to the anticipated hydrogen bonding-based fluorescence enhancement peak at 445 nm. Moreover, another intense emission peak appeared at 520 nm with molar incremental addition of AcO− (Fig. 9a). The Job plot experiment had revealed that the binding mode between M and the Y-shaped AcO− is likely a 1:1, interacting through the –OH and –NH groups (Scheme 2c), via hydrogen bonding. Hydrogen-bonding based intramolecular charge transfer mechanism, with basicity characteristics such as AcO− are mostly associated with the deprotonation process of the molecule (M), when analytes are in excess.75 Thus, the dual emission characteristics of M in both cases was ascribed to the hydrogen bonding process and the gradual deprotonation action, which eventually lead to the formation of an enol–keto tautomeric forms (Scheme 5). The titration profile (Fig. 9a inset) showed that the sensitivity and selectivity of M toward AcO− was somehow high, attained within the addition of 0.5 μM, with the limit of detection of 6 × 10−9 M and the association constant of 1.88 × 106 M−1 within 0–0.5 μM (Fig. 9b) and the R2 value of 098674 (Fig. S5a†). Similar trends were observed when CN−, F− and H2PO4− were used (Fig. 9b–d), with their respective calibration profiles (Fig. 9 insets) as well as the emission intensity vs. concentration profiles (Fig. S5b–d†), respectively, with the complete profiles summarized in Table 1.
 | ||
| Fig. 9 The fluorescence titration spectra and titration profiles (insets) of M (1 × 10−5 M) in DMSO–H2O, upon the addition of (a) 2.5 equiv. of AcO−, (b) 2.5 equiv. of CN− (c) upon the addition of 2.5 equiv. of F−, and (d) 2.5 equiv. of H2PO4−. Excitation wavelength was at 390 nm. | ||
 | ||
| Scheme 5 Proton transfer model of M detailing the ESIPT process upon the addition of anions. | ||
| Complexa | Stoichiometry | Kass (M−1) | LOD (M) | Quantum yield (Φf) |
|---|---|---|---|---|
| a The cations were added in their nitrate salts. | ||||
| M–Cu | 2:1 |
7.50 × 106 | 1.40 × 10−9 | 0.35 |
| M–Hg | 2:1 |
4.67 × 106 | 1.00 × 10−8 | 0.42 |
| M–Al | 2:1 |
4.15 × 106 | 8.00 × 10−9 | 0.81 |
| M–Ni | 1:1 |
8.33 × 106 | 8.00 × 10−9 | 0.30 |
| M–Sn | 1:1 |
2.73 × 105 | 2.00 × 10−9 | 0.77 |
| M–F | 1:1 |
3.30 × 106 | 8.00 × 10−9 | 0.34 |
| M–CN | 1:1 |
3.57 × 106 | 1.00 × 10−9 | 0.40 |
| M–AcO | 1:1 |
1.88 × 106 | 6.00 × 10−9 | 0.42 |
| M–H2PO4 | 1:1 |
2.21 × 10−6 | 8.00 × 10−9 | 0.23 |
| M–Ag | 1:1 |
2.10 × 107 | 2.00 × 10−9 | 0.33 |
| M–OCN | 1:1 |
3.65 × 106 | 4.00 × 10−9 | 0.23 |
| M–OH | 1:1 |
3.89 × 106 | 1.00 × 10−10 | 0.36 |
| M–Fe(II) | 1:1 |
7.97 × 105 | 2.00 × 10−8 | 0.33 |
Moreover, the molar addition of CN− yielded in fluorescence enhancement characterized by dual emission channels with signals centered at 445 nm and 520 nm. The emission at lower wavelength (445 nm) was ascribed to hydrogen-bonding process (Scheme 2b), while the peak at higher wavelengths (520 nm) attributed to the deprotonation of the hydroxyl group and secondary amine groups. The nature of interaction of interaction between the two species has a strong emission enhancement effect on M, showing strong association, even when smaller molar amounts (0–2.5 equiv.) were used in titration (Fig. 9b). The titration profile (Fig. 9b inset) shows that the sensitivity and selectivity of M toward CN− was high, attained within the addition of 0.5 μM, with the limit of detection of 1 × 10−9 M and the association constant of 3.57 × 106 M−1 within 0–1.0 μM (Fig. 9d) and the R2 value of 0.97 (Fig. 9d inset).
Furthermore, the molar additions of 25 equiv. of OCN−, 2.5 equiv. H2PO4− and 5 equiv. of OH− all resulted in fluorescence enhancement, with monotonous emissive characteristics (Fig. S6†). The additions of OCN−, H2PO4− and OH− resulted in the fluorescence enhancement at 445 nm, simultaneously with the formation and enhancement of a new peak at 520 nm (Fig. S6a–c & e†), due to hydrogen-bonding and deprotonation process. The two absorption bands enhanced were characteristics to tautomer forms, enol and keto, well associated with hydroxyl group-containing sensing probes.76,77 The fluorometric addition of OCN−, which induced fluorescence enhancement upon interacting with M, required about 30 equiv. of the analyte, this showed perhaps moderate selectivity and sensitivity of the probe (M) towards the analyte (OCN−), presumably due to the geometry and chemistry of two species. The model of hydrogen-bonding process which takes place on first host–guest encounter and the deprotonation process that follows once the analytes (anions) are in excess were summarized accordingly (Scheme 5).
N bond and possibly the photoinduced electron transfer mechanism of the molecule, in the absence of Al3+. However, on gradual introduction of Al3+ to M, emission enhancement was observed, characterized by two intensifying peaks at 466 nm and 493 nm respectively. Notingly, the association of M with Al3+ promotes structural rigidity while simultaneously restricting its flexibility, which boosts the emissive properties of the system. The titration profile (Fig. 10 inset) shows a linear relationship upon the introduction of Al3+ of up to 1 equiv. before the equilibrium/saturation is reached. The linear fit plot based on a 1:2 reaction interaction, with the R2 of 0.95 is presented (Fig. 10b), predicting the limit of detection extrapolated from the concentration range of 0–0.8 μM. The titration parameters, ranging from the LOD, association constant as well as the quantum yields are summarised in Table 1.
 | ||
| Fig. 10 The fluorescence titration spectra and titration profile (inset) of M (1 × 10−5 M) in DMSO–H2O, (a) with up to 10 equiv. of Al3+, excited at 390 nm, (b) linear fit plot of the conc. of Al3+ (0–0.8 μM). | ||
Similar to interactions with anions (Fig. 9), M was characterized by dual emission channels, upon interacting with Al3+, at 466 nm and 493 nm respectively, ascribed to the combinations of ICT and ESIPT mechanisms. The dual emissions were suspectedly ascribed to the to the normal excited (E*) and its tautomer (K*) forms, which resulted from an ESIPT process (E*–K*) as displayed (Scheme 6). Normally, such dual emission channels are commonly known for phenolic-containing fluorometric sensors, which undergo enol–keto tautomerization, by way of a hydroxyl proton transfer to the adjacent imine group, leading to major change in structural conformations and electronic alterations. In this case, the emissive peak at 466 nm predictably belonged to the enol form, while the keto form was reflected at 493 nm, respectively.67
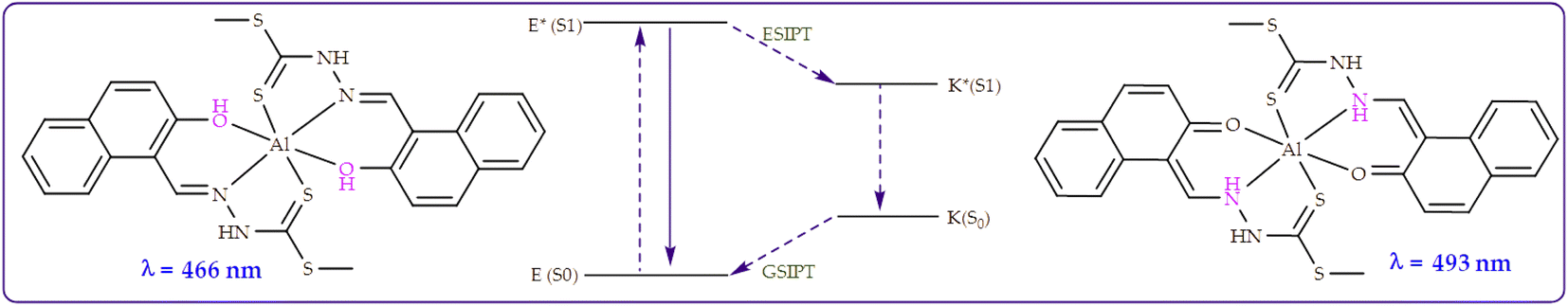 | ||
| Scheme 6 Proton transfer model of M detailing the ESIPT process upon the addition of Al3+. | ||
The fluorometric titrations of M with other cations was dominated by fluorescence quenching, unlike with Al3+, where the intensity at 445 nm of uncomplexed state decreased significantly with increasing addition of the cations (Fig. 11). Explicitly, the molar addition of cations (Cu2+, Fe2+, Ni2+) displayed fluorescence quenching, the prominent peak at 445 nm decreased in intensity with the increasing sequential addition of these cations (Fig. 11a, c & e), with titration profiles inserted. The molar titration addition of cations to M inhibit the structural fluorescence properties, ascribed to the chelation induced quenching process (CHEQ). In addition, the quenching mechanism here could also be due to collision quenching, where the excited state of M is deactivated due to contacts with cationic species, as well as heavy-atom and paramagnetic quenching mechanisms.78 The presence of cations in the matrix of M inhibited the tautomerism process, as displayed by the demeaning of the spectra, resulting from the chelation process. The calibration curves for the respective interactions were used to extrapolate the intensity vs. concentrations relationship for each cation (M–Fe, M–Ni and M–Cu), where linear fit method was used to estimate the slope of the best fits, with R2 values ranging from 0.93, 0.97 and 0.97, respectively (Fig. 11c, d and f).
 | ||
| Fig. 11 The fluorescence titration spectra and titration profiles (insets) of M (1 × 10−5 M) in DMSO–H2O, (a) upon the addition of 15 equiv. of Fe2+, (b) fluorescence signals of M to changing conc. of Fe2+ (0–15 μM) at 450 nm, (c) upon the addition of 5 equiv. of Ni2+ and (d) fluorescence signals of M to changing conc. of Ni2+ (0–5.0 μM), (e) upon the addition of 3.0 equiv. of Cu2+ and (f) fluorescence signals of M to changing conc. of Cu2+ (0–3.0 μM), at 450. | ||
However, a different behavior was noticed when Hg2+ was added to M, upon interacting, not only fluorescence quenching at 445 nm due to the chelation effect was observed, but the emergence of a new peak at 505 nm appeared concomitantly (Fig. 12a). A new peak at 505 nm was characterized by the isosbestic point at 475 nm, showing the co-existence of two species (an emissive and a non-emissive) at equilibrium. Interestingly, the initial addition of Hg2+ to M inhibited non-emissive behaviors at first ascribed by the chelation fluorescence quenching (CHEQ) effect, however, on increasing addition resulted in the formation of a new complexed compound, which is highly emissive (CHEF), presumably attributed to the CN isomerization and the enol–keto tautomerism effect (Scheme 7), resulting in CHEF, while inhibiting the ESIPT process. Similarly, the fluorometric behaviors was observed upon the addition of Ag+ and Sn2+ as displayed (Fig. 12c & e), respectively.
 | ||
| Fig. 12 The fluorescence titration spectra and titration profiles (insets) of M (1 × 10−5 M) in DMSO–H2O, (a) upon the addition of 1 equiv. of Hg2+, (b) fluorescence signals of M to changing conc. of Hg2+ (0–1 μM) at 500 nm, (c) upon the addition of 15 equiv. of Ag+ and (d) fluorescence signals of M to changing conc. of Ag+ (0–0.3 μM), (e) upon the addition of 10.0 equiv. of Sn2+ and (f) fluorescence signals of M to changing conc. of Sn2+ (0–2.0 μM), at 510 nm. | ||
 | ||
| Scheme 7 Proton transfer model of M detailing the ESIPT process upon the addition of Hg2+. | ||
 | (1) |
The limit of detection (LOD) was calculated based on the emission–titration curves of M with analytes, using eqn (2) below
| LOD = 3σ/k | (2) |
| Φs = Φs(ηs/ηr)2 × As/Ar × Fr/Fs | (3) |
| Optimized structure | HOMO (eV) | LUMO (eV) | Gap (eV) |
|---|---|---|---|
 |
 |
 |
3.37 |
 |
 |
 |
0.46 |
 |
 |
 |
0.61 |
 |
 |
 |
0.76 |
 |
 |
 |
1.36 |
 |
 |
 |
0.76 |
 |
 |
 |
2.75 |
 |
 |
 |
2.61 |
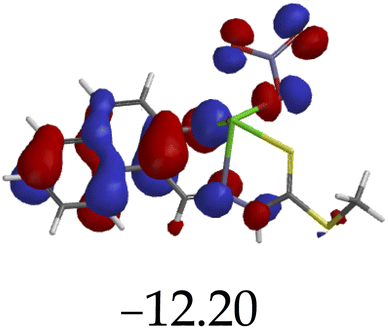
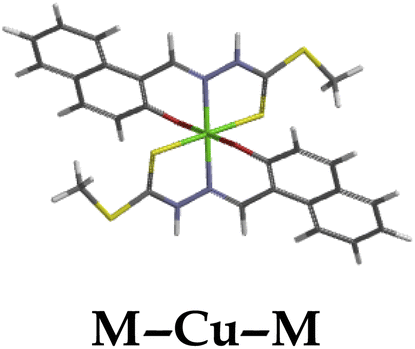
The introduction of cations to M resulted in mixed values of the energy gaps with fewer correlations, where M–Al–M (0.46 eV), M–Zn (0.61 eV), M–Hg (0.76 eV), M–Cu–M (1.36 eV), M–Ni (0.76 eV), M–Sn–M (2.75 eV) and M–Ag respectively as displayed in Table 2. The lowest energy gap was registered with M–Al–M (0.46 eV) conformation, perhaps due to highly active ICT, ESIPT and MLCT upon interaction, based on a 2:1 molar ratio. The conformation of M–Al–M showed relatively good enhancing properties (CHEF) exhibiting the highest fluorescence intensity of all cations used, going through tautomeric transformation in the process, as displayed by double emission peaks. Similar trends of CHEF were experienced by the conformations of M–Hg (0.76 eV), M–Ag (2.61 eV), and M–Sn–M (2.75 eV), in which all the band gaps conforming to responding to photon absorption across the visible light range, towards near infrared (NIR) region. The QHEF effect of M–Hg and M–Ag, even though relatively different in values in a proposed 1:1 interaction ratio based on the Job plot data (Fig. 7), were guided by the combination of ICT, ESIPT and MLCT processes. In addition, CHEF effect was also induced upon M–Sn–M, similarly to M–Al–M, thereby undergoing ESIPT and MLCT processes. The selectivity of CHEF-based effect for these cations is ascribed to their varying chemical make-up towards M, ranging from increasing atomic radii going down the group, surface plasmon resonance phenomenon and inert-pair effect.79–84
Moreover, other metal cations induced CHEQ effects upon interacting with M, ascribed to their chemical blueprints towards M. The CHEQ effect was observed on complexes such as M–Cu–M (1.36 eV) and M–Ni (0.76 eV), presumably due to their possibly square planar geometries, likely formed, thereby quenching the emission significantly. The theoretical calculations are suggestive that these complexes have lower HOMOs and LUMOs, with energy gaps varying from 1.36 eV and 0.76 eV, corresponding to photon absorption within the visible region. The variance in the energy gaps was attributed to the combination of ESIPT, ICT and MLCT, which were all likely to occur upon interaction. The HOMO distribution was clearly concentrated on the donor species (ligands) compared to the LUMO, which was located on the cation center respectively.
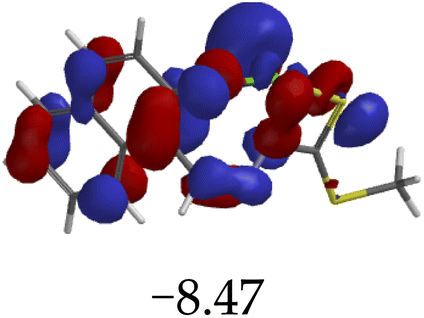
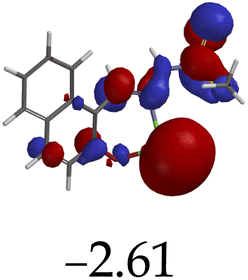
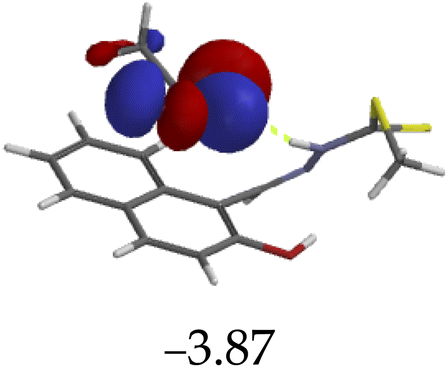
Furthermore, hydrogen bonding induced charge transfer, which guided the interaction of M with anions through the –NH group, resulted in relatively high HOMO and LUMO levels (Table 3). The energy gaps decreased in all cases studied, with M–CN (3.46 eV) the highest, while M–OH displayed the lowest of those used (1.45 eV). The fact that –OH contains a hydroxyl group interacting with another hydrogen bonding capable –NH species of M, resulted in a complementing ESIPT and ICT, thus resulting in the CHEF effects. In fact, all interactions of M with anions, resulted in CHEF effects, due to deprotonation and thus proton transfer processes, brought about by the formation of ENOL and KETO tautomers. Thus, the quenching of these compounds proved that structural deprotonation favored non-emissive properties. The distribution of HOMOs was concentrated on the analytes for most of the complexes, except for M–CN, where the cloud of HOMOs was fairly distributed along the entire complexed molecule. The distribution of the LUMOs was concentrated on the molecule, bar the analyte (Table 3). The CHEF effect, which was based on the deprotonation process, was still observed by the interaction of M–F and M–AcO, which are all well-known to form strong associations with –NH based receptors, ascribed to the high electronegative nature of F− and the potent Y-shaped structure of AcO−.
| Optimized structure | HOMO (eV) | LUMO (eV) | Gap (eV) |
|---|---|---|---|
 |
 |
 |
3.37 |
 |
 |
 |
1.45 |
 |
 |
 |
2.03 |
 |
 |
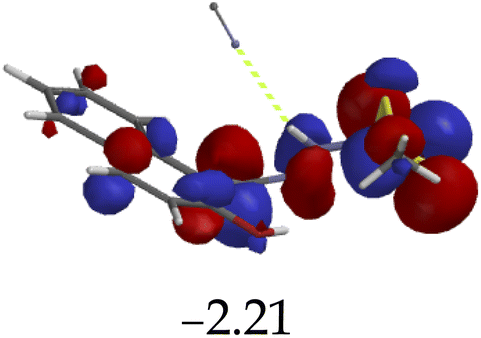 |
3.46 |
 |
 |
 |
1.83 |
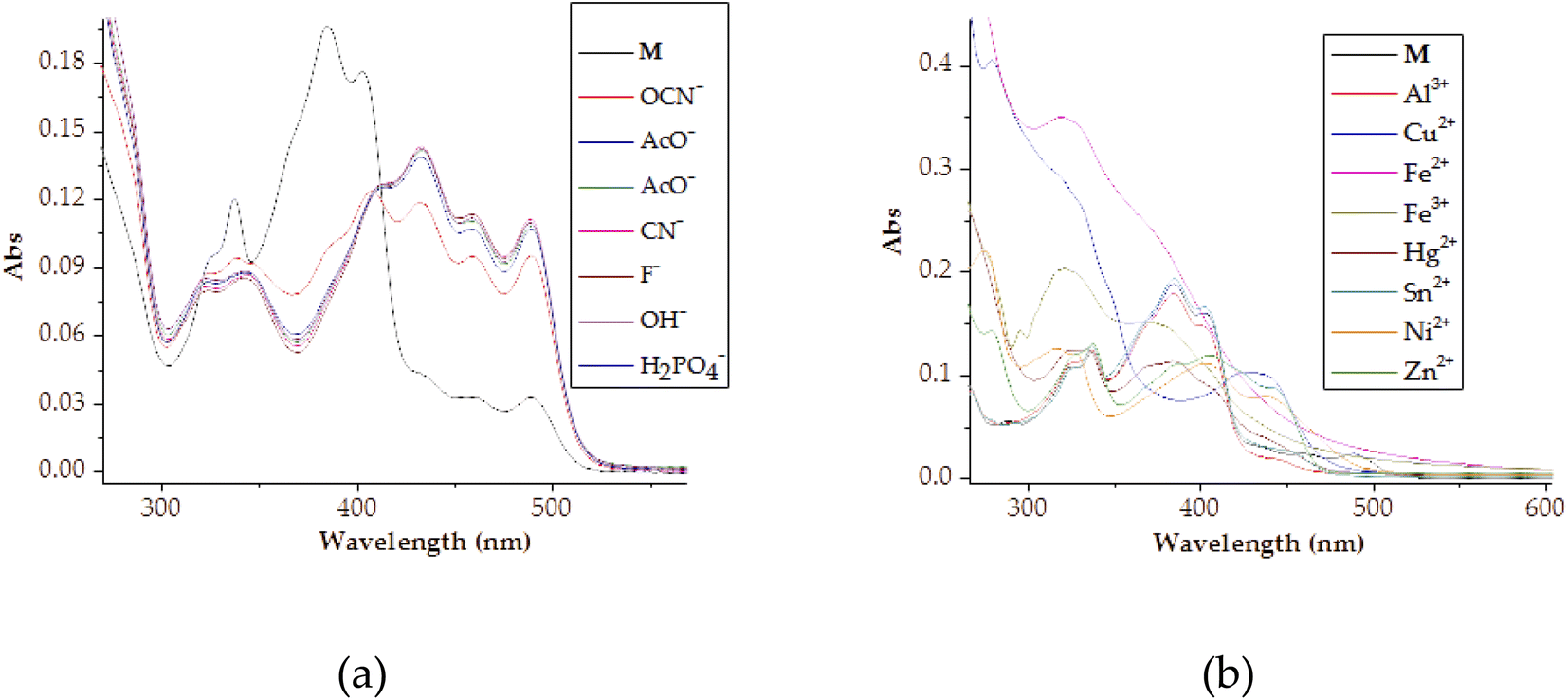 | ||
| Fig. 13 Combined absorption plots of M (1 × 10−5 M) in DMSO–H2O, upon the addition of 3 equiv. of each individual (a) anions and (b) cations. | ||
4. Conclusion
A multi-sensing fluorometric probe with dual emissive channels was synthesized, via a cost-effect and one step mechanism of Schiff base. The ESIPT-steered probe proved highly versatile in recognition of heavy metal cations as well as biological important anions. A weakly fluorescent-based probe displayed varying interaction patterns through contrasting absorption and fluorescence behaviors upon associating with specific cations or anions, presenting ratiometric behaviors for some, as well as CHEQ and CHEF effects for others. For instance, the introduction of Al3+ resulted in a significant fluorescence enhancement with the effect of more than ten folds, while displaying an insignificant, but yet unique, absorption behaviors upon titration with M. The addition of Hg2+, Ag+ and Sn2+ had uniquely displayed similar fluorescence patterns of ratiometric nature upon interacting with M, where the initial peak at lower wavelengths disappeared, with the new peak at longer wavelengths simultaneously being formed. Moreover, the interactions with other metals, such as Zn2+, Cu2+ and Ni2+ resulted in the CHEQ effect, with the inhibition of the ESIPT process. Interestingly, all anions resulted in a resounding CHEF effect with similar patterns, clearly demonstrating the tautomeric effect via two well-resolved emission bands, ascribed to the enol and keto forms. Thus, M showed versatility properties in its encounter with both cations and anions, through a variety of responsive displays in its absorption and fluorescence properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Physics, Chemistry and Material Science, University of Namibia, Namibia. The University of Cape Town for 1H NMR analysis by Mr P Shanika, The Royal Society-DFiD Africa Capacity Building Initiative, New Materials for a Sustainable Energy Future.References
- J. Shao, Y. Qiao, H. Lin and H. Lin, A C3-symmetric colorimetric anion sensor bearing hydrazone groups as binding sites, Spectrochim. Acta, Part A, 2009, 71(5), 1736–1740 CrossRef.
- C. Huang and H. Chang, Fluorescent Sensors for Detection of Mercury (II) in aqueous solution has been developed in bulk solution fluoresce weakly when they are adsorbed, Representations, 2006, 78(24), 8332–8338 CAS.
- H. A. El-Shekheby, A. H. Mangood, S. M. Hamza, A. S. Al-Kady and E. Z. M. Ebeid, A highly efficient and selective turn-on fluorescent sensor for Hg2+, Ag+ and Ag nanoparticles based on a coumarin dithioate derivative, Luminescence, 2014, 29(2), 158–167 CrossRef CAS PubMed.
- E. A. Elshehy, S. A. El-safty, M. A. Shenashen and M. Khairy, Chemical Design and evaluation of optical mesocaptor for the detection/recovery of Au (III) from an urban mine, Sens. Actuators, B, 2014, 203, 363–374, DOI:10.1016/j.snb.2014.06.055.
- R. Dhivya, A. Gomathi and P. Viswanathamurthi, Pyrene Based Fluorescent Turn-on Chemosensor for Sequential Detection of Fe3+ and Fe2+ Ions and its Application in Live Cell Imaging, J. Fluoresc., 2019, 29, 797–802, DOI:10.1007/s10895-019-02392-2.
- N. O. Laschuk, I. I. Ebralidze, S. Quaranta, S. T. W. Kerr, J. G. Egan and S. Gillis, et al, Rational design of a material for rapid colorimetric Fe2+ detection, Mater. Des., 2016, 107, 18–25, DOI:10.1016/j.matdes.2016.06.016.
- N. Bhuvanesh, K. Velmurugan, S. Suresh, P. Prakash, N. John, S. Murugan, T. Daniel Thangadurai and R. Nandhakumar, Naphthalene based fluorescent chemosensor for Fe2+-ion detection in microbes and real water samples, J. Lumin., 2017, 217–222, DOI:10.1016/j.jlumin.2017.04.026.
- G. Gao, W. Qu, B. Shi, Q. Lin, H. Yao and J. Chang, et al, Sens. Actuators, B, 2015, 213, 501–507, DOI:10.1016/j.snb.2015.02.077.
- S. Kannan Balasingam, M. Lee, M. Gu Kang and Y. Jun, Improvement of dye-sensitized solar cells toward the broader light harvesting of the solar spectrum, Chem. Commun., 2013, 49(15), 1471–1487 RSC.
- A. Mohammadi, F. S. Zabihi and N. Chaibakhsh, Chemical sensor using metal-organic complex: Preparation, characterization and application for highly selective detection of cyanide ions in mixed aqueous-organic media, J. Photochem. Photobiol., A, 2018, 367, 384–389, DOI:10.1016/j.jphotochem.2018.09.004.
- J. H. Kang, S. Y. Lee, H. M. Ahn and C. Kim, Sequential detection of copper (II) and cyanide by a simple colorimetric chemosensor, Inorg. Chem. Commun., 2016, 74, 62–65, DOI:10.1016/j.inoche.2016.10.039.
- J. Prabhu, K. Velmurugan, A. Raman, N. Duraipandy and M. S. Kiran, A simple chalcone based ratiometric chemosensor for sensitive and selective detection of Nickel ion and its imaging in live cells, Sens. Actuators, B, 2017, 238, 306–317, DOI:10.1016/j.snb.2016.07.018.
- A. Thakur, D. Mandal and S. Ghosh, A triazole based triferrocene derivative as a multiresponsive chemosensor for Hg (II) ion and a redox chemosensor for H2PO4− ion, J. Organomet. Chem., 2013, 726, 71–78, DOI:10.1016/j.jorganchem.2012.11.031.
- V. Tharmaraj and K. Pitchumani, An acyclic, dansyl based colorimetric and fluorescent chemosensor for Hg (II) via twisted intramolecular charge transfer (TICT), Anal. Chim. Acta, 2012, 751, 171–175, DOI:10.1016/j.aca.2012.09.016.
- S. Y. Lee, S. Y. Kim, J. A. Kim and C. Kim, A dual chemosensor: colorimetric detection of Co2+ and fluorometric detection of Zn2+, J. Lumin., 2016, 179, 602–609, DOI:10.1016/j.jlumin.2016.07.054.
- H. Na Kim, W. Xiu Ren, J. Seung Kim and J. Yoon, Fluorescent and colorimetric sensors for detection of lead, cadmium, and mercury ions, Chem. Soc. Rev., 2012, 41(8), 3210–3244 RSC.
- J. Briffa, E. Sinagra and R. Blundell, Heavy metal pollution in the environment and their toxicological effects on humans, Heliyon, 2020, 6, e04691, DOI:10.1016/j.heliyon.2020.e04691.
- A. Apilux, W. Siangproh, N. Praphairaksit and O. Chailapakul, Simple and rapid colorimetric detection of Hg(II) by a paper-based device using silver nanoplates, Talanta, 2012, 97, 388–394, DOI:10.1016/j.talanta.2012.04.050.
- L. Lin, S. T. Hu, Y. C. Yan, D. J. Wang, L. Fan and Y. J. Hu, et al, A highly selective chemosensor for nickel(II) based on fluorescence quenching of a bispyrazole derivative, Res. Chem. Intermed., 2017, 43(1), 283–295 CrossRef CAS.
- X. Liu, Q. Lin, T. B. Wei and Y. M. Zhang, A highly selective colorimetric chemosensor for detection of nickel ions in aqueous solution, New J. Chem., 2014, 38(4), 1418–1423 RSC.
- D. Seo, K. W. Park, J. Kim, J. Hong and K. Kwak, DFT computational investigation of tuning the electron donating ability in metal-free organic dyes featuring a thienylethynyl spacer for dye sensitized solar cells, Comput. Theor. Chem., 2016, 1081, 30–37, DOI:10.1016/j.comptc.2016.02.009.
- F. dos Santos Carlos, M. C. Nunes, L. De Boni, G. S. Machado and F. S. Nunes, A novel fluorene-derivative Schiff-base fluorescent sensor for copper(II) in organic media, J. Photochem. Photobiol., A, 2017, 348, 41–46, DOI:10.1016/j.jphotochem.2017.08.022.
- K. S. Min, R. Manivannan and Y. A. Son, Rhodamine-fluorene based dual channel probe for the detection of Hg2+ ions and its application in digital printing, Sens. Actuators, B, 2018, 261, 545–552, DOI:10.1016/j.snb.2018.01.178.
- T. A. Khattab, S. A. Aly and T. M. Klapötke, Naked-eye facile colorimetric detection of alkylphenols using Fe(III)-impregnated silica-based strips, Chem. Pap., 2018, 72(6), 1553–1559, DOI:10.1007/s11696-018-0409-7.
- S. Pawar, U. Fegade, V. K. Bhardwaj, N. Singh, R. Bendre and A. Kuwar, 2-((E)-(2-aminophenylimino)methyl)-6-isopropyl-3-methylphenol based fluorescent receptor for dual Ni2+ and Cu2+ recognition: nanomolar detection, Polyhedron, 2015, 87, 79–85, DOI:10.1016/j.poly.2014.10.034.
- F. Paquin, J. Rivnay, A. Salleo, N. Stingelin and C. Silva, Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors, J. Mater. Chem. C, 2015, 3(207890), 10715–10722 RSC . http://xlink.rsc.org/?DOI=C5TC02043C.
- Z. Salarvand, M. Amirnasr and S. Meghdadi, Colorimetric and fluorescent sensing of Al3+ by a new 2-hydroxynaphthalen based Schiff base “Off-On” chemosensor, J. Lumin., 2019, 207, 78–84 CrossRef CAS.
- P. Mohanty, R. Behura, V. Bhardwaj, P. P. Dash, S. K. Sahoo and B. R. Jali, Recent advancement on chromo-fluorogenic sensing of aluminum(III) with Schiff bases, Trends Environ. Anal. Chem., 2022, 34, e00166, DOI:10.1016/j.teac.2022.e00166.
- H. Thomas, J. Naimhwaka, P. Endjala and V. Uahengo, ESIPT-influenced C3-symmetry, disk-shaped fluorescence turn-on probe for Zn2+ based on melamine-naphthyl moiety with high affinity towards Cu2+ in CH3CN, Results Opt., 2022, 6, 100215, DOI:10.1016/j.rio.2022.100215.
- J. Naimhwaka and V. Uahengo, A naphthoquinone based colorimetric probe for real-time naked eye detection of biologically water: experimental and theoretical studies, RSC Adv., 2019, 37926–37938 RSC.
- A. Mohammadi, F. S. Zabihi and N. Chaibakhsh, Chemical sensor using metal-organic complex: Preparation, characterization and application for highly selective detection of cyanide ions in mixed aqueous-organic media, J. Photochem. Photobiol., A, 2018, 367, 384–389 CrossRef CAS.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and L. Mosca, The interaction of fluoride with fluorogenic ureas: an ON 1-OFF-ON2 response, J. Am. Chem. Soc., 2013, 135(16), 6345–6355 CrossRef CAS PubMed.
- J. Naimhwaka and V. Uahengo, A naphthoquinone based colorimetric probe for real-time naked eye detection of biologically important anions including cyanide ions in tap water: experimental and theoretical studies, RSC Adv., 2019, 9(65), 37926–37938 RSC.
- G. J. Park, G. R. You, Y. W. Choi and C. Kim, A naked-eye chemosensor for simultaneous detection of iron and copper ions and its copper complex for colorimetric/fluorescent sensing of cyanide, Sens. Actuators, B, 2016, 229, 257–271 CrossRef CAS.
- V. Uahengo, J. Naimhwaka, L. S. Daniel, A. Rahman, M. I. Elzagheid, L. Rhyman, P. Ramasami and P. Cai, A colorimetric probe for the real-time naked eye detection of cyanide and hydroxide ions in tap water: experimental and theoretical studies, Analyst, 2019, 144, 6422–6431, 10.1039/C9AN01481K.
- A. Mohammadi, Z. Dehghan, M. Rassa and N. Chaibakhsh, Colorimetric probes based on bioactive organic dyes for selective sensing of cyanide and fluoride ions, Sens. Actuators, B, 2016, 230, 388–397 CrossRef CAS . http://linkinghub.elsevier.com/retrieve/pii/S092540051630226X.
- T. D. Thangadurai, C. J. Lee, S. H. Jeong, S. Yoon, Y. G. Seo and Y.-I. Lee, A novel colorimetric and fluorescent sensor for fluoride and pyrophosphate based on fluorenone signaling units, Microchem. J., 2013, 106, 27–33, DOI:10.1016/j.microc.2012.04.012.
- X. Yang, L. Xie, R. Ning, X. Gong, Z. Liu and Y. Li, et al, A diketopyrrolopyrrole-based near-infrared sensor for selective recognition of fluoride ions, Sens. Actuators, B, 2015, 210, 784–794, DOI:10.1016/j.snb.2015.01.025.
- W. Huang, H. Su, J. Li, H. H. Lin and H. H. Lin, An acetate sensor based on azo in aqueous media, Spectrochim. Acta, Part A, 2010, 77(1), 146–149 CrossRef PubMed.
- A. Okudan, S. Erdemir and O. Kocyigit, “Naked-eye” detection of fluoride and acetate anions by using simple and efficient urea and thiourea based colorimetric sensors, J. Mol. Struct., 2013, 1048, 392–398, DOI:10.1016/j.molstruc.2013.04.077.
- E. Saikia, M. P. Borpuzari, B. Chetia and R. Kar, Experimental and theoretical study of urea and thiourea based new colorimetric chemosensor for fluoride and acetate ions, Spectrochim. Acta, Part A, 2016, 152, 101–108, DOI:10.1016/j.saa.2015.07.065.
- H. Lin, W. Huang, H. Su and H. Lin, An efficient novel receptor for sensing acetate, J. Inclusion Phenom. Macrocyclic Chem., 2011, 70(1–2), 129–133 Search PubMed.
- J. Feng, J. Zhong and X. Tang, Selective and sensitive detection of cyanate using 3-amino-2-naphthoic acid-based turn-on fluorescence probe, 2019.
- P. D. Beer and S. R. Bayly, Anion sensing by metal-based receptors, Top. Curr. Chem., 2005, 255, 125–162 CrossRef CAS.
- S. Y. Li, D. B. Zhang, J. Y. Wang, R. M. Lu, C. H. Zheng and S. Z. Pu, A novel diarylethene-hydrazinopyridine-based probe for fluorescent detection of aluminum ion and naked-eye detection of hydroxide ion, Sens. Actuators, B, 2017, 245, 263–272, DOI:10.1016/j.snb.2017.01.149.
- Y. Son, S. Gwon, B. Ananda, H. Kim, Y. Son and S. Kim, Novel styrylbenzothiazolium dye-based sensor for mercury, cyanide and hydroxide ions, Spectrochim. Acta, Part A, 2015, 144, 226–234, DOI:10.1016/j.saa.2015.02.094.
- A. Singh, S. Tom and D. R. Trivedi, Aminophenol based colorimetric chemosensor for naked-eye detection of biologically important fluoride and acetate ions in organo-aqueous medium: Effective and simple anion sensors, J. Photochem. Photobiol., A, 2018, 353 Search PubMed.
- S. M. Basheer, J. Haribabu, N. S. P. Bhuvanesh, R. Karvembu and A. Sreekanth, Naphthalenyl appended semicarbazone as “turn on” fluorescent chemosensor for selective recognition of fluoride ion, J. Mol. Struct., 2017, 1145, 347–355 CrossRef CAS.
- V. K. Gupta, A. K. Singh and N. Gupta, Chemical Colorimetric sensor for cyanide and acetate ion using novel biologically active hydrazones, Sens. Actuators, B, 2014, 204, 125–135, DOI:10.1016/j.snb.2014.07.029.
- F. Bu, B. Zhao, W. Kan, L. Ding, T. Liu and L. Wang, et al, An ESIPT characteristic “turn-on” fluorescence sensor for Hg2+ with large Stokes shift and sequential “turn-off” detection of S2 – as well as the application in living cells, J. Photochem. Photobiol., A, 2020, 387, 112165, DOI:10.1016/j.jphotochem.2019.112165.
- A. S. Yin, S. Sun, M. Pan, L. Chen, Z. Wang, Y. Hou, Y. Fan, H. Wang and C. Su, An imidazole based ESIPT molecule for fluorescent detection of explosives, J. Photochem. Photobiol., A, 2018, 355, 377–381, DOI:10.1016/j.jphotochem.2017.07.044.
- J. G. Amol and S. Nagaiyan, Substituent Modulation from ESIPT to ICT Emission in Benzoimidazolphenyl-methanones Derivatives: Synthesis, Photophysical and DFT Study, J. Solution Chem., 2017, 46(4), 777–797, DOI:10.1007/s10953-017-0602-2.
- F. S. Santos, E. Ramasamy, V. Ramamurthy and F. S. Rodembusch, Excited state chemistry of flavone derivatives in a confined medium: ESIPT emission in aqueous media, Photochem. Photobiol. Sci., 2014, 13, 992–996, 10.1039/C4PP00096J.
- Z. Kowser, U. Rayhan, P. E. Georghiou and T. Yamato, A fluorescence “turn-on” sensor for multiple analytes: OAc− and F− triggered fluorogenic detection of Zn2+ in a co-operative fashion, Tetrahedron, 2017, 73, 5418–5424 CrossRef CAS.
- J. Massue, T. Pariat, P. M. Vérité, D. Jacquemin, M. Durko, T. Chtouki, L. Sznitko, J. Mysliwiec and G. Ulrich, Natural Born Laser Dyes: Excited-State Intramolecular Proton Transfer (ESIPT) Emitters and Their Use in Random Lasing Studies, Nanomaterials, 2019, 9(8), 1093, DOI:10.3390/nano9081093.
- Y. Zhou, G. Baryshnikov, X. Li, M. Zhu, H. Ågren and L. Zhu, Anti-Kasha’s Rule Emissive Switching Induced by Intermolecular H-Bonding, Chem. Mater., 2018, 30(21), 8008–8016 CrossRef CAS.
- H. Singh, G. Bhargava, S. Kumar and P. Singh, Quadruple-signaling (PET, ICT, ESIPT, –CN– rotation) mechanism-based dual chemosensor for detection of Cu2+ and Zn2+ ions: TRANSFER, INH and complimentary OR/NOR logic circuits, J. Photochem. Photobiol., A, 2018, 357, 175–184, DOI:10.1016/j.jphotochem.2018.02.030.
- M. Shahid and A. Misra, Photoenolization via excited state proton transfer and ion sensing studies of hydroxy imidazole derivatives, J. Photochem. Photobiol., A, 2017, 335, 190–199, DOI:10.1016/j.jphotochem.2016.12.003.
- M. Mathivanan, B. Tharmalingam, C. Lin, B. V. Pandiyan, V. Thiagarajan and B. Murugesapandian, ESIPT-active multi-color aggregation-induced emission features of triphenylamine–salicylaldehyde-based unsymmetrical azine family, CrystEngComm, 2020, 22, 213–228, 10.1039/C9CE01490J.
- Y. Liu, Z. Liang, Z. Li, K. Zhao, Y. Sun and X. Zhang, Anti-solvatochromic fluorescence of thiazole [5, 4-d] thiazole by forming hydrogen bond network and its application in fast detection of trace water, Microchem. J., 2020, 154, 104640, DOI:10.1016/j.microc.2020.104640.
- S. Zeng, S. Li, T. Liu, X. Sun and Z. Xing, A significant fluorescent “turn-on” chemosensor for Al3+ detection and application in real sample, logic gate and bioimaging, Inorg. Chim. Acta, 2019, 495, 118962, DOI:10.1016/j.ica.2019.118962.
- Y. Ishibashi, M. Murakami, K. Araki, T. Mutai and T. Asahi, Excited-State Intramolecular Proton-Transfer Process of Crystalline Femtosecond Pump − Probe Microspectroscopy, J. Phys. Chem. C, 2019, 123, 11224–11232 CrossRef CAS.
- S. Jana, S. Dalapati and N. Guchhait, Excited State Intramolecular Charge Transfer Suppressed Proton Transfer Process in 4-(Diethylamino)-2-hydroxybenzaldehyde, J. Phys. Chem. A, 2013, 117(21), 4367–4376 CrossRef CAS.
- V. Uahengo, E. N. Hamukwaya, P. T. Endjala and J. H. Naimhwaka, A potential naphthyl-thiazole-based organic dye and a ditopic chromogenic probe for CN− and Fe3+ with molecular logic functions, New J. Chem., 2020, 44, 18588–18600, 10.1039/D0NJ03806G.
- A. S. Gupta, K. Paul and V. Luxami, Ratiometric fluorescent chemosensor for fluoride ion based on inhibition of excited state intramolecular proton transfer, Spectrochim. Acta, Part A, 2015, 138, 67–72, DOI:10.1016/j.saa.2014.11.026.
- Z. Mao, L. Hu, C. Zhong, H. Zhang, B. Liu and Z. Liu, A dual-mechanism strategy to design a wide-range pH probe with multicolor fluorescence, Sens. Actuators, B, 2015, 219, 179–184, DOI:10.1016/j.snb.2015.05.024.
- V. Kumar, A. Kumar, U. Diwan, S. K. Srivastava and K. K. Upadhyay, Salicylideneimines as efficient dual channel emissive probes for Al3+: Harnessing ESIPT and ICT processes, Sens. Actuators, B, 2015, 207, 650–657, DOI:10.1016/j.snb.2014.10.068.
- S. S. Razi, R. C. Gupta, R. Ali, S. K. Dwivedi, P. Srivastava and A. Misra, A new D–π—A type Intramolecular charge transfer Dyad System to detect F−: Anion induced CO2 sensing, Sens. Actuators, B, 2016, 236, 520–528, DOI:10.1016/j.snb.2016.06.016.
- F. A. S. Chipem, S. K. Behera and G. Krishnamoorthy, Enhancing Excited State Intramolecular Proton Transfer in 2-(2′-Hydroxyphenyl) benzimidazole and Its Nitrogen-Substituted Analogues by β-Cyclodextrin: The Effect of Nitrogen Substitution, J. Phys. Chem. A, 2013, 117(20), 4084–4095 CrossRef CAS PubMed.
- L. Hao, Q. Qiu, W. Wang, L. Gu and H. Li, Fluorescent Detection of Trace Water in Methanol Based on a Al (III) Chemical Sensor, Chin. J. Chem., 2016, 34, 1109–1113, DOI:10.1002/cjoc.201600358.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Artificial receptors for the recognition of phosphorylated molecules, Chem. Rev., 2011, 111(11), 6603–6782 CrossRef CAS PubMed.
- U. N. Yadav, P. Pant, D. Sharma, S. K. Sahoo and G. S. Shankarling, Quinoline-based chemosensor for fluoride and acetate: A combined experimental and DFT study, Sens. Actuators, B, 2014, 197, 73–80, DOI:10.1016/j.snb.2014.02.075.
- G. R. Suman, S. G. Bubbly, S. B. Gudennavar and V. Gayathri, Benzimidazole and benzothiazole conjugated Schiff base as fluorescent sensors for Al3+ and Zn2+, J. Photochem. Photobiol., A, 2019, 382, 111947, DOI:10.1016/j.jphotochem.2019.111947.
- A. Saravanan, S. Shyamsivappan, N. K. Kalagatur, T. Suresh, N. Maroli and N. Bhuvanesh, et al, Application of real sample analysis and biosensing: Synthesis of new naphthyl derived chemosensor for detection of Al3+ ions, Spectrochim. Acta, Part A, 2020, 241, 118684, DOI:10.1016/j.saa.2020.118684.
- B. Kuzu, M. Tan, Z. Ekmekci and N. Menges, A novel structure for ESIPT emission: Experimental and theoretical investigations, J. Photochem. Photobiol., A, 2019, 381, 111874, DOI:10.1016/j.jphotochem.2019.111874.
- D. Trotsek, J. Chem. Inf. Model., 2017, 110(9), 1689–1699 Search PubMed.
- A. C. Sedgwick, L. Wu, H. H. Han, S. D. Bull, X. P. He and T. D. James, et al, Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents, Chem. Soc. Rev., 2018, 47(23), 8842–8880 RSC.
- A. W. Varnes, R. B. Dodson and E. L. Wehry, Interactions of Transition-Metal Ions with Photoexcited States of Flavins. Fluorescence Quenching Studies, J. Am. Chem. Soc., 1972, 94(3), 946–950 CrossRef CAS PubMed.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Plasmon resonant enhancement of photocatalytic water splitting under visible illumination, Nano Lett., 2011, 11(3), 1111–1116 CrossRef CAS.
- R. Selvapriya, T. Abhijith, V. Ragavendran, V. Sasirekha, V. S. Reddy and J. M. Pearce, et al, Impact of coupled plasmonic effect with multishaped silver nanoparticles on efficiency of dye sensitized solar cells, J. Alloys Compd., 2022, 894, 162339, DOI:10.1016/j.jallcom.2021.162339.
- D. Peralta-Domínguez, M. Rodriguez, G. Ramos-Ortiz, J. L. Maldonado, D. Luna-Moreno and M. Ortiz-Gutierrez, et al, A Schiff base derivative used as sensor of copper through colorimetric and surface plasmon resonance techniques, Sens. Actuators, B, 2016, 225, 221–227, DOI:10.1016/j.snb.2015.11.013.
- Cdi T, Al C, Cdi T, By CC, Al C, Wikipedia E, et al., 8.5.2: Heavier Elements of Group 13 and the Inert Pair Effect, 1818, 1–9.
- T. Jurca, L. K. Hiscock, I. Korobkov, C. N. Rowley and D. S. Richeson, The tipping point of the inert pair effect: Experimental and computational comparison of In(i) and Sn(ii) bis(imino)pyridine complexes, Dalton Trans., 2014, 43(2), 690–697 RSC.
- Evaluation T, Effect IP. + 1.1516 2. 1985;(3).
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04568k |
| This journal is © The Royal Society of Chemistry 2022 |