
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting angled thin film vortex microfluidics for expeditious syntheses of iminosugars†
Jeremy Duczynski a,
Colin L. Rastonb and
Keith A. Stubbs*a
a,
Colin L. Rastonb and
Keith A. Stubbs*a
aSchool of Molecular Sciences, University of Western Australia, Crawley, WA 6009, Australia. E-mail: keith.stubbs@uwa.edu.au
bFlinders Institute for Nanoscale Science and Technology, College of Science and Engineering, Flinders University, Bedford Park, SA 5042, Australia
First published on 17th August 2022
Abstract
Iminosugars are important compounds in the area of carbohydrate-based therapeutics. A simple synthetic methodology utilizing the vortex fluidic thin film microfluidic reactor is effective in the synthesis of such compounds for diverse reaction types, with the optimal tilt angle of the reactor at 45° and the optimal rotational speed dependent on the nature of the liquid.
Introduction
An important underlying theme of organic chemistry is to develop new methodologies and techniques to access molecules of interest. New chemical efforts can arise from developing preparations of molecules with specific functional groups or chemical bonds. They can also come from innovative processing technologies as well as from incorporating these with the chemical efforts, in particular in being able to rapidly prepare target molecules. For the latter, an area of burgeoning research involves flow chemistry1 which is now commonplace in academia and industry.2 This is especially so in the utilization of microfluidic technology which is gaining attention as a method for the specific and controlled synthesis of organic molecules. The advantages provided by this technology as such are in controlling chemical reactions at various levels including varying reagent quantities and increased reaction surface area to drive the chemistry and automation.3A recently developed microfluidic technology is the thin film microfluidic vortex fluidic device (VFD) (Fig. 1A), which has been demonstrated to be a reliable platform for preparing molecules and for controlling chemical reactivity and selectivity.4,5 This device has advantages over other microfluidic platforms which use conventional-based processing where the liquid is confined within channels and typically under laminar flow conditions, and where scaling up of the reactions requires arrays of such channels.6 The VFD also has the advantage of not suffering from mechanical issues such as clogging which can occur in conventional flow chemistry using channel reactors.7 The VFD is based around the generation of a thin film of liquid in a rapidly rotating tube at a tilt angle above the horizontal position4 with the thickness of the film controlled by varying the speed of rotation of the tube (ω) and the tilt angle (θ). Reactions can be undertaken using two different methodologies, using the confined mode for a finite volume of liquid in the tube, and continuous flow mode which allows for higher volume processing where scaling up is required.4 Note that under confined mode of operation of the VFD, the liquid is flowing internally within the thin film. This arises from the induced mechanical energy, and as such there is a clear distinction between the confined mode of operation of the device and conventional batch processing.
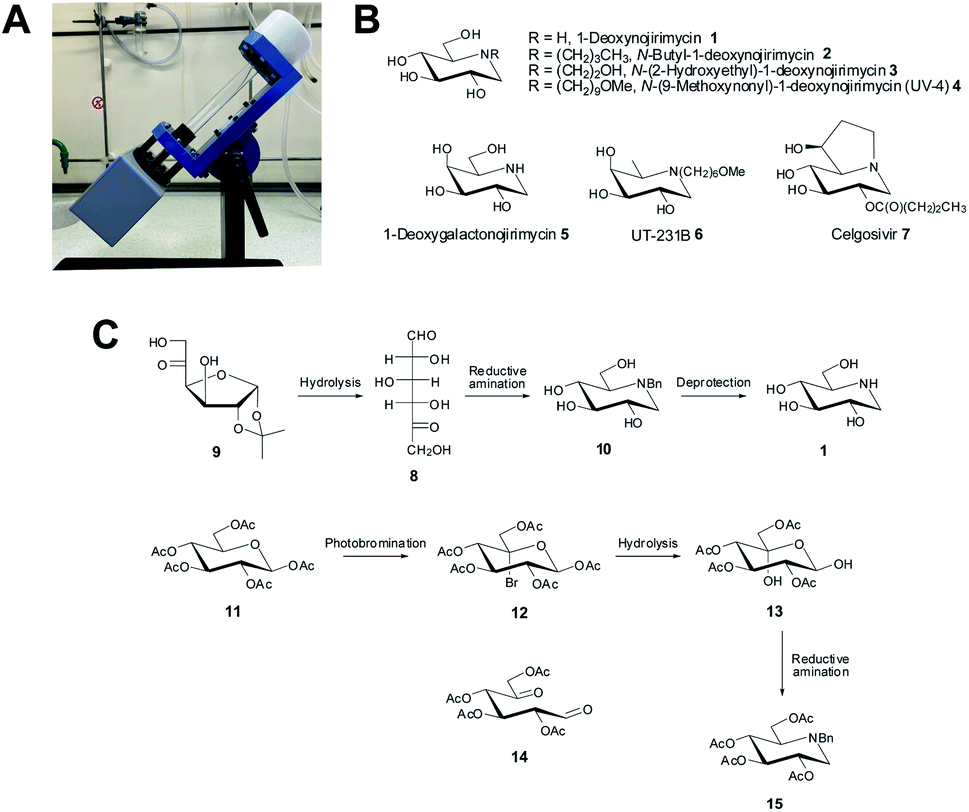 | ||
| Fig. 1 (A) Photograph of a VFD in the confined mode of operation with the device featuring a tube, being rotated at a specific angle, θ, at high speed, ω, to generate a thin film. (B) Structures of biologically and industrially relevant iminosugars. (C) Key dicarbonyl-based syntheses of iminosugars.8 | ||
One area of synthetic organic chemistry which continues to enjoy interest is in the development of strategies to prepare iminosugars, a class of carbohydrate-processing enzyme inhibitor. This class of molecule is a family of polyhydroxylated heterocycles containing an endocyclic nitrogen atom such as in 1–7 (ref. 9) (Fig. 1B), with the inhibitory activity displayed by these compounds resulting in their use as tools for inhibiting carbohydrate-processing enzymes in vitro and in vivo, as well as ligands in X-ray crystallographic studies and in the study of various disease states, which are caused by, or thought to be regulated in some way by a carbohydrate-processing enzyme.10–13 Specifically, 1-deoxynojirimycin (DNJ) 1, a naturally occurring exemplar of the iminosugars and a potent inhibitor of α-glucosidases14 (a specific class of carbohydrate-processing enzyme), has attracted interest from organic chemists in terms of developing synthetic methodologies for its preparation. In turn, these synthetic methodologies can be easily translated to other derivatives such as N-butyl-1-deoxynojirimycin 2, N-(2-hydroxyethyl)-1-deoxynojirimycin 3, N-(9-methoxynonyl)-1-deoxynojirimycin (UV-4) 4 which are examples of iminosugars that have also demonstrated clinical application.15–19 Notably 1–3 have been shown, at the cellular level, to reduce the spread of SARS-CoV-2 infection by stopping the production of virions.19 In addition UV-4 4, has been shown to also inhibit SARS-CoV-2 replication by inhibiting viral replication and also preventing SARS-CoV-2-induced cell death.18
As a result of the biological importance of iminosugars, there have been a number of syntheses published to date.9,20 Here we describe the use of the VFD in developing a microfluidic-based platform to synthesis iminosugars, using a simple yet robust synthesis, in a timely and efficient manner. A synthetic methodology that we were drawn to involves the use of 1,5-dicarbonyl compounds, which are easy to prepare, and have been shown to be a useful synthon in preparing iminosugars of various types8,21–25 and specifically the work of Baxter and Reitz who developed a synthesis of iminosugars using this approach (Fig. 1C).8 For the synthesis of 1, the desired dicarbonyl compound 8 (prepared from 9), was converted to 10 under reductive amination conditions (utilizing NaCNBH3) with subsequent removal of the protecting groups from 10, affording 1. An additional methodology explored in the study towards preparing 1 involved first a photobromination of the per-acetate 11, followed by silver-mediated hydrolysis of 12 to give the diol 13 (which is the hydrate of the desired 1,5-dicarbonyl compound 14). Disappointingly though, a very poor yield (<15%) resulted from conversion of 13 to 15, under reductive amination conditions. We hypothesised that, as the VFD has been shown to facilitate reactions, and in particular reactions involving photochemical transformations, with surprising and unusual outcomes5 that this synthetic methodology could be explored further to examine whether indeed 1 could be prepared with a greatly improved yield using this method.
Results and discussion
In the first instance, we chose to study the photobromination of 11 in the confined mode of operation of the VFD as a model reaction, given that this reaction could be directly compared to the aforementioned batch methods.26 Photobrominations of this type usually employ either N-bromosuccinimide or bromine as the bromine source.26 We felt that the use of bromine would be more amenable to use in the VFD due to the lack of solubility of N-bromosuccinimide (and the by-product of the reaction, succinimide) in the reaction solvent. Moreover, the use of elemental bromine over N-bromosuccinimide improves the percent atom economy, qualitatively, of the reaction. The confined mode of operation of the VFD was used here and below such that it is directly comparable with conventional batch processing, and to normalise all reactions including those where continuous flow mode of operation is limited for reactions requiring longer processing (residence) times than is possible using such flow.Initially, we employed a procedure using the VFD at a standard operating condition (5000 rpm, 45° tilt angle)4 in the confined mode as this has been found to be optimal in comparing the reaction to batch conditions. Gratifyingly, when employing bromine (2.4 equiv.) as by the literature26 gave the desired 5-bromo derivative 12 in a modest yield of 56% (Scheme 1), which although was lower than the yield obtained in the literature,26 the time to achieve complete consumption of 11 was reduced significantly, to only 15 minutes (from 45 minutes). With this result in hand, attention was focussed on investigating the VFD operational parameters (rotational speed and angle of inclination of the tube) as these have been shown to affect greatly the reaction yield in various reaction settings.5 Systematically, we varied these conditions for the reaction (Scheme 1) and found that optimal yields favoured medium rotational speeds and tilt angle.
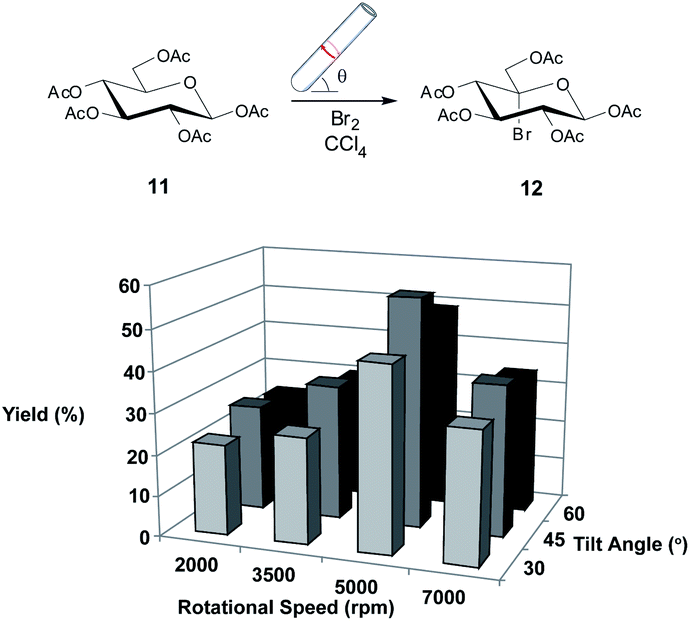 | ||
| Scheme 1 Investigating the utility of the VFD in the photobromination of 11. Variation in isolated yield for the reaction when changing the rotational speed and tilt angle of the VFD. The optimisation procedure was conducted for 15 minutes irradiation (250 W lamp at a distance of 10 cm). All reactions were conducted in triplicate with the average shown. Errors were within ±2%. | ||
We attempted to further investigate whether the amount of bromine could be reduced under these conditions as the literature procedure uses a higher stoichiometry than what is required for the reaction. Indeed, the poorer yields observed previously (due to unwanted side reactions)26 were presumably caused by the large excess of reagent used. Again gratifyingly, using optimised VFD conditions (5000 rpm, 45° tilt angle) we found that reducing the amount of bromine resulted in an increased yield (1.8 equiv., 63%; 1.2 equiv. 71%) and importantly no compromise was seen in terms of time required for conversion. This finding alone is fundamentally important in improving percent atom efficiency, qualitatively, and as discussed below this finding was a feature of other reactions studied in the VFD. With this promising result, attention was now turned to the next reaction which was a Ag(I)-mediated hydrolysis of 12 to give the diol 13.
Again, we initially employed the VFD at a standard operating condition (5000 rpm, 45° tilt angle)4 in the confined mode. Treatment of 12 with silver(I) oxide in aqueous acetone8,27 showed complete conversion of 12 after only 2 hours (reduced from 18 hours) and gratifyingly resulted in a good yield of 65% (Scheme 2). The VFD operational parameters were then varied and interestingly the best yield of 13 was obtained at 3500 rpm and 45° tilt angle (74%), which demonstrates the importance of each reaction in the VFD being systematically examined for optimal conditions. This is facilitated by the higher throughput of processing in the VFD relative to batch processing.
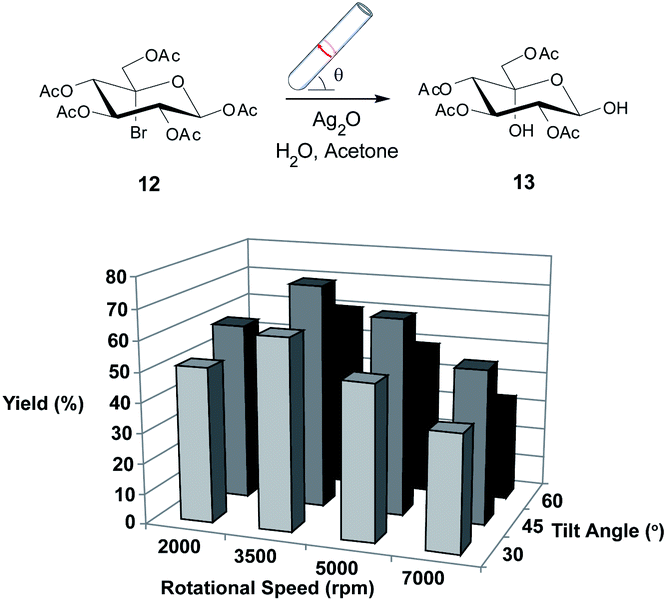 | ||
| Scheme 2 Investigating the utility of the VFD in the hydrolysis of 12. Variation in isolated yield for the reaction when changing the rotational speed and tilt angle of the VFD. The optimisation procedure was conducted for 2 hours. All reactions were conducted in triplicate with the average shown. Errors were within ±2%. | ||
With the desired synthetic intermediate in hand we now focused our attention on performing the reductive amination to prepare our desired compound DNJ, 1. We were inspired by earlier studies utilizing a Pd(0)-immobilized cellulose material that was shown to be readily prepared and easily setup within the VFD, and was efficient in the hydrogenation of various functional groups in high yields, and under ambient pressure and temperature.28 After preparing the Pd(0)-cellulose material following the published procedure28 we incubated 13 with the material in the VFD in the presence of hydrogen gas and ammonia at 5000 rpm for 60 minutes with the tube tilted at 45°.
Thereafter, to our surprise we found that there was no presence of 13 (by TLC). We presumed that the desired intermediate 16 had formed so we took the mixture and treated it with NaOMe which would facilitate the removal of the acetyl protecting groups. After purification we found that indeed DNJ 1, had been formed in a yield of 47%. VFD operational parameters were again varied and interestingly we found an improvement at the highest speed that was explored in the photobromination and hydrolysis reactions, so we extended the speed out to 9000 rpm. The best yield of 1 was obtained at 7000 rpm and 45° tilt angle (79%) (Scheme 3).
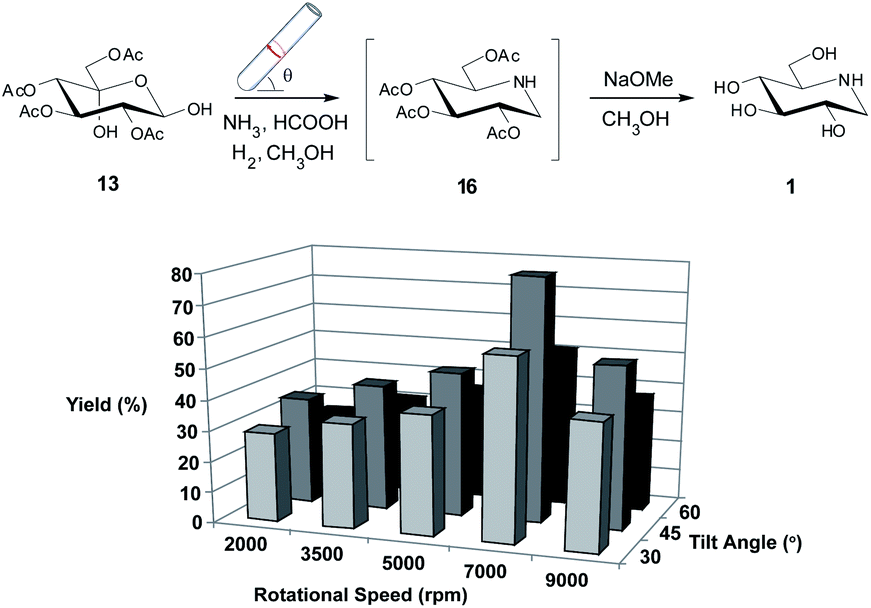 | ||
| Scheme 3 Investigating the utility of the VFD in the preparation of 1 from 13 utilizing a Pd(0)-cellulose catalyst with ammonia. Variation in isolated yield for the reaction when varying the rotational speed and tilt angle of the VFD. The optimisation procedure was conducted for 60 minutes. All reactions were conducted in triplicate with the average shown. Errors were within ±3%.‡ | ||
Overall, the yield of DNJ 1 from 11 was 41%, which is highly comparable to those obtained utilising other methods.9 We next focussed on the preparation of N-butyl-1-deoxynojirimycin 2, N-(2-hydroxyethyl)-1-deoxynojirimycin 3 as these can also be prepared from 13, and the appropriate amine. Gratifyingly, 2 and 3 were prepared from 13 in yields of 75% and 71% respectively (Scheme 4A). For N-(9-methoxynonyl)-1-deoxynojirimycin (UV-4) 4 an appropriate amine was first prepared (ESI Scheme 1†) and then upon treatment with 13 the desired compound 4 was formed in 69% yield.
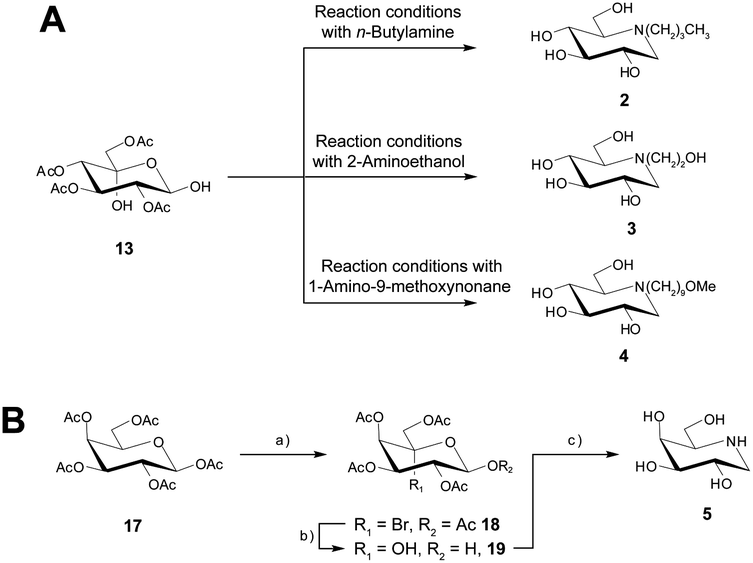 | ||
| Scheme 4 (A) Preparation of 2, 3 and 4 using the optimised VFD hydrogenation conditions. (B) Overall synthetic strategy to prepare 1-deoxygalactonojirimycin 5 employing the optimised VFD conditions for all steps. (a) Br2, CCl4 (67%); (b) Ag2O, H2O, acetone (77%); (c) (i) Pd(0)-cellulose, H2, ammonia, formic acid, CH3OH; (ii) NaOMe, CH3OH (76%). | ||
To expand the synthetic approach we decided to test our synthetic strategy and focused on the preparation of another iminosugar, 1-deoxygalactonojirimycin 5, which has shown promise as a treatment for the lysosomal storage disease, Fabry disease.29 Utilizing the appropriate starting material 17 and utilizing the optimised VFD conditions for each step we were able to prepare 5, through 18 and 19 in a good overall yield of 35% (Scheme 4B).
The optimal highly reproducible conditions for the VFD mediated reactions were established by systematically varying the rotational speed, ω, and tilt angle, θ. We note that the optimal tilt angle of 45° herein is common for a number of applications of the VFD, including those in materials processing and self-assembly.30,31 This result and the variation in the optimal rotational speed for the different reactions is consistent with recent findings on the nature of the mechanical energy induced in the thin film of liquid in the VFD.30,31 This is in the form of submicron high shear topological fluid flows in the 20 mm diameter quartz tube housed in the device and used herein, namely a spinning top (typhoon-like) arising from the Coriolis force from the hemispherical base of the tube, double helical flow associated with eddies from Faraday waves twisted by the side wall Coriolis force, and spicular flow as a combination of these when they are the same diameter. The variation in optimal rotational speed is also consistent with these studies,30,31 in its dependence on the properties of the liquids, which are different for each reaction studied. These topological fluid flows are the internal fluid flows in the VFD under confined mode of operation of the device, and also under continuous flow.
Conclusions
In summary, we have developed a simple synthetic methodology, utilizing the VFD thin film microfluidic platform, allowing for the synthesis of iminosugars, which are important compounds in the area of carbohydrate-based therapeutics. The use of the VFD in diverse organic reactions for gaining access to, in this case iminosugars, is noteworthy, further highlighting the potential of VFD processing in general in organic synthesis. The VFD methodology results in increased yields, in general, relative to conventional batch (flask) chemistry, and with significant reduction in waste generation and processing time. Overall, the utility of the methodology constitutes an important first test and sets the scene for its potential use in iminosugar-analogue preparation. Moreover, given the value of these compounds in the pharmaceutical and fine chemical industry, the findings reported herein can potentially aid in the development of new uses for this important class of bioactive compounds. This is especially so given the simplicity and low cost of the VFD.Experimental section
General
The design, construction and operation of the vortex fluidic device (VFD) used herein is detailed elsewhere.4,5 All starting materials were obtained commercially and used without further purification unless otherwise stated. Thin layer chromatography (TLC) was conducted on Merck Kieselgel 60 F254 plates and products were visualised under shortwave UV light (254 nm). 1H- and 13C nuclear magnetic resonance spectra were obtained using a Bruker AV400 spectrometer (400 MHz for 1H and 100 MHz for 13C), Bruker AV-500 spectrometer (500 MHz for 1H and 125 MHz for 13C) or Bruker Avance 600 spectrometer (600 MHz for 1H and 150 MHz for 13C). Solvents used for NMR were: deuteriochloroform (CDCl3) with CHCl3 (1H, δ 7.26) or CDCl3 (13C, δ 77.00) used as an internal standard, tetradeuteriomethanol (CD3OD) with CD2HOD (1H, δ 3.30) or CD3OD (13C, δ 49.00) used as an internal standard and deuterium oxide (D2O) with DHO (1H, δ 4.79) or CH3OH (13C, δ 49.50) used as an internal standard. High resolution mass spectra (HR-MS) were recorded with a Waters LCT Premier XE spectrometer, run in W-mode, using ESI or APCI ionisation methods as indicated, with MeCN/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a matrix. For 1-azido-9-methoxynonane mass spectrometry was performed on a Waters GCT Premier XE instrument coupled to an Agilent GC 7980A gas chromatograph (equipped with an Agilent DB-5 ms column, 30 m, 0.25 mm, ×df = 0.25 μm) for high-resolution electron ionization (EI). Infrared (IR) spectra were recorded on PerkinElmer Spectrum One FT-IR or PerkinElmer Spectrum One ATR-IR spectrometers with the matrix as specified.
:1) as a matrix. For 1-azido-9-methoxynonane mass spectrometry was performed on a Waters GCT Premier XE instrument coupled to an Agilent GC 7980A gas chromatograph (equipped with an Agilent DB-5 ms column, 30 m, 0.25 mm, ×df = 0.25 μm) for high-resolution electron ionization (EI). Infrared (IR) spectra were recorded on PerkinElmer Spectrum One FT-IR or PerkinElmer Spectrum One ATR-IR spectrometers with the matrix as specified.
Optimisation of reaction conditions for the photobromination of 11
A 20 mm OD quartz tube (17.5 ID), 18.5 cm in length with a hemispherical base was charged with 11 (195 mg, 0.5 mmol), bromine (2.4 equiv.) and CCl4 (1 mL). The tube was capped tightly then rotated in the VFD at 2000, 3500, 5000 or 7000 rpm with a tilt angle of 15, 45 or 60° for 15 minutes in the presence of a 250 W lamp (Osram Incandescent, 12 cm size) at a distance of 10 cm. The mixture was then cooled and washed with water, dried (MgSO4), filtered and concentrated. The residue was then subjected to flash chromatography (EtOAc:hexanes, 1:2) to give the desired compound 12.
1,2,3,4,6-Penta-O-acetyl-5-bromo-β-D-glucopyranose 12
Using 11, the title compound was obtained (130 mg, 56%). 1H and 13C NMR spectra were consistent with literature values.8 Rf 0.62 (1:1, EtOAc:hexanes); 1H NMR (600 MHz, CDCl3) δ 6.22 (d, J = 8.7 Hz, 1H), 5.56 (dd, J = 9.7 Hz, 1H), 5.26 (dd, J = 8.7, 9.7 Hz, 1H), 5.23 (d, J = 9.7 Hz, 1H), 4.57 (d, J = 12.3 Hz, 1H), 4.31 (d, J = 12.3 Hz, 1H), 2.12 (s, 3H), 2.10 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.00 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 169.9, 169.7, 169.4, 169.2, 168.4, 96.0, 91.5, 71.2, 69.6, 68.3, 65.8, 20.8, 20.7, 20.63, 20.6.
1,2,3,4,6-Penta-O-acetyl-5-bromo-β-D-galactopyranose 18
Using 17 (195 mg, 0.5 mmol) and the optimised conditions gave the title compound as a colourless oil (157 mg, 67%). Rf 0.66 (1:1, EtOAc:hexanes); 1H NMR (600 MHz, CDCl3) δ 6.21 (d, J = 8.5 Hz, 1H), 5.78 (dd, J = 3.3, 10.5 Hz, 1H), 5.26 (d, J = 3.3 Hz, 1H), 5.39 (dd, J = 8.6, 10.5 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H), 4.35 (d, J = 12.0 Hz, 1H), 2.12 (s, 6H), 2.06 (s, 3H), 2.05 (s, 3H), 1.97 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 169.8, 169.6, 169.5, 169.2, 168.5, 93.8, 91.5, 70.3, 68.5, 67.3, 66.3, 20.75, 20.7, 20.6, 20.5. HR-MS (APCI): m/z [M + H]+ calcd for C16H2279BrO11: 469.0345, found: 469.0350.
Optimisation of reaction conditions for the hydrolysis of 12
A 20 mm OD quartz tube (17.5 ID), 18.5 cm in length with a hemispherical base was charged with 12 (234 mg, 0.5 mmol), silver(I) oxide (2.0 equiv.) and H2O:acetone (1:5, 1 mL, v/v). The tube was capped tightly then rotated in the VFD at 2000, 3500, 5000 or 7000 rpm with a tilt angle of 15, 45 or 60° for 2 hours. The mixture was then filtered (Celite) and the residue washed with CH2Cl2 (3 × 10 mL). The combined materials were then dried (MgSO4), filtered and concentrated. The residue was then subjected to flash chromatography (EtOAc:hexanes, 3:2) to give the desired compound 13.
2,3,4,6-Tetra-O-acetyl-5-hydroxy-β-D-glucopyranose 13
Using 12, the title compound was obtained (134 mg, 74%). 1H and 13C NMR spectra were consistent with literature values.8 Rf 0.39 (7:3, EtOAc:hexanes); 1H NMR (600 MHz, CDCl3, major isomer) δ 5.52 (dd, J = 9.6 Hz, 1H), 5.26 (d, J = 8.4 Hz, 1H), 5.18 (d, J = 9.6 Hz, 1H), 4.96 (dd, J = 8.4, 9.6 Hz, 1H), 4.43 (br s, 1H), 4.15 (d, J = 12.0 Hz, 1H), 4.02 (d, J = 12.0 Hz, 1H), 2.12 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.00 (s, 3H); 13C NMR (150 MHz, CDCl3, major isomer) δ 171.3, 170.9, 170.2, 169.9, 95.4, 91.0, 73.5, 70.2, 69.8, 65.5, 20.9, 20.7.
2,3,4,6-Tetra-O-acetyl-5-hydroxy-β-D-galactopyranose 19
Using 18, and the optimised conditions gave the title compound as a colourless oil (125 mg, 69%). Rf 0.32 (3:2, EtOAc:hexanes); 1H NMR (600 MHz, CDCl3, major isomer) δ 5.46 (dd, J = 3.4, 10.5 Hz, 1H), 5.37 (d, J = 3.4 Hz, 1H), 5.21 (dd, J = 8.2 Hz, 1H), 5.12 (dd, J = 8.2, 10.5 Hz, 1H), 4.30 (s, 1H), 4.23 (d, J = 12.0 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 3.97 (d, J = 8.2 Hz, 1H), 2.16 (s, 3H), 2.10 (s, 6H), 1.98 (s, 3H); 13C NMR (150 MHz, CDCl3, major isomer) δ 171.6, 171.2, 170.2, 170.0, 96.6, 92.0, 71.1, 68.6, 68.5, 65.3, 21.0, 20.9, 20.8. HR-MS (APCI): m/z [M–H2O + H]+ calcd for C14H19O10: 347.0978, found: 347.0975.
Optimisation of reaction conditions for the reductive amination of 13
A 20 mm OD quartz tube (17.5 ID), 18.5 cm in length with a hemispherical base was charged with 13 (180 mg, 0.5 mmol), which was first co-evaporated with toluene, ammonia (from 2 M solution in MeOH, 1.0 equiv.), formic acid (1.0 equiv.), MeOH (15, 1 mL, v/v), and the Pd(0)-immobilized cellulose.28 The tube was capped tightly and put under vacuum and back-filled with hydrogen. The tube was then rotated in the VFD at 2000, 3500, 5000, 7000 or 9000 rpm with a tilt angle of 15, 45 or 60° for 60 minutes. The mixture was then removed and the Pd(0)-immobilized cellulose washed with MeOH (3 × 3 mL). The combined materials were then treated with 1 M NaOMe (1 mL) at room temperature for 1 hour. At this time the mixture was neutralized with resin (Dowex 50WX4) and the mixture filtered. The resin was further washed with methanol and finally 1 M aq. ammonia. The organic and aqueous materials were then concentrated independently and then combined and the resultant residue was then subjected to flash chromatography (CHCl3/MeOH/conc. NH3, 10:10:1, v/v/v), to give the desired compound 1.
1-Deoxynojirimycin 1
Using 13, the title compound was obtained (64 mg, 79%). 1H and 13C NMR spectra were consistent with literature values.32 Rf 0.23 (CHCl3/MeOH/conc. NH3, 10:10:1, v/v/v); 1H NMR (500 MHz, D2O) δ 3.83 (dd, J = 3.0, 11.7 Hz, 1H), 3.63 (dd, J = 6.3, 11.7 Hz, 1H), 3.51–3.46 (m, 1H), 3.31 (dd, J = 9.1 Hz, 1H), 3.23 (dd, J = 9.7 Hz, 1H), 3.12 (dd, J = 5.3, 12.4 Hz, 1H), 2.57–2.53 (m, 1H), 2.46 (dd, J = 10.9, 12.3 Hz, 1H); 13C NMR (125 MHz, D2O) δ 81.1, 74.2, 73.6, 64.1, 63.2, 51.4.
N-Butyl-1-deoxynojirimycin 2
Using 13, 1-aminobutane, the optimised conditions and flash chromatography (CHCl3/MeOH/conc. NH3, 12:8:1, v/v/v), the title compound was obtained (82 mg, 75%). 1H and 13C NMR spectra were consistent with literature values.32 Rf 0.51 (CHCl3/MeOH/conc. NH3, 10:10:1, v/v/v); 1H NMR (500 MHz, D2O) δ 3.89 (dd, J = 2.4, 12.9 Hz, 1H), 3.81 (dd, J = 2.7, 12.9 Hz, 1H), 3.55–3.50 (m, 1H), 3.36 (dd, J = 9.4 Hz, 1H), 3.24 (dd, J = 9.4 Hz, 1H), 3.01 (dd, J = 4.9, 11.4 Hz, 1H), 2.76–2.70 (m, 1H), 2.62–2.56 (m, 1H), 2.28 (dd, J = 11.0 Hz, 1H), 2.24–2.21 (m, 1H), 1.49–1.40 (m, 2H), 1.30–1.22 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, D2O) δ 81.0, 72.7, 71.5, 67.8, 60.2, 57.9, 54.5, 27.7, 22.8, 15.9.
N-(2-Hydroxyethyl)-1-deoxynojirimycin 3
Using 13, 2-aminoethanol and the optimised conditions the title compound was obtained (73 mg, 71%). 1H and 13C NMR spectra were consistent with literature values.32 Rf 0.26 (CHCl3/MeOH/conc. NH3, 10:10:1, v/v/v); 1H NMR (600 MHz, D2O) δ 3.94 (dd, J = 2.6, 12.9 Hz, 1H), 3.83 (dd, J = 3.0, 12.9 Hz, 1H), 3.78–3.71 (m, 2H), 3.57–3.52 (m, 1H), 3.37 (dd, J = 9.4 Hz, 1H), 3.27 (dd, J = 9.1 Hz, 1H), 3.09 (dd, J = 4.8, 11.8 Hz, 1H), 2.96 (dt, J = 6.4, 13.2 Hz, 1H), 2.73 (dt, J = 5.4, 13.8 Hz, 1H), 2.37 (dd, J = 11.2 Hz, 1H), 2.33 ppm (dt, J = 2.8, 9.8 Hz, 1H); 13C NMR (150 MHz, D2O) δ 78.9, 70.6, 69.3, 66.2, 58.5, 58.2, 56.7, 53.4.
1-Deoxygalactonojirimycin 5
Using 19, ammonia and the optimised conditions the title compound was obtained (62 mg, 76%). 1H and 13C NMR spectra were consistent with literature values.33 Rf 0.20 (CHCl3/MeOH/conc. NH3, 10:10:1, v/v/v); 1H NMR (500 MHz, D2O) δ 3.99 (dd, J = 1.5, 3.3 Hz, 1H), 3.76–3.71 (m, 1H), 3.63 (dd, J = 6.6, 11.2 Hz, 1H), 3.59 (dd, J = 6.7, 11.2 Hz, 1H), 3.46 (dd, J = 3.2, 9.7 Hz, 1H), 3.11 (dd, J = 5.4, 12.7 Hz, 1H), 2.75–2.72 (m, 1H), 2.37 (dd, J = 10.8, 12.7 Hz, 1H); 13C NMR (125 MHz, D2O) δ 75.7, 69.9, 68.8, 62.0, 59.4, 49.7.
N-(9-Methoxynonyl)-1-deoxynojirimycin (UV-4) 4
Using 13, 1-amino-9-methoxynonane, the optimised conditions and flash chromatography (1:9, MeOH:CHCl3) the title compound was obtained (110 mg, 69%). Rf 0.50 (2:3, MeOH:CHCl3); 1H NMR (600 MHz, CD3OD) δ 3.85 (dd, J = 2.7, 11.9 Hz, 1H), 3.82 (dd, J = 2.9, 11.9 Hz, 1H), 3.49–3.43 (m, 1H), 3.37 (dd, J = 6.4 Hz, 2H), 3.34 (dd, J = 9.4 Hz, 1H), 3.30 (s, 3H), 3.11 (dd, J = 8.8 Hz, 1H), 2.97 (dd, J = 4.9, 11.4 Hz, 1H), 2.81–2.76 (m, 1H), 2.59–2.53 (m, 1H), 2.16 (dd, J = 10.9 Hz, 1H), 2.10 ppm (dt, J = 2.5, 9.4 Hz, 1H), 1.57–1.45 (m, 4H), 1.36–1.25, (m, 10H); 13C NMR (150 MHz, CD3OD) δ 80.6, 73.9, 72.0, 70.7, 67.4, 59.4, 58.7, 57.7, 53.8, 30.7, 30.6, 30.5, 28.6, 27.2, 25.2. FT-IR (ATR): ν = 3361 (s), 2926 (s) cm−1; HR-MS (ESI): m/z [M + Na]+ calcd for C16H33NNaO5: 342.2256, found: 342.2249.
1-Azido-9-methoxynonane
To a solution of 1-iodo-9-methoxynonane34 (210 mg, 0.74 mmol) in acetone:H2O (3:1, 4 mL, v/v) was added sodium azide (144 mg, 0.22 mmol) and the resultant mixture was stirred (60 °C, 4 h). The mixture was then cooled and diluted with EtOAc, and washed with H2O, brine, dried (MgSO4), filtered and concentrated. The residue was then subjected to flash chromatography (EtOAc:hexanes, 1:19) to give 1-azido-9-methoxynonane as a colourless oil (130 mg, 88%). Rf 0.68 (1:4, EtOAc:hexanes); 1H NMR (400 MHz, CDCl3) δ 3.36 (dd, J = 6.5 Hz, 2H), 3.32 (s, 3H), 3.25 (dd, J = 7.1 Hz, 2H), 1.61–1.52 (m, 4H), 1.39–1.27 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 73.1, 58.7, 51.6, 29.8, 29.53, 29.5, 29.2, 29.0, 26.8, 26.2; FT-IR (ATR): ν = 2926 (s), 2091 (s) cm−1; HR-MS (EI): m/z [M]+ calcd for C10H21N3O: 199.1685, found: 199.1681.
1-Amino-9-methoxynonane
To a solution of 1-azido-9-methoxynonane (125 mg, 0.63 mmol) in EtOH (4 mL) was added Pd(OH)2/C (20%, 10 mg) and the resultant mixture stirred under an atmosphere of H2 gas (2 h). The mixture was then filtered (Celite) and concentrated. The residue was then subjected to flash chromatography (1:9, MeOH:CHCl3) to give 1-amino-9-methoxynonane as a colourless oil (100 mg, 92%). Rf 0.21 (1:9, MeOH:CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.51 (br s, 2H), 3.35 (dd, J = 6.2 Hz, 2H), 3.32 (s, 3H), 2.76 (dd, J = 7.3 Hz, 2H), 1.58–1.49 (m, 4H), 1.35–1.26 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 73.1, 58.7, 41.6, 31.9, 29.8, 29.6, 29.5, 29.4, 26.9, 26.2; FT-IR (ATR): ν = 3250 (w), 2923 (s) cm−1; HR-MS (ESI): m/z [M + H]+ calcd for C10H24NO: 174.1858, found: 174.1858.
Author contributions
J. D. and K. A. S. conducted experiments. All authors reviewed data. K. A. S. and C. L. R. directed the project. All authors contributed to the writing of the manuscript and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Centre for Microscopy, Characterisation and Analysis at The University of Western Australia, which is supported by University, State and Federal Government funding. C. L. R. thanks Flinders University, the Government of South Australia, and both K. A. S. and C. L. R. also thank the Australian Research Council for funding (DP200101016).Notes and references
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC.
- G. Gambacorta, J. S. Sharley and I. R. Baxendale, Beilstein J. Org. Chem., 2021, 17, 1181–1312 CrossRef CAS PubMed.
- N. Convery and N. Gadegaard, Micro Nano Eng., 2019, 2, 76–91 CrossRef.
- L. Yasmin, X. Chen, K. A. Stubbs and C. L. Raston, Sci. Rep., 2013, 3, 2282 CrossRef PubMed.
- J. Britton, K. A. Stubbs, G. A. Weiss and C. L. Raston, Chem.–Eur. J., 2017, 23, 13270–13278 CrossRef CAS PubMed.
- C. Lee, C. Chang, Y. Wang and L. Fu, Int. J. Mol. Sci., 2011, 12, 3263–3287 CrossRef CAS PubMed.
- E. Dressaire and A. Sauret, Soft Matter, 2017, 13, 37–48 RSC.
- E. W. Baxter and A. B. Reitz, J. Org. Chem., 1994, 59, 3175–3185 CrossRef CAS.
- Iminosugars: From Synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, John Wiley & Sons, Ltd., Chichester, 2007 Search PubMed.
- I. Conforti and A. Marra, Org. Biomol. Chem., 2021, 19, 5439–5475 RSC.
- F. Hossain and P. R. Andreana, Pharmaceuticals, 2019, 12, 84 CrossRef CAS PubMed.
- D. S. Alonzi, K. A. Scott, R. A. Dwek and N. Zitzmann, Biochem. Soc. Trans., 2017, 45, 571–582 CrossRef CAS PubMed.
- R. J. Nash, A. Kato, C. Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed.
- N. Asano, R. J. Nash, R. J. Molyneaux and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1645 CrossRef CAS.
- P. H. Joubert, G. N. Foukaridis and M. L. Bopape, Eur. J. Clin. Pharmacol., 1987, 31, 723–724 CrossRef CAS PubMed.
- P. H. Joubert, H. L. Venter and G. N. Foukaridis, Br. J. Clin. Pharmacol., 1990, 30, 391 CrossRef CAS PubMed.
- H. E. Lebovitz, Diabetes Rev., 1998, 6, 132 Search PubMed.
- E. C. Clarke, R. A. Nofchissey, C. Ye and S. B. Bradfute, Glycobiology, 2021, 31, 378–384 CrossRef CAS PubMed.
- A. Casas-Sanchez, A. Romero-Ramirez, E. Hargreaves, C. C. Ellis, B. I. Grajeda, I. L. Estevao, E. I. Patterson, G. L. Hughes, I. C. Almeida, T. Zech and A. Acosta-Serrano, mBio, 2022, 13, e0371821 CrossRef PubMed.
- D. Dhara, A. Dhara, J. Bennett and P. V. Murphy, Chem. Rec., 2021, 21, 2958–2979 CrossRef CAS PubMed.
- A. J. Steiner, A. E. Stutz, C. A. Tarling, S. G. Withers and T. M. Wrodnigg, Carbohydr. Res., 2007, 342, 1850–1858 CrossRef CAS PubMed.
- A. J. Steiner, G. Schitter, A. E. Stutz, T. M. Wrodnigg, C. A. Tarling, S. G. Withers, K. Fantur, D. J. Mahuran, E. Paschke and M. J. Tropak, Bioorg. Med. Chem., 2008, 16, 10216–10220 CrossRef CAS PubMed.
- A. J. Steiner, G. Schitter, A. E. Stutz, T. M. Wrondigg, C. A. Tarling, S. G. Withers, D. J. Mahuran and M. J. Tropak, Tetrahedron: Asymmetry, 2009, 20, 832–835 CrossRef CAS PubMed.
- K. A. Stubbs, J. P. Bacik, G. E. Perley-Robertson, G. E. Whitworth, T. M. Gloster, D. J. Vocadlo and B. L. Mark, ChemBioChem, 2013, 14, 1973–1981 CrossRef CAS PubMed.
- S. S. Wills, C. L. Raston and K. A. Stubbs, Bioorg. Med. Chem. Lett., 2018, 28, 3748–3751 CrossRef CAS PubMed.
- R. Blattner and R. J. Ferrier, J. Chem. Soc., Perkin Trans. 1, 1980, 1523 RSC.
- R. J. Ferrier and P. C. Tyler, J. Chem. Soc., Perkin Trans. 1, 1980, 1528–1534 RSC.
- J. M. Phillips, M. Ahamed, X. Duan, R. N. Lamb, X. Qu, K. Zheng, J. Zou, J. M. Chalker and C. L. Raston, ACS Appl. Bio Mater., 2019, 2, 488–494 CrossRef CAS PubMed.
- D. A. Hughes, K. Nicholls, S. P. Shankar, G. Sunder-Plassmann, D. Koeller, K. Nedd, G. Vockley, T. Hamazaki, R. Lachmann, T. Ohashi, I. Olivotto, N. Sakai, P. Deegan, D. Dimmock, F. Eyskens, D. P. Germain, O. Goker-Alpan, E. Hachulla, A. Jovanovic, C. M. Lourenco, I. Narita, M. Thomas, W. R. Wilcox, D. G. Bichet, R. Schiffmann, E. Ludington, C. Viereck, J. Kirk, J. Yu, F. Johnson, P. Boudes, E. R. Benjamin, D. J. Lockhart, C. Barlow, N. Skuban, J. P. Castelli, J. Barth and U. Feldt-Rasmussen, J. Med. Genet., 2017, 54, 288–296 CrossRef CAS PubMed.
- T. M. D. Alharbi, M. Jellicoe, X. Luo, K. Vimalanathan, I. K. Alsulami, B. S. Al Harbi, A. Igder, F. A. J. Alrashaidi, X. Chen, K. A. Stubbs, J. M. Chalker, W. Zhang, R. A. Boulos, D. B. Jones, J. S. Quinton and C. L. Raston, Nanoscale Adv., 2021, 3, 3064–3075 RSC.
- M. Jellicoe, A. Igder, C. Chuah, D. B. Jones, X. Luo, K. A. Stubbs, E. M. Crawley, S. J. Pye, N. Joseph, K. Vimalanathan, Z. Gardner, D. P. Harvey, X. Chen, F. Salvemini, S. He, W. Zhang, J. M. Chalker, J. S. Quinton, Y. Tang and C. L. Raston, Chem. Sci., 2022, 13, 3375–3385 RSC.
- A. L. Concia, C. Lozano, J. A. Castillo, T. Parella, J. Joglar and P. Clapes, Chem.–Eur. J., 2009, 15, 3808–3816 CrossRef CAS PubMed.
- H. Takahata, Y. Banba, H. Ouchi and H. Nemoto, Org. Lett., 2003, 5, 2527–2529 CrossRef CAS PubMed.
- S. Mohan, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 342, 901–912 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04409a |
| ‡ The yield of the reaction can be directly related to the formation of 16 as the deprotection of 16 by NaOMe, would be the same in each case. |
| This journal is © The Royal Society of Chemistry 2022 |