
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrocracking of crude palm oil to a biofuel using zirconium nitride and zirconium phosphide-modified bentonite
Hasanudin Hasanudin *ab,
Wan Ryan Asriab,
Indah Sari Zulaikhaab,
Cik Ayuab,
Addy Rachmatab,
Fahma Riyantiab,
Fitri Hadiahc,
Rahadian Zainuld and
Roni Maryanae
*ab,
Wan Ryan Asriab,
Indah Sari Zulaikhaab,
Cik Ayuab,
Addy Rachmatab,
Fahma Riyantiab,
Fitri Hadiahc,
Rahadian Zainuld and
Roni Maryanae
aDepartment of Chemistry, Faculty of Mathematics and Natural Science, Universitas Sriwijaya, Indralaya 30662, Indonesia. E-mail: hasanudin@mipa.unsri.ac.id
bBiofuel Research Group, Faculty of Mathematics and Natural Science, Universitas Sriwijaya, Indralaya 30662, Indonesia
cDepartment of Chemical Engineering, Faculty of Engineering, Universitas Sriwijaya, Indralaya 30662, Indonesia
dDepartment of Chemistry, Faculty of Mathematics and Natural Science, Universitas Negeri Padang, Padang, Indonesia
eResearch Center for Chemistry, Indonesian Institute of Sciences, Building 452 Kawasan PUSPIPTEK, Serpong, Tangerang Selatan, Banten, Indonesia
First published on 9th August 2022
Abstract
In this study, bentonite modified by zirconium nitride (ZrN) and zirconium phosphide (ZrP) catalysts was studied in the hydrocracking of crude palm oil to biofuels. The study demonstrated that bentonite was propitiously modified by ZrN and ZrP, as assessed by XRD, FTIR spectroscopy, and SEM-EDX analysis. The acidity of the bentonite catalyst was remarkably enhanced by ZrN and ZrP, and it showed an increased intensity in the Lewis acid and Brønsted acid sites, as presented by pyridine FTIR. In the hydrocracking application, the highest conversion was achieved by bentonite-ZrN at 8 mEq g−1 catalyst loading of 87.93%, whereas bentonite-ZrP at 10 mEq g−1 showed 86.04% conversion, which suggested that there was a strong positive correlation between the catalyst acidity and the conversion under a particular condition. The biofuel distribution fraction showed that both the catalysts produced a high bio-kerosene fraction, followed by bio-gasoline and oil fuel fractions. The reusability study revealed that both the catalysts had sufficient conversion stability of CPO through the hydrocracking reaction up to four consecutive runs with a low decrease in the catalyst activity. Overall, bentonite-ZrN dominantly favored the hydrocracking of CPO than bentonite-ZrP.
Introduction
Energy is essential in the current life and is an elementary human necessity due to its application in automobiles, industry, and so forth. As yet, petroleum-based energy, such as gasoline and kerosene fuels provenance, have been known as a primary non-renewable energy, and its supply on Earth in the near future will inevitably run out.1 Fossil fuel consumption also emits greenhouse gases, such as carbon dioxide, carbon monoxide, nitrogen oxides, and other toxic gases. Amid the current energy crisis as well as concerns regarding environmental issues, the notion of developing different energy sources linked to renewable alternative energy with nontoxicity and environmental benefits, such as biofuels, arises.2,3 Many kinds of biofuels have been extensively produced with various adequate methods.4 Transesterification is known as a method that is often used in producing biofuels. However, this method is ineffective due to inflexible feedstocks, poor infrastructure compatibility, and lower processing costs; when using a homogeneous catalyst, transesterification results in high energy consumption and separation costs.5 The pyrolysis method is also employed, but due to the product generating a high oxygen content and a high acidity level, it is corrosive and unsuitable for use in engines.6 In particular, hydrocracking and catalytic cracking are preferred processes due to their high conversion performance, and these processes produce a wide range of carbon–hydrocarbons that correspond to the boiling point ranges of gasoline, kerosene, and fuel oil, which can be directly used.7 This method provides several advantages such as using a lower reaction temperature than the pyrolysis temperature, offering a cheaper route for energy consumption,8 and generating an oxygen-free and high heating value product. In this process, the double bonds in triglycerides are cracked into smaller compound molecules in the presence of hydrogen and a suitable catalyst.9 Complex reactions such as dehydration, dehydrogenation, deoxygenation, and decarboxylation are also involved in the hydrocracking process.10The feedstocks of the hydrocracking reaction are one of the critical parameters for sustainable and feasible biofuel production. Currently, various vegetable and non-edible feedstocks such as nyamplung oil,11 palm oil,12 and soybean oil13 have been developed in the hydrocracking process. Crude palm oil (CPO) is the most favored vegetable oil feedstock for the production of biofuels through hydrocracking due to its high long-chain hydrocarbon content,14 the absence of sulfur and nitrogen, and also wide availability throughout the world, particularly in Indonesia and Malaysia, with its low cost and highest yield per hectare among all vegetable oil feedstocks.15
Various catalysts and hydrotreating reaction conditions using CPO as the feedstock have been reported. Subsadsana and Ruangviriyachai16 successfully converted CPO to biofuels through hydrocracking using an NiW-modified zeolite-based catalyst and provided high conversion efficiency. Subsadsana et al.17 showed that the hydrocracking of CPO generated the corresponding hydrocracking fractions such as gasoline, kerosene, and diesel catalyzed by NiW-modified ZSM-5 zeolite. Presently, the use of several modified aluminosilicates such as bentonite,18 MCM-41,19 and zeolite,20 as catalysts for hydrocracking various feedstocks has shown remarkably improved outcomes in several aspects, most notably, increased catalytic activity, which inherently promotes high biofuel yield.21 Specifically, the modified bentonite catalyst for the hydrocracking process showed relatively superior performance compared to commercial carbon-based catalysts. These features are strongly related to the efficient adsorption of large molecules in the pores of bentonite; it is also thermally stable, and the presence of strong acid sites of modified bentonite is highly suitable for catalyzing the hydrocracking reactions.22 Modified bentonite, in addition, has recently gained popularity as a solid catalyst due to its acidity, which can be modulated according to clay minerals,23 relative abundance, low cost, the process of modification or inclusion of metal species, thus showing versatility in various applications.24 Moreover, compared to other solids such as zeolites, it is evident that their qualities generate fewer environmental issues during the synthesis and recovery or disposal after usage.
The modification of bentonite using various metals such as Ni,25 Ni/Al2O3,26 Ni-modified sulfated ZrO2,27 NiMoS and NiWS,28 and Ni–Cu/ZrO2 (ref. 21) have been extensively employed for the hydrocracking reaction. Along with the expanding interest and exploration in the use of heterogeneous catalysts, it appears that metal phosphide29,30 and metal nitride-based31,32 catalysts have attracted deep interest due to their distinctive properties with various potential applications. Some of the appealing aspects of adopting this active phase include the fact that it is less expensive than noble metal catalysts and less prone to catalyst deactivation than non-noble catalysts.33 Phosphide catalysts have bifunctional features (acid/metal feature), which promote acidity. On the other hand, metal nitride-based catalysts have a lot of potential because of their ceramic-like physical qualities, combined with the chemical features akin to noble metals that are good for hydrotreating processes.34 These materials are also in charge of the distinct catalytic pathway that results in the necessary product selectivity and high catalytic activity. Several metal phosphides and nitrides were conducted for hydrogen-involvedreactions.de Souza et al.35 employed NiP combined with various support oxide catalysts and showed that the phosphide phase provided a high activity for the hydrodeoxygenation of phenol. A sequence of NiP, CoP, and CuP catalysts has been studied by Ruangudomsakul et al.,36 in order to generate green diesel through the hydrodeoxygenation of palm oil. Zhao et al.37 used Ni2P, Fe2P, MoP, Co2P, and WP-supported SiO2 catalysts for the hydrodeoxygenation reaction as promising metal phosphide-based catalysts. Molybdenum nitride34,38 supported with various catalysts and unsupported catalysts have also been employed for the other hydrodeoxygenation reaction.
Among metals that have been well studied, specifically, zirconium nitride appears to be worth glancing at since it provides a high surface area,39 high activity, and superior stability,40 and showed remarkable thermal conductivity, good abrasive resistance, and good corrosion resistance.41 Besides, some researchers reported that zirconium phosphide has good catalytic properties in the performance of the nitrogen evolution reaction42 and electrochemical application.43 Taking the above discussion into account, the comparison or investigative studies on the catalytic ability of zirconia nitride and zirconia phosphide-modified bentonite in the context of their application in the hydrocracking reactions of CPO are either adequately limited, and to the best of knowledge, have not been reported yet. In this study, the modification of bentonite using zirconium phosphide and nitride would provide a potential synergetic effect due to the increase in the acidic active site properties, which intrinsically affect the hydrocracking reaction as well as the biofuel fraction, and broaden and offer the prospective of the bentonite-based catalyst. In this context, the catalysts were employed for the hydrocracking of CPO for producing bio-gasoline, bio-kerosene, and fuel oil with different metal loadings, and the catalysts were assessed using XRD, FTIR spectroscopy, and SEM-EDX. The reusability of the catalysts was investigated within the four-cycle period. Gravimetric pyridine adsorption and pyridine FTIR were used to determine the catalyst acidity, and the product of hydrocracking was examined using GC-MS.
Experimental
Catalyst synthesis
Natural bentonite (Al2O3·4SiO2·xH2O) from Bayan, Central Java, was treated using saturated NaCl based on the previously reported procedure,30 and identified as Na-bentonite. Zirconyl chloride octahydrate (ZrOCl2·8H2O, ≥99.0% purity, Merck) was used as a Zr precursor, whereas ammonium nitrate (NH4NO3, 95.0% purity, Merck) and ammonium phosphate ((NH4)3PO4, 95.0% purity, Merck) were employed as nitrate and phosphate sources, respectively. Briefly, 5 g of the as-prepared Na-bentonite 200-mesh was mixed with a 0.1 M ZrOCl2·8H2O solution by varying the predetermined volume in order to get 2, 4, 6, 8, and 10 mEq g−1 concentrations. Afterward, the solution was stirred for 1 h at room temperature and dripped with a 1 M NH4NO3 solution with a predetermined volume at a 1 mL min−1 flow rate to obtain bentonite-zirconium nitrate, followed by further stirring for 24 h at 80 °C until a paste is formed. Similarly, bentonite-zirconium phosphate was synthesized using 1 M ((NH4)3PO4) solution mixed with bentonite and Zr precursor with a similar procedure, as previously mentioned. Subsequently, both pastes were dried in an oven at 105 °C and calcined using a muffle furnace at 550 °C for 3 h. Prior to obtaining the as-synthesized bentonite-zirconium nitride and bentonite-zirconium phosphide, the catalysts were reduced under an H2 atmosphere by gradually increasing temperature from 350 up to 600 °C and held for 2 h.Catalyst characterization
The phase structure of Na-bentonite and modified bentonite were assessed using a Rigaku Mini Flex 600 Powder Diffractometer with a Cu (1.54060 Å) X-ray tube and recorded in the 2θ range from 5 to 80°. Shimadzu FTIR 8201 (KBr pellet method) was employed to investigate the functional groups of the catalysts. The morphological surfaces of the catalyst were captured using SEM JSM 650 with 20 kV acceleration combined with an EDX instrument. The acidity of the catalyst was measured via the gravimetric method44 with pyridine vapor adsorption. Pyridine FTIR was also conducted to observe the properties of catalyst acidity.Hydrocracking of CPO
The hydrocracking of CPO was conducted using a reactor according to the previously reported procedure.45 First, the reactor was saturated using H2 atmosphere in order to eliminate oxygen gas. In this process,32 11.94 g min−1 CPO flow rate was used and controlled by a peristaltic pump, and 12 g of the catalyst weight was employed. Hydrocracking was conducted at 450 °C for 0.12 h with a gas flow of H2 of 2 mL s−1. The hydrocracking reaction products that leave the reactor are directed to the condenser pot, where they are separated into liquid and gas. The hydrocracking product liquid was vacuum distilled at 200 °C to obtain the biofuels and was later analyzed using GC-MS (Thermo Scientific, equipped with TG-5MS column). The bio-gasoline fraction consisted of C5–C12, the bio-kerosene fraction was C13–C16, whereas the fuel oil was > C17. The conversion of CPO (X) was determined according the eqn (1) as follows:
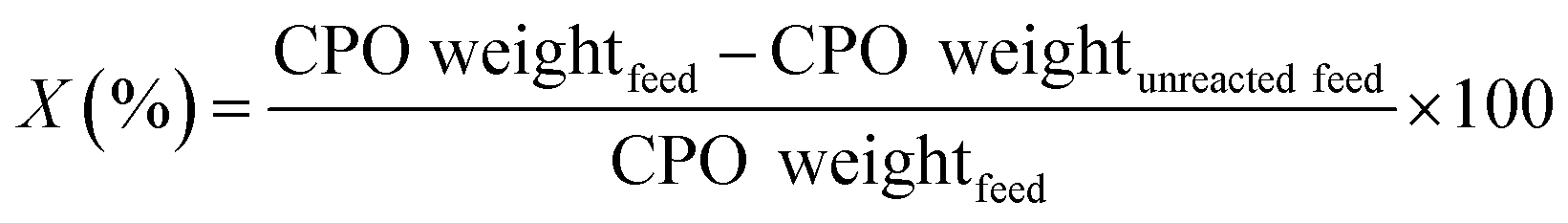 | (1) |
In order to investigate the reusability of the modified catalyst, the catalyst and hydrocracking product were separated by centrifugation and subsequently washed with hexane. Afterward, the powder was dried at 105 °C for 1 h. The catalyst was then calcined and reduced similar to the previously described in the experimental section. All of the coke was thermally degraded in this stage, producing fresh catalytic sites.
Results and discussion
Catalyst characterization
Na-bentonite, bentonite-zirconium phosphide, and bentonite-zirconium nitride were analyzed employing XRD, FTIR spectroscopy, SEM-EDX, and acidity measurements using the gravimetric method. The diffractograms for Na-bentonite prior to and after modification by ZrP and ZrN are shown in Fig. 1. The smectite minerals were clearly recognized in Fig. 1a at 2θ diffraction peaks of 5.80°, 19.93°, and 20.67°.46 A strong peak at a low 2θ value was mainly attributed to the representative of montmorillonite in bentonite,47 and this finding was consistent with other reports.48 The calcite (ICDD 47-1743) and quartz (ICDD 00-046-1045) minerals also appeared at 29.16° and 26.23°, respectively, and were coherent with other reported profound clay-based materials.49 The 2θ peaks at 33.09°, 35.19°, and 40.03° (Fig. 1b) corresponded to the zirconium nitride phase.50 The lack of strong peaks was presumably related to the amorphous structure.51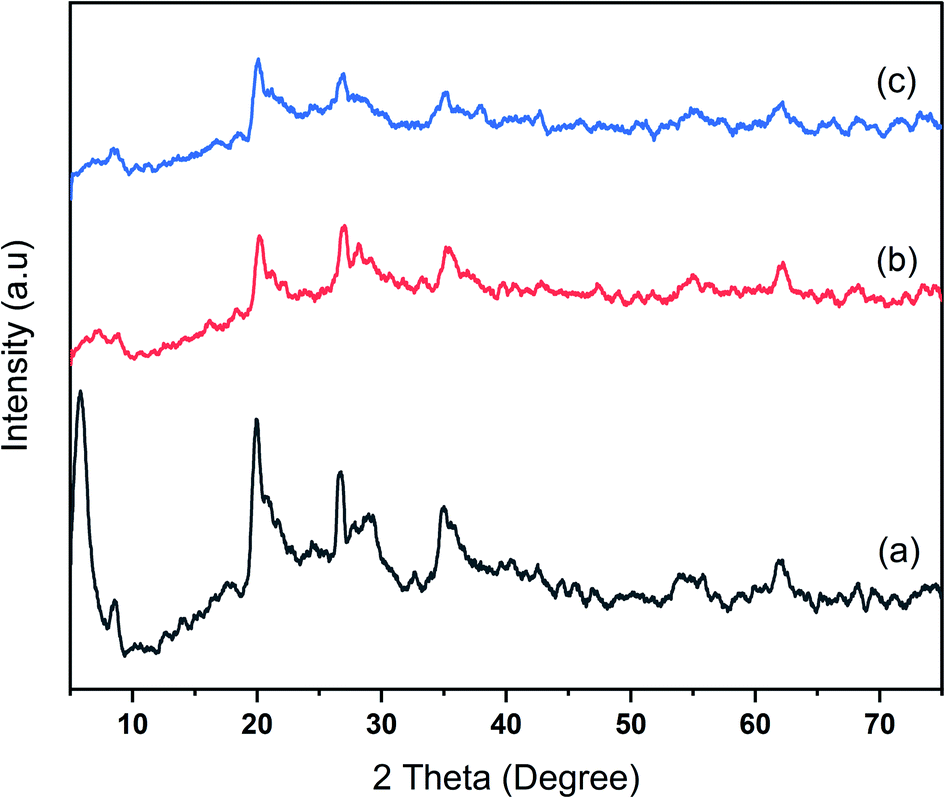 | ||
| Fig. 1 XRD pattern of (a) Na-bentonite, (b) bentonite-ZrN, and (c) bentonite-ZrP catalyst. | ||
The zirconium phosphide phase in Fig. 1c was likely unobserved, which could be attributed to the possibility that zirconium phosphide was highly irregular on the bentonite's surface or, due to a low percentage concentration, was employed with high dispersions, which lay below the detection limit of the analysis.52 A similar finding was reported by Amaya et al.22 who modified the acid properties of dealuminated bentonite using AlZr or AlCe. The broadband 2θ peak of Na-bentonite at 5.80° was less noticeable after modification, indicating the less-ordered bentonite-ZrP and bentonite ZrN. This peak nearly vanished for the modified bentonite due to the disordered nature of the bentonite layer structure. A similar finding was also reported consistently by Huo et al.53 who studied the zirconium-modified natural clay. These XRD patterns demonstrated that the bentonite had been successfully modified.
Fig. 2 represents the FTIR spectra of Na-bentonite and the modified bentonite. The FTIR spectra of Na-bentonite in Fig. 2a revealed two peaks at 3391.4 cm−1 and 1632.6 cm−1, which were associated with the –OH stretching vibration of the water molecules that exist in the interlayer and the structural hydroxyl group in bentonite as well.54 The absorption band at 3615.5 cm−1 was attributed to the strain vibration of the octahedral layer's Al-coordinated O–H bond.55 There bands were also presented in the modified bentonite. However, as shown in Fig. 2b and c, the intensity bands were relatively decreased due to the effect of the modification.56 This condition presumably indicated that Zr was likely linked to Al–O in the alumina octahedral sheets and Si–O in the silica tetrahedron plates.57 Another study consistently reported this condition as well.58 The peaks at 976 cm−1, 909 cm−1, and 1105 cm−1 corresponded to Si–O–Si and Si–O stretching vibrations,59 whereas the peaks of 842 cm−1 and 797 cm−1 indicated the deformation stretching of Al–Al–OH and Al–Mg–OH related to the bentonite framework.60 It can be revealed that these peaks were shifted to higher wavenumbers in the modified bentonites, which was consistent with the finding of another study.58 Furthermore, the bands at 689 cm−1 and 782 cm−1 were observed for modified bentonite, which indicated the presence of Zr–O bonds.61 In brief, FTIR analysis corroborated the successful bentonite modification.
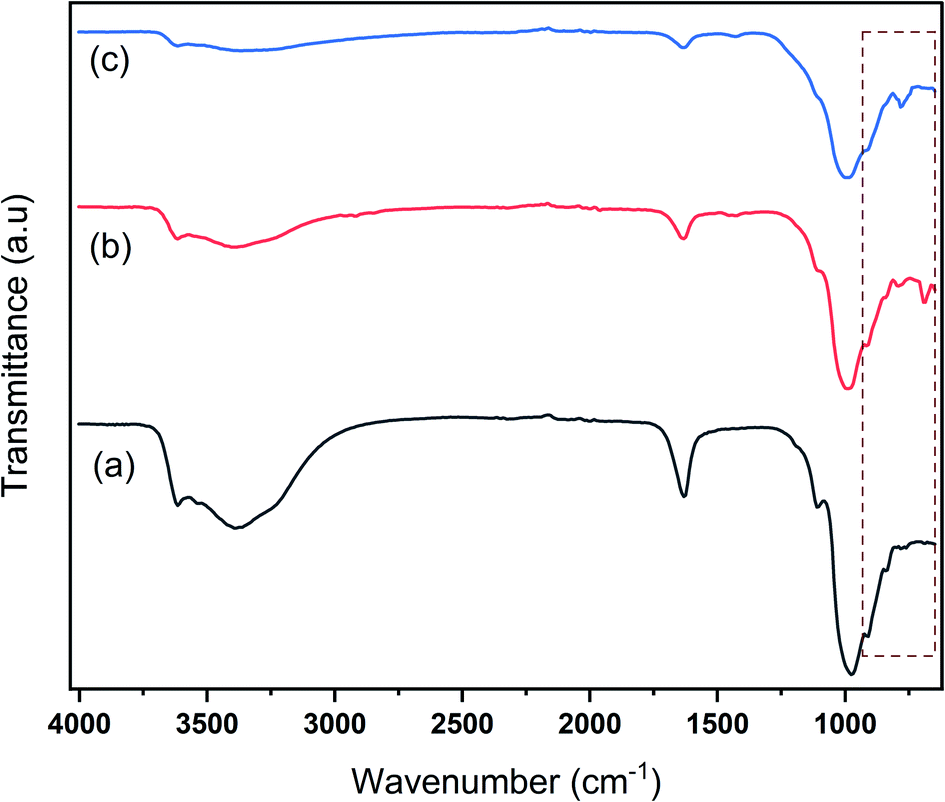 | ||
| Fig. 2 FTIR spectra of (a) Na-bentonite, (b) bentonite-ZrN, and (c) bentonite-ZrP catalyst. | ||
Fig. 3a shows that the surface of Na-bentonite has a sheet-like layered structure. Kadeche et al.62 reported that the Na-bentonite had a sharp-edged lamellar structure. Some studies revealed that the bentonite was granular with an irregular structure,63 which was consistent with the as-prepared Na-bentonite, as shown in Fig. 3a. Similar morphological characteristics have been reported in other studies.64 As shown in Fig. 3b and c, the Na-bentonite surface thoroughly changed after modification.
 | ||
| Fig. 3 SEM micrographs of (a) Na-bentonite (b) bentonite-ZrP (c) bentonite-ZrN. | ||
The micrograph in Fig. 3b shows a particle of ZrP, which was suitably dispersed on the bentonite surface. Similarly, the surface of Na-bentonite changed after being modified by ZrN (Fig. 3c). Mudzielwana et al.63 stated that the modification of bentonite could change the surface morphology of bentonite. Mahadevan et al.23 showed that the modification of bentonite using Zr changed its surface morphology with the ability to promote a large surface area with more porosity. It can be seen that the modification using zirconium nitride gave different results than that with zirconium phosphide, whereas ZrP showed a sharp multiangle granule-like structure, and ZrN exhibited a needle-like shape with a distinctive structure. This condition could facilitate the efficient interaction of the catalytic site of modified bentonite, which can result in an effective hydrocracking reaction.
The EDX analysis of all the catalysts is presented in Table 1. It can be seen that the Zr content increased after the modification of ZrP and ZrN from 0 to 4.67 and 12.28 wt%, respectively. Furthermore, the concentration of N and P were also increased from 0 to 0.19 and 0 to 6.45 wt%, respectively, which indicated that the modification of bentonite using ZrN and ZrP was successfully employed towards Na-bentonite. After modification, the relative amounts of Si and Al remained almost constant in all the modified bentonites. These results indicated that the composition of the bentonite layer is preserved in the modified bentonite catalyst.54 Some elements such as C, O, Ca, Ti, Fe, K, and Mg existed in the Na-bentonite, and this condition was also reported by another study.65
| Elements | Atomic (wt%) | ||
|---|---|---|---|
| Na-bentonite | Bentonite-ZrP | Bentonite-ZrN | |
| Na | 1.97 | 1.30 | 1.62 |
| Mg | 2.73 | 2.03 | 0 |
| Al | 8.33 | 6.07 | 6.27 |
| Si | 25.18 | 19.08 | 19.51 |
| K | 0.47 | 0.24 | 0.37 |
| C | 16.32 | 16.42 | 14.65 |
| O | 40.05 | 40.14 | 35.91 |
| Cl | 0.45 | 0.56 | 1.12 |
| Ca | 0.26 | 0.23 | 0 |
| Ti | 0.24 | 0.18 | 0.41 |
| Fe | 3.10 | 2.24 | 5.84 |
| Zn | 0.90 | 0 | 0 |
| Zr | 0 | 4.67 | 12.28 |
| P | 0 | 6.45 | 0 |
| N | 0 | 0 | 0.19 |
The acidity value of Na-bentonite and modified bentonite were calculated using the gravimetric method by pyridine adsorption, which can be seen in Fig. 4. The acidity of the catalyst is an essential aspect since it impacts the catalytic activity of the hydrocracking process. This was mainly attributed to the linear relationship between the number of accessible acid sites and the catalytic activity as that was where the hydrocracking reactions took place.66 As presented in Fig. 4, it can be seen that Na-bentonite had a low acidity value of 0.054 mmol g−1, which was generated by the aluminosilicate framework bound to the pyridine. The acidity value of Na-bentonite gradually increased after being modified due to the existence of a vacant p orbital derived from the metal, i.e., zirconium, which would act as the Lewis acid active site.67 In regards to the bentonite-ZrN trend, the highest acidity value was achieved up to 1.822 mmol g−1 at the metal loading of 8 mEq g−1 and had showed further decrease at higher metal loading. At high metal loading, the distribution and amount of MoP in the matrix of bentonite presumably reached its maximum point, resulting in a drop in the adsorbed pyridine, thereby reducing the acidity of the catalyst.68
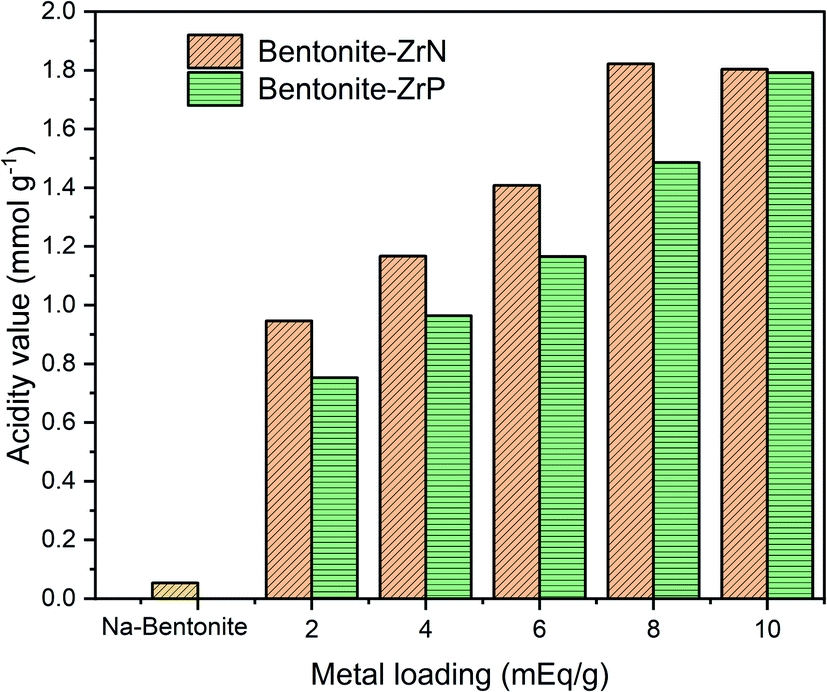 | ||
| Fig. 4 Effect of metal loading on the acidity of the catalysts. | ||
Noticeably, as can be seen in Fig. 4, a prolonged metal loading could increase the catalyst acidity of bentonite-ZrP up to 1.792 mmol g−1 at a metal loading of 10 mEq g−1 due to high metal loading, which would provide higher active acid site species, leading to an increase in the catalyst acidity. Furthermore, the high catalyst acidity was attributed to the Brønsted acid site, which was associated with the presence of a phosphate species bonded to a zirconia group.69 Sinhamahapatra et al.70 stated that the increase in the catalyst acidity could also be attributed to the availability of geminal P(OH) groups from the zirconium framework. A particular species concentration was correlated to the amount of acid sites.71 In this context, the metal loading as well as the type of zirconium metal clearly influenced the catalyst acidity since it provided alternative acid sites derived from ZrP and ZrN species. Furthermore, Fig. 4 showed that the bentonite-ZrN exhibited higher catalyst acidity than bentonite ZrP. This condition might be associated with the higher reactant adsorption of bentonite ZrN or perhaps a change in the metal structure, which provide highly active sites that affect the acidity of the catalyst.31
The acidity of the catalyst was further investigated using pyridine FTIR to determine the Brønsted and Lewis acid sites.72 As can be seen in Fig. 5, the representative pyridine FTIR spectra showed a distinctive peak, whereas that at ∼1640 cm−1 was attributed to the pyridinium ion, which corroborated the existence of Brønsted acid,73 and at ∼1423 cm−1, indicated the presence of the Lewis acid site.67 Marianou et al.74 stated that the coordinated pyridine to the Lewis acid site could be observed at 1435–1470 cm−1.
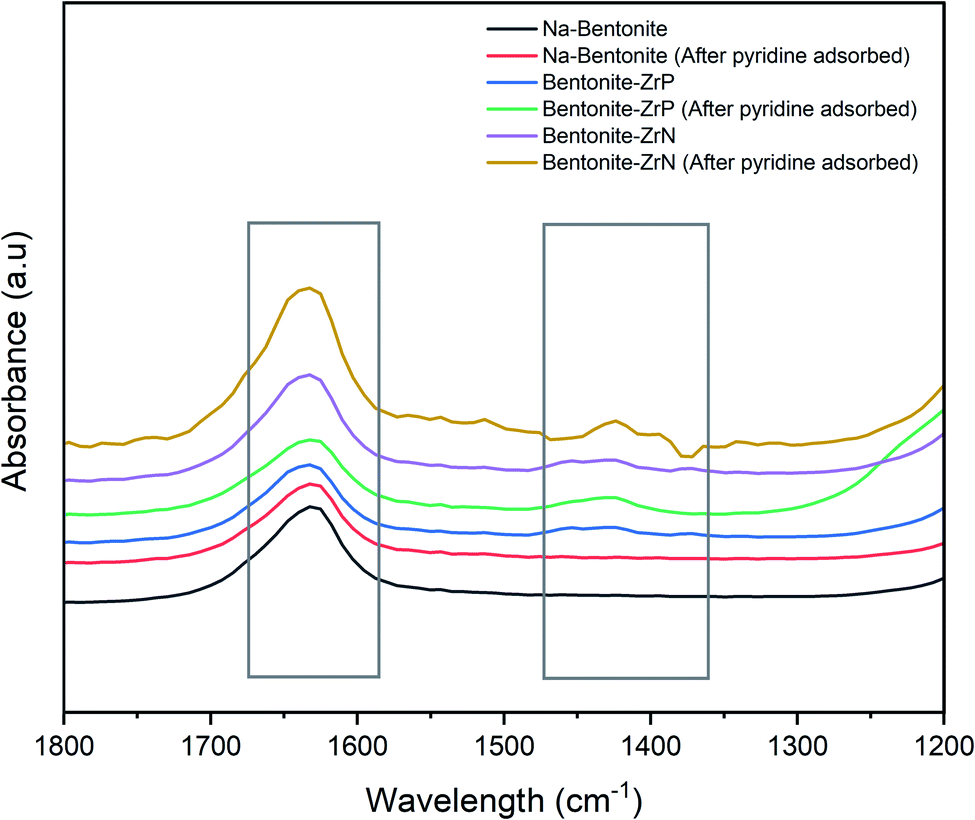 | ||
| Fig. 5 Pyridine FTIR spectra of the catalysts. | ||
The spectra of the parent Na-bentonite after adsorbed pyridine had only one prominent high intensity band at ∼1640 cm−1 and a weak intensity band at ∼1423 cm−1, indicating a low concentration of Lewis acid and a dominant amount of Brønsted acid sites. The Lewis acid site intensity was remarkably increased after incorporating ZrP and ZrB into Na-bentonite, indicating that the modification successfully enhanced the acidity of the bentonite catalyst. The intensity of the Brønsted acid site on the modified bentonite was relatively increased, presumably due to the presence of the P(OH) (ref. 70) and N(OH) groups in ZrP and ZrN, respectively. Furthermore, it was found that the highest intensity was generally achieved by bentonite-ZrN, which was consistent with the acidity analysis assessed by the gravimetric method.
Hydrocracking process
CPO was used as the feedstock during the hydrocracking reaction catalyzed by bentonite-ZrN and bentonite-ZrP. The effect of metal loading on the conversion of CPO was evaluated, as shown in Fig. 6. CPO conversion over bentonite-ZrN following a metal loading of 2, 4, 6, and 8 mEq g−1 revealed a continuous rise, with 77, 79, 83.59, and 87.93% conversion, respectively. Likewise, bentonite-ZrP metal loading of 2, 4, 6, and 8 mEq g−1 showed a conversion of 75.82, 77.33, 80.42, and 83%, respectively. Afterward, the conversion tended to increase at a maximum conversion of 86.04% when a metal loading of 10 mEq g−1 was employed. These results indicated that a prolonged metal loading of the modified bentonite gradually increased the CPO conversion. An increase in the metal loading caused a high catalyst density; as a consequence, the interaction of the active site of the catalyst with the adsorbed hydrocarbon species increased.75 As a result, hydrocracking took place efficiently, thus leading to high CPO conversion.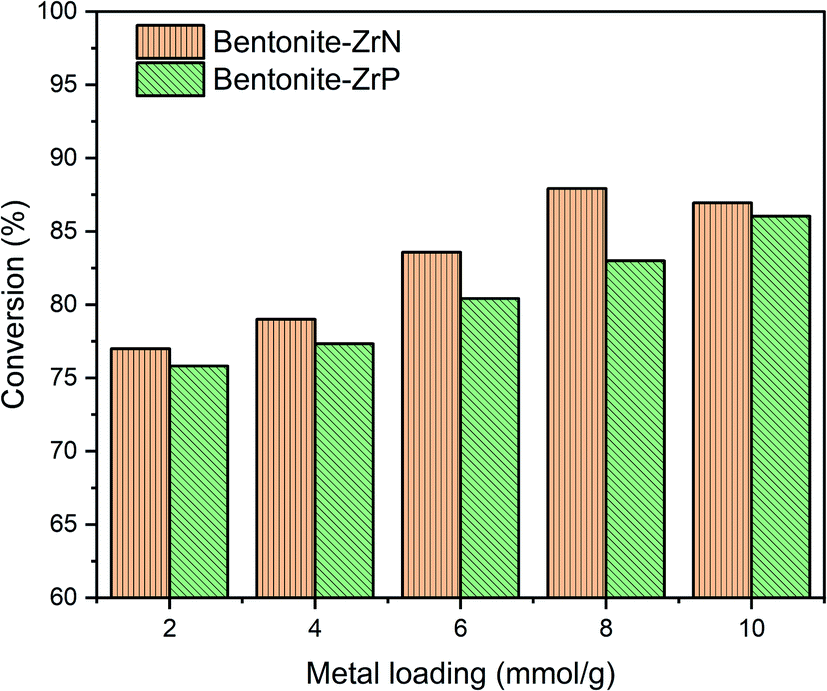 | ||
| Fig. 6 Effect of metal loading on the conversion of CPO. | ||
According to Utami et al.76 the transition metal, in this case, zirconium, had a Lewis acid feature that could promote the cracking process of longer chain hydrocarbons into shorter ones. The active site that adsorbed the hydrogen atom of H2 gas was subsequently transferred into the compound to be cracked down and later would eventually be replaced with a hydrocarbon molecule through the cracking reaction catalyzed by the Brønsted acid site on the catalyst. Hence, the more the Lewis and Brønsted acid site availability, the more effective the hydrocracking reaction. Srihanun et al.77 stated that a high catalyst concentration could promote high catalyst acidity. A similar finding was consistently reported by another study.78
Fig. 6 also showed that bentonite-ZrN provided higher conversion than bentonite-ZrP, which was consistent with the determined acidity values of the catalyst, as previously described. Despite the fact that increased catalyst loading was directly related to a higher availability of the active catalyst sites, however, a relative decrease in the conversion was noticeable at a higher catalyst loading of bentonite-ZrN, presumably due to the agglomeration, hence reducing the contact of the active catalytic site with the reactant,79 which directly reduces CPO conversion. Susi et al.80 stated that agglomeration could decrease the acidity of the catalyst, which consequently lowered the conversion. Similar findings were also reported by other studies.68
As a comparison, several reports on the conversion of palm oil-based feedstocks through hydrocracking catalyzed by various alumina-silicate are presented in Table 2. For instance, the ZSM-5/MCM-41 catalyst reached ≥ 62.60% conversion after introducing the bimetallic NiMoW as an external active site of the catalyst. It can be noticed that bentonite-ZrP and bentonite-ZrN showed adequate performance in the CPO conversion relative to other alumina silicate-based catalysts as well as bentonite-MoN, as previously reported.
| Catalyst | Feedstock | Condition | Conversion (%) | Ref. |
|---|---|---|---|---|
| NiMoW-ZSM-5/MCM-41 | CPO | 400 °C for 2 h | 62.60–71.40 | 81 |
| NiW-HZSM-5 | CPO | 400 °C for 2 h | 48.18 | 16 |
| CoMo/Al2O3 | CPO | 350 °C for 1 h, 500 psi | 64.21 | 82 |
| Bentonite-molybdenum nitride | Palm oil | 458.79 °C, 0.12 h of contact time | 78.33 | 32 |
| Bentonite-zirconium phosphide | CPO | 450 °C, 0.12 h of contact time | 86.04 | This work |
| Bentonite-zirconium nitride | CPO | 450 °C, 0.12 h of contact time | 87.93 | This work |
The GC-MS analysis of CPO and hydrocracking products is presented in Fig. 7. According to Fig. 7a, CPO had a long hydrocarbon chain in the range of C16–C22 with triacyl glycerol compounds as the constituent, with particularly a saturated and unsaturated fatty acid, as well as other compounds.
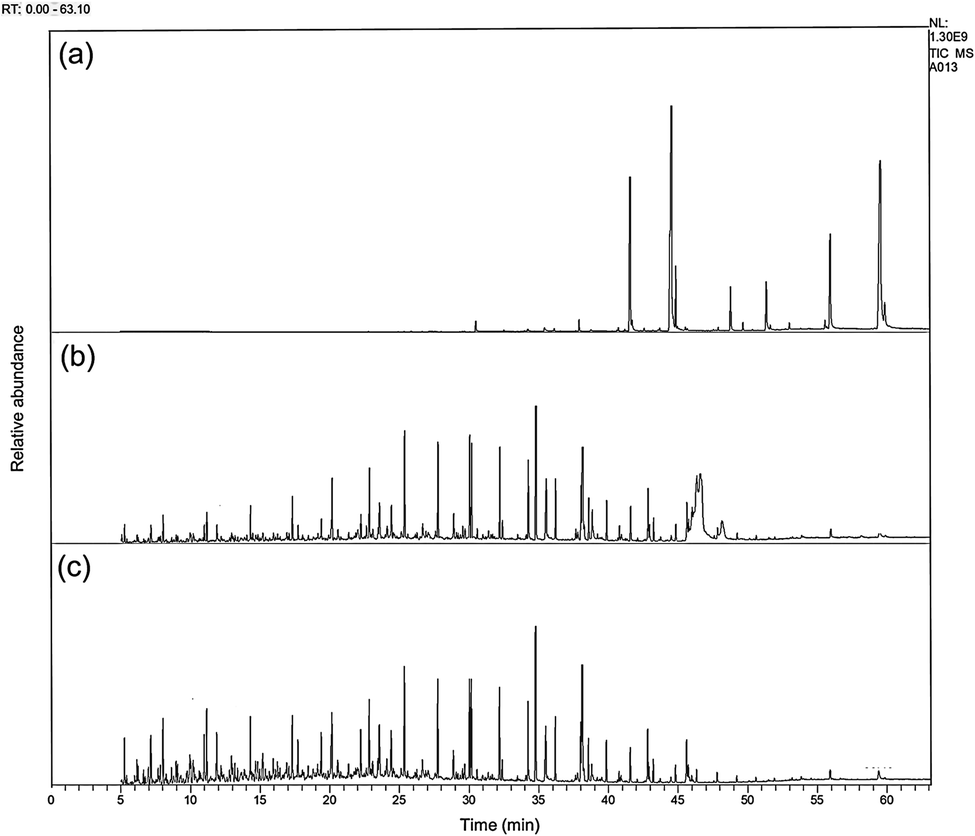 | ||
| Fig. 7 Representative GC-MS chromatograms of (a) CPO, (b) hydrocracking product catalyzed by bentonite-ZrN, and (c) bentonite-ZrP. | ||
As revealed in Fig. 7b and c, the hydrocracking products catalyzed by bentonite-ZrN and bentonite-ZrB, respectively, showed a perspicuous peak change on the chromatogram, especially a shift towards shorter retention time, indicating that the triglyceride from CPO successfully underwent the hydrocracking reaction and formed a short carbon chain consisting of biofuel.
The fraction of bio-gasoline, bio-kerosene, and fuel oil of the hydrocracking product catalyzed by bentonite-ZrN and bentonite-ZrP is presented in Fig. 8. It can be observed that there was no significant change in the bio-gasoline, bio-kerosene, and oil fuel fraction, either at low or high metal loading. Bentonite-ZrN catalyst (Fig. 8a) could generate a bio-gasoline, bio-kerosene, and oil fuel fraction in the range of 72.84–75.09%, 21.02–22.97%, and 3.89–5.22%, respectively, whereas bio-gasoline, bio-kerosene, and oil fuel fraction in the range of 74.73–83.09%, 6.84–13.96%, and 2.91–18.43, respectively, were generated by bentonite-ZrP (Fig. 8b). The biofuel fraction distribution emerges to be influenced by the strength of the site type rather than the catalyst concentration.83 As a result, the distribution of the biofuel fraction with various catalyst loadings was almost insignificantly different and close to each other.
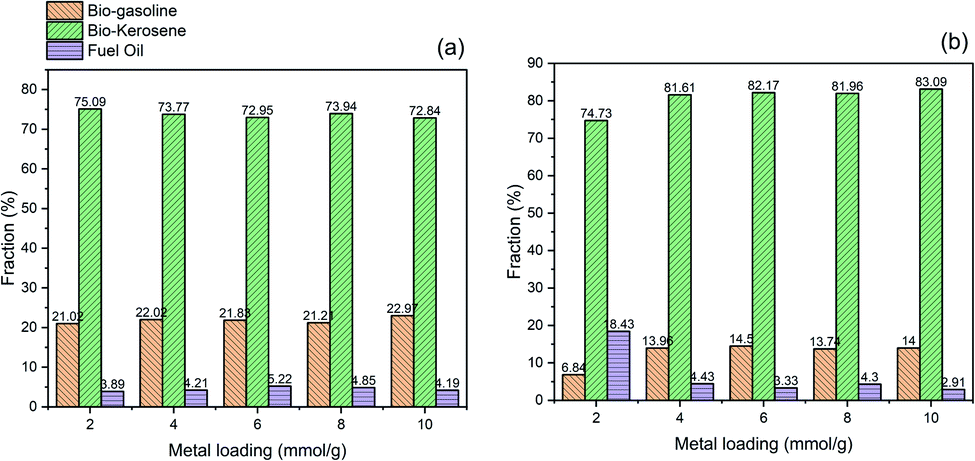 | ||
| Fig. 8 Effect of metal loading on the biofuel fraction of the hydrocracking product catalyzed by (a) bentonite-ZrN and (b) bentonite-ZrP. | ||
It was also revealed that the highest fraction was found to be bio-kerosine, followed by bio-gasoline and fuel oil. In this hydrocracking process, bentonite-ZrN and bentonite-ZrP generated a dominant fraction of bio-kerosene, indicating that the possibility of effective hydrocracking and decarboxylation reaction was likely more prominent. The reaction began with cracking the glycerol ester bond of the fatty acids to form free fatty acids. Bio-kerosene was produced when hydrogenation took place. Simultaneously, the decarboxylation reaction began with the adsorption of fatty acids on the catalyst surface, which was accomplished through the adsorption of oxygen atoms with the carboxyl groups of the fatty acids. As a result, the bio-kerosene fractions were produced in high amounts. Fig. 8 showed that a low oil fuel fraction was produced by both the catalysts either due to insufficient hydrocracking process or coupling reaction, hence producing the heavy fraction compound (>C17), whereas the further hydrocracking of the C13–C16 hydrocarbon, i.e., bio-kerosene, would produce a bio-gasoline fraction.21 Hence, it was reasonable that more bio-kerosene was generated than bio-gasoline and oil fuel. Utami et al.76 generated 36.51% of the gasoline fraction and 45.81% of the diesel fraction when employing Cr/ZrO2–SO4 in the hydrocracking of palm oil. Marini et al.26 reported that Ni/Al2O3 and H/Bentonite could exhibit 60.37% and 39.83% of bio-gasoline fraction, respectively, in the hydrocracking of nyamplung seed, whereas Wijaya et al.27 produced 70.28% of the gasoline fraction and 6.20% of the diesel fraction when employing 1% Ni-sulfated zirconia for the hydrocracking of waste cooking oil. However, with regard to the biofuel distribution fraction, it was necessary to evaluate the hydrocracking process by varying the process parameter, such as temperature, hydrogen feed, and pressure as a means to get an adequate understandable influence on the biofuel distribution fraction.
The reusability of bentonite-ZrN and bentonite-ZrP catalysts were evaluated under the same identical conditions of CPO hydrocracking. Bentonite-ZrN with a metal loading of 8 mEq g−1 and bentonite-ZrP with 10 mEq g−1 metal loading were employed as they generated high conversion in CPO hydrocracking. The effect of the fresh catalyst and the reused bentonite-ZrN and bentonite-ZrP catalyst up to 4 cycles in the conversion is presented in Fig. 9.
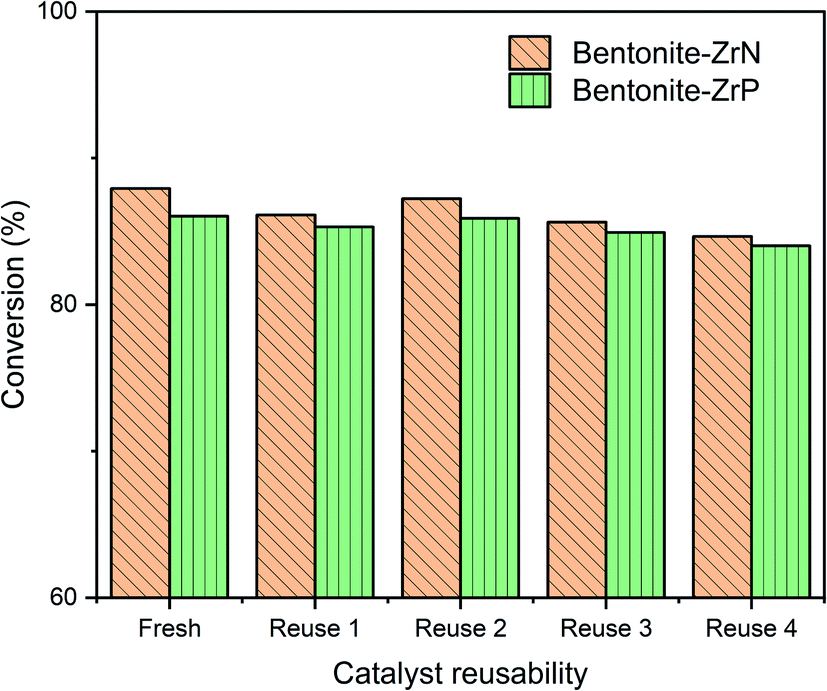 | ||
| Fig. 9 Reusability of bentonite-ZrN and bentonite-ZrP catalyst. | ||
The bentonite-ZrN catalyst showed a slight decreased in the catalyst activity up to 2.06% in the first cycle, with the conversion decreasing from 87.93 to 86.12%. Afterward, there was an increase up to 1.29% in the catalyst activity in the second cycle, as indicated by the increase in the conversion from 86.12 to 87.23%. This condition indicated that the conversion was relatively stable at two consecutive runs. Similarly, the bentonite-ZrP catalyst revealed a slight decrease in the catalyst activity (only ∼1%) after one cycle and tended to be stable, producing up to 85.89% conversion in the second cycle.
Furthermore, in the third cycle, the bentonite-ZrN decreased the catalyst activity up to 1.83%, generated 85.63% conversion, and gradually decreased up to 84.64% when the catalyst was reused up to four cycles. Likewise, the bentonite-ZrP revealed a decrease in the catalyst activity up to 1.12%, which produced 84.93% conversion and moderately decreased to 84.02% at four consecutive runs. The decrease in the conversion at four consecutive runs was presumably due to the deactivation of the catalyst through the undesired coke formation84 The catalyst's surface was covered by coke and clogged the catalyst's active site.85 Under these conditions, the reactant and catalyst sites had limited accessibility, which inhibited the hydrocracking reaction's effectiveness,86 thereby reducing the CPO conversion. Ayodele et al.87 stated that the reduction in the catalyst activity was probably due to the loss of catalyst acidity.
Nevertheless, the bentonite-ZrN catalyst revealed an only 3.73% decrease in the catalyst activity towards conversion at four consecutive cycles relative to the fresh catalyst, whereas only 2.34% decreased in the catalyst activity by bentonite-ZrP. Papageridis et al.88 reported that the nitride-based catalyst had more stability than the sulfide catalyst towards the conversion of guaiacol through the hydrodeoxygenation reaction after 4 h on stream during continuous operation, whereas de Souza et al.35 showed that the phosphide-based catalyst had an insignificant deactivation towards phenol conversion through the hydrodeoxygenation reaction during 20 h on stream. According to the reusability study (Fig. 9), the bentonites modified by ZrP and ZrN were promising and comparable catalysts, which had adequate conversion stability of CPO through the hydrocracking reaction up to four consecutive runs.
Conclusions
Herein, bentonite was appropriately modified by zirconium nitride and zirconium phosphide for the hydrocracking of crude palm oil to biofuels. The modification of bentonite using zirconium nitride and zirconium phosphide was successfully achieved, as confirmed by XRD, FTIR spectroscopy, and SEM-EDX analysis. The acidity of bentonite increased sufficiently due to the modification by zirconium nitride and zirconium phosphate. The hydrocracking study revealed that bentonite-ZrN had higher conversion than bentonite-ZrP due to high catalyst acidity. The hydrocracking product exposed that both the catalysts showed a high distribution fraction of bio-kerosene, followed by bio-gasoline and fuel oil. The reusability study of the catalyst showed that the catalyst could generate more than 85% conversion for both the catalysts at four consecutive cycles with an insignificant decrease in the catalyst activity. Overall, this study offered the potential of bentonite modified with zirconium nitride and zirconium phosphide catalyst for the hydrocracking of CPO with high stability towards conversion and high bio-kerosene fraction. In particular, it was imperative to optimize the hydrocracking parameter in order to understand the adequate influence on the biofuel distribution fractions.Conflicts of interest
There are no conflicts to declare.References
- P. Dujjanutat and P. Kaewkannetra, Renewable Energy, 2020, 147, 464–472 CrossRef CAS.
- Z. Fanani, H. Hasanudin, A. Rachmat and M. Said, Molekul, 2021, 16, 244–252 CrossRef.
- E. P. Sari, K. Wijaya, W. Trisunaryanti, A. Syoufian, H. Hasanudin and W. D. Saputri, Int. J. Energy Environ. Eng., 2021, 1–12 Search PubMed.
- J. L. Sihombing, A. N. Pulungan, H. Herlinawati, M. Yusuf, S. Gea, H. Agusnar, B. Wirjosentono and Y. A. Hutapea, Catalysts, 2020, 10, 1–14 CrossRef.
- A. A. Mancio, K. M. B. da Costa, C. C. Ferreira, M. C. Santos, D. E. L. Lhamas, S. A. P. da Mota, R. A. C. Leão, R. O. M. A. de Souza, M. E. Araújo, L. E. P. Borges and N. T. Machado, Ind. Crops Prod., 2016, 91, 32–43 CrossRef CAS.
- T. Burimsitthigul, B. Yoosuk, C. Ngamcharussrivichai and P. Prasassarakich, Renewable Energy, 2021, 163, 1648–1659 CrossRef CAS.
- M. Ameen, M. T. Azizan, S. Yusup, A. Ramli and M. Yasir, Renewable Sustainable Energy Rev., 2017, 80, 1072–1088 CrossRef CAS.
- S. Wang, Z. Guo, Q. Cai and L. Guo, Biomass Bioenergy, 2012, 45, 138–143 CrossRef CAS.
- Z. Eller, Z. Varga and J. Hancsók, Fuel, 2016, 182, 713–720 CrossRef CAS.
- N. Taufiqurrahmi and S. Bhatia, Energy Environ. Sci., 2011, 4, 1087–1112 RSC.
- R. Rasyid, R. Malik, H. S. Kusuma, A. Roesyadi and M. Mahfud, Bull. Chem. React. Eng. Catal., 2018, 13, 196–203 CrossRef CAS.
- T. Li, J. Cheng, R. Huang, W. Yang, J. Zhou and K. Cen, Int. J. Hydrogen Energy, 2016, 41, 21883–21887 CrossRef CAS.
- A. Ishihara, N. Fukui, H. Nasu and T. Hashimoto, Fuel, 2014, 134, 611–617 CrossRef CAS.
- I. Istadi, T. Riyanto, E. Khofiyanida, L. Buchori, D. D. Anggoro, I. Sumantri, B. H. S. Putro and A. S. Firnanda, Bioresour. Technol. Rep., 2021, 14, 100677 CrossRef CAS.
- B. Boonrod, C. Prapainainar, P. Narataruksa, A. Kantama, W. Saibautrong, K. Sudsakorn, T. Mungcharoen and P. Prapainainar, J. Cleaner Prod., 2017, 142, 1210–1221 CrossRef CAS.
- M. Subsadsana and C. Ruangviriyachai, Orient. J. Chem., 2016, 32, 839–844 CrossRef CAS.
- M. Subsadsana, P. Sangdara and C. Ruangviriyachai, Asia-Pac. J. Chem. Eng., 2017, 12, 147–158 CrossRef CAS.
- M. E. Gyftopoulou, M. Millan, A. V. Bridgwater, D. Dugwell, R. Kandiyoti and J. A. Hriljac, Appl. Catal., A, 2005, 282, 205–214 CrossRef CAS.
- Z. M. El-Deeb, W. A. Aboutaleb, R. S. Mohamed, A. S. Dhmees and A. I. Ahmed, J. Energy Inst., 2022, 103, 84–93 CrossRef CAS.
- T. Kaka khel, P. Mäki-Arvela, M. Azkaar, Z. Vajglová, A. Aho, J. Hemming, M. Peurla, K. Eränen, N. Kumar and D. Y. Murzin, Mol. Catal., 2019, 476, 110515 CrossRef CAS.
- A. Suseno, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 509, 012005 CAS.
- J. Amaya, N. Suarez, A. Moreno, S. Moreno and R. Molina, New J. Chem., 2020, 44, 2966–2979 RSC.
- H. Mahadevan, K. Anoop Krishnan, R. R. Pillai and S. Sudhakaran, Res. Chem. Intermed., 2020, 46, 639–660 CrossRef CAS.
- J. Lin, B. Jiang and Y. Zhan, J. Environ. Manage., 2018, 217, 183–195 CrossRef CAS PubMed.
- V. M. Abbasov, H. C. Ibrahimov, G. S. Mukhtarova, M. I. Rustamov and E. Abdullayev, Energy Fuel., 2017, 31, 5840–5843 CrossRef CAS.
- A. T. Marini, K. Wijaya and N. A. Sasongko, IOP Conf. Ser. Earth Environ. Sci., 2018, 124, 6 Search PubMed.
- K. Wijaya, M. Utami, A. K. Damayanti, I. Tahir, A. D. Tikoalu, R. Rajagopal, A. Thirupathi, D. Ali, S. Alarifi, S. W. Chang and B. Ravindran, Fuel, 2022, 322, 124152 CrossRef CAS.
- J. C. Cortés, C. Rodríguez, R. Molina and S. Moreno, Fuel, 2021, 295, 120612 CrossRef.
- Y. Sun, H. Jiu, J. Tian, L. Zhang, T. Han, F. Guo and M. Qiu, J. Solid State Chem., 2020, 284, 121182 CrossRef CAS.
- H. Hasanudin, W. R. Asri, K. Tampubolon, F. Riyanti, W. Purwaningrum and K. Wijaya, Pertanika J. Sci. Technol., 2022, 30, 1739–1754 Search PubMed.
- A. B. Dongil, Nanomaterials, 2019, 9, 1111 CrossRef CAS PubMed.
- H. Hasanudin, W. R. Asri, M. Said, P. T. Hidayati, W. Purwaningrum, N. Novia and K. Wijaya, RSC Adv., 2022, 12, 16431–16443 RSC.
- V. O. O. Gonçalves, P. M. de Souza, T. Cabioc’h, V. T. da Silva, F. B. Noronha and F. Richard, Appl. Catal., B, 2017, 219, 619–628 CrossRef.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, L. R. Radovic, J. L. G. Fierro, W. J. Desisto and N. Escalona, Appl. Catal., A, 2012, 439, 111–124 CrossRef.
- P. M. de Souza, C. V. M. Inocêncio, V. I. Perez, R. C. Rabelo-Neto, V. O. O. Gonçalves, G. Jacobs, F. Richard, V. T. da Silva and F. B. Noronha, Catal. Today, 2020, 356, 366–375 CrossRef CAS.
- M. Ruangudomsakul, N. Osakoo, J. Wittayakun, C. Keawkumay, T. Butburee, S. Youngjan, K. Faungnawakij, Y. Poo-arporn, P. Kidkhunthod and P. Khemthong, Mol. Catal., 2021, 523, 111422 CrossRef.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- M. Zhou, H. A. Doan, L. A. Curtiss and R. S. Assary, J. Phys. Chem. C, 2021, 125, 8630–8637 CrossRef CAS.
- S. Zhao, J. Song, R. Xu, L. Nie, J. Ma, C. Deng, X. Cheng, X. Zhao, S. Hao and J. Li, Ceram. Int., 2021, 47, 23267–23274 CrossRef CAS.
- Y. Yuan, J. Wang, S. Adimi, H. Shen, T. Thomas, R. Ma, J. P. Attfield and M. Yang, Nat. Mater., 2020, 19, 282–286 CrossRef CAS PubMed.
- S. Zhao, J. Ma, R. Xu, X. Lin, X. Cheng, S. Hao, X. Zhao, C. Deng and B. Liu, Sci. Rep., 2019, 9, 1–9 CrossRef PubMed.
- Z. Li, N. Chen, J. Wang, P. Li, M. Guo, Q. Wang, C. Li, C. Wang, T. Guo and S. Chen, Sci. Rep., 2017, 7, 1–7 CrossRef PubMed.
- T. Kokulnathan, T. J. Wang, N. Duraisamy, E. A. Kumar and A. N. Sung, J. Hazard. Mater., 2021, 412, 125257 CrossRef CAS PubMed.
- K. Wijaya, A. R. Putri, S. Sudiono, S. Mulijani, A. Patah, A. C. Wibowo and W. D. Saputri, Catalysts, 2021, 11, 1492 CrossRef CAS.
- H. Hasanudin, A. Rachmat, M. Said and K. Wijaya, Period. Polytech., Chem. Eng., 2020, 64, 238–247 CrossRef CAS.
- K. Wijaya, M. A. Kurniawan, W. D. Saputri, W. Trisunaryanti, M. Mirzan, P. L. Hariani and A. D. Tikoalu, J. Environ. Chem. Eng., 2021, 9, 105399 CrossRef CAS.
- R. Huang, C. Hu, B. Yang and J. Zhao, Desalin. Water Treat., 2016, 57, 10646–10654 CrossRef CAS.
- K. Wijaya, A. D. Ariyanti, I. Tahir, A. Syoufian, A. Rachmat and H. Hasanudin, Nano Hybrids Compos., 2018, 19, 46–54 Search PubMed.
- K. Wijaya, M. Utami, A. Syoufian and A. I. Murifal, Adv. Mater. Res., 2021, 1162, 21–26 Search PubMed.
- W. Wei, H. Wang and Y. H. Hu, J. Mater. Chem. A, 2013, 1, 14350–14357 RSC.
- A. H. Ali, Sep. Sci. Technol., 2018, 53, 2284–2296 CrossRef CAS.
- J. Lee, S. Hwang, S. B. Lee and I. K. Song, Korean J. Chem. Eng., 2010, 27, 1755–1759 CrossRef CAS.
- J. Huo, X. Min and Y. Wang, Environ. Res., 2021, 194, 110685 CrossRef CAS PubMed.
- F. Tomul, Appl. Surf. Sci., 2011, 258, 1836–1848 CrossRef CAS.
- A. M. Elfadly, I. F. Zeid, F. Z. Yehia, M. M. Abouelela and A. M. Rabie, Fuel Process. Technol., 2017, 163, 1–7 CrossRef CAS.
- F. T. Basoglu, Chem. Pap., 2016, 70, 933–945 CAS.
- J. Zhou, P. Wu, Z. Dang, N. Zhu, P. Li, J. Wu and X. Wang, Chem. Eng. J., 2010, 162, 1035–1044 CrossRef CAS.
- T. Chauhan, M. Udayakumar, M. Ahmed Shehab, F. Kristály, A. Katalin Leskó, M. Ek, D. Wahlqvist, P. Tóth, K. Hernadi and Z. Németh, Arabian J. Chem., 2022, 15, 103706 CrossRef CAS.
- M. Mirzan, K. Wijaya, I. I. Falah and W. Trisunaryanti, Asian J. Chem., 2019, 31, 229–234 CrossRef CAS.
- B. Daroughegi Mofrad, M. Hayati-Ashtiani and M. Rezaei, Asia-Pac. J. Chem. Eng., 2018, 13, 1–11 CrossRef.
- M. Sakti La Ore, K. Wijaya, W. Trisunaryanti, W. D. Saputri, E. Heraldy, N. W. Yuwana, P. L. Hariani, A. Budiman and S. Sudiono, J. Environ. Chem. Eng., 2020, 8, 104205 CrossRef CAS.
- A. Kadeche, A. Ramdani, M. Adjdir, A. Guendouzi, S. Taleb, M. Kaid and A. Deratani, Res. Chem. Intermed., 2020, 46, 4985–5008 CrossRef CAS.
- R. Mudzielwana, M. W. Gitari, S. A. Akinyemi and T. A. M. Msagati, Appl. Surf. Sci., 2017, 422, 745–753 CrossRef CAS.
- M. H. Ravari, A. Sarrafi and M. Tahmooresi, S. Afr. J. Chem. Eng., 2020, 31, 1–6 Search PubMed.
- A. T. Babu and R. Antony, Appl. Clay Sci., 2019, 183, 105312 CrossRef CAS.
- W. Trisunaryanti, K. Wijaya, T. Triyono, A. R. Adriani and S. Larasati, Results Eng., 2021, 11, 100258 CrossRef CAS.
- A. Nadia, K. Wijaya, I. I. Falah, S. Sudiono and A. Budiman, Waste Biomass Valorization, 2022, 13, 2335–2347 CrossRef CAS.
- K. Wijaya, A. Nadia, A. Dinana, A. F. Pratiwi, A. D. Tikoalu and A. C. Wibowo, Catalysts, 2021, 11, 1150 CrossRef CAS.
- R. Weingarten, Y. T. Kim, G. A. Tompsett, A. Fernández, K. S. Han, E. W. Hagaman, W. C. Conner, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 123–134 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, B. Roy, A. Tarafdar, H. C. Bajaj and A. B. Panda, Appl. Catal., A, 2010, 385, 22–30 CrossRef CAS.
- A. Aneu, K. Wijaya and A. Syoufian, Silicon, 2021, 13, 2265–2270 CrossRef CAS.
- N. A. S. Ramli and N. A. S. Amin, Energy Convers. Manage., 2015, 95, 10–19 CrossRef CAS.
- W. Ni, D. Li, X. Zhao, W. Ma, K. Kong, Q. Gu, M. Chen and Z. Hou, Catal. Today, 2019, 319, 66–75 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, Appl. Catal., A, 2018, 555, 75–87 CrossRef CAS.
- A. Alkhlel and H. De Lasa, Catalysts, 2019, 9, 542 CrossRef CAS.
- M. Utami, R. Safitri, M. Fajar Pradipta, K. Wijaya, S. Woong Chang, B. Ravindran, D. Ovi, J. R. Rajabathar, N. Poudineh and R. Moonsamy Gengan, Mater. Lett., 2022, 309, 131472 CrossRef CAS.
- N. Srihanun, P. Dujjanutat, P. Muanruksa and P. Kaewekannetra, Catalysts, 2020, 10, 241 CrossRef CAS.
- S. Cheng, L. Wei, J. Julson and M. Rabnawaz, Energy Convers. Manage., 2017, 150, 331–342 CrossRef CAS.
- Y. Utubira, K. Wijaya, T. Triyonol and E. S. Kunarti, Orient. J. Chem., 2018, 34, 1484–1491 CrossRef CAS.
- E. P. Susi, K. Wijaya, W. Wangsa, R. A. Pratika and P. L. Hariani, Asian J. Chem., 2020, 32, 2773–2777 CAS.
- M. Subsadsana, S. Sansuk and C. Ruangviriyachai, Energy Sources, Part A, 2018, 40, 237–243 CrossRef CAS.
- C. T. Tye, P. Y. Looi and T. L. Meow, Adv. Mater. Res., 2012, 560–561, 538–543 CAS.
- A. Tavasoli, E. Taghizadeh Yusefabad and Y. Zamani, Energy Sci. Eng., 2021, 9, 1103–1114 CrossRef CAS.
- G. Y. Nazarova, E. N. Ivashkina, E. D. Ivanchina and M. Y. Mezhova, Catalysts, 2022, 12, 98 CrossRef CAS.
- N. Hosseinpour, A. A. Khodadadi, Y. Mortazavi and A. Bazyari, Appl. Catal., A, 2009, 353, 271–281 CrossRef CAS.
- R. Hamidi, R. Khoshbin and R. Karimzadeh, Adv. Powder Technol., 2021, 32, 524–534 CrossRef CAS.
- O. B. Ayodele, O. S. Togunwa, H. F. Abbas and W. M. A. W. Daud, Energy Convers. Manage., 2014, 88, 1104–1110 CrossRef CAS.
- K. N. Papageridis, N. D. Charisiou, S. Douvartzides, V. Sebastian, S. J. Hinder, M. A. Baker, A. A. AlKhoori, S. I. AlKhoori, K. Polychronopoulou and M. A. Goula, RSC Adv., 2021, 11, 8569–8584 RSC.
| This journal is © The Royal Society of Chemistry 2022 |