
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiclofenac derivatives as concomitant inhibitors of cholinesterase, monoamine oxidase, cyclooxygenase-2 and 5-lipoxygenase for the treatment of Alzheimer's disease: synthesis, pharmacology, toxicity and docking studies†
Muhammad Aamir Javeda,
Saba Bibia,
Muhammad Saeed Jan b,
Muhammad Ikramc,
Asma Zaidia,
Umar Farooqa,
Abdul Sadiq*d and
Umer Rashid*a
b,
Muhammad Ikramc,
Asma Zaidia,
Umar Farooqa,
Abdul Sadiq*d and
Umer Rashid*a
aDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, 22060 Abbottabad, Pakistan. E-mail: umerrashid@cuiatd.edu.pk
bDepartment of Pharmacy, University of Swabi, Swabi, 23430, KP, Pakistan
cDepartment of Pharmacy, COMSATS University Islamabad, Abbottabad Campus, 22060 Abbottabad, Pakistan
dDepartment of Pharmacy, Faculty of Biological Sciences, University of Malakand, Chakdara, 18000 Dir (L), KP, Pakistan. E-mail: sadiquom@yahoo.com
First published on 11th August 2022
Abstract
Targeting concomitantly cholinesterase (ChEs) and monoamine oxidases (MAO-A and MAO-B) is a key strategy to treat multifactorial Alzheimer's disease (AD). Moreover, it is reported that the expression of cyclooxygenase-2 (COX-2) and lipoxygenase (LOX) is increased significantly in the brain of AD patients. Using the triazole of diclofenac 12 as a lead compound, we synthesized a variety of analogs as multipotent inhibitors concomitantly targeting COX-2, 5-LOX, AChE, BChE, MAO-A and MAO-B. A number of compounds showed excellent in vitro inhibition of the target biological macromolecules in nanomolar concentration. Compound 39 emerged as the most potent multitarget ligand with IC50 values of 0.03 μM, 0.91 μM, 0.61 μM, 0.01 μM 0.60 μM and 0.98 μM towards AChE, BChE, MAO-A, MAO-B, COX-2 and 5-LOX respectively. All the biologically active compounds were found to be non-neurotoxic and blood–brain barrier penetrant by using PAMPA assay. In a reversibility assay, all the studied active compounds showed reversibility and thus were found to be devoid of side effects. MTT assay results on neuroblastoma SH-SY5Y cells showed that the tested compounds were non-neurotoxic. An in vivo acute toxicity study showed the safety of the synthesized compounds up to a 2000 mg kg−1 dose. In docking studies three-dimensional construction and interaction with key residues of all the studied biological macromolecules helped us to explain the experimental results.
1 Introduction
Alzheimer's disease (AD) is a progressive neurological age-related disorder that begins slowly, worsens with time, and causes loss of memory as well as cognitive impairments. Formation of neurofibrillary tangles and free radicals, oxidative stress, excessive beta-amyloid peptide accumulation, irregular bio metal homeostasis, dystrophic neurites, dysfunctional microglia, reactive astrocytes, and reduced cholinergic functions are all pathological factors that contribute to the onset and progression of disease.1–4 It primarily affects persons over the age of 65, and it is caused by a lack of acetylcholine (ACh), a neurotransmitter, in the neocortex and hippocampus. As a result, AD is marked by severe cholinergic neuron damage and decreased cholinergic functioning.5,6 Increasing evidence revealed that pathologically AD is not only confined to the neuronal dysfunction but is also associated with the immunological processes of the brain which triggers the secretion of proinflammatory agents, involved in the development and severity of disease.7,8 Neuroinflammation (NI) is a defensive mechanism in the brain that develops in response to neuronal stress and has been associated to a variety of chronic neurodegenerative diseases, including Alzheimer's disease. Toxins, infections, and certain medicines can all induce NI. Glia cells in the brain, such as microglia and astroglia, create proinflammatory mediators such cytokines and chemokines, that trigger neuroinflammatory processes.9–11COX-1 is expressed in the brain on a constant basis, but COX-2 expression in neurons and glial cells is increased in neurodegenerative disorders. COX-2 is an enzyme that converts arachidonic acid to prostaglandins, that are important inflammatory mediators. COX-2-produced prostaglandins such as PGD2, PGE2, and PGJ2 have been found to increase in the frontal cortex of Alzheimer's disease patients, coupled with microglia and astrocyte activation in senile plaques. Inhibition of COX-2 can help to alleviate inflammation and pain symptoms. COX-2 expression is dramatically enhanced in the brains of AD patients. COX-2 inhibitors could thus be beneficial in the treatment of neurological diseases.5,12,13
LOX-5 is a central nervous system enzyme that catalyzes the conversion of arachidonic acid to leukotrienes. It was also discovered that LOX-5 inhibitors can protect against β-amyloid mediated neurotoxicity. The AD brain has a higher amount of lipoxygenase (LOX) expression, which has been linked to increased Aβ production and tau phosphorylation. Studies on animal models have shown that 5- or 12- or 15-LOX inhibitors can reduce β-amyloid and tau pathology while also improving cognitive impairment. Experiments have also indicated a relationship between oxidative stress and LOXs and their metabolites in Alzheimer's patients. These data suggest that LOX-5 could be a promising target for future anti-NI medicines.14,15
Furthermore, nonsteroidal anti-inflammatory medications (NSAIDs) have been demonstrated to have protective benefits in lowering or delaying the progression of Alzheimer's disease in various epidemiologic and observational investigations. Randomized controlled trials, on the other hand, refute this theory. This discrepancy could be owing to the disease's multidimensional character and/or a failure to choose a proper cohort of patients, or due to a lack of relevant biomarkers for accurate patient stratification, or due to a lack of appropriate biomarkers.16 Another essential enzyme associated with the etiology of AD is MAO, a vital flavoenzyme, which has two isoforms, MAO-A and MAO-B. MAO-B is involved in the metabolism of dopamine and performs a vital function in oxidative stress as it alters the redox state of glial cells and neurons. MAO-B expression is shown to be much higher in the hippocampus and cerebral cortex of Alzheimer's patients, accompanied with an excess of reactive oxygen species and hydrogen peroxide.17–19 AD is currently untreatable and presently available anti-cholinesterase FDA approved drugs like donepezil, rivastigmine, galantamine and memantine, are single-target drugs that improve memory loss and cognitive functions whereas these are unable to completely cure dementia. In AD several relevant targets in various cell signaling pathways interact to form a network, thus making it difficult for single target drugs to cure the disease effectively. Therefore, multi-target strategies are being adopted to regulate the disease progression.20–23 In designing MTDLs, to obtain maximum efficiency and superior outcomes for the treatment of AD, cholinesterase inhibitors are used to reduce the expression of AChE which is also accompanied by controlling other disease modifying factors which are involved in pathogenesis of AD.4
Our research group previously synthesized pharmacological vital pyrimidine based dual binding site concomitant inhibitors for AChE and BChE. Compound (1) exhibited inhibition potential of 0.17 μM and 2.37 μM for AChE and BChE.24 We recently reported the pyrrolidine-sulfonamide derivative as potent multitarget inhibitors for the treatment of cholinergic deficit and neuroinflammation in AD. The potent compounds (2) showed inhibition of COX-2, 5-LOX, AChE and BChE in nano molar range.5 Tacrine-sertraline derivative (3) emerged as the most potent inhibitor of AChE, BChE, MAO-A and MAO-B with IC50 values of 0.008 μM, 0.174 μM, 0.311 and 0.031 μM respectively.25 We also reported thiazolidine derivatives as potent multi target inhibitor for treatment of AD. Compound (4) exhibits potential inhibition of AChE, BChE, MAO-A, MAO-B with IC50 of 0.042 μM, 0.630 μM, 1.93 μM and 0.33 μM respectively.26 Askarani et al., reported triazole based chromenone derivatives as multitarget inhibitor for treatment of AD, the potent inhibitor (5) showed IC50 of 3.4 μM (AChE) and 1.1 μM (BChE).27 Yamali et al., reported phenothiazine based chalcones as AChE/BChE and MAO-A/MAO-B inhibitors via base catalyzed Claisen Schmidt condensation reaction. The potent compound (6) presented excellent inhibition of AChE and MAO-B with IC50 of 0.053 ± 0.001 μM and 0.048 ± 0.001 μM respectively (Fig. 1).28
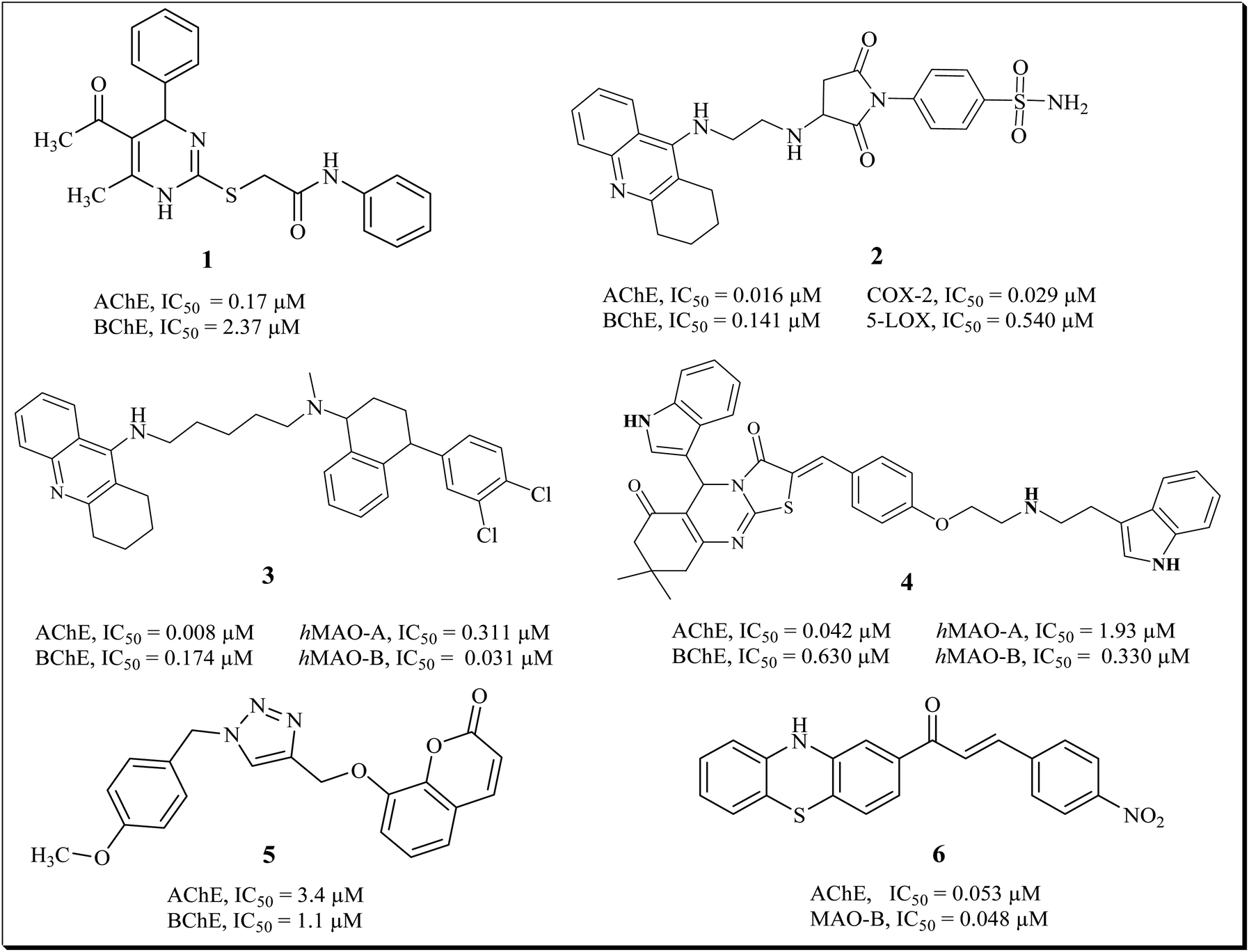 | ||
| Fig. 1 Structures of some reported multitargeted compounds. | ||
2 Results and discussion
2.1 Design strategy
Diclofenac is considered as an important anti-inflammatory agent. The presence of phenylacetic acid, secondary amine and a phenyl ring with two chloro-groups at ortho-position make it widely used for structural modification. In current study, we decided to modify the diclofenac structure to obtain compounds with multiple targeting properties related to AD including neuroinflammation. As described in Introduction section, previous reports from our laboratory identified pyrrolidine, pyrimidine and sulfonamide structural motifs endowed with multitarget inhibition for the treatment of AD. Moreover, it is also identified that the volume of active sites of AChE/BChE and MAO isoforms support bulky compounds for excellent inhibition. In current research, the bulk of the compounds were increased by the incorporation of various rigid (triazole, pyrazoline) and flexible (alkyl) linkers. Hydrophobic moieties such as phenyl rings, bicyclic (pyrimidine) and tricyclic fused ring systems (phenothiazine) at one or at both ends of the molecules to enhance the interactions with hydrophobic Trp279 and Trp84 (TcAChE numbering). In our previous reports, incorporation of pyrrolidine 2,5-dione framework showed multitarget anti-inflammatory and anti-Alzheimer activities. Hence, we also decide to incorporate this framework to enhance the multitarget inhibition.2.2 Chemistry
The synthesis of 5-(2-(2,6-dichlorophenylamino) benzyl)-4-(3,4-dichlorophenyl)-4H-1,2,4-triazole-3-thiol (12) is illustrated in Scheme 1. Acyl halide of diclofenac acid was synthesized by using thionyl chloride in dry acetone. The synthesized derivative was further treated with hydrazine hydrate in the presence of absolute ethanol to obtain 2-(2-(2,6-dichlorophenylamino)phenyl)acetohydrazide 9. Further, hydrazide 9 on treatment with 1,2-dichloro-4-isothiocyanatobenzene (10) in dry acetone gave thiosemicarbazide derivative 11. The synthesized thiosemicarbazide 11 when treated with 5% NaOH solution, produced target compound 12 (Scheme 1).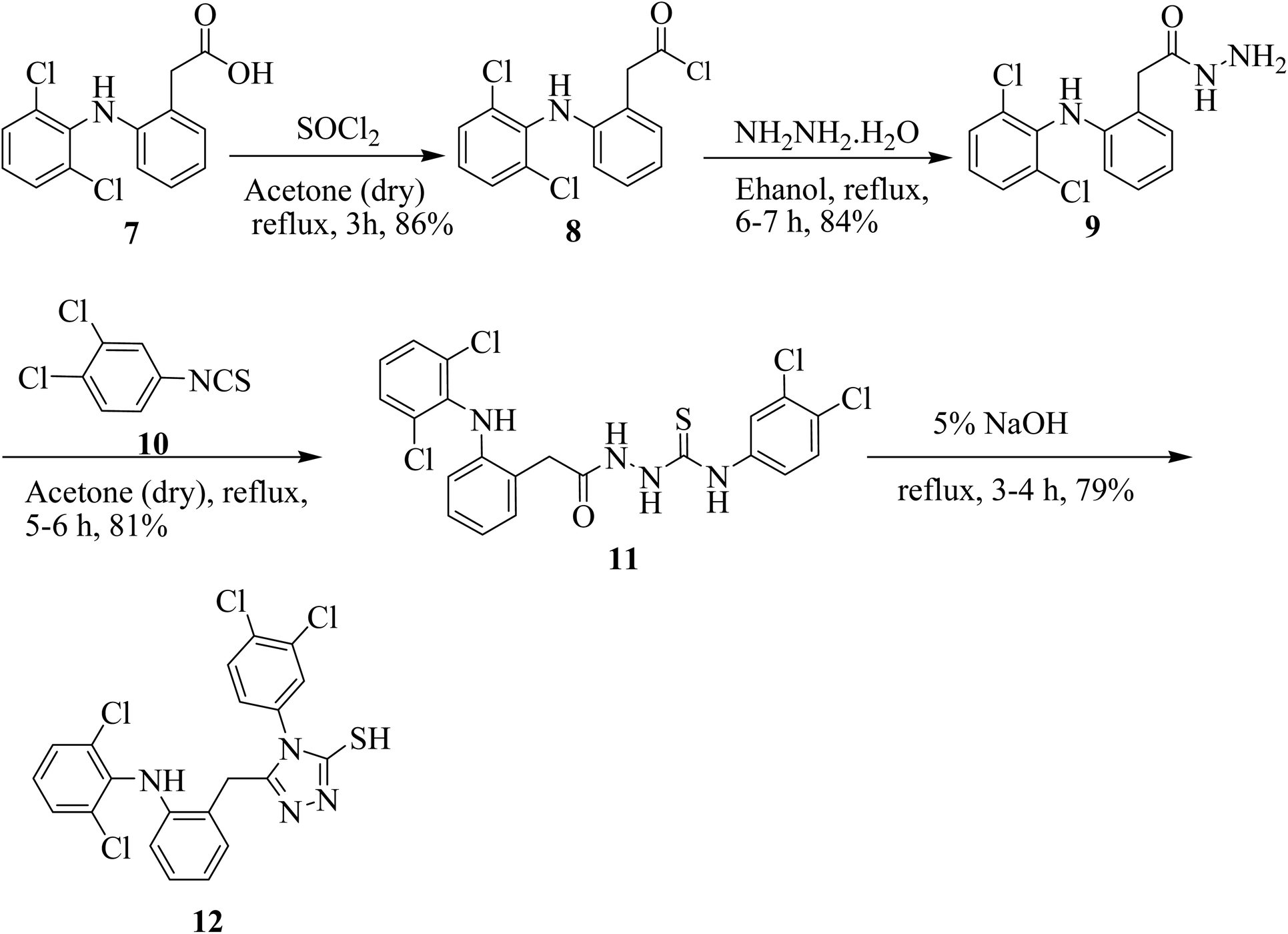 | ||
| Scheme 1 Synthesis of triazole analog 12. | ||
A synthetic strategy in hand allowed us to explore the SAR of 5-substituted-phenyl-4H-1,2,4-triazole-3-thiol derivative compound 15. First, we synthesized Mannich base by the reaction of 3-thiol, formaldehyde and sulfanilamide in methanol under reflux conditions (Scheme 2).
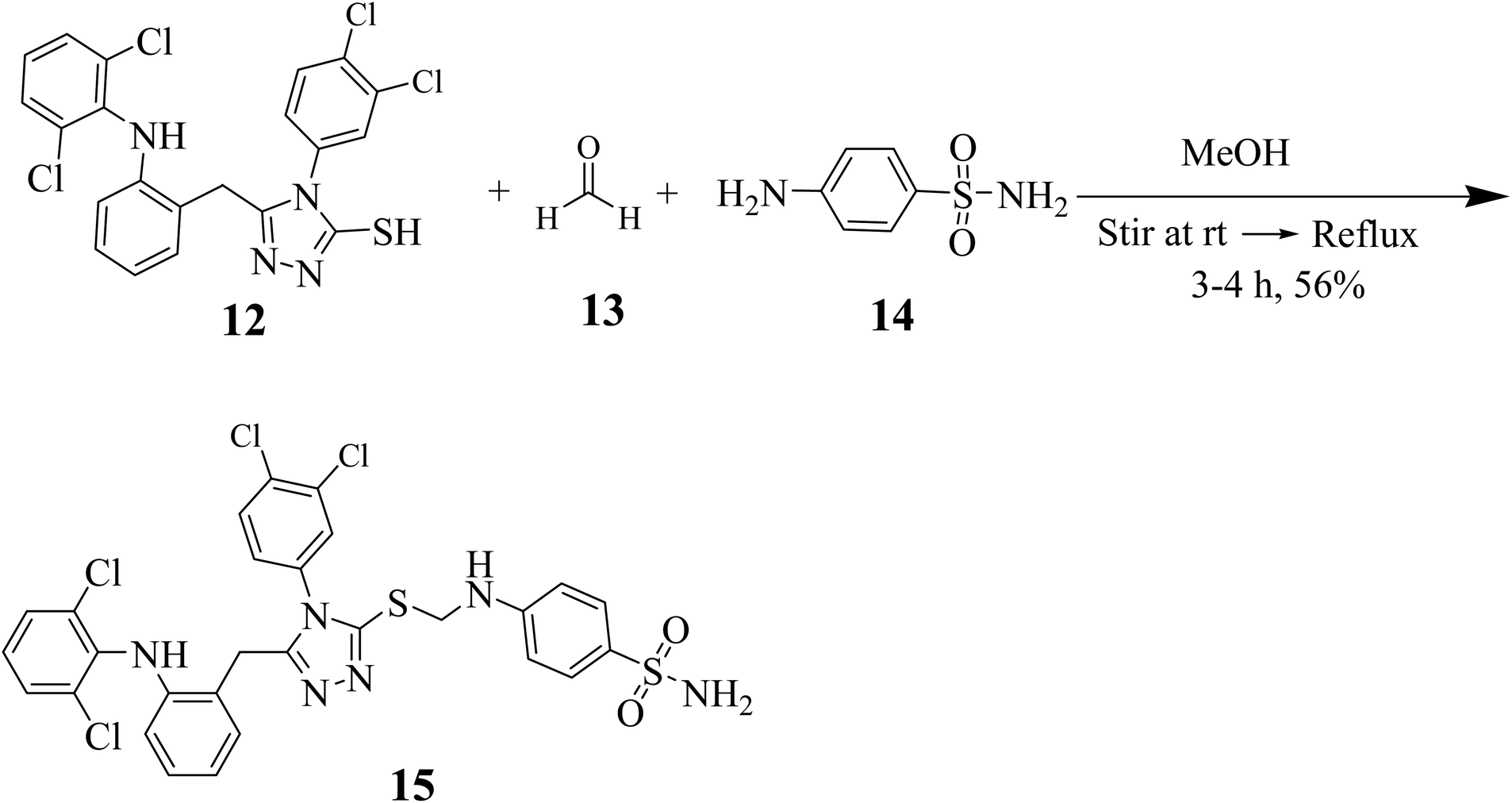 | ||
| Scheme 2 Synthesis of Mannich base 15. | ||
Furthermore, we synthesized derivative of 12 with thiols and secondary amines (R-XH, 18–25) having four methylene units. Compound 12 was treated with 1,4-dibromo butane in the presence of TEA in dichloromethane (DCM) as solvent. The synthesized intermediate was further reacted with dihydropyrimidine-2-thiones (18–22), succinimide derivatives 23, 24 and tricyclic phenothiazine 25 to obtain the target compounds 26–33 (Scheme 3).
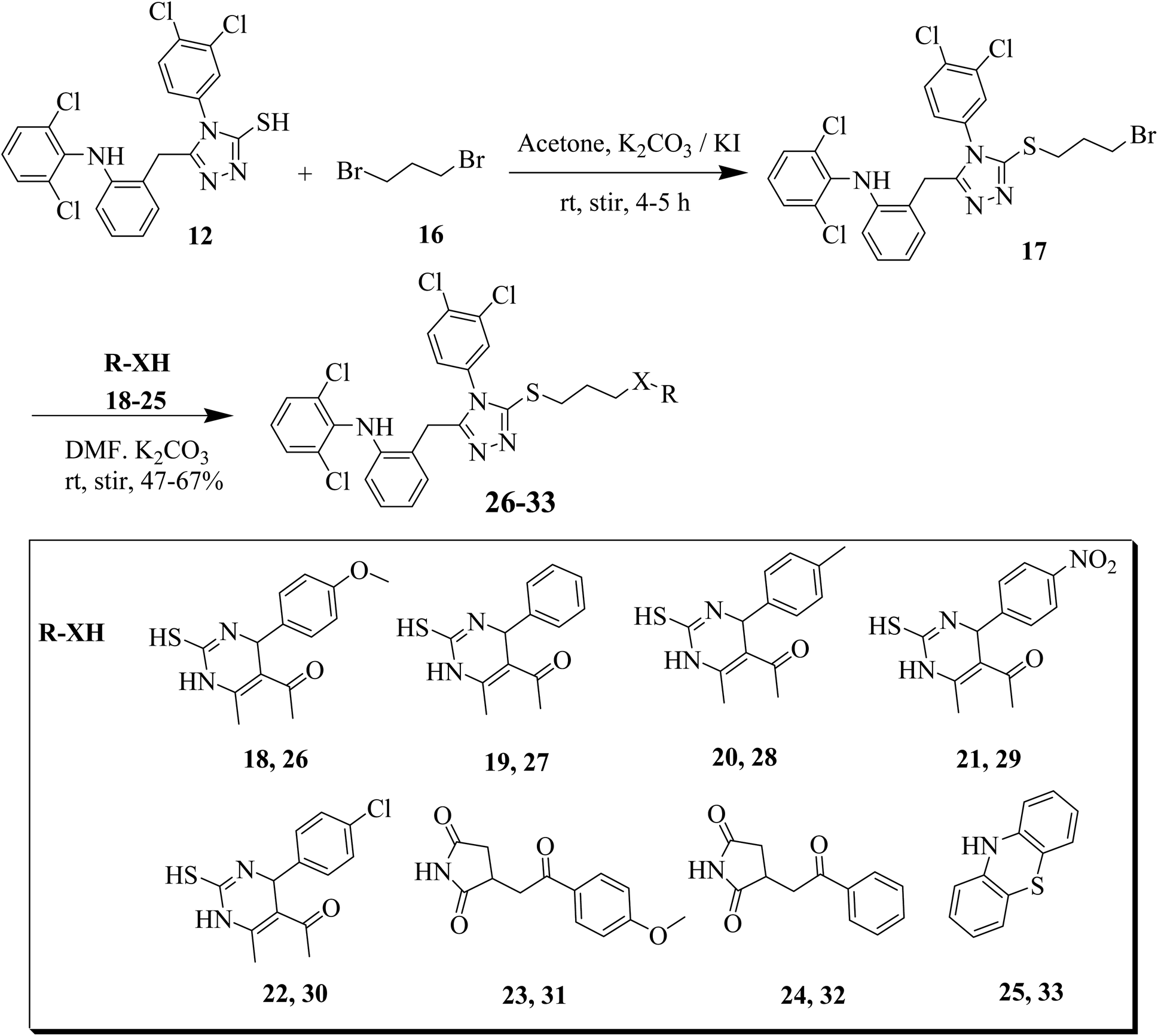 | ||
| Scheme 3 Synthesis of derivatives of 26–33. | ||
Subsequently, we synthesized the pyrazoline based sulfonamide derivative 39 of diclofenac acid by following a multi-steps protocol. In the first step, 4-aminoacetophenone (34), was coupled with diclofenac acid by using (DCC/DMAP) to obtain amide (35). The amide intermediate was then reacted with 4-chlorobenzaldehyde (36) to form chalcone derivative 37. The chalcone based compound (37) finally treated with 4-hydrazinylbenzenesulfonamide (38) in the presence of base NaOH and stir in ethanol at room temperature for 2 h and obtained pyrazoline based compound 39 (Scheme 4).
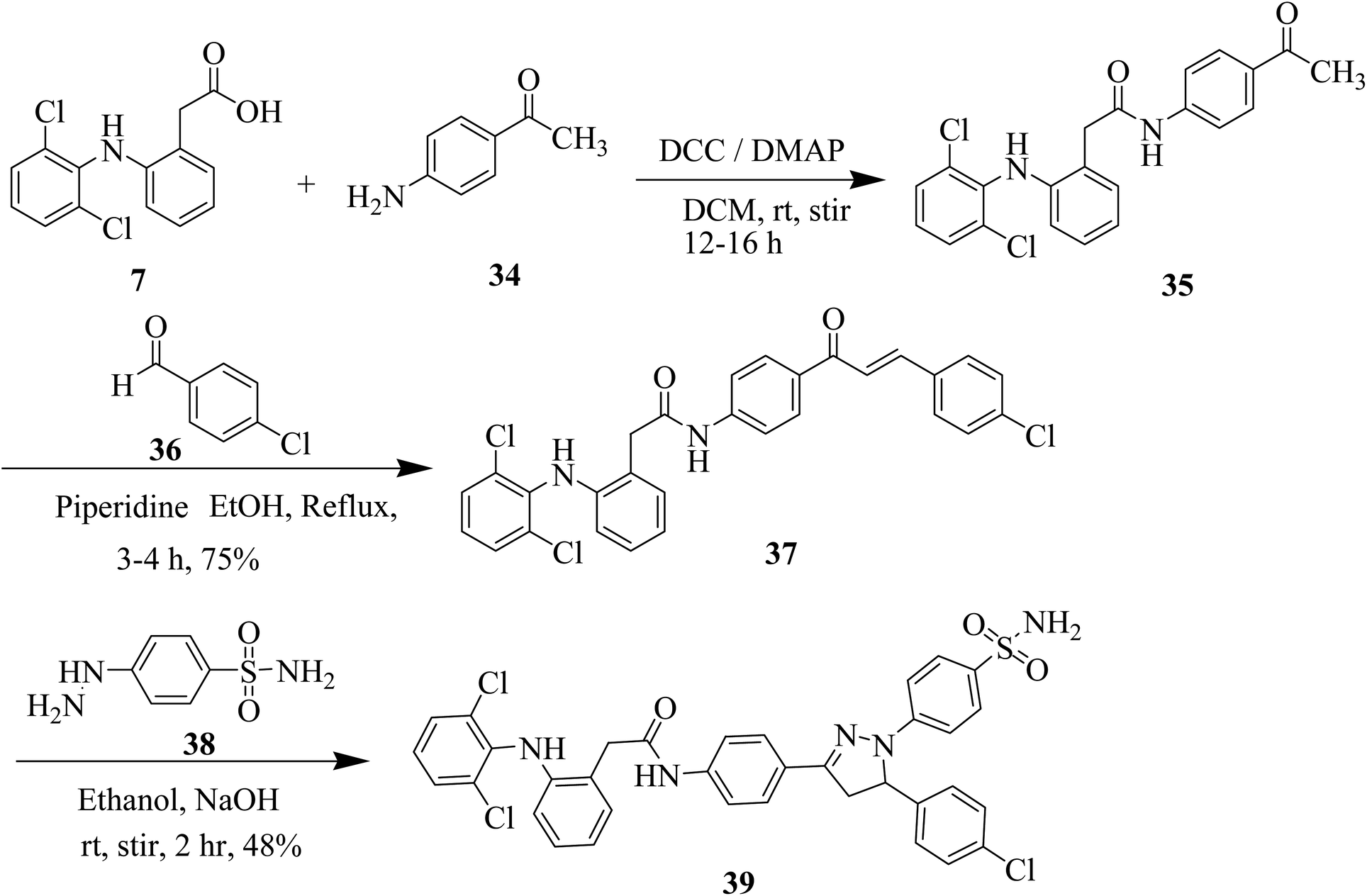 | ||
| Scheme 4 Synthesis of pyrazoline derivative of diclofenac 39. | ||
2.3 In vitro pharmacology
Diclofenac is a non-selective NSAID used for the inhibition of cyclooxygenase (COX) as a pain reliever. In current study, we modified the diclofenac structure to obtain compounds with multiple targeting properties related to AD including neuroinflammation. The standard reported literature screening assay procedures were used for all inhibition bioassays. The inhibitory activities of the synthesized derivatives toward macromolecules inhibition are listed in Tables 1 and 2. Diclofenac sodium, celecoxib, zileuton, donepezil and safinamide were used as reference drugs. Conversion of 4-phenylthiosemicarbazide (11) to 1,2,4-triazole-3-thiol (12) was inspired by the typical 1,2-disubstituted pattern of celecoxib like selective COX-2 inhibitors. Interestingly as per our expectations, 1,2,4-triazole-3-thiol (12) showed improved COX-2 inhibition profile (IC50 = 0.69 μM) better than reference diclofenac (Table 1). However, as compared with triazole 12, intermediate thiosemicarbazide derivative (11) showed good inhibition toward eeAChE, eqBChE, MAO-A and MAO-B (Table 2). Continued with triazole 12 as lead compound, we modified it by considering the 3D structures of macromolecules. Synthesis of Mannich base 15 containing sulfonamide moiety exhibited improved COX-2 and 5-LOX inhibition compared to lead 12 (Table 1). Incorporation of 3-methylene unit linker and pyrimidine/pyrrolidine/phenothiazine cores resulted in no inhibition of COX-2 in tested concentration. This showed that COX-2 binding pocket can accommodate only compounds with less bulky substituents. Compared with lead compound 12, pyrazoline-sulfonamide derivative 39 was able to show a comparable COX-2 inhibition profile (IC50 = 0.60 μM). However, compound 39 showed good 5-LOX inhibition with IC50 value of 0.98 μM.| Comp. | IC50 (μM) ± SEM | |
|---|---|---|
| COX-2 | 5-LOX | |
| a NA = no activity found in tested concentration. | ||
| 11 | 1.29 ± 0.11 | 11.38 ± 1.03 |
| 12 | 0.69 ± 0.13 | 75.51 ± 1.57 |
| 15 | 0.16 ± 0.04 | 3.42 ± 0.21 |
| 26 | NAa | 35.03 ± 1.71 |
| 27 | NA | 29.81 ± 1.03 |
| 28 | NA | 21.64 ± 1.19 |
| 29 | NA | 14.21 ± 1.08 |
| 30 | NA | 39.54 ± 1.55 |
| 31 | NA | 51.29 ± 1.37 |
| 32 | NA | 48.33 ± 1.42 |
| 33 | 23.01 ± 0.92 | 9.11 ± 0.08 |
| 39 | 0.60 ± 0.03 | 0.98 ± 0.01 |
| Diclofenac | 4.80 ± 0.11 | — |
| Celecoxib | 0.05 ± 0.01 | — |
| Zileuton | 0.65 ± 0.04 | |
| Comp. | IC50 (μM) ± SEM | SI (IC50 eqBChE/IC50 eeAChE) | IC50 (μM) ± SEM | |||
|---|---|---|---|---|---|---|
| eeAChE | eqBChE | hMAO-A | hMAO-B | SI (IC50 hMAO-A/IC50 hMAO-B) | ||
| 11 | 0.79 ± 0.08 | 29.55 ± 1.04 | 37.4 | 20.28 ± 0.11 | 0.081 ± 0.009 | 250.4 |
| 12 | 2.58 ± 0.41 | 56.39 ± 1.28 | 21.8 | 11.28 ± 1.01 | 13.07 ± 1.22 | 0.8 |
| 15 | 0.11 ± 0.01 | 2.99 ± 0.33 | 27.2 | 1.21 ± 0.07 | 0.89 ± 0.33 | 1.3 |
| 26 | 0.53 ± 0.01 | 0.94 ± 0.11 | 1.8 | 2.36 ± 0.73 | 8.28 ± 0.01 | 0.3 |
| 27 | 0.47 ± 0.02 | 11.22 ± 0.98 | 23.9 | 1.44 ± 0.04 | 10.08 ± 0.07 | 0.1 |
| 28 | 0.10 ± 0.07 | 36.01 ± 1.31 | 360.1 | 2.39 ± 0.21 | 15.21 ± 0.01 | 0.1 |
| 29 | 0.09 ± 0.01 | 31.89 ± 1.33 | 354.3 | 1.26 ± 0.09 | 9.13 ± 0.03 | 0.1 |
| 30 | 0.06 ± 0.01 | 9.55 ± 1.01 | 159.1 | 0.91 ± 0.03 | 1.32 ± 0.01 | 0.7 |
| 31 | 0.08 ± 0.01 | 48.10 ± 1.51 | 601.2 | 4.63 ± 0.31 | 0.024 ± 0.001 | 193 |
| 32 | 0.39 ± 0.08 | 3.39 ± 0.83 | 8.7 | 6.48 ± 0.39 | 0.106 ± 0.004 | 61.1 |
| 33 | 0.05 ± 0.00 | 5.89 ± 1.00 | 117.8 | 8.55 ± 0.26 | 0.13 ± 0.01 | 65.7 |
| 39 | 0.03 ± 0.01 | 0.91 ± 0.16 | 30.33 | 0.61 ± 0.08 | 0.010 ± 0.001 | 61 |
| Donepezil | 0.05 ± 0.01 | 5.4 ± 0.27 | 108.0 | — | — | — |
| Safinamide | — | — | — | 6.75 ± 0.44 | 0.025 ± 0.003 | 270 |
The Ellman method was used to assess the inhibitory potential of synthesized derivatives against eeAChE and eqBChE. Structural modification on diclofenac derivative 12 further increases eeAChE/eqBChE activity. Intermediate thiosemicarbazide 11 emerged as improved and selective eeAChE inhibitor compared with lead derivative 12. Excellent inhibition and selectivity results towards eeAChE were obtained by increasing the linker length and with bulky scaffolds. Pyrimidine derivatives 28–30 inhibited eeAChE with a nanomolar concentration of 0.10 μM (100 nM) and 0.09 μM (90 nM) and 0.06 μM (60 nM) respectively. The selectivity index for these compounds were 360, 354 and 159.3 respectively. Pyrrolidine derivative 31 with IC50 value of 0.08 μM for eeAChE and 48.10 μM for eqBChE showed highest selectivity of 601.2 toward eeAChE. While phenothiazine derivative 33 showed the best eeAChE inhibitory capacity with IC50 value of 0.05 μM (Table 2). The data in Table 2 showed the enhancement of inhibitory potentials of modified derivatives of triazole 12. However, pyrazoline derivative 39 synthesized from modification of starting diclofenac 7 emerged as most potent eeAChE inhibitor with IC50 value of 0.03 μM. Pyrazoline derivative 39 was also found most potent towards eqBChE with IC50 of 0.91 μM.
All the synthesized compounds were also assessed for their MAO-A and MAO-B inhibitory activity. Structural analysis of MAO-B showed that the binding pocket is a bipartite (two sub-pockets) elongated hydrophobic cavity divided by Ile199. In vitro enzyme inhibition results presented in Table 2 showed that thiosemicarbazide (11) and pyrrolidine derivatives 31 and 32, phenothiazine analog 33 and pyrazoline derivative 39 inhibit MAO-B in nanomolar concentration. We believe that high activity of these compounds is due to their linear conformation like safinamide (docking section) into the elongated binding site of MAO-B. Compound 31 with IC50 value of 4.63 μM and 0.024 μM toward MAO-A and MAO-B respectively showed excellent selectivity index of 193. Pyrazoline derivative 39 emerged as most potent MAO-B inhibitor with IC50 value of 0.01 μM. However, 39 also inhibited MAO-A in low submicromolar concentration. The selectivity index calculated for 39 is 61.
Good inhibition of MAO-A by dihydropyrimidine derivatives 26–29 is supposed to be due to its curved binding site. Reference drug safinamide is 265 times more potent toward MAO-B compared with MAO-A.
2.4 Toxicity studies
Before submitting the samples for animal studies (in vivo toxicity studies), first of all we predicted the toxicity profile of the selected compounds by using online AdmetSAR software. We found toxicity profiles in Category III. Then we proceeded for in vitro neurotoxicity assay.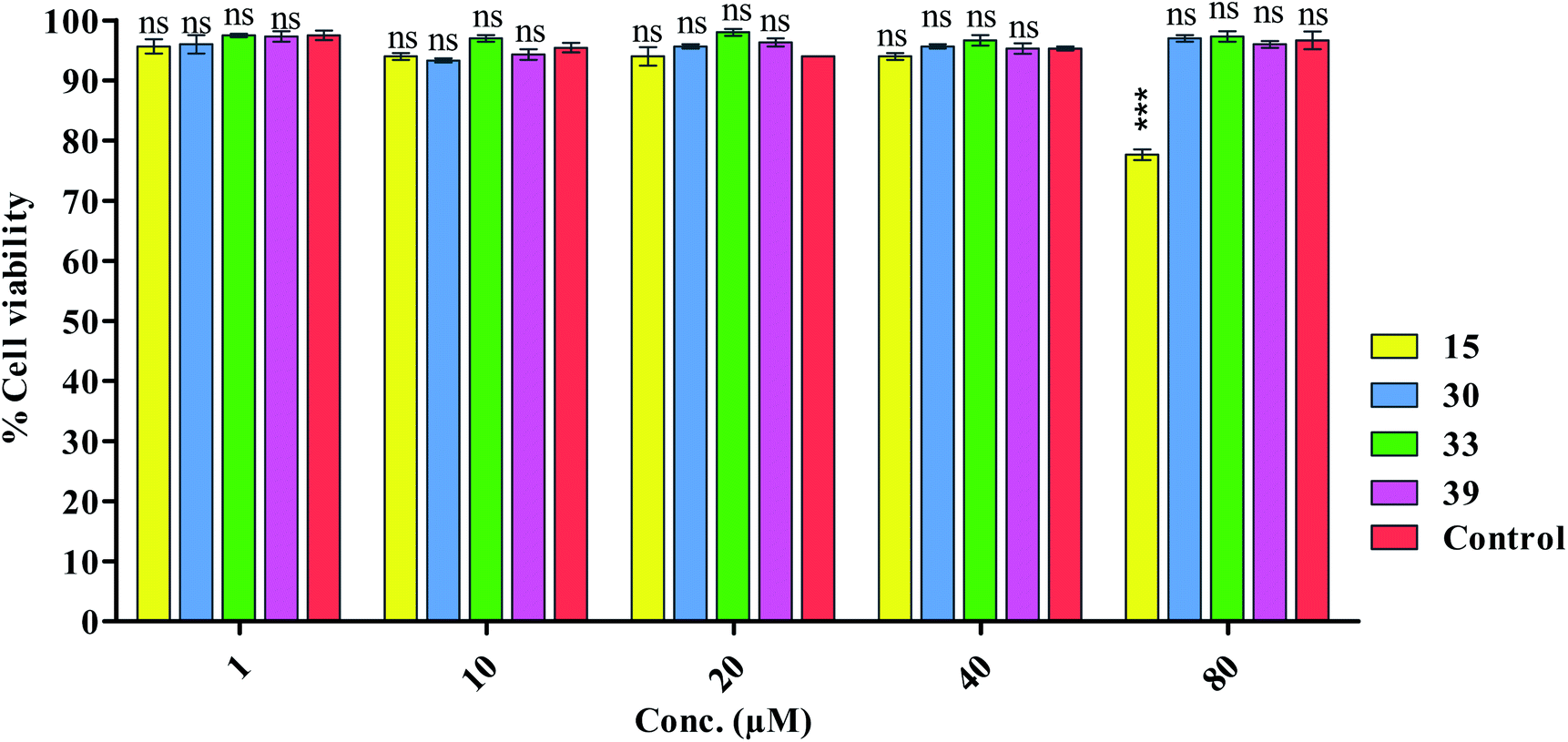 | ||
| Fig. 2 The MTT assay was used to determine the cell viability of the tested compounds at varied doses in the neuroblastoma SH-SY5Y cell line. The Bonferroni test was used after the two-way ANOVA. *P = 0.05, **P = 0.01, ***P = 0.001, ns; non-significant when compared to the control group; n = 3, *P = 0.05, **P = 0.01, ***P = 0.001, ns; non-significant when compared to the control group. | ||
| No. of groups | Animals | Tested synthesized compounds (15, 30, 33 and 39) |
|---|---|---|
| 1 | 8 | 50 |
| 2 | 8 | 100 |
| 3 | 8 | 200 |
| 4 | 8 | 300 |
| 5 | 8 | 400 |
| 6 | 8 | 500 |
| 7 | 8 | 1000 |
| 8 | 8 | 2000 |
2.5 PAMPA BBB assay
The penetration of the blood–brain barrier (BBB) is a crucial concern in the development of Alzheimer's disease (AD) therapies. Using previously reported procedure, we performed a parallel artificial membrane permeation assay (PAMPA).26,31,32 We selected all the four compounds i.e. 15, 30, 33 and 39 for this purpose. All the selected compounds concomitantly inhibited the tested enzymes in nanomolar range. Table 4 summarizes the results of the PAMPA BBB penetration, and all the tested compounds were found to be BBB penetrant. The existence of hydrophobic functional groups could explain these observations.| Compounds label | Permeability (PAMPA-BBB)a Pe(tested) (10−6 cm s−1) | Prediction (PAMPA-BBB) (CNS+b, CNS−)c |
|---|---|---|
| a Data represent are the assay mean for the marketed drugs (n = 3).b ‘CNS+’ (prediction of high BBB permeation); Pe (10−6 cm s−1) > 4.39.c ‘CNS−’ (prediction of low BBB permeation); Pe (10−6 cm s−1) < 1.78. | ||
| Evaluation of Pe (10−6 cm s−1) for the test compounds and standard | ||
| 15 | 6.36 ± 0.30 | CNS+ |
| 30 | 8.21 ± 0.15 | CNS+ |
| 33 | 9.05 ± 0.20 | CNS+ |
| 39 | 7.55 ± 0.14 | CNS+ |
| Donepezil | 6.55 ± 0.10 | CNS+ |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Validation of the model by seven commercial drugs | ||
| Diazepam | 15.30 ± 0.12 | CNS+ |
| Atenolol | 0.75 ± 0.10 | CNS− |
| Alprazolam | 5.60 ± 0.21 | CNS+ |
| Lomefloxacin | 1.12 ± 0.09 | CNS− |
2.4 Molecular docking studies
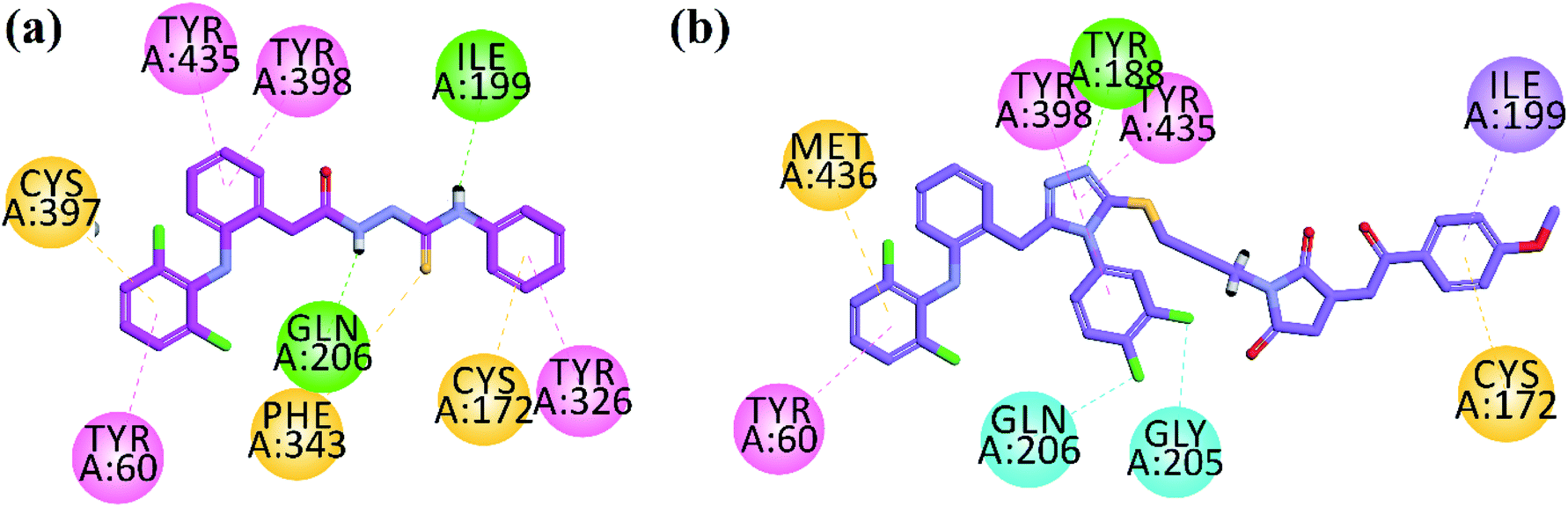
To explore binding interaction of synthesized compounds with the active site of target enzymes, docking experiments were performed. All the synthesized compounds were docked into the active sites of AChE, BChE, MAO-A, MAO-B, COX-2 and 5-LOX with accession code 1EVE, 4BDS, 2Z5V, 2V5Z, 1CX2 and 6N2W. We assessed docking reliability by re-docking native ligands prior to predicting the docking poses of synthesized compounds. The measured RMSD values were within acceptable ranges (<2.0 Å).Dual binding site inhibitors of AChE interact with the catalytic active site (CAS) and peripheral anionic site (PAS). Trp84, Tyr130, Phe330 and Phe331 are important amino acid residues present in CAS. While Tyr70, Asp72, Tyr121, Trp279 and Tyr334 are important residues in PAS. Ser200, Glu327, His440 are present in catalytic triads, Phe288 and Phe290 in acyl pocket and Gly118, Gly119 and Ala201 in oxyanion hole of AChE. Compound 33 (IC50 = 0.05 μM) is supposed to inhibit AChE by interacting with active site via formation of six hydrophobic interactions (π–π and π–sulfur type) and one halogen interaction between chlorine atom and Tyr334 (Fig. 3a). Potent compound 39 showed bifurcated π–π, H-bond and π–sulfur interactions with important residues present in CAS (Trp84, Phe330, Phe331) and PAS (Tyr121, Trp279, Tyr334) of AChE (Fig. 3b).
 | ||
| Fig. 3 2D interaction plot of synthesized compounds 33 (a) and 39 (b) in the active site of AChE. | ||
MAO-B binding pocket is a bipartite (two sub-pockets) elongated hydrophobic cavity divided by Ile199. The entrance cavity contains Pro102, Pro104, Trp119, Leu164, Ile198, Ile199, Thr201, Ile316, and Tyr326. While substrate cavity is formed by Ser59, Tyr60, Cys172, Gln206, Tyr398 and Tyr435.33,34
In vitro MAO-B inhibition results (Table 2) demonstrated that the high inhibition potentials of thiosemicarbazide analog 11 and pyrrolidine derivatives 31 and 32 may be due to their linear conformation like safinamide (docking section) into the elongated binding site of MAO-B. The three-dimensional (3D) overlaid diagrams of 11 and 31 and reference drug safinamide are shown in Fig. 4a and b. Two-dimensional (2D) interaction plots shown in Fig. 5 revealed that the ligand (11)–enzyme complex is stabilized by forming hydrogen bonds, π–π and π–sulfur, interactions. The entrance cavity residues (Ile199/Tyr326) and substrate cavity residues Tyr60, Cys172, Gln206, Tyr398 and Tyr435 are involved in these types of interactions (Fig. 5a). While residues Tyr188, Ile199, Tyr60, Cys172, Gly205, Gln206, Tyr398 and Tyr435 are involved in stabilizing ligand (31)–enzyme complex (Fig. 5b).
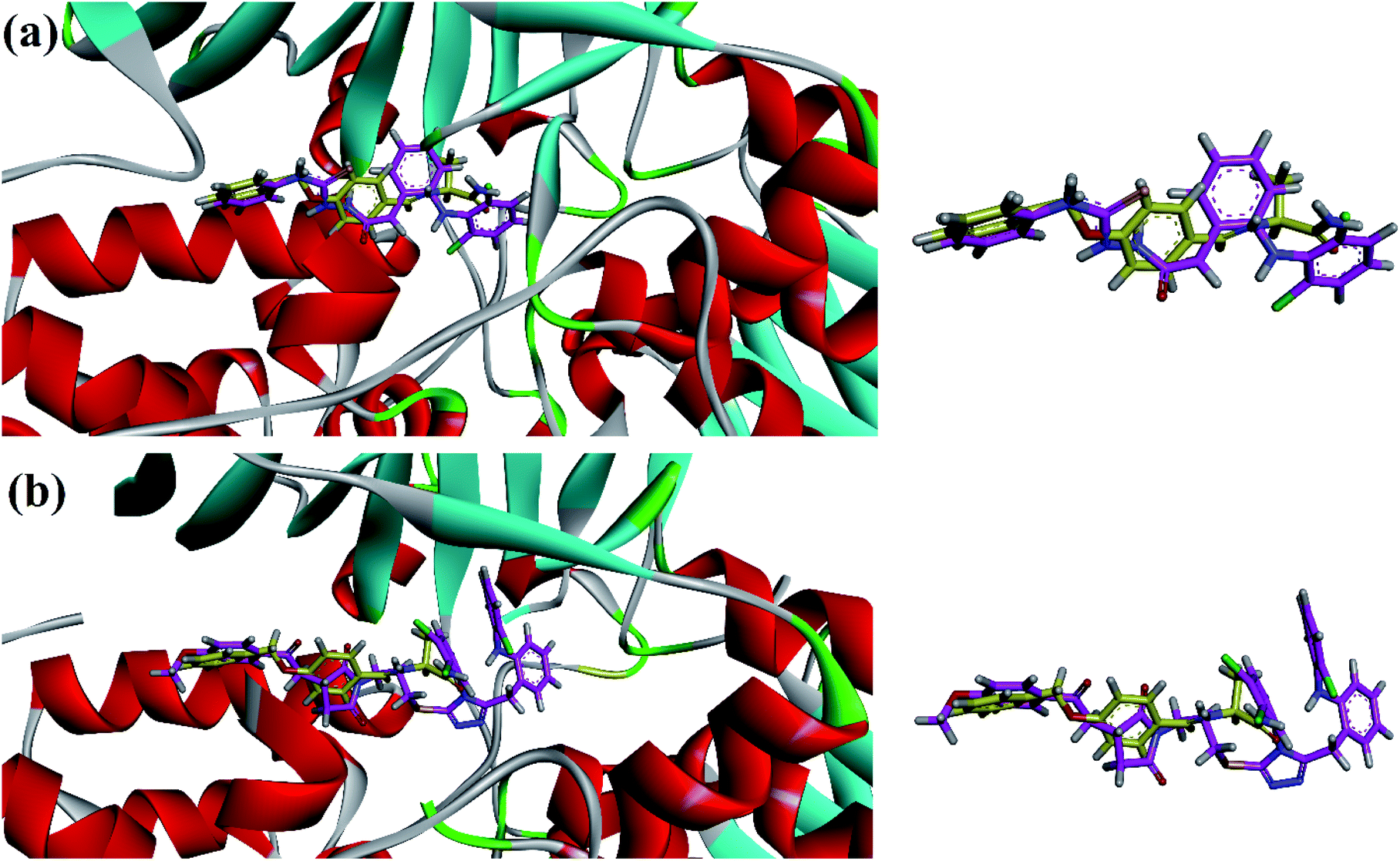 | ||
| Fig. 4 3-D superposed diagram of compound 11 (a), 31 (b) and reference drug safinamide (yellow sticks) into the binding site of MAO-B. | ||
 | ||
| Fig. 5 2D interaction plots of synthesized compound 11 (a) and 31 (b) in the active site of MAO-B (PDB ID = 2V5Z). | ||
Superposed binding poses of most active MAO-B pyrazoline analog 39 over safinamide demonstrated that pyrazoline core oriented toward entrance cavity while diclofenac part toward substrate cavity (Fig. 6a). Very high inhibition potential may be due six hydrogen bond interactions with Arg42, Cys197, Ile19, 9Gln206, Tyr326 and Trp388 (Fig. 6b).
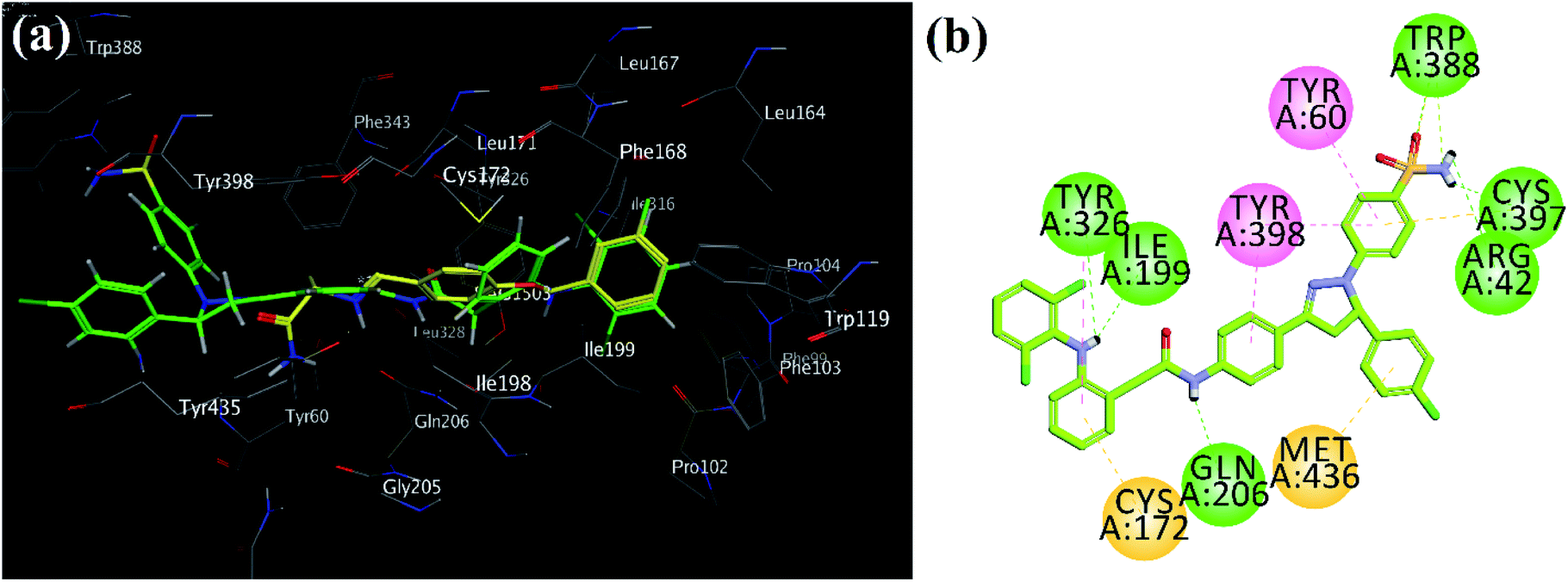 | ||
| Fig. 6 (a) MOE generated 3D superpose diagram of compound 39 (green) and reference drug safinamide (yellow sticks) into the binding site of MAO-B; (b) 2D interaction plots of synthesized compound 39 in the active site of MAO-B (PDB ID = 2V5Z). | ||
3 Experimental
3.1 General
All reagents and solvents were purchased from Sigma Aldrich and were used without any purification. Diclofenac acid was purchased from Alfa Aesar. 1H NMR and 13C NMR spectra were recorded in deuterated solvent on Bruker 400 MHz spectrometers, with TMS as an internal reference. Melting points were determined in open capillary using Stuart melting point apparatus (SMP10). TLC plates pre-coated with silica gel 60F254 were used to monitor the progress of all reactions. The synthesized compounds that were more than 95% pure by HPLC (Schimadzu) were submitted for biological evaluation. MeOH/Water (1% CH3COONH4, w/v) was used as isocratic solvent system in the 70:30 ration with a flow rate of 1.0 mL min−1.
3.2 General method for the synthesis of diclofenac methyl ester (8)
Diclofenac acid 7 (20 mmol) was stirred at 0 °C in dry acetone followed by addition of 1.5 equivalent of thionyl chloride. Reaction mixture was stirred initially for two hours at 0 °C and then 1 hour at an ambient temperature then refluxed for three hours to yield acid chloride (8).White crystalline solid; Rf = 0.69 (n-hexane:ethyl acetate 4:1); yield 86%; mp 98–99 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.44 (d, J = 8.1 Hz, 2H, Ar–H), 7.25 (dd, J = 7.4, 2.4 Hz, 1H, Ar–H), 7.08 (t, J = 8.1 Hz, 1H, Ar–H), 6.98 (td, J = 7.7, 3.8 Hz, 1H, Ar–H), 6.94 (s, 1H, NH), 6.76 (t, J = 7.5 Hz, 1H, Ar–H), 6.22 (d, J = 7.8 Hz, 1H, Ar–H), 3.86 (s, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 172.41, 145.05, 143.37, 130.89, 130.61, 129.44, 127.30, 125.46, 123.93, 119.26, 116.29, 52.20, 37.93.
3.3 General method for the synthesis of diclofenac hydrazide (9)
A mixture of diclofenac acid chloride (8, 30 mmol) and hydrazine hydrate (50 mmol) was refluxed in absolute ethanol (30 mL) for 6–7 h. Completion of reaction was monitored through TLC, the solid product was collected through filtration, dried and recrystallized from ethanol.White amorphous solid; Rf = 0.16 (n-hexane:ethyl acetate 4:1); yield 84% mp 134–136 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H, NH), 7.44 (d, J = 8.1 Hz, 2H, Ar–H), 7.19–7.09 (m, 2H, Ar–H), 6.98 (t, J = 7.6 Hz, 1H, Ar–H), 6.94 (s, 1H, NH), 6.79 (t, J = 7.5 Hz, 1H, Ar–H), 6.22 (d, J = 7.8 Hz, 1H, Ar–H), 4.28 (s, 2H, NH2), 3.44 (s, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.2, 143.6, 143.3, 130.6, 129.4, 129.1, 127.8, 125.4, 123.9, 123.2, 116.9, 38.39.
3.4 General method for the synthesis of thiosemicarbazide (11)
An equimolar mixture of diclofenac hydrazide (20 mmol) and aryl isothiocyanate (10, 20 mmol) was refluxed in dry acetone for 5–6 h. After completion of reaction, the reaction mixture was cooled, solid product was thus separated out, dried and was recrystallized from ethanol.White solid; Rf = 0.46 (n-hexane:ethyl acetate 4:1); yield 81%; mp 195–197 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (br s, 1H, NH), 9.97 (br s, 1H, NH), 9.51 (br s, 1H, NH), 7.97 (br s, 1H, NH), 7.81 (s, 1H, Ar–H), 7.60 (d, J = 8.7 Hz, 1H, Ar–H), 7.53 (d, J = 8.1 Hz, 2H, Ar–H), 7.40 (d, J = 9.0 Hz, 1H, Ar–H), 7.25 (d, J = 7.1 Hz, 1H, Ar–H), 7.18 (t, J = 8.1 Hz, 1H, Ar–H), 7.07 (t, J = 7.1 Hz, 1H, Ar–H), 6.88 (t, J = 7.2 Hz, 1H, Ar–H), 6.31 (d, J = 7.9 Hz, 1H, Ar–H), 3.70 (s, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 181.9, 169.8, 143.6, 143.4, 140.1, 131.8, 130.6, 129.4, 129.1, 128.9, 127.8, 125.5, 124.8, 124.0, 123.9, 123.3, 118.8, 116.9, 38.2. HRMS (EI) calcd for C21H16Cl4N4OS [M+]: 511.9799 found 511.9783.
3.5 General method for the synthesis of 5-(2-(2,6-dichlorophenylamino)benzyl)-4-(3,4 dichlorophenyl)-4H-1,2,4-triazole-3-thiol (12)
Thiosemicarbazide 11 (15 mmol) was added dropwise to the stirred solution of NaOH (5%, 30 mL). The reaction mixture was allowed to reflux for 3–4 h. After cooling, the reaction mixture was filtered, and the filtrate was acidified with 6 N HCl to pH 2–3. The precipitated solid was filtered, washed thoroughly with water and recrystallized with ethanol.Peach solid; Rf = 0.59 (n-hexane:ethyl acetate 4:1); yield 79%; mp 254–256 °C. 1H NMR (500 MHz, DMSO-d6) δ 13.89 (s, 1H, SH), 8.13 (s, 1H, NH), 7.79 (d, J = 9.1 Hz, 1H, Ar–H), 7.71 (d, J = 2.3 Hz, 1H, Ar–H), 7.51 (d, J = 8.0 Hz, 2H, Ar–H), 7.40 (dd, J = 8.5, 2.4 Hz, 1H, Ar–H), 7.21 (t, J = 8.0 Hz, 1H, Ar–H), 7.01 (td, J = 9.0, 1.2, Hz, 1H, Ar–H), 6.86 (dd, J = 8.6, 1.3 Hz, 1H, Ar–H), 6.73 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 6.14 (d, J = 8.0 Hz, 1H, Ar–H), 3.97 (s, 2H, CH2). 13C NMR (125 MHz, DMSO-d6) δ; 168.4, 150.8, 143.3, 137.5, 133.9, (132.9)2, 132.0, 131.8, 131.6, 131.1, (129.5)3, 128.3, (126.8)2, 123.1, 120.6, 115.9, 28.6. HRMS (EI) calcd for C21H14Cl4N4S [M+]: 493.9693 found 493.9679.
3.6 General method for the synthesis of 4-((5-(2-(2,6-dichlorophenylamino)benzyl)-4-(3,4-dichlorophenyl)-4H-1,2,4-triazol-3-ylthio)methylamino)benzenesulfonamide (15)
At 0 °C, sulfanilamide (14) was stirred for 30 minutes in methanol in the presence of 5 mmol formalin (7, 1 equiv.). Compound 12 in methanol was then added and the reaction mixture was refluxed for three hours after some time. Using thin layer chromatography, reaction progress was noted. After the completion of reaction, the reaction mixture was kept in the refrigerator overnight. The formed product was filtered, washed, and recrystallized using ethanol.Off-white solid; Rf = 0.52 (DCM:MeOH 10:1); yield 56%; 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.80 (brs, 1H, NH), 7.92 (d, 1H, J = 8 Hz, ArH), 7.81 (d, 2H, J = 8 Hz, ArH), 7.69 (brs, 1H, NH), 7.50–7.46 (m, 4H, ArH), 7.43–7.39 (m, 2H, ArH), 7.31–7.26 (m, 3H, ArH), 7.21–7.19 (m, 2H, ArH), 5.95 (brs, 2H, NH2), 4.86 (s, 2H, CH2), 3.97 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6), δ. 166, 150.2, 144.3, 138.6, 136.3, 134.6, (133.4)2, 130.7, 130.0, (129.7)3, 128.9, (127.8)3, (126.9)2, 126.0, 125.7, 122.1, 120.4, 119.2, (112.7)2, 57.4, 21.5. LC-MS: m/z = 681.4 [M + H]+; analysis calculated for C28H22Cl4N6O2S2. C, 49.42; H, 3.26; Cl, 20.84; N, 12.35; O, 4.70; S, 9.42. Observed; C, 49.49; H, 3.24; N, 12.32.
3.7 General method for the synthesis of intermediate 17
Diclofenac intermediate 12 (20 mmol) and 1,3 dibromopropane 16 were stirred in the presence of K2CO3/KI in acetone, the mono bromo diclofenac triazole intermediate 17 was obtained. On completion of reaction, the residue left after evaporation of solvent was dissolved in NaHCO3 solution and extraction was carried out with ethyl acetate (4 × 30 mL) followed by washing the combined extract with brine (2 × 20 mL) then dried over anhydrous Na2SO4. Solvent was removed under reduced pressure. The compound 17 obtained was used without further purification in next step.3.8 General method for the synthesis of compounds 26–33
A solution of monobromo intermediate 17 (5 mmol), R-XH (18–25, 10 mmol) and K2CO3 in DMF were stirred for 3 h. After completion of reaction, the mixture was poured into water and extracted with ethyl acetate (4 × 20 mL), washed with water then brine and dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude products (26–33) were purified by using silica gel chromatography (DCM/MeOH; 10:1).
:1); yield 61%; mp 195–197 °C. HPLC purity = 97.9% (C18 RP, acetonitrile/H2O 97:3), TR = 13.7 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.73 (brs, 1H, NH), 8.32 (brs, 1H, NH), 7.89 (d, 1H, ArH), 7.62 (s, 1H, ArH), 7.43 (d, 1H, ArH), 6.98 (d, 2H, ArH), 6.85 (d, 2H, ArH), 6.65 (t, 1H, ArH), 6.61 (d, 2H, ArH), 6.57 (m, 4H, ArH), 5.17 (s, 1H, CH), 4.04 (s, 2H, CH2), 3.83 (s, 3H, OCH3), 2.95 (t, 2H, CH2), 2.85 (t, 2H, CH2), 2.43 (m, 2H, CH2), 2.31 (s, 3H, CH3), 2.35 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6), δ. 1.97.1, 164.3, 161.6, 158.8, 155.2, 146.5, 137.7, 135.3, 134.7, (132.2)2, 131.4, (130.9)2, 130.1, (129.8)3, 128.8, 128.1, (127.2)2, 126.9, 126.2, 120.4, 119.5, 118.3, (114.4)2, 111.5, 55.5, 51.3, 36.3, 29.3, 27.7, 25.4, 21.1, 15.6. LC-MS: m/z = 813.6 [M + H]+; analysis calculated for C38H34Cl4N6O2S2. C, 56.16; H, 4.22; Cl, 17.45; N, 10.34; O, 3.94; S, 7.89. Observed; C, 56.21; H, 4.21; N, 10.31.:1); yield 63%; mp 189–195 °C. HPLC purity = 96.3% (C18 RP, acetonitrile/H2O 97:3), TR = 7.1 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.69 (brs, 1H, NH), 8.28 (brs, 1H, NH), 7.86 (d, 1H, ArH), 7.61 (s, 1H, ArH), 7.45 (d, 1H, ArH), 7.04 (m, 5H, ArH), 6.95 (d, 2H, ArH), 6.63 (t, 1H, ArH), 6.58 (m, 4H, ArH), 5.19 (s, 1H, CH), 4.02 (s, 2H, CH2), 2.93 (t, 2H, CH2), 2.86 (t, 2H, CH2), 2.44 (m, 2H, CH2), 2.33 (s, 3H, CH3), 2.37 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6), δ. 195.3, 164.7, 162.5, 158.2, 155.5, 146.6, 137.3, 136.3, 135.5, (132.4)2, 130.7, (129.5)2, (128.8)2, (128.1)3, (127.3)2, 126.2, 125.3, 121.3, 120.5, 117.9, 115.5, 112.3, 52.5, 37.3, 29.6, 27.2, 25.1, 21.5, 15.1. LC-MS: m/z = 783.63 [M + H]+; analysis calculated for C37H32Cl4N6OS2. C, 56.78; H, 4.12; Cl, 18.12; N, 10.74; O, 2.04; S, 8.19. Observed; C, 56.84; H, 4.10; N, 10.71.:1); yield 59%; mp 183–189 °C. HPLC purity = 97.7% (C18 RP, acetonitrile/H2O 97:3), TR = 13.2 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.69 (brs, 1H, NH), 8.30 (brs, 1H, NH), 7.88 (d, 1H, ArH), 7.60 (s, 1H, ArH), 7.41 (d, 1H, ArH), 6.94 (d, 2H, ArH), 6.82 (d, 2H, ArH), 6.67 (t, 1H, ArH), 6.63 (d, 2H, ArH), 6.56 (m, 4H, ArH), 5.18 (s, 1H, CH), 4.01 (s, 2H, CH2), 2.94 (t, 2H, CH2), 2.83 (t, 2H, CH2), 2.44 (m, 2H, CH2), 2.37 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.34 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6), δ. 197.3, 164.7, 162.5, 158.2, 155.5, 146.6, 137.3, 136.3, (135.5)2, (132.4)2, 131.7, 130.7, (129.7)2, (128.8)3, 128.1, (127.3)2, 126.2, 121.3, 120.5, 117.9, 115.5, 112.3, 52.5, 37.3, 29.6, 27.2, 25.1, 21.5, 15.1. LC-MS: m/z = 797.65 [M + H]+; analysis calculated for C38H34Cl4N6OS2. C, 57.29; H, 4.30; Cl, 17.80; N, 10.55; O, 2.01; S, 8.05. Observed: C, 57.34; H, 4.28; N, 10.52.:1); yield 67%; mp 192–198 °C. HPLC purity = 99.2% (C18 RP, acetonitrile/H2O 98:2), TR = 15.5 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.72 (brs, 1H, NH), 8.33 (brs, 1H, NH), 7.89 (d, 1H, ArH), 7.64 (s, 1H, ArH), 7.43 (d, 1H, ArH), 6.96 (d, 2H, ArH), 6.86 (d, 2H, ArH), 6.68 (t, 1H, ArH), 6.64 (d, 2H, ArH), 6.57 (m, 4H, ArH), 5.19 (s, 1H, CH), 4.05 (s, 2H, CH2), 2.96 (t, 2H, CH2), 2.84 (t, 2H, CH2), 2.46 (m, 2H, CH2), 2.34 (s, 3H, CH3), 2.33 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6), δ. 196.6, 165.2, 163.1, 157.2, 155.5, 146.6, 137.5, 136.1, 135.9, (133.4)2, 132.7, 130.2, (129.2)3, 128.8, (128.0)3, (127.8)2, 126.0, 125.6, (122.8)2, 121.5, 118.2, 116.3, 112.3, 55.3, 36.3, 29.1, 27.6, 26.1, 20.5, 16.3. LC-MS: m/z = 828.62 [M + H]+; analysis calculated for C37H31Cl4N7O3S2. C, 53.70; H, 3.78; Cl, 17.13; N, 11.85; O, 5.80; S, 7.75. Observed; C, 53.62; H, 3.80; N, 11.88.:1); yield 65%; mp 190–196 °C. HPLC purity = 99.4% (C18 RP, acetonitrile/H2O 97:3), TR = 11.5 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.70 (brs, 1H, NH), 8.29 (brs, 1H, NH), 7.85 (d, 1H, ArH), 7.61 (s, 1H, ArH), 7.42 (d, 1H, ArH), 6.91 (d, 2H, ArH), 6.85 (d, 2H, ArH), 6.67 (t, 1H, ArH), 6.63 (d, 2H, ArH), 6.54 (m, 4H, ArH), 5.15 (s, 1H, CH), 4.03 (s, 2H, CH2), 2.93 (t, 2H, CH2), 2.80 (t, 2H, CH2), 2.42 (m, 2H, CH2), 2.32 (s, 3H, CH3), 2.30 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6), δ. 1.95.8, 165.5, 163.4, 157.2, 156.4, 146.4, 138.2, 136.8, 135.3, (133.5)2, 132.2, 131.4, (130.9)2, 130.2, (129.6)3, (128.5)2, (127.7)2, 126.5, 125.3, 120.2, 119.4, 118.5, 116.3, 113.1, 51.8, 36.6, 30.6, 27.7, 25.5, 20.3, 15.9. LC-MS: m/z = 817.07 [M + H]+; analysis calculated for C37H31Cl5N6OS2. C, 54.39; H, 3.82; Cl, 21.70; N, 10.29; O, 1.96; S, 7.85. Observed; C, 54.33; H, 3.84; N, 10.32.:1); yield 51%; mp 182–188 °C. HPLC purity = 96.8% (C18 RP, acetonitrile/H2O 98:2), TR = 13.8 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 8.18 (brs, 1H, NH), 7.77 (d, 2H, ArH), 7.44 (d, 1H, ArH), 7.61 (s, 1H, ArH), 7.41 (d, 1H, ArH), 6.92 (d, 2H, ArH), 6.85 (d, 2H, ArH), 6.57 (t, 1H, ArH), 6.54 (m, 4H, ArH), 4.03 (s, 2H, CH2), 3.72 (s, 3H, OCH3), 3.32 (m, 1H, CH), 3.13 (dd, J = 18.4 Hz, 5.3 Hz, 1H), 2.93 (t, 2H, CH2), 2.80 (t, 2H, CH2), 2.73 (dd, J = 18.4 Hz, 9.2, Hz, 1H), 2.42 (m, 2H, CH2), 2.29 (dd, J = 5.72, 18.2 Hz, 1H), 2.17 (dd, J = 8.2, 18.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6), δ. 202.2, 176.5, 167.2, 163.5, 139.7, 137.4, 136.2, (133.5)2, 131.1, (130.3)2, (129.7)3, 129.3, 128.9, 128.4, (127.8)2, 127.5, 127.1, 126.7, 126.2, 125.9, 121.3, 119.9, 118.2, (113.3)2, 56.5, 41.2, 38.7, 33.1, 32.6, 26.3, 20.5. LC-MS: m/z = 784.55 [M + H]+; analysis calculated for C37H31Cl4N5O4S. C, 56.72; H, 3.99; Cl, 18.10; N, 8.94; O, 8.17; S, 4.09. Observed; C, 56.78; H, 3.97; N, 8.92.:1); yield 47%; mp 195–199 °C. HPLC purity = 99.3% (C18 RP, acetonitrile/H2O 98:2), TR = 13.3 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 8.18 (brs, 1H, NH), 7.86 (d, 2H, ArH), 7.44 (d, 1H, ArH), 7.61 (s, 1H, ArH), 7.49 (t, 1H, ArH), 7.41 (d, 1H, ArH), 7.31 (d, 2H, ArH), 6.85 (d, 2H, ArH), 6.57 (t, 1H, ArH), 6.54 (m, 4H, ArH), 4.03 (s, 2H, CH2), 3.32 (m, 1H, CH), 3.13 (dd, J = 18.4 Hz, 5.3 Hz, 1H), 2.93 (t, 2H, CH2), 2.80 (t, 2H, CH2), 2.73 (dd, J = 18.4 Hz, 9.2, Hz, 1H), 2.42 (m, 2H, CH2), 2.29 (dd, J = 5.72, 18.2 Hz, 1H), 2.17 (dd, J = 8.2, 18.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6), δ. 201.4, 175.3, 165.9, 162.4, 138.4, 137.3, 135.5, (133.2)2, 130.7, (129.3)2, 129.0, 128.9, 128.6 (128.2)2, (127.9)2, 127.1, 126.9, 126.6, 126.0, 125.3, 120.5, 119.3, 118.8, (113.6)2, 41.6, 36.2, 33.8, 32.3, 25.5, 20.2. LC-MS: m/z = 753.52 [M + H]+; analysis calculated for C36H27Cl4N5O3S. C, 57.38; H, 3.88; Cl, 18.82; N, 9.29; O, 6.37; S, 4.26. Observed; C, 57.45; H, 3.86; N, 9.26.:MeOH 10:1); yield 49%; mp 191–197 °C. HPLC purity = 98.3% (C18 RP, acetonitrile/H2O 97:3), TR = 15.8 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 7.70 (brs, 1H, NH), 7.52–7.49 (m, 4H, ArH), 7.41–7.39 (m, 1H, ArH), 7.31–7.26 (m, 3H, ArH), 7.21–7.19 (m, 2H, ArH), 7.11–7.08 (m, 4H, ArH), 6.94–6.89 (m, 4H, ArH), 4.34 (s, 2H, CH2), 3.51 (t, 2H, J = 4.8 Hz, CH2), 3.25 (t, 2H, J = 5.6 Hz, CH2), 1.77–1.75 (m, 2H, CH2), 1.45–1.43 (m, 2H, CH2), 13C NMR (100 MHz, DMSO-d6), δ. 167, 149.5, 143.8, 139.7, 137.2, 136.1, (132.8)2, (129.3)2, 128.9, 128.1, (127.7)3, 127.0, (126.9)2, 126.1, 125.8, 125.2, 124.9, 123.7, 123.0, 121.5, 118.7, 118.3, 117.8, 117.1, 114.3, 66.5, 49.3, 47.6, 37.5, 29.0, 28.1, 19.3. LC-MS: m/z = 736.57 [M + H]+; analysis calculated for C36H27Cl4N5S2. C, 58.78; H, 3.70; Cl, 19.28; N, 9.52; S, 8.72. Observed: C, 58.86; H, 3.68; N, 9.49.3.9 General method for the synthesis of N-(4-acetylphenyl)-2-(2-(2,6-dichlorophenylamino)phenyl)acetamide (35)
The diclofenac 7 (10 mmol) were diluted in DCM 4 mL using an inert condition of nitrogen gas. Subsequent additions of N,N′-dicyclohexylcarbodiimide (DCC, 12 mmol), 1-hydroxy-benzotriazole (HOBt, 1.2 equiv.) and DMAP (2.5 equiv.) were performed and the was stirred for 5 minutes at normal laboratory temperature. 4-Amino acetophenone 34 (10 mmol) was added to the reaction and continued stirring at laboratory temperature for 12–16 h. After 24 h, the reaction was diluted with ethyl acetate and washed with aq. HCl (0.5 M, 20 mL). The extraction with ethyl acetate was repeated three times (50 mL each time). The three organic layers were combined and was added saturated sodium bicarbonate and brine (each 20 mL). The mixture was dried, filtered and the organic solvents were removed with reduced pressure of a rotary evaporator. The crude product 35 was purified by gravity column chromatography eluting with hexane and ethyl acetate solvent system.3.10 General method for the synthesis (E)-N-(4-cinnamoylphenyl)-2-(2-(2,6-dichlorophenylamino)phenyl) acetamide (37)
Compound 37 was synthesized by stirring the mixture of ketone derivative 35, (20 mmol) and para chlorobenzaldehyde (36, 20 mmol) in ethanol using piperidine as a base via Claisen Schmidt condensation reaction to yield chalcone based compound 37.3.11 General method for the synthesis of N-{4-[5-(4-chlorophenyl)-1-(4-sulfamoylphenyl)-4,5-dihydro-1H-pyrazol-3-yl]phenyl}-2-{2-[(2,6-dichlorophenyl)amino]phenyl}acetamide (39)
In 15 mL of ethanol, chalcones-based compound 37 (5 mmol) and 4-hydrazinyl benzene sulfonamide 38 (5 mmol) were stirred for two hours to obtain pyrazoline compound. When the reaction was completed, precipitate was formed, and recrystallization with ethanol afford compound 39.Yellow solid; Rf = 0.54 (DCM:MeOH 10:1); yield 48%; melting point 239–241 °C, HPLC purity = 99.1% (C18 RP, acetonitrile/H2O 98:2), TR = 12.4 min. 1H NMR (400 MHz, DMSO-d6, ppm) δ: 1H NMR (400 MHz, DMSO-d6, ppm) δ: 9.51 (brs, 1H, NH), 8.12 (brs, 1H, NH), 7.90 (d, 2H, J = 8.4 Hz, ArH), 7.69 (d, 2H, J = 8.0 Hz, ArH), 7.61 (d, 2H, J = 8.4 Hz, ArH), 7.55 (d, 2H, J = 8.0 Hz, ArH), 7.40 (d, 2H, J = 7.6 Hz, ArH), 7.21 (d, 2H, J = 8.0 Hz, ArH), 7.16–7.12 (m, 4H, ArH), 7.09 (d, 2H, J = 8.0 Hz, ArH), 6.69 (d, 1H, J = 8.0 Hz, ArH), 5.98 (s, 2H, NH2), 4.66 (dd, 1H, J = 11.7 Hz, 7.5 Hz, CH), 3.79 (s, 2H, CH2), 3.41 (dd, J = 12.0 Hz, 17.4 Hz, 1H, CH), 2.89 (dd, J = 7.8 Hz, 17.1 Hz, 1H, CH); 13C NMR (100 MHz, DMSO-d6), δ. 171.2, 152.3, 147.5, 142.5, 140.9, 139.4, 136.8, 133.1, 130.5, (129.1)4, (128.9)3, (128.4)2, (127.8)2, (127.4)2, (126.8)2, 126.2, (122.4)2, 121.5, 119.9, 118.1, (113.5)2, 54.4, 41.2, 32.5. LC-MS: m/z = 706.05 [M + H]+; analysis calculated for C35H28Cl3N5O3S. C, 59.62; H, 4.00; Cl, 15.09; N, 9.93; O, 6.81; S, 4.55. Observed value: C, 59.56; H, 4.02; N, 9.95.
3.12 In vitro enzyme inhibition studies
| Inhibition (%) = (Abs of control – Abs of test comp/Abs of control) × 100 |
3.13 Blood–brain barrier (BBB) permeation assay
For the BBB penetration experiment, we used the PAMPA-BBB in vitro model. In this test, the synthesized compounds were dissolved in DMSO at a concentration of 10 mM and subsequently diluted with 100 M phosphate buffer at pH 7.4. The donor plate's filter membrane was coated with PBL in dodecane. The plate was then filled with 0.2 mL of test chemical solution. Similarly, the acceptor plate was filled with 0.2 mL of pH 7.4 PBS buffer and gently placed beneath the donor plate to form a “sandwich.” For 20 hours, the plate was maintained at room temperature. The drug deliberation in acceptor, donor, and reference wells was observed using a UV plate reader. The synthesized compound's effective permeability (Pe) was calculated using the formula.1| Pe = [C × In {1 − ((drug)acceptor)/((drug)equilibrium)}] |
3.14 MTT assay for the evaluation of neurotoxicity
The MTT assay was used to examine the effect of the potent synthesized compounds on SH-SY5Y neuroblastoma cells (purchased from the ATCC). The SH-SY5Y cells were first cultivated in 96-well plates at a density of 1 × 105 cells per well in MEM/F12 supplemented with 10% fetal bovine serum, medium (200 μL), for one day at 37 °C under CO2 (5 percent). The cells were then treated for 24 hours with varied doses of the test compounds (ranging from 1–40 μM). Following treatment of the cells with various doses of synthesized compounds and the standard, 20 μL of MTT reagent (5 mg mL−1) was added to each well and incubated for 180 minutes at 37 °C (5 percent CO2). Finally, under the microscope, the crystallization of a purple coloured precipitates were seen, which were solubilized by 1 mL of DMSO. The viability of the cells was measured using a microplate reader at 570 nm. The results were expressed as a percentage growth in each well in comparison to control cells cultivated without the standard/synthesized compounds.2 The following equation was used to compute relative cell viability:| Cell viability (%) = (Abs. of sample)/(Abs. of control) × 100 |
3.15 In vivo acute toxicity studies
We followed the Ethical Guidelines for Animal Testing. Transgenic animals were purchased from the Jackson Laboratory USA; The Ethical Committee (Ref. No. DREC/20200405/06) of the Department of Pharmacy, University of Swabi, Pakistan, approved all the research protocols. The animals were euthanized humanely after the experimental protocol, following the AVMA Guidelines for the Euthanasia of Animals. To induce anaesthesia, the animals were slowly fed halothane fumes. The animals were euthanized as a result of the dosage and the prolonged time period.Acute toxicity was determined by using our previously reported method.35
3.16 Docking studies
To explore the binding modes of newly synthesized compounds in the active site of LOX enzyme, all the compounds were docked into the binding pocket of COX-2, AChE and 15-LOX using Molecular Operating Environment (MOE). The crystal structure of human 15-lipoxygenase (15-LOXh) with PDB code 4NRE was downloaded from protein data bank. The protein structure was prepared using the preparation module of MOE and was subjected for 3-D protonation and finally was energy minimized to get a stable conformation of the target protein. All compounds were subjected to MOE for protonation and energy minimization using the default parameters (gradient: 0.05, Force Field: MMFF94X). The default parameters of MOE were used for molecular docking purpose, i.e., Placement: Triangle Matcher, Rescoring-1: London dG, Refinement: Forcefield, Rescoring-2: GBVI/WSA. For each ligand total, ten conformations were allowed to generate, and the top-ranked conformations based on docking score were selected for protein-ligand interaction profile analysis. Ligand interaction and visualization was carried out via Pymol.4 Conclusions
Present work described an extensive work to design a framework of multitarget scaffolds by using 3D crystal structures of the target enzymes. Continued with triazole 12 as lead compound, we modified it by considering the 3D structures and volumes of the active sites of target enzymes. The volume of active sites of AChE/BChE and MAO isoforms support bulky compounds for excellent inhibition. Hence in SAR study, incorporation of 3-methylene unit linkers resulted in loss of COX-2 inhibition. However, excellent inhibition and selectivity results towards eeAChE were obtained by increasing the linker length and with bulky scaffolds. Pyrimidine and pyrrolidine derivatives 28–31 inhibited eeAChE with a nanomolar concentration form 0.10 μM to 0.06 μM. Pyrazoline derivative 39 synthesized from modification of starting diclofenac 7 emerged as most potent eeAChE inhibitor (IC50 = 0.03 μM) and MAO-B inhibitor with IC50 value of 0.01 μM. All the biological active compounds were found to be non-neurotoxic. In a reversibility assay, all the studied active compounds showed reversibility and thus found to be devoid of side effects. In docking studies three-dimensional construction and interaction with key residues of all the studied biological macromolecules helped us to explain the experimental results.Author contributions
UR conceived, designed, and supervised this study. He was involved in all the phases (from synthesis to pharmacological evaluation and manuscript writing/editing) that led to the completion of the manuscript. MAJ and SB synthesized the compounds. MAJ performed computational study and HPLC. In vitro and in vivo studies were supervised by AS and performed by MSJ. AZ and UF helped in pharmacological evaluation (in vitro) with AS and MSJ. MI performed cell viability/neurotoxicity assay and MAJ helped him. All the authors have read the manuscript and approved it for publication. The authors declare that there is no conflict of interest.Conflicts of interest
There is no conflict of result between the authors.Acknowledgements
Dr Umer Rashid (PI) is also thankful to Higher Education Commission (HEC) of Pakistan for the purchase of MOE 2016.0802 license under NRPU project (5291/Federal/NRPU/R&D/2016).Notes and references
- U. Rashid and F. L. Ansari, Challenges in designing therapeutic agents for treating Alzheimer’s disease-From serendipity to rationality, Drug Design and Discovery in Alzheimer’s Disease, Elsevier, 2014, vol. 6, ch. 2, pp. 40–141 Search PubMed.
- M. Campora, C. Canale, E. Gatta, B. Tasso, E. Laurini, A. Relini, S. Pricl, M. Catto and M. Tonelli, ACS Chem. Neurosci., 2021, 12, 447–461 CrossRef CAS.
- B. P. Imbimbo and M. Watling, Expert. Opin. Investig. Drugs, 2019, 28, 967–975 CrossRef CAS PubMed.
- Y. Xu, J. Zhang, H. Wang, F. Mao, K. Bao, W. Liu, J. Zhu, X. Li, H. Zhang and J. Li, ACS Chem. Neurosci., 2018, 10, 482–496 CrossRef PubMed.
- M. A. Javed, N. Ashraf, M. Saeed Jan, M. H. Mahnashi, Y. S. Alqahtani, B. A. Alyami, A. O. Alqarni, Y. I. Asiri, M. Ikram, A. Sadiq and U. Rashid, ACS Chem. Neurosci., 2021, 12, 4123–4143 CrossRef CAS PubMed.
- H. Akıncıoğlu and İ. Gülçin, Mini Rev. Med. Chem., 2020, 20(8), 703–715 CrossRef.
- K. Pape, R. Tamouza, M. Leboyer and F. Zipp, Nat. Rev. Neurol., 2019, 15(6), 317–328 CrossRef PubMed.
- A. Webers, M. T. Heneka and P. A. Gleeson, Immunol. Cell Biol., 2020, 98(1), 28–41 CrossRef.
- V. Calsolaro and P. Edison, Alzheimer's Dementia, 2016, 12, 719–732 CrossRef PubMed.
- L. J. Van Eldik, M. C. Carrillo, P. E. Cole, D. Feuerbach, B. D. Greenberg, J. A. Hendrix and K. K. Bales, Alzheimer's Dementia, 2016, 2, 99–109 CrossRef PubMed.
- O. Hidalgo-Lanussa, E. Baez-Jurado, V. Echeverria, G. M. Ashraf, A. Sahebkar, L. M. Garcia-Segura and G. E. Barreto, J. Neuroendocrinol., 2020, 32, e12776 CrossRef CAS.
- C. Patrono, Br. J. Clin. Pharmacol., 2016, 82(4), 957–964 CrossRef CAS PubMed.
- A. Zarghi and S. Arfaei, Iran. J. Pharm. Res., 2011, 10, 655–683 CAS.
- R. G. Biringer, Int. J. Environ. Res. Publ. Health, 2019, 16(14), 2560 CrossRef CAS.
- S. E. Desale and S. Chinnathambi, J. Neuroinflammation, 2020, 17(1), 1–14 CrossRef.
- J. C. Udeochu, J. M. Shea and S. A. Villeda, Clin. Exp. Neuroimmunol., 2016, 7(2), 114–125 CrossRef PubMed.
- S. Manzoor and N. Hoda, Eur. J. Med. Chem., 2020, 206, 112787 CrossRef CAS.
- M. H. Elsherbeny, J. Kim, N. A. Gouda, L. Gotina, J. Cho, A. N. Pae, K. Lee, K. D. Park, A. Elkamhawy and E. J. Roh, Antioxidants, 2021, 10, 1641 CrossRef CAS PubMed.
- S. Ilgın, D. Osmaniye, S. Levent, B. N. Sağlık, U. Acar Çevik, B. K. Çavuşoğlu, Y. Özkay and Z. A. Kaplancıklı, Molecules, 2017, 22, 2187 CrossRef.
- M. R. Bronzuoli, A. Iacomino, L. Steardo and C. Scuderi, J. Inflamm. Res., 2016, 9, 199 CrossRef CAS PubMed.
- A. Delgado, C. Cholevas and T. C. Theoharides, Biofactors, 2021, 47, 207–217 CrossRef CAS.
- F. Zemek, L. Drtinova, E. Nepovimova, V. Sepsova, J. Korabecny, J. Klimes and K. Kuca, Expet Opin. Drug Saf., 2014, 13, 759–774 CAS.
- P. Zhang, S. Xu, Z. Zhu and J. Xu, Eur. J. Med. Chem., 2019, 176, 228–247 CrossRef CAS PubMed.
- S. Ahmad, F. Iftikhar, F. Ullah, A. Sadiq and U. Rashid, Bioorg. Chem., 2016, 69, 91–101 CrossRef CAS PubMed.
- M. S. Nadeem, J. A. Khan and U. Rashid, Int. J. Biol. Macromol., 2021, 193, 19–26 CrossRef CAS PubMed.
- M. S. Nadeem, J. A. Khan, I. Kazmi and U. Rashid, ACS Omega, 2022, 7(11), 9369–9379 CrossRef PubMed.
- H. K. Askarani, A. Iraji, A. Rastegari, S. N. A. Bukhari, O. Firuzi, T. Akbarzadeh and M. Saeedi, BMC Chem., 2020, 14(1), 1–13 CrossRef.
- C. Yamali, F. S. Engin, S. Bilginer, M. Tugrak, D. Ozmen Ozgun, G. Ozli and H. I. Gul, J. Heterocycl. Chem., 2021, 58(1), 161–171 CrossRef CAS.
- P. Sharma, A. Tripathi, P. N. Tripathi, S. S. Singh, S. P. Singh and S. K. Shrivastava, ACS Chem. Neurosci., 2020, 11, 2782 CrossRef CAS.
- A. Seth, P. A. Sharma, A. Tripathi, P. K. Choubey, P. Srivastava, P. N. Tripathi and S. K. Shrivastava, Med. Chem. Res., 2018, 27, 1206–1225 CrossRef CAS.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38(3), 223–232 CrossRef CAS.
- N. Augustin, V. K. Nuthakki, M. Abdullaha, Q. P. Hassan, S. G. Gandhi and S. B. Bharate, ACS Omega, 2020, 5(3), 1616–1624 CrossRef CAS PubMed.
- C. F. Jin, Z. Z. Wang, K. Z. Chen, T. F. Xu and G. F. Hao, J. Med. Chem., 2020, 63(23), 15021–15036 CrossRef CAS PubMed.
- R. K. Tripathi, O. Goshain and S. R. Ayyannan, ChemMedChem, 2013, 8(3), 462–474 CrossRef CAS PubMed.
- M. S. Jan, S. Ahmad, F. Hussain, A. Ahmad, F. Mahmood, U. Rashid, F. Ullah, M. Ayaz and A. Sadiq, Eur. J. Med. Chem., 2020, 186, 111863.5 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04183a |
| This journal is © The Royal Society of Chemistry 2022 |