
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoles of cysteine in the structure and metabolic function of Mycobacterium tuberculosis CYP142A1†
Yun Lua,
Lilan Suna,
Jing Panga,
Congran Lia,
Xiukun Wanga,
Xinxin Hua,
Guoqing Lia,
Xue Lia,
Youwen Zhanga,
Hao Wangb,
Xinyi Yang‡
*a and
Xuefu You‡ *a
*a
aBeijing Key Laboratory of Antimicrobial Agents, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. E-mail: yangxinyi1976@hotmail.com; xuefuyou@hotmail.com
bSchool of Pharmacy, Minzu University of China, Beijing, China
First published on 30th August 2022
Abstract
CYP142A1 is a cytochrome P450 (CYP) enzyme expressed in Mycobacterium tuberculosis (Mtb), which supports the growth of Mtb H37Rv relying on cholesterol, in the absence of CYP125A1. Since cysteine residues usually play a fundamental role in maintaining the structure and function of CYP enzymes, in this study, we aimed to determine the potential biochemical functions of six cysteine residues except for the heme-binding cysteine in the amino acid sequence of recombinant Mtb CYP142A1 by replacing each one using site-directed mutagenesis. Recombinant CYP142A1 mutants were heterologously expressed, purified, and analyzed using ESI-MS, far-UV CD spectroscopy, UV-vis spectrophotometric titration, and metabolic function assays. Substitution of the cysteine residues caused various effects on the structure and function of CYP142A1. Separate substitution of the six cysteine residues resulted in numerous changes in the secondary structure, expression level, substrate-binding ability, inhibitor-binding ability, thermal stability and oxidation efficiency of the enzyme. These results contribute to our understanding of the biochemical roles of cysteine residues in the structure and function of Mtb CYP enzymes, especially their effects on the structure and function of CYP142A1.
Introduction
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb), which kills millions of patients every year. According to the global tuberculosis report published by the World Health Organization (WHO) in 2020, an estimated 10 million people were infected with TB globally in 2019, making it one of the top ten causes of death worldwide.1 The widespread occurrence of multidrug- or rifampicin-resistant TB (MDR/RR-TB) has exacerbated this situation owing to decreased effectiveness of the traditional front-line anti-TB drugs, which include rifampicin, isoniazid, streptomycin and ethambutol.2,3 Therefore, novel anti-TB drugs with unique mechanisms of action and new targets are now the main strategies required to overcome increasing resistance to anti-TB drugs.Cytochrome P450 (CYP) enzymes belong to the monooxygenase superfamily of possessing heme iron, which are widely distributed in various life forms and play an essential role in biocatalytic reactions. Genomic sequencing has shown that Mtb H37Rv encodes 20 CYP enzymes over the entire length of the chromosomal DNA comprising 4.4 million base pairs, whereas some bacteria such as Escherichia coli (E. coli) lack CYP.4,5 The dense distribution of coding genes in the genome and their susceptibility to azole drugs suggest that CYPs are promising targets for the development of anti-mycobacterial drugs. Subsequently, some researchers studied the physiological functions of Mtb CYPs and found that they play critical roles in diverse biochemical processes of the pathogen, including lipid metabolism and synthesis, cholesterol utilization and the electron transport chain (ETC).6 Mtb abundantly synthesizes complex lipids that help this bacterium resist various chemicals, stimulate the host immune response during pathogenesis, and survive in the harsh environment of macrophages for long periods. The cell wall lipids of Mtb are unusually complex and genome sequencing predicted that at least 250 genes of Mtb are involved in processes related to lipid synthesis and metabolism. Cholesterol is widely distributed in animals, not only as a cell membrane component, but also as an important anabolic precursor of biosynthesized bile acids, vitamin D, and steroid hormones. Although cholesterol is mainly synthesized in animal cells, the cell membranes of plants and fungi also contain small amounts of cholesterol. However, the biosynthesis of sterols in bacteria has remained controversial because it has been confirmed only in Methylococcus capsulatus and Gemmata obscuriglobus; Mtb lacks the squalene monooxygenase and epoxy squalene cyclase necessary for sterol biosynthesis.7 Therefore, like most bacteria, Mtb can only obtain cholesterol through host cells if necessary. Studies on Mtb survival inside macrophages and mouse models of infection with defective mutants have revealed a series of genes related to cholesterol uptake and metabolism; by activation of the corresponding genes and their translation products, Mtb can obtain carbon and energy sources by degrading the cholesteric ring and side chains of cholesterol in the dormant, persistent or chronic infection stage of the host.8–14 Some studies have also shown that high levels of cholesterol in foods up-taken by the host can greatly increase the Mtb load in the lungs and damage the immune system,15,16 and the level of cholesterol in the host lesion tissue is positively correlated with the Mtb infection to some extent.17 Inhibition of cholesterol metabolism can cause 4-cholesten-3-one accumulation, leading to cell death and bacteriostasis.7 These laboratory data show that cholesterol plays an important role in Mtb at various stages of infection and in dormant retention. Among the Mtb CYP enzyme systems, CYP125A1, CYP124A1 and CYP142A1 have been reported to be involved in cholesterol metabolism. CYP125A1 participates in cholesterol metabolism in Mtb CDC1551 and acts as a steroid C26 monooxygenase. In the absence of CYP125A1, CYP142A1, rather than CYP124A1,18 supplemented the cholesterol catabolism function of CYP125A1 to maintain Mtb H37Rv growth. Both CYP142A1 and CYP125A1, which are considered as promising drug targets,19 oxidize cholesterol side chains and use cholesterol to provide energy for pathogens through redox reactions. CYP142A1 can oxidize cholesterol propionate in addition to cholest-4-en-3-one and cholesterol. However, CYP125A1 cannot oxidize cholesterol propionate and has lesser ability to oxidize cholesterol sulfate than CYP142A1. Perhaps the unique three-dimensional structure of CYP142A1 confers the ability to oxidize cholesterol esters, providing an additional carbon source for the growth of Mtb under specific conditions.20 Given the importance of CYP142A1 in Mtb cholesterol metabolism, it might be considered as a potential target for screening anti-TB drugs.
Cysteine residues play an important role in maintaining the basic structure and enzymatic function of CYPs. First, the thiol group of cysteine residues is very active and easily oxidized to form disulfide bonds, which are necessary for stabilizing the secondary and tertiary structures of a protein.21 Second, a heme–thiolate complex, the catalytic active center for CYP enzymes, is composed of a Fe(III) heme prosthetic group and a cysteine thiol group, through which the bound molecular oxygen is reduced in CYP450-catalyzed redox reaction. When the cysteine residues constituting the active central axis are mutated to other amino acid residues, the redox state of the active center of the enzyme changes, consequently affecting the state of Fe(III)/Fe(II) complexes and the progress of redox reactions.22 In addition, in some proteins containing heme–thiolate complex other than CYPs, cysteine residues work as heme sensing sites to help control the aggregation and disintegration of the heme complex. Moreover, structurally important hydrogen bonds and activity regulation via posttranslational modification are also related to cysteine in some specific proteins such as membrane proteins on the Golgi apparatus.23 In summary, the contribution and role of cysteine residues in the structural stability and/or enzymatic activity of Mtb CYP deserves a comprehensive study. The amino acid sequence of the CYP142A1 monooxygenase contains seven cysteine residues. One is thought to reside in the active center, while six might have regulatory effects on enzymatic structure and activity, but their specific functions need to be further explored through experimental methods such as mutations. Therefore, we conducted a systematic functional study of the six cysteine residues except for the heme binding one using mutational analysis. To the best of our knowledge, this is the first description of roles that cysteine residues play in the regulation of the structure and function of Mtb CYP142A1.
Experimental
Bacterial strains, plasmids and DNA isolation
Mtb H37Rv genomic DNA was obtained from the Department of Pharmacology, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing Chest Hospital. The expression plasmid pET30a for transformation into E. coli BL21 DE3 (TransGen Biotech, Beijing, China) was amplified in E. coli DH5α (TransGen Biotech). E. coli strains were cultured in Luria–Bertani medium (Affymetrix, Cleveland, OH, USA) with appropriate antibiotics. Plasmid DNA was isolated using EasyPure HiPure Plasmid MiniPrep Kits (TransGen Biotech). Fragments of DNA generated by PCR were extracted from agarose gels using EasyPure Quick Gel Extraction Kits (TransGen Biotech).Chemicals
All solvents and reagents were of the highest purity commercially available, unless noted otherwise. Isopropyl-β-D-thiogalactopyranoside (IPTG), acrylamide, N,N′-methylenebisacrylamide, ammonium persulfate, 30% acrylamide, TEMED, glycine, Tris, DTT, ampicillin sodium, kanamycin sulfate, chloramphenicol, hydroxypropyl NADH, NADPH, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, catalase and ammonium sulfate were all purchased from Sangon Biotech (Shanghai, China). Cholesterol, cholesterol propionate, NADPH, 5-aminolevulinic acid, spinach ferredoxin (spFDX) and spinach ferredoxin reductase (spFDR) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 4-Cholesten-3-one and methyl-β-cyclodextrin were purchased from J&K Scientific Ltd. (Beijing, China). Taq DNA polymerase, PrimeSTAR HS DNA Polymerase (with GC buffer), T4 DNA ligase, restriction endonucleases Nde I and Xho I, T-A cloning kits and DNA MW markers were purchased from Takara (Dalian, China). Cholesterol sodium sulfate was purchased from Ark Pharm Inc. (Libertyville, IL, USA).Cloning and site-directed mutagenesis of CYP142A1
The CYP142A1 gene (Rv3518c) was amplified by PCR from Mtb H37Rv genome DNA (NC_000962.2) using PrimeSTAR® HS DNA Polymerase with GC buffer (Takara Bio Inc., Kusatsu, Japan). The oligonucleotide PCR primers were designed according to the Mtb genomic sequence (ESI Table S2†). The forward primer contained an Nde I site permitting cloning into the pET30a, and the reverse primer contained a Xho I site. The amplification conditions were 98 °C for 3 min, then 30 cycles at 98 °C for 15 s, 56 °C for 15 s and 72 °C for 20 s, then 72 °C for 10 min. Amplicons were cloned into a similarly restricted pET30a vector and expressed via a T7 promoter under IPTG induction to produce a recombinant CYP protein with a C-terminal 6-His tag. The recombinant plasmid was then transformed into E. coli BL21 (DE3), and kanamycin-resistant colonies containing cloned cyp142, were confirmed by PCR and sequencing. A positive clone was selected for plasmid extraction, and cysteine-to-serine mutants were created using a Fast MultiSite Mutagenesis System (Transgen Biotech, Beijing, China). The mutations were verified using DNA sequencing.Expression and purification of recombinant proteins
Recombinant CYP142A1, C110S, C118S, C123S, C281S, C296S, C316S and C340T proteins were heterologously expressed in the E. coli BL21 (DE3) strain (Takara) as described.24 Transformed E. coli BL21 (DE3) cells containing wild-type or cysteine mutants of pET30a-cyp142 were incubated in LB medium supplemented with 50 μg mL−1 of kanamycin at 37 °C until the OD600 reached 0.6. Protein expression was induced by shaking the cells with 0.1 mM IPTG with 0.5 mM 5-aminolevulinic acid at 150 rpm for 30 h at 16 °C. Pelleted cells harvested by centrifugation at 4000×g (10 min, 4 °C) were washed twice with 0.85% NaCl, resuspended in binding buffer (200 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH 7.4), then again sedimented by centrifugation at 4000×g (10 min, 4 °C). The cells were resuspended in phosphate buffer (pH 7.4) and sonicated on ice (4 s rest/2 s sonication, 38%). Cell debris was removed and the sonicates were subsequently separated by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 30 min at 4 °C. The supernatants containing crude enzyme extracts were passed through a 0.22 μm filter (Merck KGaA, Darmstadt, Germany) for subsequent purification. The filtrates were loaded onto a HisTrap™ HP column (GE Healthcare, Madison WI, USA) using an AKTA purification system (GE Healthcare), and fractions were eluted with a linear gradient of 20∼500 mM imidazole. Fractions containing relatively pure samples were pooled, desalinated and concentrated to ≤500 μL using a 10 kDa ultrafiltration tube (Merck KGaA) at 4 °C. Proteins were then fractionated by molecular size exclusion chromatography using Superdex 75 10/300 GL. Fractions were concentrated in 50 mM Tris-Cl with 30% glycerin (pH 7.4) by ultrafiltration.
000×g for 30 min at 4 °C. The supernatants containing crude enzyme extracts were passed through a 0.22 μm filter (Merck KGaA, Darmstadt, Germany) for subsequent purification. The filtrates were loaded onto a HisTrap™ HP column (GE Healthcare, Madison WI, USA) using an AKTA purification system (GE Healthcare), and fractions were eluted with a linear gradient of 20∼500 mM imidazole. Fractions containing relatively pure samples were pooled, desalinated and concentrated to ≤500 μL using a 10 kDa ultrafiltration tube (Merck KGaA) at 4 °C. Proteins were then fractionated by molecular size exclusion chromatography using Superdex 75 10/300 GL. Fractions were concentrated in 50 mM Tris-Cl with 30% glycerin (pH 7.4) by ultrafiltration.
Evaluation of molecular weight
Purified proteins (2 μM) were injected directly via an injection pump at a rate of 6 μL min−1 into an Orbitrap Fusion Lumos mass spectrometer with an ESI ion source. The experimental parameters comprised: Intect Protein mode, MS Scan, Orbitrap detector, resolution 30000, scan range 150–2000 m/z, 60% RF lens and 10 microscans. Raw data were analyzed by using Protein Deconvolution software.
Far-UV circular dichroism (CD) spectroscopic and UV-visible spectroscopic assays
The buffer (0.1% formic acid) containing proteins was exchanged with water using an Ultra 10,000-MWCO filter (Thermo Fisher Scientific, Waltham, MA, USA). Circular dichroism of the enzymes in solutions was measured at 190–260 nm using a Jasco J-815 CD spectropolarimeter (Jasco Corp., Hachioji, Japan). Heat-induced denaturation of recombinant CYP142A1 and all cysteine mutants was determined in duplicate at 222 nm with a temperature increase from 10 °C to 85 °C in 1 °C min−1 increments. The UV-visible (UV-vis) absorbance of the proteins was measured using a PerkinElmer EnSpire® 2300 Multimode Plate Reader (PerkinElmer Life and Analytical Sciences Inc., Waltham, MA, USA) a 96-well UV plate (Corning Inc., Corning, NY, USA) and spectra were recorded between 350 and 700 nm at 25 °C. Samples were diluted to 2.0 μM with phosphate buffer (pH 7.4). Econazole was dissolved in DMSO and added to samples to a final concentration of 100 nM.25Spectrophotometric binding assays
Spectrophotometric titration of the recombinant CYP142A1 and cysteine mutants with the substrates, cholesterol, cholest-4-en-3-one, cholesteryl propionate and cholesteryl sulfate and inhibitor econazole nitrate proceeded at 25 °C as described.20 All ligand binding assay titrations were conducted in triplicate in 50 mM potassium phosphate buffer (pH 7.4) containing 150 mM NaCl using an EnSpire® 2300 Multimode Plate Reader (PerkinElmer). Cholesterol and cholest-4-en-3-one stock solutions (5.0 mM) were prepared in warm 10% (w/v) methyl-β-cyclodextrin (MβCD) dissolved in water, sonicated for 10 min, and finally diluted to 1.0 mM in 2% MβCD. Cholesteryl propionate was dissolved in chloroform–acetone (1:10 v/v) containing 0.05% Tween 20 (v/v), dried and resuspended in reaction buffer. Cholesteryl sulfate and econazole nitrate were dissolved in methanol. Proteins (0.2 mL; 2.0 mM) in the buffer were placed in 96-well plates. The same volume of buffer without proteins served as the blank control to account for the absorbance of each tested compound. After background scanning, equal volumes of ligand solution were added to wells containing buffer and proteins and mixed well. The equivalent volume of titrant was added to blank wells to correct for solvent effects. Difference spectra were recorded between 350 and 700 nm. Titration data points were fitted to the quadratic equation using GraphPad Prism 5.0 to determine the KD values In eqn (1) shown below: Aobs is the absorption shift determined at any ligand concentration; Amax is the maximum change in absorbance; KD in the Hill equation is the apparent dissociation constant; [S] is the ligand concentration; n is the Hill coefficient. The results represent mean ± SD of three replicate experiments.| Aobs = Amax[([S] + [E] + [KD]) − (([S] + [E] + KD)2 − (4[S][E]))0.5]/2[Et] | (1) |
Steady-state kinetic studies
Reactions of six mutants were carried out as previously reported.26 Briefly, CYP142A1 (0.2 μM) were preincubated for 5 min with choles-4-en-3-one of gradient concentration in 50 mM potassium phosphate buffer (pH 7.5) containing 0.45% (w/v) MβCD, 150 mM NaCl, and 10 mM MgCl2. Reactions were started by adding 1 μM spinach ferredoxin, 0.2 U mL−1 spinach ferredoxin-NADP+ reductase, 0.3 mM NADPH, 0.1 mg mL−1 bovine liver catalase, 5 mM glucose-6-phosphate, and 0.4 U mL−1 glucose-6-phosphate dehydrogenase. Aliquots of 50 μL were taken at 20 min and mixed with 150 μL of acetonitrile and 50 μM ergosterol as an internal standard (Johnston et al., 2010). Reactions were analyzed using an Agilent Series 1200 HPLC system and a reverse phase C18 column (Waters Xterra C18 column, 3.5 μm, 2.1 × 50 mm) at a flow rate of 0.5 mL min−1 (solvent A, H2O + 0.1% formic acid; solvent B, CH3CN + 0.1% formic acid) monitoring at 240 nm. Gradient elution started from 70% solvent B for 1 min and ramped up to 100% solvent B over 12 min. Then the solvent was maintained at 100% solvent B up to 14 min and then decreased to 70% solvent B within 1 min. The enzymatic reaction rate was calculated using the reduced amount of substrate. To determine the Km and kcat values, the data points were fitted to the Michaelis–Menten equation using GraphPad Prism. All data represent mean ± SD of three replicate experiments.Statistical analysis
Results are presented as means ± standard error (SE). Paired comparisons were analyzed by Student's t-tests. All data were statistically analyzed using GraphPad Prism 5.0, values with P < 0.05 were considered as significant.Results
Purification and basic properties of recombinant CYP142A1 and cysteine mutants
We constructed a set of mutants with replaced serine residues to determine whether any of the six non-heme binding cysteine residues are structurally or functionally important for CYP142A1. We cloned the cyp142 gene from the Mtb H37Rv genome (Fig. 1A), and expressed CYP142A1 using a vector as described in the Materials and methods section. | ||
| Fig. 1 (A) Gel electrophoresis of cyp142 gene cloned by PCR. Lane 1, DL2000 DNA marker. Lane 2, cyp142. The whole gel electrophoresis was shown in Fig. S8.† (B) Amino acid sequence of CYP142A, with cysteine residues marked in red. | ||
The Cys110, Cys118, Cys123, Cys281, Cys296 and Cys316 residues were separately mutated to serine using the Fast MultiSite Mutagenesis System (Fig. 1B). Recombinant CYP142A1 and mutants were expressed in the T7-based vector pET30a with a C-terminal His-tag, and transformed into E. coli BL21 (DE3). The expression levels of mutants in whole cell lysates were tentatively evaluated by the absolute coefficient for the extensively low-spin (LS) substrate-free CYP142A1 of ε418 = 140 mM−1 cm−1 (ref. 18) (Fig. S1†). All His-tagged proteins were obtained in a soluble form in E. coli lysates, and the purified fractions displayed a brown-red-color, which indicates the presence of heme center.
All the mutated cysteine residues were marked in 3D structure of CYP142A1 by Pymol 2.4 (Fig. 2A). According to the crystal structure of Mtb CYP142A1,18 the Cys123 and Cys281 are located on the β-sheets, while Cys118 located prior to β-sheets and Cys296 located next to β-sheets. Cys110 is located on the α-helix and Cys 316 is located on the loop. And Cys281 is located next to the Arg282, which is the binding site of HEM. We further investigated changes in the mutants using ESI-MS and far-UV CD spectroscopy and by determining the molecular weight (MW) of all the mutants. Table S2† shows that the MW of all mutants was decreased by serine replacement, and that the decrease was minimal in C123S. Far-UV CD spectroscopy showed similar curves among the mutated proteins, with prominent peaks at 222 and 208 nm (Fig. 3A). These findings indicated that the secondary structures of the mutants remained almost identical to that of the wild-type.
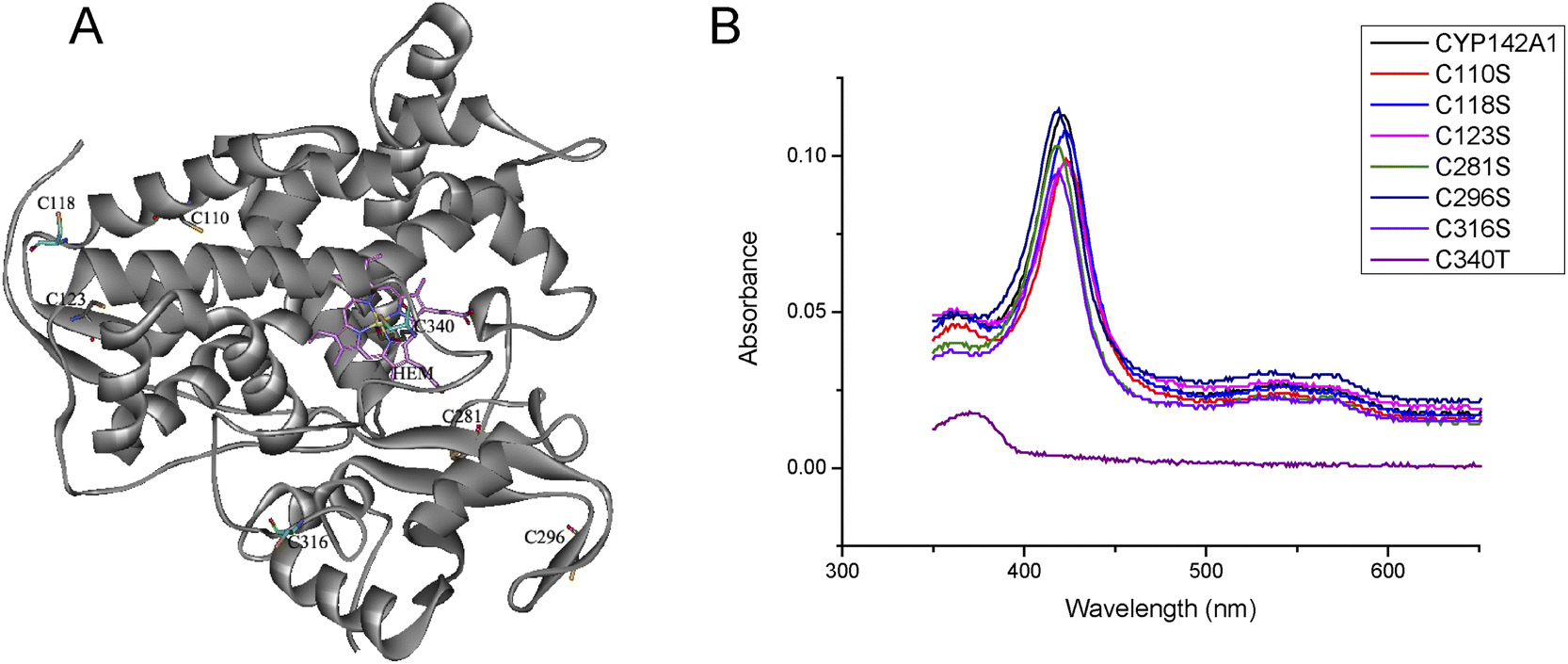 | ||
| Fig. 2 (A) Three-dimensional model of CYP142A1 (PDB ID: 2XKR) 18, the cysteine residues and Heme (HEM) are represented in sticks model, and the Fe atom is represented in ball model. (B) UV-vis spectra of recombinant CYP142A1 and mutants recorded at 2.0 μM with Soret band at 418 nm and smaller bands at 566 and 535 nm. | ||
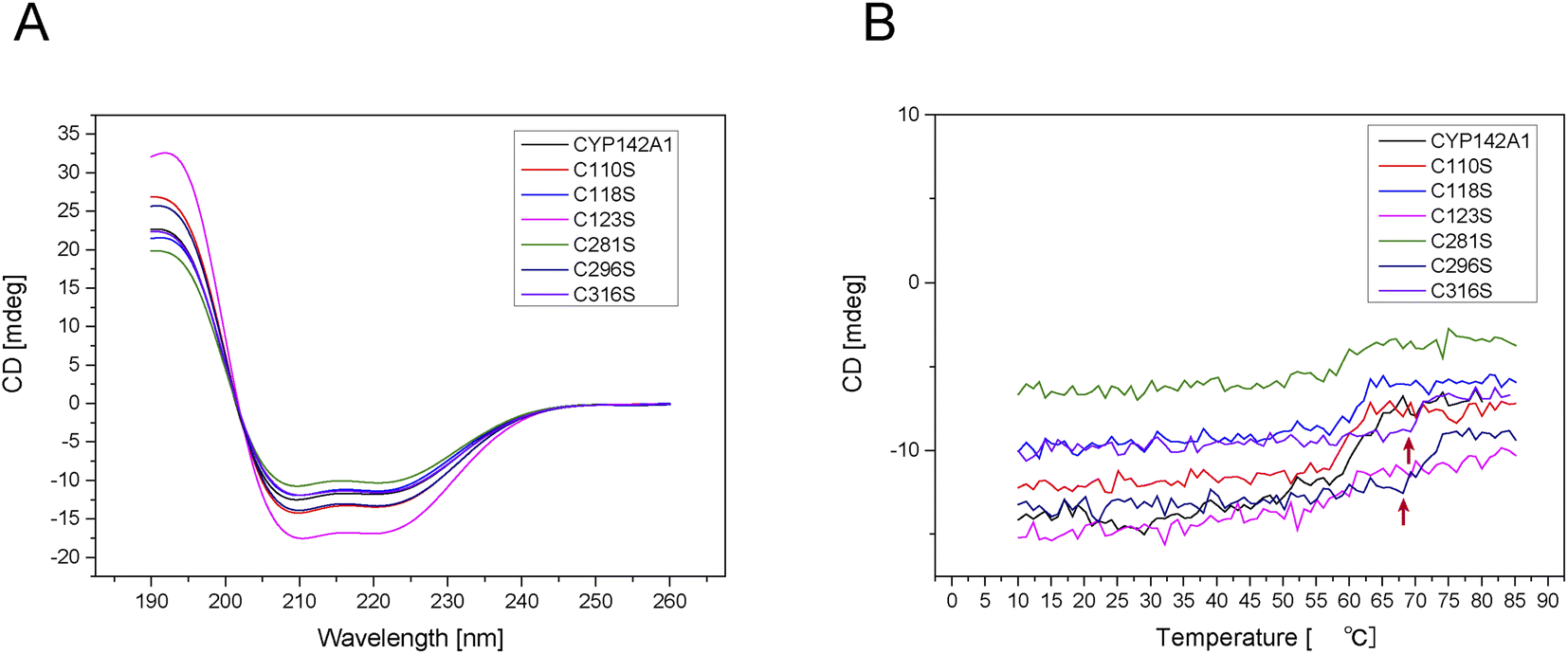 | ||
| Fig. 3 (A) Far-UV CD spectra of recombinant CYP142A1 and cysteine-to-serine mutants. (B) Heat-induced changes in secondary structure of recombinant CYP142A1 and cysteine-to-serine mutants determined by far-UV CD spectroscopy at 222 nm. | ||
The ratios of Helix, Turn, and Random were increased and that of Beta was decreased in C118S, C123S, C281S, C296S and C316S proteins, whereas the Helix and Random ratios were respectively increased and decreased in C110S. The far-UV CD spectrum of the enzyme solution at 222 nm, representing α-helices, was monitored over a temperature increase from 10 °C to 85 °C. Fig. 3B shows that the secondary structure of C296S and C316S decomposed at ∼70 °C, whereas that of the other mutants and the wild-type decomposed at 60 °C.
Spectral features of recombinant CYP142A1 and cysteine mutants
The electronic spectrum of purified CYP142A1 indicates an extensively low-spin (LS) ferric P450 enzyme, with a Soret band at 418 nm and smaller bands at 566 and 535 nm.18 Fig. 2B shows that C118S, C281S, C296S, and C316S were LS ferric P450 enzymes, which is consistent with the wild-type CYP142A1. Moreover, CYP142A1 undergoes a heme optical Soret shift to 423 nm, which is typical of P450 enzymes, upon binding the heme-coordinating inhibitor econazole nitrate. The Soret bands of all cysteine mutants except C110S and C123S shifted toward the red end of the spectrum to 423 nm after inhibition with econazole nitrate (Fig. S2†). C110S and C123S have a Soret band at 423 nm at a state of inhibitor free. And the maximum absorbance of C110S and C123S slightly changed to 424 nm when econazole nitrate was added.Spectrophotometric binding assays
Substrates (including cholest-4-en-3-one, cholesterol, Fig. 1 cholesteryl propionate, and cholesteryl sulfate) and inhibitor (econazole nitrate) binding were determined by measuring changes in optical absorption spectra. The absolute Soret region in the absorption spectra of mutants after incubation with cholest-4-en-3-one or cholesterol were consistent with that of CYP142A1.18 Adding cholest-4-en-3-one or cholesterol led to the complete conversion of enzymes to the high spin (HS) form due to displacement of the water molecule coordinated to the heme iron atom, as evidenced by a clear Type I shift of the Soret band from 418, to 393 nm, and a clear LS/HS isosbestic point at 406 nm (Fig. S3–S4†). We also monitored cholesteryl sulfate and cholesteryl propionate binding as substrate-induced spectral changes of heme iron. Binding by both cholesterol esters produced a Type I absorption shift Fig. S5–S6.†20 CYP142A1 was converted to the LS state by econazole nitrate in all proteins, with a Type II shift of the Soret band to 423 nm (Fig. S2†).The KD values of CYP142A1 and the cysteine mutants for cholest-4-en-3-one, cholesterol, cholesteryl propionate and cholesteryl sulfate and econazole nitrate were determined from spectral titration curve-fitting using GraphPad Prism. Plots of the induced spectral change versus steroid and the azole concentrations were fitted to a quadratic tight binding equation (eqn (1)) to generate KD values (Table 2).
| Fraction | Ratio | ||||||
|---|---|---|---|---|---|---|---|
| CYP142A1 | C110S | C118S | C123S | C281S | C296S | C316S | |
| Helix | 34.9 | 39 | 43.1 | 44.5 | 45 | 45.6 | 47.3 |
| Beta | 42.5 | 42.1 | 21.3 | 21.5 | 17.2 | 16.9 | 13.1 |
| Turn | 0 | 0 | 8.7 | 8.2 | 9.6 | 9.9 | 11.2 |
| Random | 22.6 | 18.9 | 27 | 25.7 | 28.2 | 27.7 | 28.4 |
| Total | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Protein | KD | ||||
|---|---|---|---|---|---|
| Cholest-4-en-3-one (μM) | Cholesterol (μM) | Cholesteryl propionate (μM) | Cholesteryl sulfate (μM) | Econazole nitrate (μM) | |
| CYP142A1 | 0.032 ± 0.0036 | 0.0039 ± 0.0028 | 45 ± 7.0 | 27 ± 6.9 | 2.1 ± 0.17 |
| C110S | 0.22 ± 0.018 | 0.34 ± 0.082 | 35 ± 4.9 | 37 ± 8.7 | 4.3 ± 1.2 |
| C118S | 0.033 ± 0.0022 | 0.067 ± 0.030 | 43 ± 10 | 33 ± 4.3 | 7.2 ± 1.6 |
| C123S | 0.17 ± 0.012 | 0.11 ± 0.012 | 23 ± 5.0 | 35 ± 9.3 | 39 ± 8.6 |
| C281S | 0.032 ± 0.0053 | 0.012 ± 0.0051 | 47 ± 7.6 | 37 ± 8.8 | 0.52 ± 0.081 |
| C296S | 0.14 ± 0.046 | 0.0042 ± 0.0023 | 48 ± 5.9 | 25 ± 9.6 | 0.56 ± 0.086 |
| C316S | 0.020 ± 0.00047 | 0.019 ± 0.0099 | 46 ± 6.8 | 26 ± 8.0 | 0.36 ± 0.064 |
The binding affinity of the C118S, C281S, and C316S to cholest-4-en-3-one did not significantly differ, whereas that of C110S, C123S, and C296S was much lower than that of wild-type CYP142A1, with KD values of 0.22 ± 0.0187, 0.17 ± 0.012, and 0.14 ± 0.046 μM respectively. And the binding affinity of all the mutants to cholesterol except for C296S was much lower than that of wild-type CYP142A1. The binding affinity of C110S and C123S to cholesteryl propionate was higher than other mutants, with KD values of 35 ± 4.9 and 23 ± 5.0 μM, whereas that of C118S, C123S, C296S, and C316S was similar with that of wild-type CYP142A1. The binding affinity of C110S, C118S, C123S, and C281S to cholesteryl sulfate were lower than that of wild-type CYP142A1. Fig. S7† shows that the C110S, C118S and C123S peaks for inhibitor binding at 435 nm were not as sharp as those of the other mutants, leading to higher KD. Furthermore, the binding affinity of C281S, C296S, and C316S improved at KD values of 0.52 ± 0.081, 0.56 ± 0.086, and 0.36 ± 0.064 μM, respectively.
Steady-state kinetic studies
We analyzed turnover rates by reconstituting CYP142A1 and the cysteine mutants with spFDR and spFDX and an NADPH regenerating system over a concentration gradient of substrates to determine the effect of the cysteine residues on CYP142A1 enzyme activity. Steady-state kinetics was measured, and the parameters were calculated using a nonlinear regression of the Michaelis–Menten equation. Fig. 4 shows curves of enzyme kinetic for cholest-4-en-3-one. Table 3 shows the parameters.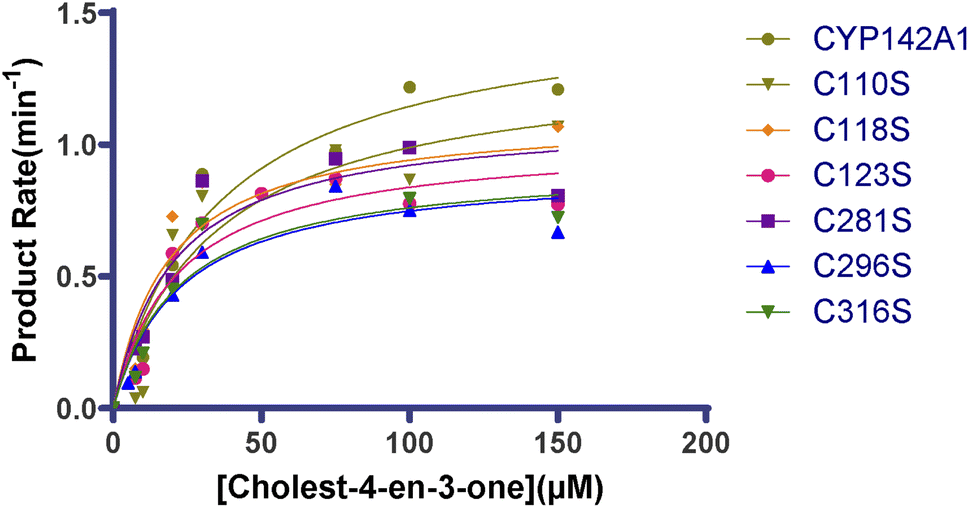 | ||
| Fig. 4 Enzyme kinetic curve of cholest-4-en-3-one by recombinant CYP142A1 and cysteine-to-serine mutants. Statistical analysis was performed between CYP142A1 and mutants by t-test, p < 0.05. | ||
| Enzyme | Km (μM) | kcat (min−1) | kcat/Km (min−1 μM−1) |
|---|---|---|---|
| CYP142A1 | 35 ± 5.4 | 7.7 ± 0.43 | 0.22 |
| C110S | 34 ± 9.7 | 6.6 ± 0.68 | 0.20 |
| C118S | 18 ± 4.3 | 5.6 ± 0.38 | 0.30 |
| C123S | 22 ± 5.1 | 5.1 ± 0.37 | 0.23 |
| C281S | 20 ± 4.4 | 5.5 ± 0.37 | 0.27 |
| C296S | 23 ± 5.6 | 4.6 ± 0.35 | 0.20 |
| C316S | 22 ± 5.3 | 4.7 ± 0.37 | 0.21 |
The values calculated from the oxidation of cholest-4-en-3-one after incubation with CYP142A1 and the cysteine mutants differed. The Km range for the oxidation of cholest-4-en-3-one was 18–23 μM for most cysteine mutants, while that for C110S was 34 μM, which was similar with CYP142A1. The Km values of all the mutants were lower than the wild-type, and those of C118S, C123S, C281S, C296S and C316S were much lower. Table 3 also lists kcat values that directly reflect catalytic production under specific conditions. The second-order rate constants kcat/Km provided parameters for comparing the cysteine mutants, and the data in Table 3 show that replacing cysteine residues at various sites caused changes in enzyme activity.
Discussion
Cysteine is a nucleophilic amino acid residue with low abundance but usually highly conserved in proteins. Since the thiol group of cysteine renders its unique biochemical properties at functional sites, such as the formation of disulfide bonds and binding to high-affinity metals, the amino acid residue plays an irreplaceable role in maintaining structural stability and activating the catalytic, binding and regulatory functions of many proteins.27,28 CYP142A1 is one of the major monooxygenases in Mtb that catalyze the oxidation of cholesterol and fatty acid from host cells, by which the pathogen can obtain carbon source and energy during chronic or latent phases of infection.18 Similar to CYP125A1 (a primary P450 enzyme responsible for the growth of Mtb on cholesterol), CYP142A1 can oxidize the aliphatic side-chain of cholesterol or cholest-4-en-3-one at C-26 to the carboxylic acid, which is a necessary step for Mtb to utilize cholesterol and survive in macrophages.19,26 And catalytic assays revealed that the ability of CYP142A1 enzyme to oxidize cholesteryl propionate and cholesteryl sulfate was much more efficient than CYP125 enzymes.20 In previous studies, the ability of CYP142A1 to bind and oxidize the cholest-4-en-3-one, cholesterol, cholesteryl propionate, and cholesteryl sulfate was analyzed by spectral titration and enzyme kinetics along with the ability to bind inhibitors such as azole-drugs, because of which, it is considered to be a promising anti-TB drug target.18,20,29 When comparing the amino acid sequences of CYP125A1 and CYP142A1, it was found that there were 3 cysteine residues in CYP125A1, while up to 7 cysteine residues (Cys110, Cys118, Cys123, Cys281, Cys296, Cys316 and Cys340) were observed in CYP142A1. Given the importance of CYP142A1 for the lipid metabolism and survival of Mtb during host infection, it is reasonable to believe that the seven cysteine residues may play different roles in the structure and catalytic activity of the P450 enzyme. Thus in this study, we focused on these cysteine residues to discover their potential roles in the structure and function of Mtb CYP142A1. Given the similarity in the structural framework,30 cysteine residues were replaced with serine residues, individually, to obtain mutant proteins.Among the seven cysteine residues, Cys340 locates at the active center in the C-terminal part of CYP142A1 and has been revealed to coordinate the heme iron by forming a Fe–S bond to support the monooxygenase activity.18,31 Previous studies have found that the replacement of the axial cysteine residue led to the collapse of the substrate binding site28 and the other six cysteine residues were located out of the letterbox-shaped entry-exit channel formed by the FG-loop, the BC-loop and the I-helix N-terminal region.18 However, very limited information on the contribution of the cysteine residues beyond Cys340 to the structure and function of CYP142A1 has been known. Thus in this study, cysteine-to-serine mutations were performed and a series of experiments were followed to majorly explore the roles of the six cysteine residues. For the six cysteine mutants, as measured using ESI-MS, substitution of cysteine with serine led to a slight decrease in the molecular weight relative to the wild-type (Table S2†), which was consistent with the lower molecular weight of serine than cysteine. The level of expression of CYP142A1 was affected by single cysteine mutations. There was a 60% decrease in the level of C123S mutant expression and 10–40% for others (Fig. S1†). The UV-Vis spectra of all the six cysteine mutants are almost identical to the wild-type (Fig. 2B), which means that any substitution of these cysteines does not produce a noticeable impact on the structure of the active core center of the enzyme. In the resting state of CYPs, water molecules usually act as distal ligands in the active center of CYP. When a substrate binds to the active site, it will replace the water ligand and result in a specific UV-vis spectrum called type I shift. Inhibitors can also replace water ligands, but they usually coordinate with the heme iron through the lone pairs of electrons from heteroatoms, which produces a characteristic UV-vis spectrum called type II shift. Typical type I and type II shift32 were observed in all cysteine-to-serine mutants.
Studies have shown that the substitution of C-terminal cysteine increased the thermal stability of argininosuccinate lyase from Mtb.33 According to the far-UV CD spectra (Fig. 3B), the secondary structure of C296S and C316S decomposed at about 70 °C, 10 °C higher than wild-type, representing the enhancement in thermal stability. In terms of the parameters calculated from far-UV CD spectra in present study (Table 1), except that C110S had a secondary structure very similar to the wild-type, the remaining 5 cysteine mutants (C118S, C123S, C281S, C296S and C316S) all possessed significantly higher composition ratios of Helix, Turn and Random, but a lower ratio of Beta relative to the wild-type, with C316S owning the highest proportion of helix-turn-random and the lowest proportion of beta motif. All the results were basically consistent with the locations of the cysteine residues. To our surprise, the replacement of Cys316, which is located on the loop, brought apparent changes on the secondary structure and thermal stability. Overall, it appeared that the closer the C-terminal cysteine substitution resulted in a higher proportion of folding changes, suggesting that Cys118, Cys123, Cys281, Cys296 and Cys316 contribute notably to the correct folding state of CYP142A1 and show a “polarity-like” effect on the secondary structure of the enzyme.
In order to analyze the potential roles of the six cysteines on the enzymatic functions of CYP142A1, the classical spectrophotometric titration experiments were utilized to determine and compare the dissociation constants from the cysteine mutants and wild-type that interacted with several substrates and an inhibitor, econazole nitrate, in vitro. Summarized from Table 2, higher KD values (about >4 times of the wild-type) were observed from C110S-cholest-4-en-3-one, C110S-cholesterol, C118S-cholesterol, C123S-cholest-4-en-3-one, C123S-cholesterol, C296S-cholest-4-en-3-one, and C316S-cholesterol, while a lower KD value (63% of the wild-type) was obtained from C316S-cholest-4-en-3-one, which means that each of these cysteines may play different but moderately regulating roles in the direct binding strength of CYP142A1 to the cholest-4-en-3-one and cholesterol. To cholesterol derivatives, lower KD values were observed in C110S-cholesteryl propionate and C123S-cholesteryl propionate, while higher KD values were observed in C110S-cholesterol sulfate, C118S-cholesterol sulfate, C123S-cholesterol sulfate, and C281S-cholesterol sulfate. The allosteric effect may explain these phenomenon, but more verification is needed.
It seems that Cys296 and Cys316 have limited effects on the binding of the enzyme to some of the substrates, while Cys110, Cys118, Cys123, and Cys281 contributes more to the dissociation of the enzyme from the substrates, except for Cys110 and Cys123 in the binding to cholesteryl propionate. As shown in Fig. S2,† when econazole nitrate was added, the type II shifts of C110S, C118S, and C123S, toward 423 nm was smaller than that of wild-type. In the titration analysis of econazole nitrate, the absorption changes between 435 nm and 416 nm from C110S, C118S or C123S binding with econazole were not as abrupt as that from the wild-type CYP142A1 binding with econazole (Fig. S7†), which meant higher KD values and the relatively lower affinity of the three cysteine-to-serine mutants to econazole. Whereas, the KD value of C281S, C296S or C316S with econazole decreased significantly (about 17–27% of the wild-type) in comparison to that of the wild-type. Thus, it can be presumed that the cysteine residues at the C-terminus (Cys281, Cys296, and Cys316) may contribute to the binding of the enzyme with the inhibitor, while the cysteine residues at the N-terminus (Cys110, Cys118, and Cys123) may play the opposite roles.
In order to investigate the possible effects of the cysteines on this metabolic function of the P450 enzyme, we further analyzed and compared the steady-state kinetic constants (Km, kcat, and kcat/Km) of the cysteine mutants and the wild-type through reconstituting the enzymatic reaction system with spFDR/spFDX as redox partners and monitoring product formation in vitro. Steady state kinetic analysis of cholest-4-en-3-one binding reveals that the Km values of C118S, C123S, C281S, C296S and C316S were reduced to 53–65% relative to that of the wild-type, while the Km values of C110S were not significantly altered, which suggests that Cys118, Cys123, Cys281, Cys296 and Cys316 have a relative obvious improvement on the affinity of CYP142A1 to the substrate. All the mutants showed decreased kcat values, while for kcat/Km values, C118S and C281S increased. Thus, it can be speculated that Cys118 and Cys281 have a more obvious improvement on the CYP142A1 oxidation efficiency of cholest-4-en-3-one than the other cysteine residues. According to the location of these cysteine residues in the 3D structure, we can speculate that they may facilitate the enzymatic reaction to a certain extent by promoting the association of the enzyme to the substrate as the influence on the β-sheet. In general, it can be recognized that when CYP142A1 catalyzes cholest-4-en-3-one, the cysteine residues in different positions may play different regulatory roles.
Conclusions
In summary, the seven cysteine residues at various positions in CYP142A1 have distinct biochemical functions. Among them, Cys340 is indispensable for the active center of the enzyme, and its mutation or deletion could lead to the complete destruction of the enzyme structure and loss of activity. The six cysteine residues outside the active center play roles in maintaining the correct folding and function of the enzyme. Since the crystal structure of CYP142A1 (PDB ID 2XKR) shows that no disulfide bond was identified among the seven cysteine residues, functional roles of the cysteine residues majorly depend on their positions and the local environment, as well as whether it is interior or solvent-exposed.18 In present study, separate substitution of the six cysteine residues resulted in numerous changes in the secondary structure, expression level, substrate-binding ability, inhibitor-binding ability, thermal stability and oxidation efficiency of the enzyme. Although most substitutions in cysteine residues led to a relative mild effect on the interactions between the enzyme and cholesterol-like substrates, some of the substitutions at C-terminal have a pronounced effect on the interactions between enzymes and inhibitors, which will provide some clues for the design of Mtb CYP inhibitors in the future. And substitutions of cysteine residues located near or on the β-sheet resulted in changes in enzyme activities. At the same time, the results of this study also improve our understanding of the structure and function of CYP142A1, especially the function of cysteine residues in the P450 enzyme and the consequences of their substitution. These findings will eventually encourage the development of inhibitors against Mtb CYP142A1.Author contributions
Conceptualization, X. Y. (Yang), X. Y. (You) and Y. L.; methodology, Y. L., L. S. and J. P.; software, Y. L., H. W. and J. P.; validation, Y. L.; formal analysis, Y. Z.; investigation, X. L.; resources, X. H. and X. W.; writing—original draft preparation, Y. L.; writing—review and editing, X. Y. (Yang), X. Y. (You) and C. L.; visualization, G. L.; supervision, Y. L.; project administration, Y. L.; funding acquisition, X. Y. (Yang), X. Y. (You) and Y. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare there is no conflicts of interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant number 81803593, 81273427, 32141003), the CAMS Innovation Fund for Medical Sciences (CIFMS) (grant number 2021-1-12M-039), the National Science and Technology Infrastructure of China (No. National Pathogen Resource Center-NPRC-32), and the Fundamental Research Funds for the Central Universities (grant number 3332018094, 2021-PT350-001). We are very grateful to Dr Yu Lu and Jian Xu from Beijing tuberculosis and thoracic tumour research institute for supply of the Mycobacterium tuberculosis H37Rv genomic DNA. We also appreciate the manuscript editing services provided by Elsevier.References
- W. H. Organization, Global Tuberculosis Report, 2020 Search PubMed
.
- M. Lauzardo and C. A. Peloquin, Expert Opin. Pharmacother., 2012, 13, 511–526 CrossRef CAS PubMed
- G. Riccardi, M. R. Pasca and S. Buroni, Future Microbiol., 2009, 4, 597–614 CrossRef CAS PubMed
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS PubMed
- A. W. Munro, K. J. McLean, K. R. Marshall, A. J. Warman, G. Lewis, O. Roitel, M. J. Sutcliffe, C. A. Kemp, S. Modi, N. S. Scrutton and D. Leys, Biochem. Soc. Trans., 2003, 31, 625–630 CrossRef CAS PubMed
- H. Ouellet, J. B. Johnston and P. R. Ortiz de Montellano, Arch. Biochem. Biophys., 2010, 493, 82–95 CrossRef CAS PubMed
- H. Ouellet, J. B. Johnston and P. R. O. d. Montellano, Trends Microbiol., 2011, 19, 530–539 CrossRef CAS PubMed
- R. Van der Geize, K. Yam, T. Heuser, M. H. Wilbrink, H. Hara, M. C. Anderton, E. Sim, L. Dijkhuizen, J. E. Davies, W. W. Mohn and L. D. Eltis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1947–1952 CrossRef CAS PubMed
- W. W. Mohn, R. van der Geize, G. R. Stewart, S. Okamoto, J. Liu, L. Dijkhuizen and L. D. Eltis, J. Biol. Chem., 2008, 283, 35368–35374 CrossRef CAS PubMed
- J. C. Chang, M. D. Miner, A. K. Pandey, W. P. Gill, N. S. Harik, C. M. Sassetti and D. R. Sherman, J. Bacteriol., 2009, 191, 5232–5239 CrossRef CAS PubMed
- N. M. Nesbitt, X. Yang, P. Fontan, I. Kolesnikova, I. Smith, N. S. Sampson and E. Dubnau, Infect. Immun., 2010, 78, 275–282 CrossRef CAS PubMed
- K. C. Yam, I. D'Angelo, R. Kalscheuer, H. Zhu, J. X. Wang, V. Snieckus, L. H. Ly, P. J. Converse, W. R. Jacobs Jr, N. Strynadka and L. D. Eltis, PLoS Pathog., 2009, 5, e1000344 CrossRef PubMed
- A. K. Pandey and C. M. Sassetti, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4376–4380 CrossRef CAS PubMed
- J. K. Capyk, R. Kalscheuer, G. R. Stewart, J. Liu, H. Kwon, R. Zhao, S. Okamoto, W. R. Jacobs, L. D. Eltis and W. W. Mohn, J. Biol. Chem., 2009, 284, 35534–35542 CrossRef CAS PubMed
- G. Schafer, R. Guler, G. Murray, F. Brombacher and G. D. Brown, PLoS One, 2009, 4, e8448 CrossRef PubMed
- G. W. Martens, M. C. Arikan, J. Lee, F. Ren, T. Vallerskog and H. Kornfeld, Infect. Immun., 2008, 76, 3464–3472 CrossRef CAS PubMed
- M. J. Kim, H. C. Wainwright, M. Locketz, L. G. Bekker, G. B. Walther, C. Dittrich, A. Visser, W. Wang, F. F. Hsu, U. Wiehart, L. Tsenova, G. Kaplan and D. G. Russell, EMBO Mol. Med., 2010, 2, 258–274 CrossRef CAS PubMed
- M. D. Driscoll, K. J. McLean, C. Levy, N. Mast, I. A. Pikuleva, P. Lafite, S. E. J. Rigby, D. Leys and A. W. Munro, J. Biol. Chem., 2010, 285, 38270–38282 CrossRef CAS PubMed
- S. Ortega Ugalde, M. Boot, J. N. M. Commandeur, P. Jennings, W. Bitter and J. C. Vos, Appl. Microbiol. Biotechnol., 2019, 103, 3597–3614 CrossRef CAS PubMed
- D. J. Frank, Y. Madrona and P. R. Ortiz de Montellano, J. Biol. Chem., 2014, 289, 30417–30425 CrossRef CAS PubMed
- H. Qiu, D. M. Honey, J. S. Kingsbury, A. Park, E. Boudanova, R. R. Wei, C. Q. Pan and T. Edmunds, Protein Sci., 2015, 24, 1401–1411 CrossRef CAS PubMed
- T. Shimizu, J. Inorg. Biochem., 2012, 108, 171–177 CrossRef CAS PubMed
- X. Xia, L. M. Longo and M. Blaber, J. Pharm. Sci., 2015, 104, 566–576 CrossRef CAS PubMed
- Y. Lu, F. Qiao, Y. Li, X. H. Sang, C. R. Li, J. D. Jiang, X. Y. Yang and X. F. You, Appl. Microbiol. Biotechnol., 2017, 101, 7201–7212 CrossRef CAS PubMed
- K. J. McLean, P. Lafite, C. Levy, M. R. Cheesman, N. Mast, I. A. Pikuleva, D. Leys and A. W. Munro, J. Biol. Chem., 2009, 284, 35524–35533 CrossRef CAS PubMed
- E. Garcia-Fernandez, D. J. Frank, B. Galan, P. M. Kells, L. M. Podust, J. L. Garcia and P. R. Ortiz de Montellano, Environ. Microbiol., 2013, 15, 2342–2359 CrossRef CAS PubMed
- L. B. Poole, Free Radical Biol. Med., 2015, 80, 148–157 CrossRef CAS PubMed
- S. Yoshioka, S. Takahashi, H. Hori, K. Ishimori and I. Morishima, Eur. J. Biochem., 2001, 268, 252–259 CrossRef CAS PubMed
- J. B. Johnston, H. Ouellet and P. R. Ortiz de Montellano, J. Biol. Chem., 2010, 285, 36352–36360 CrossRef CAS PubMed
- S. Konig, E. Romp, V. Krauth, M. Ruhl, M. Dorfer, S. Liening, B. Hofmann, A. K. Hafner, D. Steinhilber, M. Karas, U. Garscha, D. Hoffmeister and O. Werz, Cell Chem. Biol., 2019, 26, 60–70 CrossRef PubMed
- H. Ouellet, J. B. Johnston and P. R. Ortiz de Montellano, Arch. Biochem. Biophys., 2010, 493, 82–95 CrossRef CAS PubMed
- H. Ouellet, P. M. Kells, P. R. Ortiz de Montellano and L. M. Podust, Bioorg. Med. Chem. Lett., 2011, 21, 332–337 CrossRef CAS PubMed
- A. Mishra and A. Surolia, IUBMB Life, 2017, 69, 896–907 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04257f |
| ‡ These corresponding authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |