DOI:
10.1039/D2RA04032H
(Paper)
RSC Adv., 2022,
12, 30605-30610
Regulating the pyrolysis process of cation intercalated MnO2 nanomaterials for electrocatalytic urea oxidation performance†
Received
30th June 2022
, Accepted 20th October 2022
First published on 26th October 2022
Abstract
Exploring an efficient way to enhance electron/ion transport behavior of nanomaterials plays an important role in the study of energy storage & conversion. However, the evolution rules of lattice and electronic structure during the pyrolysis process of low-dimensional nanomaterials, which further regulate its electron/ion transport properties, have not been effectively elucidated. Here we study the pyrolysis process of cation intercalated MnO2 as a case for realizing optimized electron/ion transport behavior. In our case, thermogravimetry-mass spectrometry (TG-MS) was adopted for tracking the remaining products in pyrolysis and decomposition products, further finding out the evolution law of the manganese–oxygen polyhedron structure during the pyrolysis. Moreover, the internal relations between the crystal structure and the electronic structure during the pyrolysis process of low-dimensional manganese oxide are revealed by fine structure characterization. As expected, partially treated 2D MnO2 nanosheets with controlled pyrolysis displays ultrahigh UOR performance with the overpotential of 1.320 V vs. RHE at the current density of 10 mA cm−2, which is the best value among non-nickel-based materials. We anticipate that studying the mechanism of the pyrolysis process has important guiding significance for the development of high electron/ion transport devices.
Introduction
The most efficient way to convert and store energy is to achieve the mutual conversion between electrical energy and chemical energy.1–3 As a carrier for energy conversion and storage, electron/ion transport properties determine the expression of functional behavior.4 Therefore, finding efficient material systems and effective research methods is the key to improving the performance of electron/ion transport. So far, the mainstream systems for studying electron/ion transport performance materials are perovskite materials,5 inorganic 3d transition metal oxides/chalcogenides/nitrides,6–8 metal organic frameworks (MOF), etc.9–15 These material systems have made great progress in the study of electron/ion transport properties, providing us with extensive experience in optimizing the structure of electron/ion transport properties through the development and in-depth study of one of the material systems. Of note, manganese oxide provides a good material platform for the regulation of novel electronic states due to its easy regulation property.16 Manganese oxide is usually composed of manganese oxygen polyhedron, while different Mn valence and manganese oxygen coordination form determines its special physicochemical behavior.17–25 For example, the trivalent manganese ion may have the Jahn–Teller effect, causing Jahn–Teller distortion in the system, resulting in a special electronic structure.26 Besides, when trivalent manganese ions and tetravalent manganese ions coexist in the MnO6 octahedron system, double exchange effect may be generated, resulting in novel magnetic and electrical transport properties.27,28 Moreover, when divalent manganese ions are arranged in a face-centered cubic manner in MnO, super-exchange structure would be produced, which can be used to explain the phenomenon of anti-ferromagnetic spontaneous magnetization.29 Therefore, regulating the valence state of manganese ion and manganese oxygen coordination form of manganese oxide provides an ideal model for studying the electronic structure of the material and finding out the influencing factors that affect the electron/ion transport performance. As an effective route of in-depth analysis of materials, pyrolysis has made considerable progress in recent years, especially qualitative research on as-pyrolysis products. In general, pyrolysis of materials is carried out by controlled temperature heating in a furnace or CVD/PVD/ALD method.30 However, pyrolysis is generally viewed as a means to obtain the target product through high-temperature chemical reactions, without a thorough understanding of materials in the pyrolysis process. In order to analyze the pyrolysis process, the method of TG analysis is usually used to analyze the quality variation of the material during the pyrolysis process, which combined with the differential scanning calorimetry (DSC) to monitor thermodynamic process that takes place on pyrolysis. Moreover, the development of TG-MS coupled with mass spectrometry (MS) provides abundant information for further studying the pyrolysis process. Currently, TG-MS is mostly used to analyze well-defined materials such as graphene,31 carbon nanotubes,32 iron oxide33 and so on. TG-MS combines TG and MS to detect very low levels of impurities. During the pyrolysis of the sample, the sample thermally decomposes and volatilizes out of the gas, which is transmitted to the mass spectrometer for identification.34 Therefore, TG-MS can be used to pyrolyze materials with well-defined structure, and the pyrolysis residual products and decomposition products can be regulated by different pyrolysis temperature and holding time to further study the pyrolysis reaction process of materials to obtain functional materials with different electronic structures. Here we take advantage of TG-MS tracking of pyrolysis processes in real-time. In our case, the evolution low of their lattice and electronic structure of cation intercalated 2D MnO2 nanomaterials were successfully elucidated, further successfully realizing the controllable synthesis of low-dimensional MnOx with controlled specific surface area, more active sites, significant improved conductivity. As expected, 2D-MnO2-550 nanosheet with controlled pyrolysis displays ultrahigh UOR performance with the overpotential of 1.320 V vs. RHE at the current density of 10 mA cm−2 (Scheme 1).
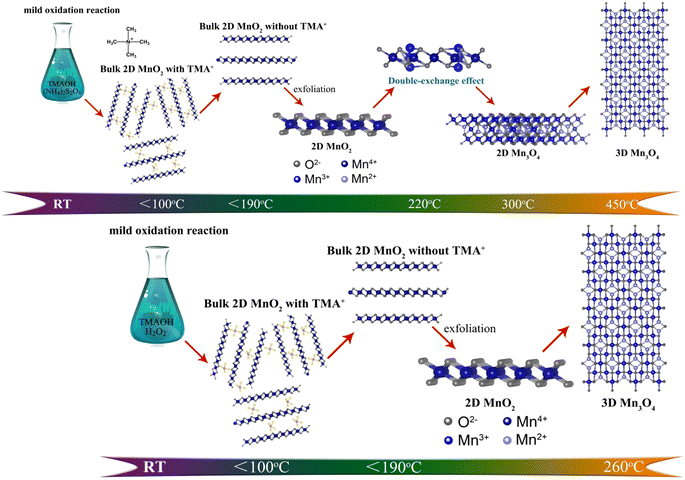 |
| | Scheme 1 The schematic illusion of evolution process of two different types of 2D MnO2 nanomaterials during pyrolysis process. | |
Results and discussions
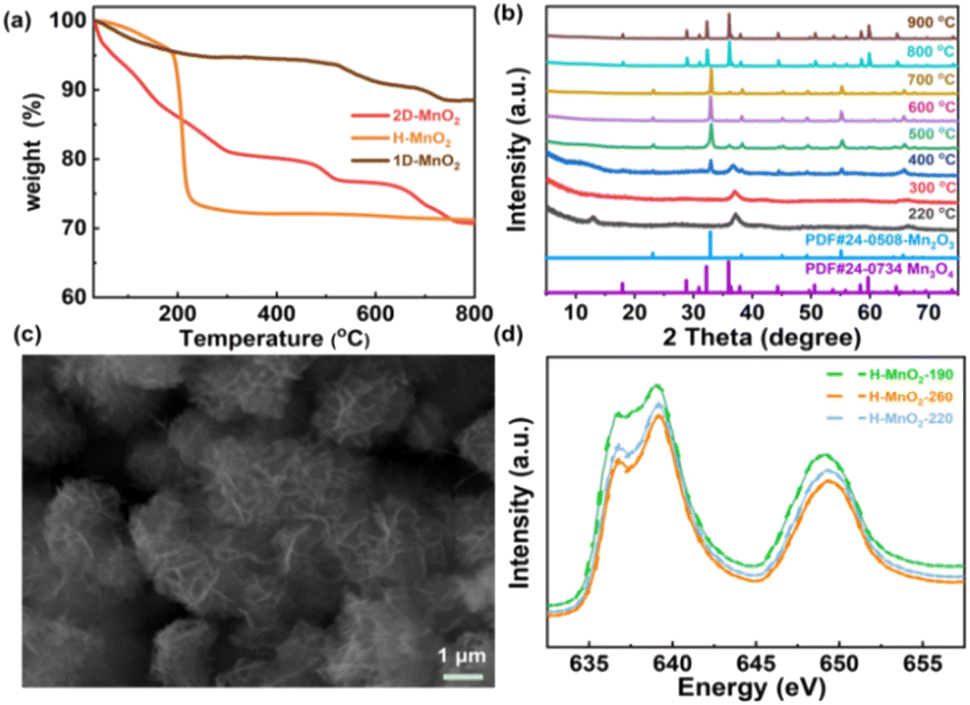
In order to further clarify the pyrolysis process and mechanism of MnO2, we chose three typical low-dimensional manganese dioxides as reference: Two kinds of 2D birnessite MnO2, colloidal birnessite MnO2 (H-MnO2) and bulk MnO2 (1D-MnO2), as we previously reported. And one-dimensional MnO2 nanowire as reported in literature (Fig. 1). Notably, the 1D MnO2 nanowire has no cation intercalated behavior, which can be seen as typical MnO2. While two kinds of 2D birnessite MnO2 both have TMA+ intercalated in the layers of 2D MnO2. The difference between these two kinds of 2D birnessite MnO2 is the physical characteristics of materials, of which one is colloidal and other is powder-like. As shown in Fig. 2a, the pyrolysis process is first studied via TG analysis underwent helium atmosphere. As expected, the 1D MnO2 nanowire with no cation intercalated has typical MnO2 decomposition behavior in TG curves. The remaining weight percentage is 90%, which can be assigned to the transformation to Mn3O4 (Fig. S1†), where the pyrolysis in a low oxygen partial pressure environment can lead to the escape of O elements. In contrast, the two kinds of the cation intercalated 2D MnO2 with tetramethylammonium hydroxide (TMA+) ions more distinct TG curves. For H-MnO2, there is significant weight loss around 200 °C, and the weight loss rate is 11%. The remaining weight percentage is 72%. Meanwhile, for 2D-MnO2, have obvious transition characteristics in thermogravimetric analysis. The weight loss area is more rich than other materials, there are three weight loss parts, and the remaining weight percentage is 70%.
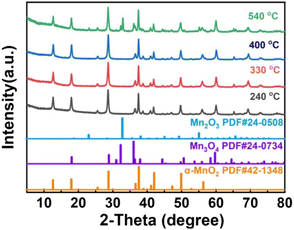 |
| | Fig. 1 PXRD pattern of 1D-MnO2 at different pyrolysis temperatures. | |
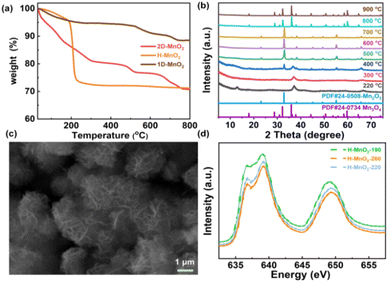 |
| | Fig. 2 (a) TG curves of three types of MnO2, (b) XRD patterns of 2D-MnO2 at 220–1000 °C, (c) SEM images of 2D-MnO2 at 550 °C, (d) Mn L3 edge XAS of H-MnO2, -190, -220, -260. | |
We first analyzed the two-dimensional MnO2 nanomaterials with different pyrolysis temperatures by X-ray diffraction (XRD) (Fig. 2b). It should be noted that when the pyrolysis temperature of 2D-MnO2 is higher than 400 °C, only Mn3O4 can be observed in the resulting material (JCPDS No. 24-0734). The diffraction peaks at 17.7°, 28.7°, 32.1°, 35.8°, 44.2°, 59.6° and 64.4° can assign to (101), (112), (103), (211), (220), (224) and (400) lattice plane. clearly shows that 2D-MnO2 nanomaterials still maintain two-dimensional morphology at 500 °C, and are nanoflowers formed by the aggregation of nanosheets (Fig. S2†). Under the condition of small temperature range, the phase of H-MnO2 has changed greatly, which leads to further exploration of the compounds (Fig. S3†). In this paper, we hope to study the fine electronic states of H-MnO2-190, -220, -260, especially in the Mn state, to observe the changes in pyrolysis. We have carried out Mn L3 side XAS tests on compounds H-MnO2-190, -220, -260 (Fig. 2d). It can be seen from the spectrum that the edge of Mn L3 is divided into two parts because the Mn 3d orbital of Oh symmetry is divided into two groups: eg and t2g. The edge spectra of Mn L3 show that the intensity of the transition peak increases at 2p → t2g, and the valence states of H-MnO2-260, -220 increase. This fact shows that compared with compound H-MnO2-190, the 3d orbital of Mn in compounds H-MnO2-260 and H-MnO2-220 with higher pyrolysis temperature has the phenomenon of electron transfer, and the electron injection process occurs.35 At the same time, the lower valence state is also reflected in the absorption shift of Mn L3 edge. Compared with compound H-MnO2-190, the absorption edge of compound H-MnO2-260, -220 shifts in the low energy direction.
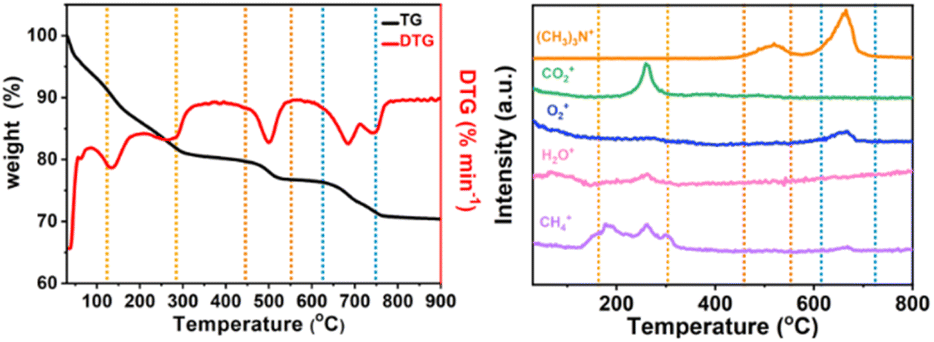
In order to analyze the structure of low-dimensional MnO2 nanomaterials during pyrolysis, thermogravimetric analysis and mass spectrometry (TG-MS) were used. With a constant flow rate of helium gas of 5 mL min−1, the sample was heated from room temperature (RT) to 800 °C, and then the thermally separated entities were analyzed by an EI mass spectrometer. As shown in Fig. 3, the three main steps of mass loss detection are Area A: 100 to 310 °C, Area B: 470–580 °C and Area C: 620–780 °C. The first step (Area A) starts with showing only the molecular fragments of m/z 16 and 18, because the residues of water and TMA+ have been inserted between the δ-MnO2 nanosheet layers. It was found that the structure of δ-MnO2 nanosheets was consistent, and TMA+ molecules began to decompose at 140 °C. At around 270 °C, m/z 16, 18 and 44 show a series of different peaks. These facts indicate that δ-MnO2 nanosheets are converted into Mn3O4 nanosheets together with oxygen atoms from octahedral MnO6 structure from δ-MnO2 octahedral MnO6 structure to Mn3O4 tetrahedral MnO4 structure. In the second step (region B), only m/z 64 molecular fragments were detected, indicating that the (CH3)3N molecules generated by TMA+ molecular pyrolysis had been intercalated with δ-MnO2 nanosheets before 470 °C, and maintained a two-dimensional structure. In the third step (region C), the molecular fragments of m/z 64 and m/z 32 were detected (Fig. S4†). At this time, MnO2 had been completely transformed into Mn3O4 nanosheets, and with the loss of intercalated (CH3)3N molecules, the two-dimensional structure collapsed. The possible reactions include:
| | |
(CH3)4N+ + OH− → CH4 + (CH3)3N + ½O2
| (2) |
| | |
CH4 + 2O2 → CO2 + 2H2O
| (3) |
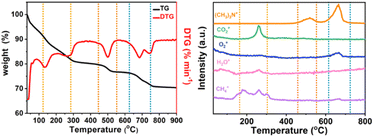 |
| | Fig. 3 TG-MS analysis of low-dimensional MnO2 nano-materials. TG curves of (a) 2D-MnO2, (b) MS trace of 2D-MnO2 at RT ∼ 800 °C. | |
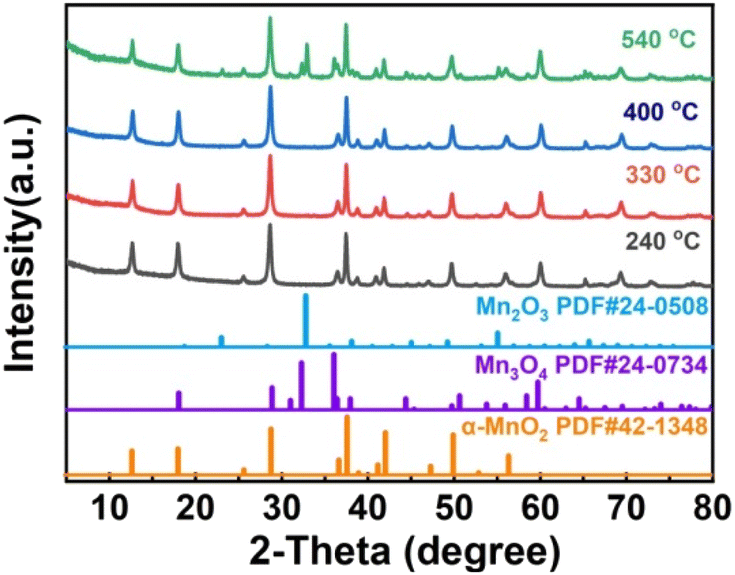
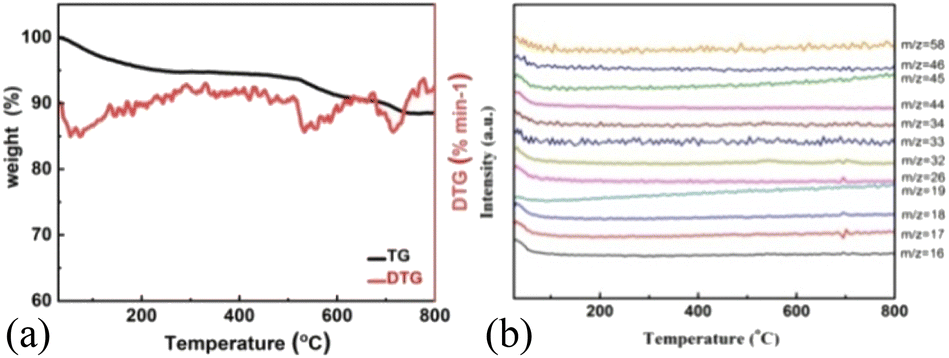
As shown in Fig. 3a, H-MnO2 only has a significant weightless area, ie 200 °C to 300 °C, while MS only detected a significant peak range in this area (Fig. S5†). These peaks can be explained by the decomposition of TMA+ molecules and the conversion of δ-MnO2 to Mn3O4. The structural evolution of MnO2 nanosheets during pyrolysis was also confirmed by XRD patterns of MnO2 nanosheets at different pyrolysis temperatures. The change of phase during pyrolysis was explored by XRD (Fig. 2). The diffraction peaks of 240 °C and 400 °C α-MnO2 are still clear and sharp on its pyrolysis process (Fig. S5†). When the temperature rises to 400 °C, two new diffraction peaks appear. The diffraction peaks at 32.3° are in line with the (103) crystal surface of Mn3O4 (JCPDS Card No. 24-0734), and the diffraction peaks at 32.9° match with (222) crystal surface of Mn2O3 (JCPDS Card No. 24-0508). This indicates that the phase changes at 400 °C, and it is likely to contain three manganese oxides. In order to further understand the pyrolysis process of different MnO2, one-dimensional manganese dioxide was also analyzed by TG-MS (Fig. 4). From the TG curve, it can be seen that the compound is stable δ-MnO2 the mass percent loss of MnO2 is not much. At 800 °C, the total loss is 10%. The trend of the curve is similar to that of typical MnO2, indicating that there is no intercalation structure. No other special molecular fragments were found in MS. After 500 °C, there are two slight weightlessness, with a weight loss of 6%. This part also shows the change of biological phase in XRD analysis. This weightlessness may due to the loss of oxygen in the lattice of manganese oxide, resulting in the change of valence state.36 To further explore the structural changes of MnO2/NF composites, we conducted X-ray photoelectron spectroscopy (XPS) to effectively study the chemical composition, as shown in Fig. S7–S8.†
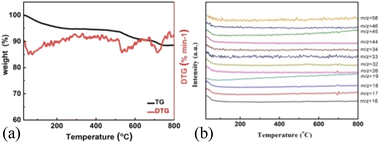 |
| | Fig. 4 TG-MS analysis of low-dimensional MnO2 nano-materials. TG curves of (a) 1D-MnO2, (b) MS trace of 1D-MnO2 at RT ∼ 800 °C. | |
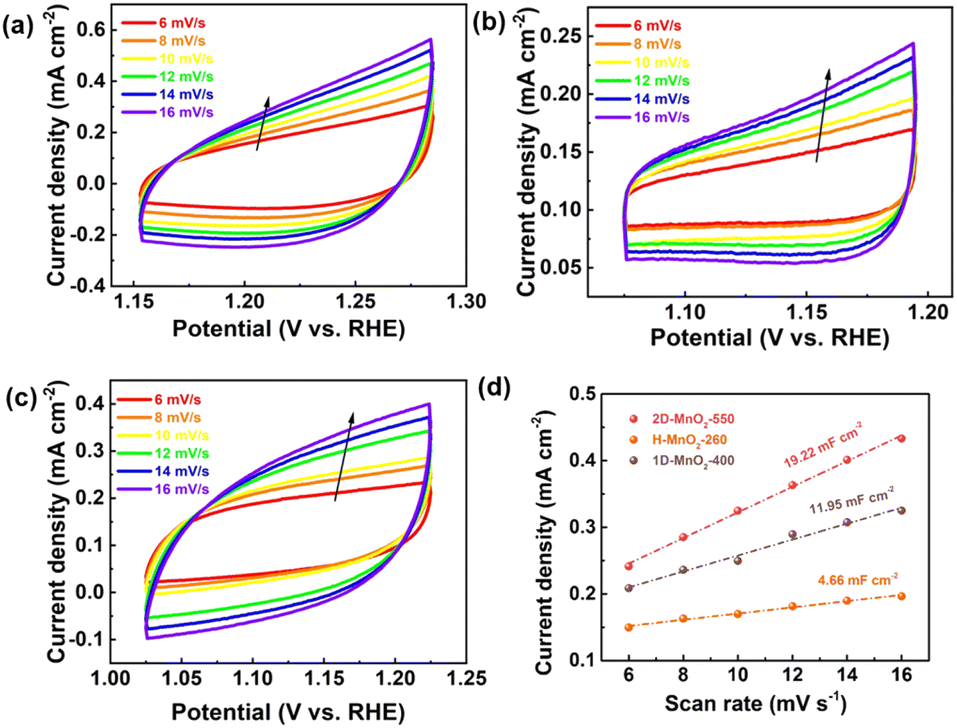
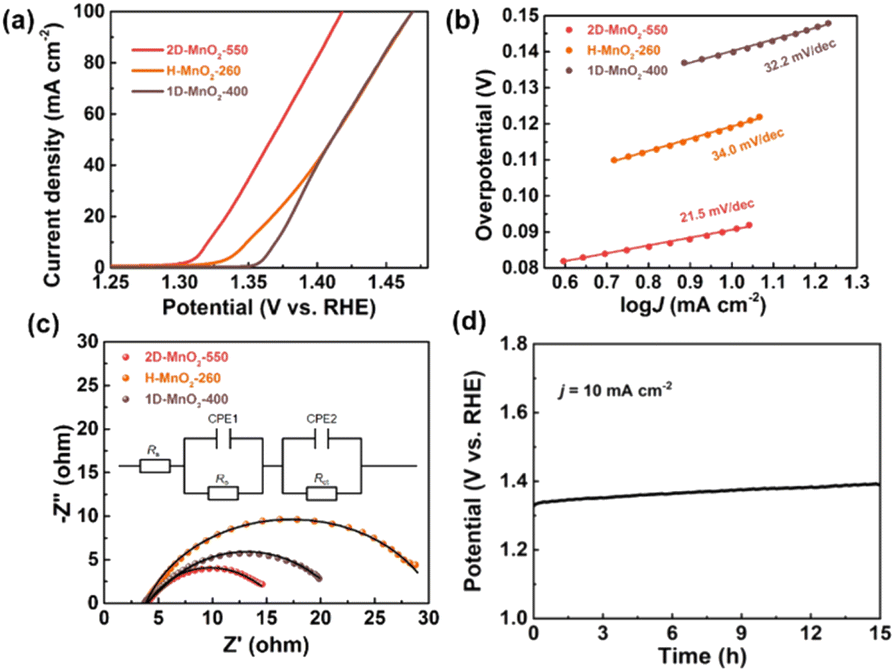
We used a typical three-electrode configuration in 1 M KOH electrolyte and 0.33 M urea to perform electrochemical tests on three different MnO2 pyrolyzed at different temperatures. The loading of the catalyst on the nickel foam was 1.68 mg cm−2. As shown in Fig. 6a and Fig. S9–S11† in a typical linear sweep voltammogram (LSV) plot, the anodic current of all samples simultaneously increases as the potential becomes more positive. The 2D-MnO2-550 electrode displayed the smaller overpotential of 1.320 V (at the current density of 10 mA cm−2) than other samples such as H-MnO2-260 (1.383 V), 1D-MnO2-400 (1.393 V) and nickel foam (1.391 V). The UOR kinetics of the catalyst was studied by the corresponding Tafel diagram. As shown in Fig. 6b, the Tafel slope of 2D-MnO2-550 is calculated to be 21.5 mV dec−1. It is lower than the Tafel slope of H-MnO2-260 (25 mV dec−1). This also proves that two-dimensional materials are better than one-dimensional materials in UOR. This also proves the advantages of two-dimensional MnO2 over one-dimensional MnO2 in terms of UOR. In order to further prove that 2D-MnO2-550 has the most efficient electrocatalytic urea oxidation performance, we evaluated the electrochemical double-layer capacitance (CdI) and electrochemical surface area (ECSA) indexes of 2D-MnO2-550, H-MnO2-260 and 1D-MnO2-400 by cyclic voltammetry (Fig. 5).37–39 Fig. 5 shows the linear fitting curve of Δj with different scanning rates (6, 8, 10, 12, 14 and 16 mV s−1). The ECSA of 2D-MnO2-550 sample has a maximum value of 19.22 mF cm−2. The ECSA of other samples 1D-MnO2-400 was 11.95 mF cm−2 and that of H-MnO2-260 was 4.66 mF cm−2 (Fig. 5d). Therefore, this shows that 2D-MnO2-550 materials can provide more exposed electrocatalytic active sites and have more catalytic activity in the electrolysis process.40
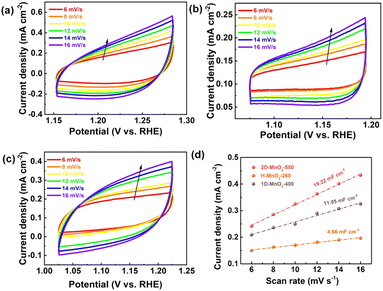 |
| | Fig. 5 (a) CVs of 2D-MnO2-550. (b) CVs of H-MnO2-260. (c) CVs of 1D-MnO2-400. (d) Electrocatalytic specific surface area of 2D-MnO2-550, H-MnO2-260 and 1D-MnO2-400. | |
At the same time, the UOR kinetics of the prepared electrode was further evaluated by electrochemical impedance spectroscopy (EIS). The semicircle of H-MnO2-260 sample is the largest, and the charge transfer resistance is also the largest, which may be related to the state of the sample. Because H-MnO2 is colloidal and difficult to disperse, the resistance is too large. We use zview software to fit the measured AC impedance, and the black line is the fitting result (Fig. 6c). According to the fitting results, the pattern of the equivalent circuit diagram is Rs (CPE1, Ro) (CPE2, Rct),41 in which the charge transfer resistance (Rct) at the catalyst/electrolyte interface of sample 2D-MnO2-550 is the smallest, with a value of 7.69 Ω. The Rct of samples H-MnO2-260 and 1D-MnO2-400 are 18.44 Ω and 8.08 Ω, respectively. The solution resistance (Rs) of the three samples is about 3.8 Ω. CPE1 and CPE2 describe the electron transport at the electrode material/catalyst interface and catalyst/electrolyte interface respectively, and Ro represents the oxide film resistance at the catalyst/electrolyte interface.42 Because 3D macroporous nickel foam has been widely used as an electrocatalyst carrier, we have tested the stability of the sample 2D-MnO2-550 loaded with nickel foam in 1 M KOH and 0.33 M urea solutions. In this experiment, the chronopotentiometry was used to test under the current density of 10 mA cm−2. As shown in Fig. 6d, sample 2D-MnO2-550 lasted for 15 hours at a current density of 10 mA cm−2, with a voltage increase of 4.5%, indicating that the sample can stably carry out a long-term catalytic reaction.43
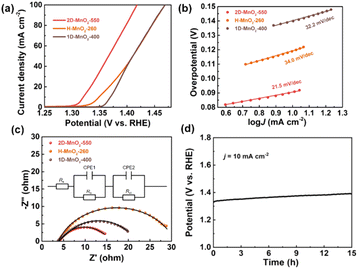 |
| | Fig. 6 UOR performance of three types of MnO2. (a) LSV plots of 2D-MnO2-550, H-MnO2-260 and 1D-MnO2-400. (b) Tafel plots of 2D-MnO2-550, H-MnO2-260 and 1D-MnO2-400. (c) Electrochemical impedance spectroscopy (EIS) of 2D-MnO2-550, H-MnO2-260 and 1D-MnO2-400. (d) Long-term durability measured at a constant current density of 10 mA cm−2 for 15 h in 1 M KOH + 0.33 M Urea solution, respectively. | |
Conclusions
In summary, we have shown how to the cation intercalated 2D MnO2 featuring TMA+ cation underwent TG-MS controlled pyrolysis can be realized as promoted electrocatalytic urea oxidation performance with distinct MnOx nanostructures. We directly confirm the evolution law of residual of cation intercalated MnO2 nanomaterials, which is responsible for the evolution law of composition of cation intercalated MnO2 nanomaterials by TG-MS, and thus clarifying the high-performance with distinct MnOx structure. As expected, partially treated 2D MnO2 nanosheet with controlled pyrolysis displays ultrahigh UOR performance with the overpotential of 1.320 V vs. RHE at the current density of 10 mA cm−2. This novel strategy of controlled pyrolysis by TG-MS offering a potential to become a bifunctional energy nanodevice with high electron/ion transport behavior.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Science Fund for NSFC (Grants 21805074), and NSFC of Hubei (Grants 2021CFB420).
Notes and references
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- X. Peng, L. Peng, C. Wu and Y. Xie, Chem. Soc. Rev., 2014, 43, 3303 RSC.
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48 CrossRef CAS PubMed.
- E. Dagotto, Science, 2005, 309, 257 CrossRef CAS PubMed.
- L. Peng, P. Xiong, L. Ma, Y. Yuan, Y. Zhu, D. Chen, X. Luo, J. Lu, K. Amine and G. Yu, Nat. Commun., 2017, 8, 15139 CrossRef PubMed.
- M. Chhowalla, H. S. Shin and G. Eda, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- Y. Zhang, B. Ouyang and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef CAS PubMed.
- X. Lu, P. Liao and J. Wang, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- J. Q. Zhao, D. D. Cai, J. Dai, M. Kurmoo, X. Peng and M. H. Zeng, Sci. Bull., 2019, 64, 1667–1674 CrossRef CAS.
- B. X. Pan, X. Peng, Y. F. Wang, Q. An, X. Zhang, Y. X. Zhang, T. S. Teets and M. H. Zeng, Chem. Sci., 2019, 10, 4560–4566 RSC.
- Y. Wang, Y. Liang, Y. Wu, J. Yang, X. Zhang, D. Cai, X. Peng and M. H. Zeng, Angew. Chem., Int. Ed., 2020, 59, 13232–13237 CrossRef CAS PubMed.
- Q. An, B. X. Pan, L. Li, X. Peng and M. H. Zeng, Chin. J. Chem., 2021, 39, 2529–2535 CrossRef CAS.
- X. Zhang, K. Zhao, X. Peng, M. Kurmoo and M. H. Zeng, Nano Res., 2022, 15, 346–351 CrossRef CAS.
- L. Li, L. Wang, S. Tao, X. Peng and M. H. Zeng, Inorg. Chem. Front., 2022, 9, 1973–1983 RSC.
- F. Qi, K. Hirofumi and O. Kenta, J. Mater. Chem., 1999, 9, 319–333 RSC.
- Y. Hu, Y. Wu and J. Wang, Adv. Mater., 2018, 30, 1802569 CrossRef PubMed.
- S. V. Streltsov and D. I. Khomskii, Phys. Rev. X, 2020, 10, 031043 CAS.
- L. S. Suib, J. Mater. Chem., 2008, 18, 1623–1631 RSC.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CrossRef CAS.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745–2755 RSC.
- K. L. Pickrahn, S. W. Park and Y. Gorlin, Adv. Energy Mater., 2012, 2(10), 1269–1277 CrossRef CAS.
- A. Ramírez, P. Hillebrand and D. Stellmach, J. Phys. Chem. C, 2014, 118, 14073–14081 CrossRef.
- S. V. Streltsov and D. I. Khomskii, Phys. Rev. X, 2020, 10, 031043 CAS.
- X. Peng, Y. Guo and Q. Yin, J. Am. Chem. Soc., 2017, 139, 5242–5248 CrossRef CAS PubMed.
- S. Jin, T. H. Tiefel, M. McCormack, R. A. Fastnacht, R. Ramesh and L. H. Chen, Science, 1994, 264, 413 CrossRef CAS PubMed.
- Y. Zhou, S. Sun and S. Xi, Adv. Mater., 2017, 1700874 Search PubMed.
- T. J. Kempa, E. C. Sadler and T. Chowdhury, Chem. Rev., 2020, 120(22), 12563–12591 CrossRef PubMed.
- Z. Sebestyén, E. Barta-Rajnai and Z. Czégény, Energy Fuels, 2016, 30(10), 7982–7993 CrossRef.
- F. Hof, S. Bosch and S. Eigler, J. Am. Chem. Soc., 2013, 135, 18385–18395 CrossRef CAS PubMed.
- S. G. Kwon, Y. Piao and J. Park, J. Am. Chem. Soc., 2007, 129, 12571 CrossRef CAS PubMed.
- R. Schäfer, D. Dasler and U. Mundloch, J. Am. Chem. Soc., 2016, 138, 1647–1652 CrossRef PubMed.
- J. C. Wu, X. Peng and Y. Q. Guo, Front. Phys., 2018, 13(3), 138110 CrossRef.
- X. Zhang, W. Yang and J. Yang, J. Cryst. Growth, 2008, 310, 716–722 CrossRef CAS.
- Q. Liu, L. Xie and F. Qu, Inorg. Chem. Front., 2017, 4, 1120 RSC.
- C. Wei, S. N. Sun and X. Wang, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- G. F. Li, Y. N. Chen and M. Pan, Sustainable Energy Fuels, 2018, 2, 237–251 RSC.
- Y. Tong, P. Chen and M. Zhang, ACS Catal., 2018, 8(1), 1–7 CrossRef.
- L. Wang, L. Ren and X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 4750–4756 CrossRef CAS PubMed.
- J. Zhang, F. Li and W. Chen, Electrochim. Acta, 2019, 300, 123–130 CrossRef CAS.
- D. Zhu, C. Guo and J. Liu, Chem. Commun., 2017, 53, 10906–10909 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a