
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe diversity and utility of arylthiazoline and aryloxazoline siderophores: challenges of total synthesis
Karolina Kamińska *ab,
Andrzej Mulara,
Evgenia Olshvangc,
Nils Metzler Noltec,
Henryk Kozłowskiad,
Elżbieta Wojaczyńska*b and
Elżbieta Gumienna-Konteckaa
*ab,
Andrzej Mulara,
Evgenia Olshvangc,
Nils Metzler Noltec,
Henryk Kozłowskiad,
Elżbieta Wojaczyńska*b and
Elżbieta Gumienna-Konteckaa
aFaculty of Chemistry, University of Wrocław, Fryderyka Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: karolina.kaminska@pwr.edu.pl; andrzej.mular@chem.uni.wroc.pl; henryk.kozlowski@chem.uni.wroc.pl; elzbieta.gumienna-kontecka@chem.uni.wroc.pl
bFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: elzbieta.wojaczynska@pwr.edu.pl
cInorganic Chemistry I-Bioinorganic Chemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitaetsstrasse, 44801 Bochum, Germany. E-mail: genia.ols@gmail.com; nils.metzler-nolte@rub.de
dDepartment of Health Sciences, University of Opole, Katowicka 68, 45-060 Opole, Poland
First published on 7th September 2022
Abstract
Siderophores are unique ferric ion chelators produced and secreted by some organisms like bacteria, fungi and plants under iron deficiency conditions. These molecules possess immense affinity and specificity for Fe3+ and other metal ions, which attracts great interest due to the numerous possibilities of application, including antibiotics delivery to resistant bacteria strains. Total synthesis of siderophores is a must since the compounds are present in natural sources at extremely small concentrations. These molecules are extremely diverse in terms of molecular structure and physical and chemical properties. This review is focused on achievements and developments in the total synthesis strategies of naturally occurring siderophores bearing arylthiazoline and aryloxazoline units.
1 Introduction
Iron is an exceptionally versatile cofactor that is indispensable for a plethora of biochemical reactions in both mammalian hosts and pathogenic microorganisms. Its acquisition and regulation is crucial for efficacious microbial growth and survival. Deficiency can hinder vital cellular processes such as respiration and proliferation, while an excess may cause toxic effects by inducing oxidative damage through reactive oxygen species produced in the Fenton and Haber–Weiss reactions.1 In the pre-oxygen era, microorganisms were able to exploit highly soluble Fe(II). Although iron is still abundant in the Earth's crust, in the aerobic environment Fe(III) exists as insoluble ferric hydroxide, largely inaccessible to microorganisms.To supply and regulate iron, microorganisms have evolved sophisticated iron acquisition and trafficking systems.2,3 Under iron depleted conditions, bacteria and fungi produce siderophores,4 low molecular weight (MW under 1500 Da) molecules with high binding affinity for ferric iron, capable of solubilizing the ferric hydroxide polymers and sequestering iron from a host's proteins and ligands. Low iron concentrations trigger a “signal” to start biosynthesis of the appropriate siderophores and the proteins involved in siderophore uptake machinery. As soon as sufficient amount of iron is transported and accumulated inside the cell, its acquisition system is turned off.5
Since the early 1950s, when the first three siderophores were isolated and identified as growth factors of bacteria, 270 different siderophores have been structurally characterized (from over 500 identified) and this number is growing.6
The process of isolation of siderophores from natural sources is very tedious, inefficient, and thus expensive. The unique chemical structure diversity and numerous applications both in medicine and industry, prompted scientists to prepare these molecules by total synthesis (more in Sections 3.1 and 3.2). Over the last decades only two reviews concerning the synthesis of naturally occurring siderophores were published.7,8
Total synthesis allows for obtaining analogs of naturally occurring siderophores – mimics, which are extremely important in biomedical applications, for example in the Trojan horse strategy (antibiotic therapy). This approach allows for a deeper investigation of the relationship between structure and metal complexing properties or bioactive function. In the retrosynthetic analysis essential to plan the total synthesis of siderophores, genetic and biochemical knowledge of siderophore biosynthesis is extremely useful. Siderophore retrosynthesis is based on finding the structure of target compound synthons that would correspond to the following stages of biosynthesis.8,9
The review covers total synthesis strategies of naturally occurring siderophores with arylthiazoline and aryloxazoline subunits. The keynote of this work is to present known strategies for preparation of this particular group of siderophores with a detailed analysis of the newest and most efficient approaches. This overview allows tracking the progress in this area made over the years, and identifying the possibilities for further development. In addition, the review describes the synthesis of analogues of some siderophores with arylthiazoline and aryloxazoline subunits, thus showing the impact of structural variations on the activity of these compounds.
Main classification of naturally occurring siderophores is based on the type of functional groups involved in the chelation of iron ions includes three basic groups such as catecholate, hydroxamate, and α-hydroxycarboxylate (Fig. 1).10 To date more than 100 siderophores bearing analogs of this type of donors have been isolated.11 In our review, we would like to focus on siderophores that have an aromatic phenolate and/or catecholate fragment in their structure linked directly to a thiazoline/oxazoline moiety, which will be called arylthiazolines/aryloxazolines (this nomenclature is not commonly used in siderophores classification).
 | ||
| Fig. 1 The iron-binding moieties in various types of siderophores. | ||
Numerous reviews have been published in the literature describing the types of siderophores produced by microbes, their properties, functions, as well as their acquisition machineries; for a comprehensive discussion, the readers are referred to selected reviews and the references therein.3–12 Arylthiazoline and aryloxazoline-type siderophores, so far have been only rarely discussed as a group,8,13 showing the diversity of the structures and the total synthesis approach, all illustrated by most prominent examples.
2 Origins and total synthesis of arylthiazoline and aryloxazoline siderophores and analogues
2.1 Arylthiazoline siderophores: origin and total synthesis
The group of arylthiazoline-based siderophores are characterized by the presence of a 2-thiazoline framework in the structure of a substituted aromatic ring and two or three linearly connected heterocyclic subunits. Phenolic hydroxyl group, the nitrogen atom of the thiazoline ring and usually a terminal carboxyl group are involved in iron coordination. The group members differ mainly in substituents of the phenolic ring, and modifications of the terminal thiazolidine fragment. Moreover, the configuration of the stereogenic centres should be also taken into account (Fig. 2). | ||
| Fig. 2 Two typical structures of thiazoline-based siderophores. Fragments involved in iron coordination marked blue. Stereogenic centres indicated by asterisks. | ||
Pyochelin is produced mainly by Gram-negative Pseudomonas18 and Burkholderia,19 highly aggressive opportunistic pathogenic bacteria that are common causative agents for severe respiratory infections affecting patients with compromised immunity. Moreover, P. aeruginosa is responsible for highly lethal hospital-acquired infections as it is unaffected by many disinfecting agents and its strains are often resistant to the majority of antibiotics.20 However, pyochelin was also isolated from Gram-positive bacteria, Streptomyces.21
This unusual siderophore is built of one phenol moiety connected to thiazoline ring which is attached to thiazolidine part with a methyl-substituted nitrogen atom and a carboxylic group attached to the adjacent position. It has three chiral centres at positions C-4′, C-2′′ and C-4′′ with absolute configurations (R,R,R) (pyochelin I), but the C-2′′-centre readily isomerizes to the (S) form (pyochelin II) yielding a equimolar mixture of two epimers.22 On the other hand, Schalk et al.23 reported about four diastereoisomers which were obtained by organic synthesis: pyochelin I (4′R,2′′R,4′′R), pyochelin II (4′R,2′′S,4′′R), neopyochelin I (4′S,2′′R,4′′R) and neopyochelin II (4′S,2′′S,4′′R) with varying proportions depending on starting compound (Fig. 2).
The synthesis of pyochelin was first described in 1988 by Cox and co-workers in a three-step non-stereoselective procedure.16 The acid intermediate 1 was prepared by method of Mathur et al.24 and converted to the corresponding aldehyde 2, which was condensed further with L-N-methylcysteine 3, providing a mixture of pyochelin isomers 4, with an overall yield of approximately 5% (Scheme 1).
 | ||
| Scheme 1 Non-stereoselective synthesis of pyochelin.16 | ||
In a following paper,25 by optimising the reaction conditions, the overall yield was improved to 65%. The performed synthesis allowed to obtain four out of eight possible stereoisomers in the 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:4 ratio (Fig. 3). Based on the comparison of NMR spectra to the one of 4-methylpyochelin I methyl ester, whose structure was resolved by X-ray crystallography, Cox assigned the absolute configuration of four stereoisomers, i.e., pyochelins I and II and neopyochelins I and II.
:1:1:4 ratio (Fig. 3). Based on the comparison of NMR spectra to the one of 4-methylpyochelin I methyl ester, whose structure was resolved by X-ray crystallography, Cox assigned the absolute configuration of four stereoisomers, i.e., pyochelins I and II and neopyochelins I and II.
 | ||
| Fig. 3 Synthesized stereoisomers of pyochelin 4.25 | ||
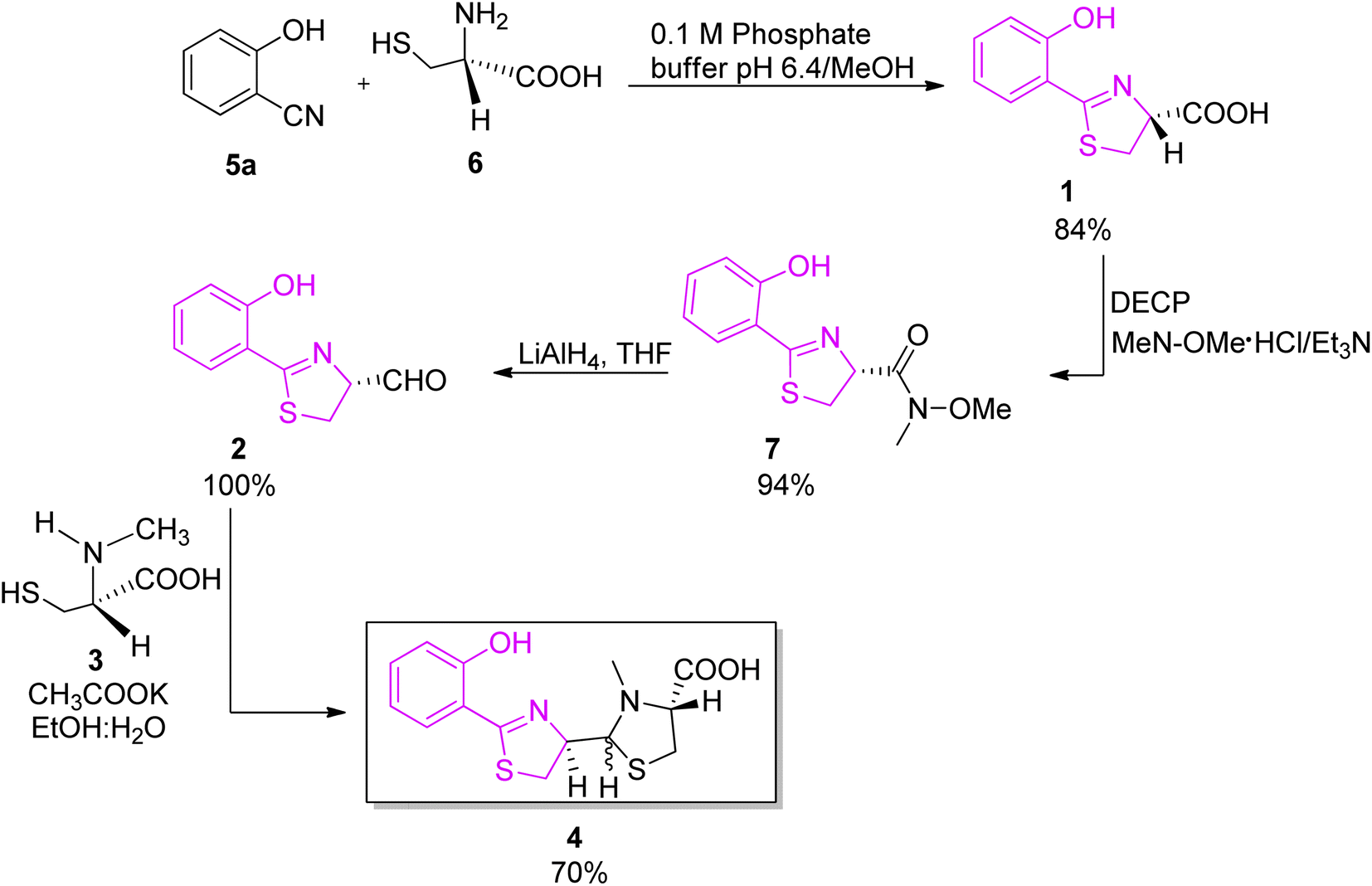
An improved synthesis of pyochelin was described by Zamri and Abdallah, who proposed a stereocontrolled synthetic pathway and obtained a mixture of four diastereoisomers in the 2:1:2:5 ratio (Scheme 2).26,27 In the new proposed synthesis, the method of Mathur et al.24 for acid preparation (1, Scheme 2) was replaced by a protocol described by Bergeron et al. to avoid epimerization of the C-4′ stereogenic centre.28 The acid 1 was converted to N-methoxy-N-methyl hydroxamate intermediate 7; N,O-dimethylhydroxylamine was coupled to compound 1 using diethyl cyanophosphonate (DECP) as coupling reagent, providing hydroxamate 7 in excellent yield (94%); reduction of the hydroxamate 7 by excess LiAlH4 afforded an aldehyde 2. The final condensation step was accomplished by the known methodology between aldehyde 2 and L-N-methylcysteine hydrochloride 3 in a 4:1 mixture of ethanol and water in the presence of potassium acetate, and yielded 70% of pyochelin 4 as a mixture of four diastereoisomers. In natural pyochelin, the absolute configurations are (4′R) and (4′′R). During the synthesis of pyochelin, partial epimerization occurs at C-4′ while the absolute configuration at C-4′′ remains unaffected thus providing a mixture of pyochelin isomers: pyochelin I and II, and neopyochelin I and II.
 | ||
| Scheme 2 Stereoselective synthesis of pyochelin proposed by Zamri and Abdallah.26,27 | ||
Synthetic analogs of pyochelin can enrich the library of biologically active molecules, to tune the chemical or physical properties of the pyochelin, as well as to provide a tool for the investigation of the structure–function relationship of the transporters. An appropriate architecture of ligands and the geometry of complexes allow a control of efficient iron coordination and bacterial recognition. However, the epimerization at the C-2′′ chiral centre shows the difficulty in preparing enantiopure synthetic pyochelin analogs.
The first pyochelin analogs were only accessed by mutasynthesis (Fig. 4).29 The mutasynthetic pyochelin analogs as mixtures of two rapidly interconverting isomers I (4′R,2′′R,4′′R) and II (4′R,2′′S,4′′R) were compared to pyochelin in their ability to transport iron in P. aeruginosa. Isomeric forms were transformed into methyl esters and separated (in case of 8c only one form I was obtained). 4-Methylpyochelins 8b and epi-8b were more active than pyochelin in 55Fe(III) transport, while the other two analogs 5-fluoropyochelin 8a and epi-8a and 6-azapyochelin 8c demonstrated a decreased iron uptake.
 | ||
| Fig. 4 Pyochelin analogs accessed by mutasynthesis.29 | ||
Extensive work in the field of synthetic pyochelin analogs was performed by the group of Schalk, Mislin and their co-workers. The Schalk group focused on studying the properties of several biologically active analogs and conjugates of pyochelin 9a–c, as will be described on several examples. In the first generation, the effect of the configuration on bacterial uptake was tested (Fig. 7).30 Uptake rates of 55Fe(III), tested on P. aeruginosa (ATCC 15692) as well on its pyoverdine defective mutant CDC5 (pPVR2), suggested that the configuration at carbon C-4′ has no effect on the biological properties of pyochelin. Zinc complexation of the mixture (4′R,2′′R,4′′R) and (4′R,2′′S,4′′R) pyochelins provided exclusively (2′′R) diastereomer, which indicated template direction effect. Moreover, substitution in the aromatic part (R1, Fig. 7) or amine group in thiazolidine part (R2, Fig. 5) did not affect dramatically the biological properties of the corresponding analogs compared to pyochelin.
 | ||
| Fig. 5 Structure of first generation of synthetic pyochelin analogs.30 | ||
In an additional series of pyochelin analogs, the thiazoline ring was replaced by a thiazole moiety (Fig. 6).31 The series was prepared using the synthetic protocol for pyochelin analogs through Weinreb amide intermediate,32 and tested for 55Fe uptake by P. aeruginosa, strains PAO1 and PAD07. As phenol function is crucial for the coordination of Fe(III),33 analogs bearing a methylated hydroxyl group failed to transport Fe(III), and only analogs 10a, 10c, 10e and 10f were able to transfer iron inside the bacteria.
 | ||
| Fig. 6 Pyochelin analogs based on the thiazole ring.31 | ||
 | ||
| Fig. 7 Retrosynthetic analysis of micacocidin.39–41 | ||
Total synthesis of micacocidin was performed by Ino and co-workers.39–41 There are five stereogenic carbon atoms in its structure. Cysteine was utilised as a chiral source for C-9, C-12 and C-18 centres, and for generation of the (S) secondary alcohol on C-14 by a stereoselective reduction of ketone. C-10 chirality was generated at a final stage by formation of a Zn complex.
Arylthiazoline part was synthesised starting from 3-methoxy-N,N-dimethylbenzylamine 11 which was converted to intermediate 12 in 5 steps and a total yield of 70%, using the procedure described by Kamikawa.42 Next, condensation of 12 with p-methoxybenzyl (PMB)-protected D-cysteine provided compound 13 which was cyclized using PCl5 to obtain thiazoline derivative in 95% yield. Methyl deprotection of both ether and ester using boron tribromide provided carboxylic acid 14, which was subsequently converted to Weinreb amide 15 using bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOP-Cl) coupling reagent. Following phenol protection by tert-butyldiphenylsilyl (TBDPS) group, the Weinreb amide was reduced by LiAlH4 to afford aldehyde 16, the desired segment of micacocidin (Scheme 3).
 | ||
| Scheme 3 Synthesis of the thiazoline synthon of micacocidin.39,40 | ||
The second synthon41 was prepared starting from thiazolidine 18 which could be obtained from L-cysteine hydrochloride 17 in a two-step route,43 followed by the reaction with carbonyldiimidazole (CDI) and a subsequent condensation with methyl isobutyrate, leading to keto-ester 19. Reduction of the ketone function and construction of an oxazolidinone ring provided compound 20. Condensation of 20 with 2-methyl-(S)-cysteine methyl ester hydrochloride using CDI yielded 21. Reaction with TFA in refluxing toluene resulted in cyclization of the N-acylcysteine. Treatment with (trimethylsilyl)diazomethane restored the methyl ester moiety which was hydrolyzed during the reaction and provided 22. Cleavage of the oxazolidinone ring was achieved in several steps and provided Boc-protected segment 23. A subsequent hydroxyl group protection and N-methylation led to intermediate 24, which was further deprotected yielding building block 25 with free hydroxyl and thiol residues (Scheme 4). Hydrolysis provided compound 26 which was condensed with the first synthon 16 as shown in Scheme 5. The TBDPS-protected aryl thiazoline fragment 16 was fairly labile, it was stored as Weinreb amide 15 which was treated with LiAlH4 shortly prior the condensation with 16. Desilylation with TBAF afforded micacocidin methyl ester 27 as a mixture of diastereomers. Hydrolysis of the terminal ester 27 by LiOH provided the corresponded acid. Treatment with zinc chloride resulted in isomerisation at C-10 and provided the desired micacocidin 29 (Scheme 5).
 | ||
| Scheme 4 Synthesis of second building block of micacocidin.39,41 | ||
 | ||
| Scheme 5 Final assembly of the two segments of micacocidin.39,41 | ||
To understand structure-antymicoplasma activity relationship of micacocidin, Ino and co-workers prepared several derivatives by modifying hydroxyl and carboxylic groups in the skeleton of the siderophore isolated from the fermentation of Pseudomonas sp. No. 57-250 (Fig. 8).44 MIC values were determined for three strains: Mycoplasma gallisepticum, M. pneumoniae, M. hyopneumoniae. Compounds 27 and 30g were active, while a bulky ester group PCB 30a, amide derivatives 30b–d as well as methyl or MOM derivatives at the phenolic position 30f–h, 30j–k resulted in a reduced activity. According to the authors, the lack of bulky groups and the presence of hydrogen bonds are required for activity, and the active derivatives have to possess a similar spatial conformation. The relationship between configuration and activity was examined as well. Regardless of the C-9 configuration, C-10 (R) isomers presented a similar activity to micacocidin 29, while C-10 (S) isomers exhibited a reduced activity. These results supported the authors' proposal that the potency of siderophores depends on the ability of the derivatives to adopt a folded conformation.
 | ||
| Fig. 8 (A) Structural variations of micacocidin analogs, (B) a proposed folded conformation of micacocidin.44 | ||
In an additional work,45 micacocidin analogs 31a–f were generated by exploiting precursor-directed biosynthesis approach. The series varied in alkyl side chain, thus did not induce significant changes in the spatial folding. Almost all gallium complexes showed promising activities against M. pneumoniae ATCC 29342, and comparable to the native micacocidin. Derivative 31b was an exception, and exhibited a significantly reduced activity. A minimum alkyl chain length might be important for the interaction with the biological target (Fig. 9).
 | ||
| Fig. 9 Structural variations of micacocidin analogs, prepared by precursor-directed biosynthesis method.45 | ||
Yersiniabactin was first isolated from Yersinia bacterial strain, a causative agent of a broad range of diseases such as plague, bowel infections and reactive arthritis.46 Afterwards it was found that this strong chelating agent is not limited to one particular bacteria species and can be utilized by different kinds of microbes such as Enterobacteriaceae (E. coli and S. enterica).47–49 The yersiniabactin system is the most frequently carried, genetically non-conserved siderophore system in uropathogenic E. coli.50 The siderophore contains a phenol and a thiazolidine ring, as well as two thiazoline rings. Its stereochemistry is striking for the existence of five chiral centres, with a mixture of two C-10 epimers being isolated during structure elucidation studies.37,38
Following the preparation of micacocidin, Ino and coworkers accomplished a total synthesis of yersiniabactin (Fig. 10).51
 | ||
| Fig. 10 Retrosynthetic analysis of yersiniabactin.51 | ||
The first building block was prepared starting from condensation of Weinreb amide 32 (ref. 52) and 2-methoxybenzoyl chloride 33, which provided ester 34. Next, in a 5-step route thioamide 36 was obtained which was converted to 7 by the reaction with the Burgess reagent. Further hydroxyl group protection and reduction of Weinreb amide 7 yielded aldehyde 37 (Scheme 6).
 | ||
| Scheme 6 Synthesis of thiazoline part of yersiniabactin.51 | ||
The second synthon for yersiniabactin was prepared according to the reaction conditions used in the total synthesis of micacocidin (Scheme 4).41 Compound 23 which was an intermediate in the synthesis of micacocidin was fully deprotected to provide derivative 38, and condensation with the first building block 37 afforded thiazolidine 39. Ester 39 hydrolysis provided the desired yersiniabactin siderophore 40 (Scheme 7).
 | ||
| Scheme 7 Final assembly of the two segments of yersiniabactin 40.51 | ||
Piscibactin, a siderophore isolated from subspecies Piscidia of Photobacterium damselae, is known as a causative agent of fish pseudotuberculosis, a malady that causes large economic losses in marine environment.53 High mortality rate and wide geographical distribution makes it a major problem in the mediterranean fishing industry. Synthesis of piscibactin is also considered as a virulence factor in other bacteria, such as Yersinia enterocolitica responsible for severe enteric inflammation in humans.53 Piscibactin isolation was first reported in 2012 together with an additional metabolite, prepiscibactin, presumably a possible intermediate of piscibactin biosynthesis.53 The piscibactin structure was determined by Souto et al.53 and the studies performed allowed to establish the stereochemistry of piscibactin as (9R,10S,12R,13S).
A successful approach for piscibactin synthesis (close analogue of yersiniabactin) was accomplished by Jiminez et al. in 2021.54 Before, Segade and co-workers proposed a total synthesis of prepiscibactin.55,56 Retrosynthetic analysis of piscibactin was based on three synthons, and in all three cysteine was utilized as starting material (Fig. 11).
 | ||
| Fig. 11 Rethrosynthetic analysis of piscibactin. | ||
Piscibactin was prepared from two key synthons, and D- and L-cysteine were used as chiral sources for C-9 (S) and C-12 (S), respectively (Fig. 11). Applying a similar methodology as for pyochelin preparation,26 the first synthon was obtained through the formation of Weinreb amide, and reduction of the resulting hydroxamic ester with LiAlH4 (Scheme 2).
The second synthon, thiazoline based amino thiol was prepared utilizing multi-functionalized carboxylic acid 41 and (S)-α-azido-methylcysteine.57 Synthetic route for full protected carboxylic acid 41 was accomplished through protection of L-cysteine 6 and a subsequent transformation in a stereoselective manner leading to final building block formation 41 in 20% of overall yield (8 steps). For preparation of thiazoline fragment 46 it was necessary to couple the activated acid of the protected statine 39 with a freshly prepared thiol 42 using EDCl. In the Staudinger reduction and a subsequent intramolecular aza-Wittig reaction, thioester 41 was transformed into the thiazoline 44 in good yield. Further deprotection of amino thiol 44 led to the second building block 46 in 42% yield. Condensation of thiazolinic aldehyde 2 with thiazoline 46 in dichloromethane afforded an epimeric mixture of methyl esters of piscibactin which were hydrolyzed and submitted without purification to complexation with gallium using Ga(acac)3. Final HPLC separation gave piscibactin Ga3+ complex 47 and its 9S epimer in ratio 2:1 and 10% yield (Scheme 8).
 | ||
| Scheme 8 Total synthesis of enantiomerically pure piscibactin gallium complex 47. | ||
The structure of anguibactin can be described as being built of two planar sections, one containing the phenyl-thiazole system with its substituents and another with the imidazole ring. It was first elucidated in 1986,58 and the initial suggestion was later confirmed by van der Helm et al. solving an X-ray structure in 1989.61
The total synthesis of angiubactin was carried out using a convergent approach.60 It was initiated by preparation of the phenylthiazole core in a similar synthetic pathway as for pyochelin,26 by condensation of L-cysteine and aryl nitrile 5a (Scheme 2). The imidazole fragment 49a was prepared in two ways. In 2010 Takeuchi et al.62 proposed its synthesis through histamine dihydrochloride 48 transformations into 4-(2-chloroethyl)imidazole hydrochloride 49 using SOCl2 and a subsequent coupling reaction with N-tert-butoxycarbonyl (Boc)-O-benzyloxyamine 50,63 followed by deprotection,64 yielding O-benzyloxyhistamine 51a in 66% total yield (Scheme 9, part A). In the second approach proposed in 2015 by Kim et al.,65 a terminal olefin 52a was first synthesized from N-Boc-O-benzyloxyamine 50 and 4-bromobut-1-ene. Dihydroxylation under Ujohn conditions afforded diol 53 which was transformed into α-hydroxyketone 54 through the selective oxidation of the secondary hydroxyl group with NaBrO3/NaHSO3.66 In the final step, the Cu(II)-promoted imidazole formation with ammonia and formaldehyde was performed followed by deprotection with TFA which resulted in desired histamine N-oxide derivative 51a in 40% overall yield (Scheme 9, part B).
 | ||
| Scheme 9 Two synthetic routes for the formation of the imidazole part 51a: (A) from histamine hydrochloride,62 (B) from N-Boc-O-benzyloxyamine and 4-bromobut-1-ene.65 | ||
A stereoselective coupling of the intermediate 57 to a free amine of N,O-imidazole 58 mediated by HATU in dichloromethane provided product 59 (er = 98:2). The use of tetrabutylammonium iodide (TBAI) and boron trichloride (BCl3) led to a clean removal of both benzyl and methyl groups and successfully generated compound 61. Along with 61, its 3-deoxy derivative 62 was also prepared in an analogous manner (Scheme 10).
 | ||
| Scheme 10 Total synthesis of angiubactin 61 and its analog 62.60 | ||
:1, respectively) from marine bacterial genus of Brevibacillus67 extracted from an unidentified sponge found near Iwate, Japan. Although Brevibacillus sp. is not considered to be a dangerous microbe, the structures of identified compounds are captivating because its core is also shared by Pseudomonas aeruginosa siderophore pyochelin, though a peculiar tricyclic ring system is present.The compounds possess an unusual heterocyclic structure in which two thiazolidine rings are fused to form a tricyclic ring system. After successful isolation and chromatographic purification, ulbactin F was obtained as pale yellow crystals and its structure was elucidated by Igarashi et al.67 A series of studies on the structure of ulbactin F allowed its identification and determination of the absolute configuration as (4′R,3′′R,6′′R,9′′R, and 9′′R). Ulbactin G with the same molecular composition is the epimeric form of ulbactin F with absolute configuration (4′R,3′′S,6′′R,9′′R, and 9′′R).68 A peculiar feature of ulbactin F and G, namely the 6,9-imino-1H,3H,5H-thiazolo[4,3-c][1,4]thiazepin-5-one tricyclic ring system containing two sulfur atoms and two nitrogen atoms makes it very intriguing. This ring system, but lacking N-methyl group, was also reported for ulbactin D.67
As ulbactin F is the major metabolite (vide supra), this diastereomer was targeted in the total synthesis.68 Convergent synthetic pathways were applied, with several building blocks prepared. Synthesis of the phenylthiazolidine core followed the pathway used for pyochelin, converting salicylnitril to aldehyde 2.26 The thiazolidine synthons were prepared from protected enantiomeric cysteines. Reaction of the two fragments in ethanol/water with NaOAc, and then heating in an ethyl acetate/methanol mixture, provided the product 69 in total yield of 12% and its epimer, epi-69, as a side product (Scheme 11).
 | ||
| Scheme 11 Total synthesis of ulbactin 69.68 | ||
2.2 Aryloxazoline siderophores: origin and total synthesis
Oxazolines are a large family of heterocyclic imino ethers possessing a five-membered ring structure. Within the oxazoline family, 2-oxazolines have been most extensively studied so far and found broad applications in various fields. This is an important structural motif and pharmacophore of natural products and bioactive compounds (including siderophores, Fig. 12).69 The aryloxazoline/oxazole moiety is present in many siderophores and plays a crucial role in coordination mode and iron transport machinery in microorganisms. | ||
| Fig. 12 A general structure of oxazoline-based siderophores. Fragments involved in iron coordination are marked in bold. | ||
To investigate the bactericidal activities of yanglingmycin, the Zhang group70 decided to synthesize this molecule and its analogs. In order to achieve the intended goal, an efficient synthetic pathway for the preparation of 2-aryl/heteroaryloxazolines from nitriles under metal- and catalyst-free condition was used.73 Benzonitrile and its substituted analogs 5a–c were applied as the starting materials. In the initial step, a Pinner reaction was performed to afford the corresponding methyl benzimidate hydrochlorides 70a–c,74 which were then reacted with various amino acid methyl ester hydrochlorides 71–73.75 The final products were obtained by an efficient reduction of esters into alcohols using LiAlH4, yielding (−)-yanglingmycin 74a and its analogs 74b–c, ent-74a–c, 75, ent-75, 76 and ent-76 in total yields of 62% and 52–65%, respectively (Scheme 12).76
 | ||
| Scheme 12 The synthetic route to yanglingmycin enantiomers and its analogs.76 | ||
For a deeper exploration of the antibacterial potency, a series of hydroxyl ester rac-77a–z and hydroxyl ether rac-78a–o derivatives of the racemic (±)-yanglingmycin and its phenyl substituted analogs rac-74a–h were prepared (Schemes 13 and 14).77 To obtain 2-aryl-substituted 4,5-dihydrooxazole analogs rac-74a–h, arylnitriles 5a–h were reacted with serinol in the presence of sodium carbonate (Scheme 13). Hydroxyl ester derivatives of (±)-yanglingmycin rac-77a–z were synthesized through esterification reactions using a catalytic amount of EDC, and hydroxyl ether derivatives 74a–o were obtained by etherifications of the alcoholic hydroxyl group in the presence of a base. Additionally, a fluorinated analog rac-77 was afforded via (±)-yanglingmycin rac-74a treatment with DAST (68% yield) (Scheme 14). In vitro antibacterial studies against four Gram-negative and three Gram-positive bacteria strains revealed that some derivatives are more active than (±)-yanglingmycin rac-74a. Structure–activity relationship analysis clearly showed that antimicrobial activity was lost after most derivatizations of alcoholic hydroxyl group and in the absence of phenolic hydroxyl substituent. On the other hand, incorporation of a short-chain ester group, an electron-deficient ether moiety and fluorination of alcoholic hydroxyl group were found beneficial for the antibacterial potency.
 | ||
| Scheme 13 Synthesis of (±)-yanglingmycin and its phenyl-substituted analogs.77 | ||
 | ||
| Scheme 14 Synthesis of esterified, etherified and fluorinated analogs of (±)-yanglingmycin rac-77a–z, rac-78a–o and rac-79.77 | ||
In 2019 Sun et al. proposed a mild catalytic approach to 2-oxazolines via oxetane ring-opening reaction (a model substrate was synthetized by acylation of commercially available 3-amino oxetane).78 This synthetic protocol is a useful tool for preparation of a diverse family of natural products with 2-oxazoline unit including spoxazomycin C rac-74a and its analogs, such as spoxazomycin D 82 and madurastatin B1 81 in racemic form (Scheme 15). It is worth emphasizing that 2-aryl-substituted 4,5-dihydrooxazole derivatives are crucial building blocks in total syntheses of many natural products such as mycobactin type siderophores etc.
 | ||
| Scheme 15 New catalytic protocol for synthesis of oxazolines from oxetanes.78 | ||
 | ||
| Scheme 16 Structures of isomeric siderophores produced by A. baumannii and P. fluorescens.86 Potential iron chelating motifs are highlighted using blue background. | ||
Apart from acinetobactin, strains of human pathogenic A. baumannii may produce two other siderophores: baumannoferrin87 (a hydroxamate siderophore) and fimsbactin.88 Production of acinetobactin and baumannoferrin is highly conserved among clinical isolates while fimsbactin production appears to be less common. Fimsbactin is structurally related to acinetobactin because of the presence of the catechol and hydroxamate moieties together with an oxazoline ring; it possesses additionally oxazoline and 1,3- or 1,4-diaminopropane fragments.88
Preacinetobactin 86a and prepseudomonine 86f consist of three significant structural subunits, responsible for metal chelation, namely catechol/phenol oxazoline, hydroxamate and imidazole fragments. The proposed synthetic routes for the formation of these siderophores are based on two condensations of key heterocyclic motifs, i.e. aryloxazoline and imidazole part. The imidazole fragment 51a was prepared in the same manner as described for anguibactin (Scheme 9). The synthesis of the second building block, aryloxazoline moiety, was initiated with preparation of O-protected benzoic acid 83a–b precursors.86,89 Crucial steps for the synthesis of phenol oxazoline moiety include amide formation via coupling of L-threonine methyl ester with protected acid 83a–b using various coupling reagents (EDC/HOBt, TBTU) and Ishihara's dehydrative cyclization using Mo(VI) oxide catalyst90 (49–70% total yield). Takeuchi et al. proposed a different route to aryloxazoline part applying methyl imidate of 2,3-dihydroxybenzoic acid and L-threonine derivative (15% total yield).62
In order to identify the key structural motifs of preacinetobactin 86a and prepseudomonine 86f that influence their Fe(III) binding and cellular delivery, various analogs were synthesized.86 Novel analogs were designed based on the modification of the iron-binding parts, i.e. aromatic, hydroxamate and imidazole fragments (Scheme 17). Proposed iron-binding mode for preacinetobactin revealed that catechol dihydroxyl and imidazole groups play crucial roles in iron binding. What is more interesting, phenolic oxazoline derivatives including prepseudomonine indicate that the 3′-hydroxyl group in the aromatic part is not crucial for metal binding. Further modification shows that N-methylation of the imidazole ring in both N1 and N3 positions did not decrease the iron-binding tendency and N1 position is a promising drug conjugation site because modifications at this site did not interfere with the iron delivery function. Additionally, all hydroxamate bridge modifications abolished iron delivery in the cellular uptake machinery. Wencewicz et al.91 prepared a rigid preacinetobactin analog via oxidation of oxazoline moiety to oxazole in order to block siderophore utilization in the pathogen growth process (Scheme 18). The strategy including rigid siderophore analogs preparation can be crucial in the synthesis of new antivirulence agents.
 | ||
| Scheme 17 The synthesis of preacinetobactin 86a and analogs 86b–h.86 | ||
 | ||
| Scheme 18 The synthesis of oxidized preacinetobactin.91 | ||
The stereoselective preparation of fimsbactins was proposed by Kim's group.92,93 Their strategy for the synthesis was based on retrosynthetic analysis in which the framework construction was accomplished via amide bond formation in condensation reaction of two building blocks, aryloxazoline carboxylate and polyamine derivatives followed by catecholate part addition through ester formation (Fig. 13).
 | ||
| Fig. 13 Structures of fimsbactins A–F. | ||
The preparation of the oxazoline carboxylates started from the coupling of o-xylyl-protected 3,4-dihydroxyl benzoic acid 83a90 with L-Ser-OMe, L-Thr-OMe or D-Thr-OMe in the presence of EDC and HOBt reagents. The obtained precursors 91, 84a and ent-84a were transformed into oxazoline forms via Ishihara's dehydrative cyclization catalyzed by a Mo(VI) oxide catalyst94 followed by mild saponification using potassium trimethylsilanolate95 which afforded the desired products 92, 85a and ent-85a in good yields (Scheme 19, part A). The synthesis of functionalized polyamine building blocks was accomplished by amide bond formation between putrescine92 or N-monoacetyl-1,3-propanediamine96 derivatives 93a–b and N,O-protected L- or D-serine 94, ent-94 using EDCl, HOBt coupling reagents (Scheme 19, part B). After derivatization of functional groups and a subsequent global deprotection, fimsbactins 97, 99 and derivatives epi-97, dia-99, epi-99, ent-99 were obtained in 12–22% total yield (Scheme 20).
 | ||
| Scheme 19 Syntheses of the (A) aryloxazoline carboxylates 85a, ent-85a and 92; (B) polyamine 95a, 95b and ent-95b fragments.92,93 | ||
 | ||
| Scheme 20 Syntheses of fimsbactin A 97, B 99 and their stereoisomers92,93 | ||
For the first time parabactin was isolated in 1975 by Tait from Paracoccus denitrificans.97 In the first approach the structure of parabactin was not well defined, because of the unstable nature of oxazoline ring. Several years later, Neilands and co-workers98,99 demonstrated that not an N-(2-hydroxybenzoyl)-L-threonyl fragment (parabactin A) but rather a (2-hydroxyphenyl)-4-carboxyl-5-methyl-2-oxazoline moiety is connected to the central nitrogen atom of the spermidine backbone in the original siderophore. Under the acidic conditions of Tait's isolation, the oxazoline ring was opened to the threonyl derivative (acid labile oxazoline ring). Moreover, Neilands' group showed that the hydrogen atoms of the oxazoline ring were trans to each other (Scheme 21).99
 | ||
| Scheme 21 The acid labile oxazoline ring.98,99 | ||
Agrobactin was isolated from iron-deficient cultures of Agrobacterium tumefaciens, the organism known to induce crown-gall in higher plants. A. tumefaciens not only produces a cancer-causing plasmid but is also interesting as a vector for incorporating foreign DNA into plants.100 Exposure of agrobactin to acid opened the oxazoline ring giving agrobactin A, in which the UV spectra undergo a characteristic redshift, same like in the case of parabactin.99
Fluvibactin was isolated and purified from Vibrio fluvialis in 1993. It contains only one catechol-oxazoline unit and is structurally related to agrobactin from which it differs only in its triamine-backbone.101 A stereochemically modified version of fluvibactin efficiently removed iron without increasing microbial growth.102
Vibriobactin (produced by Vibrio cholerae) is structurally highly similar to agrobactin with three minor alterations: (i) the polyamine backbone is norspermidine, (ii) it contains two L-threonines per molecule, forming two oxazoline rings, and (iii) it contains three DHB moieties as aryl caps. The discovery of the second oxazole ring in vibriobactin suggested some interesting possibilities in its metal-binding capacity.103 Although V. cholerae can use heme directly as an iron source, it has been shown that vibriobactin is also an important virulence factor.104
Vulnibactin, which was isolated from culture supernatants of Vibrio vulnificus M-2799 grown in a low-iron medium, is possessing two salicylate caps that are tethered to the oxazoline ring.105 Virulent Vibrio vulnificus is a halophilic estuarine bacterium that causes fatal septicemia and necrotizing wound infections in humans with high serum iron levels and vulnibactin enables it to acquire iron from highly iron saturated host proteins.106 Vulnibactin is structurally related to vibriobactin, from which it differs only in its salicyloyl moieties on two oxazoline rings, while fluvibactin contains only one oxazoline ring consisting of L-threonine and 2,3-dihydroxybenzoic acid attached to the central nitrogen of norspermidine.105
Serratiochelin A was isolated from Serratia sp. V4 and discovered in S. marcescens in 1974.107 Furthermore, in 2020 serratiochelin A was isolated from an iron-dependent co-culture of Shewanella sp. and the mechanism of its degradation to serratiochelin C was proposed.108 Serratiochelins are bis-catecholate siderophores that could be tetra- or hexadentate, moreover, serratiochelin A and B contain a propane-1,3-diamine backbone.109
So far total synthesis of six aryloxazoline iron chelators based on a polyamine backbone were described, namely the trisamines parabactin, agrobactin, fluvibactin, vibriobactin and vulnibactin, and the diamine – serratiochelin A. The pioneering works in polyamine-based siderophores synthesis and explorations were performed by Bergeron's group. They proposed total syntheses of parabactin, agrobactin, fluvibactin, vibriobactin and vulnibactin. At the turn of years, this research field was supplemented by other scientists.
Based on performed research connected with structure exploration, in 1982 Bergeron's group synthesized parabactin 106 employing a stereospecific procedure under acid free conditions to preserve the oxazoline ring.110 A proposed synthesis employed N1,N8-bis(2,3-dimethoxybenzoyl)spermidine 100 as a starting material, a unique reagent for the generation of spermidine phenolamides.111 In the first step, N-Cbz-L-threonine 101 was condensed with N1,N8-bis(2,3-dimethoxybenzoyl)spermidine 100 in the presence of coupling reagents such as NHS and DCC under basic conditions. Afterwards, both carbobenzoxy and methyl protecting groups were removed from threonylamide 102. In the last step, the most critical one, coupling of 2-hydroxybenzimido ethyl ester 104, obtained before in a Pinner reaction112 with the deprotected threonyl amide 103 was involved. As a result of this synthetic route, parabactin 106 was obtained in 18% total yield (Scheme 22).
 | ||
| Scheme 22 Synthesis of parabactin 106 and agrobactin 107 proposed by Bergeron and co-workers.110,115 | ||
Another synthetic route to parabactin 106 was proposed by Fujita in 1984.113 The researchers decided to attach the catechol moieties in the final step of the synthesis (“inside-out” approach) in contrast to Bergeron's pathway.110 The key intermediates for parabactin formation were dihydro-1,3-oxazole derivative (obtained in 7 steps, 40% total yield) and N1,N10-bis(benzyloxycarbonyl)-spermidine.114 In the final step, condensation of both intermediates and deprotection of terminal amine groups of the spermidine backbone followed by incorporation of the catechol fragment led to parabactin 106 in 5% total yield.
In the 1984 Bergeron's group obtained another spermidine-based aryloxazoline chelator, agrobactin 107.115 Due to the structural similarity to parabactin 106, a previously developed methodology was applied,110 with ethyl 2,3-dihydroxybenzimidate 105 (ref. 116) used in the stereospecific formation of the acid-sensitive trans-oxazoline ring subunit. The oxazoline-forming condensation between N-functionalized spermidine 100 and 2,3-dihydroxybenzimidate ester 105 afforded agrobactin 107 in 21% overall yield (Scheme 22).
The protocol for the synthesis of parabactin 106 (ref. 110) and agrobactin 107 (ref. 115) developed by Bergeron's group was utilized for further synthesis of vibriobactin 124,117 fluviabactin 120 (ref. 118) and vulnibactin 113 (ref. 119) (shown for the latter in Scheme 23). All these siderophores contain the symmetrical spermidine analog, N-(3-aminopropyl)-1,3-diaminopropane (norspermidine) scaffold. The selective activation of norspermidine 108,120 its N-acylation with 2,3-dimethoxybenzoic acid 107 and then with the activated L-threonine ester 111 was followed by threonyl fragment condensation with the ethyl imidate of mono- or dihydroxybenzoic acid 104 or 105. The target siderophores vibriobactin, fluviabactin and vulnibactin 113 were formed in 29%, 23%, 25% total yields, respectively.
 | ||
| Scheme 23 The synthesis of vulnibactin 113.119 | ||
A new concept of norspermidine based synthesis of oxazoline-containing siderophores was proposed by Ishihara and co-workers.90 The basis of this methodology lies in the oxazoline core construction at an early stage in total synthesis. Based on previously investigated approach for oxazoline and thiazoline rings formation via Mo(VI) oxides,94,121 preparation of fluvibactin 120 and vibriobactin 124 was reported. In the synthesis, three building blocks were used: norspermidine 117, 2,3-dialkoxybenzoate 116, and 2-(o,m-dialkoxyphenol)oxazoline 115 prepared from N-(o,m-dialkoxybenzoyl)-L-threonine epi-82a via the Mo(VI) oxide catalyzed method. Dehydrative cyclization of N-(o,m-dialkoxybenzoyl)-L-threonine epi-84a was performed using MoO2(TMHD)2 (Scheme 24). Afterwards, a selective amide formation was conducted using Sb(III) alkoxide-catalyzed ester-amide transformation122 of primary amine groups in norspermidine 117. The amidation of the secondary amine group in products 118 and 122 was achieved using the respective carboxylic acid 115 and EDC, HOAt coupling reagents in the presence of base. In the course of this transformation fluvibactin 120 and vibriobactin 124 were obtained in overall yields of 65% and 50%, respectively (Scheme 25). This improved methodology eliminates the most inefficient step in the Bergeron methodology involving 2,3-dihydroxybenzimidate formation for oxazoline scaffold construction.
 | ||
| Scheme 24 The dehydrative cyclization of N-(o,m-dialkoxybenzoyl)-L-threonine epi-84a via MoO2 (TMHD)2.90 | ||
 | ||
| Scheme 25 Total synthesis of fluvibactin 120 and vibriobactin 124.90 | ||
The most recent synthesis of fluvibactin 120 and vibriobactin 124 was described in 2013 by Raymond's group.101 In their approach, 2-mercaptothiazoline was used to prepare a building block necessary to install the catechol-amide units in all siderophores. The 1,3-thiazolodine-2-thione functionality reacted with the primary amine group in norspermidine 117 in a very selective way. The central amine group in norspermidine 117 due to low reactivity90 was functionalized using HATU as a coupling reagent followed by benzyloxycarbonyl-N-Cbz-L-threonine treatment. In the final step, a catechol-oxazoline building block obtained via Bergeron's methodology110,115 was used leading to the desired siderophores in total yields of 20% (compound 120) and 15% (compound 124).
In the group of synthesized polyamine siderophores, equipped with aryloxazoline moiety serratiochelin A 129 containing the 1,3-diaminopropane chain is also found. The synthetic route reported by Ehlert et al. for serratiochelin A 129 formation107 was based on Bergeron's procedure described for agrobactin 105.115 In the initial stage, the catechol-amide fragment of 126 was formed through N-(3-aminopropyl)benzylamide123 125 coupling with 2,3-dimethoxybenzoic acid 109. Subsequently, benzyl protection from the terminal amine group was removed, followed by N-(L-N-t-butoxycarbonylthreonyloxy)succinimide 127 incorporation. At the end all protecting groups were removed and the oxazoline core was formed using ethyl imidate of 2,3-dihydroxybenzoic acid 105 in total yield 10% (8 steps) (Scheme 26).
 | ||
| Scheme 26 The synthetic route for serratiochelin 129 synthesis.107 | ||
Many research groups have been interested in the synthesis of analogs of naturally occurring polyamine-based siderophores. The main goal for this trend is to investigate the relationship between structure and iron transporting ability.124 Moreover, there is an increased interest in developing new antibiotic treatments by linking antibiotics to siderophore moieties (Trojan horse strategy). Many efforts were put into the structural modifications of spermidine-containing catechol-type siderophore, parabactin 106 and its natural analog agrobactin 107.124,125 All modifications of these siderophores had to be performed in such a way as to preserve the ability for iron binding and microbial growth under conditions of limited iron access. The aryloxazoline part directly linked to the central amino group in the polyamine chain was mainly modified (Fig. 14). The performed research showed that the original structure of polyamine siderophore is still favoured as compared to analogs in coordination and iron transport system study.
 | ||
| Fig. 14 Examples of parabactin analogs 130a–p.124,125 | ||
In the previously cited work of Bergeron et al.118 on the synthesis of L-fluvibactin 120, the synthesis of unnatural enantiomer D-fluvibactin ent-120 and L-homofluvibactin 145 were also presented (Scheme 27). This research revealed that modifications of the polyamine chain of the fluviabctin 120 (L-homofluvibactin 145) do not affect bacterial growth and iron uptake machinery of P. denitricans. However, an invertion of the configuration of two carbon atoms in the oxazoline ring (D-fluvibactin, ent-120) inhibits microbial growth. D-Fluvibactin ent-120 still functions as deferration agent with iron clearing efficiency similar to L-fluvibactin 120, but due to the stereochemistry of the created ferric complex it cannot be utilized by the microorganisms.
 | ||
| Scheme 27 Synthesis of L-fulvibactin 120 and its unnatural analogs.118 | ||
 | ||
| Fig. 15 A general structure of mycobactin type siderophores. Fragments engaged in iron coordination marked blue, amide and ester linkages shown in red. Possible chiral centres indicated by asterisks. | ||
The mycobactins are now quite well-known siderophores since mycobactin P was first isolated from Mycobacterium phlei in 1949.128 Due to the position of the hydrophobic alkyl tail (R5 or R6), mycobactins are classified into two types: P-type (after mycobactin P) or M-type (after mycobactin M), respectively.129 Mycobactin J is one of the P-type mycobactins and is produced by M. avium ssp. paratuberculosis which is the causative agent of Johne's disease in cattle.130
The carboxymycobactins are structurally related to the mycobactins except that their R5 or R6 alkyl side chain (Fig. 15) is shorter and terminates in a carboxylic acid, resulting in much greater hydrophilicity.131 Pathogenic mycobacteria strains like M. tuberculosis and M. bovis express carboxymycobactins as the sole extracellular siderophores.132,133
Nocobactins NA1 and NA2 were isolated from Nocardia asteroides grown under conditions of iron deficiency. They are related in their structure to the mycobactins what was described by Ratledge and Snow in 1974.134
Amamistatin B (isolated from the actinomycete Nocardia – Nocardia asteroides135) is also structurally related to mycobactins siderophore as it contains lysine-derived N-formyl hydroxylamine. Amamistatin A has anti-proliferative, but not cell-killing, effects against several kinds of human tumor cell lines.135 Amamistatin's anti-cancer activity is probably due to histone deacetylase enzymes inhibition mediated by the N-formyl hydroxylamine ligand.136
Brasilibactin A, a siderophore found in Nocardia brasiliensis137 contains oxazoline moiety and a pentadecyl substituent. Brasilibactin A is a membrane-bound siderophore with structural similarity to the mycobactin class of siderophore in mycobacteria and possesses a nearly identical molecular nucleus, which includes a hydroxamic acid, an N-hydroxyformamide, and a 2-(2-hydroxyphenyl)-Δ2-1,3-oxazoline.138
Nocardichelins A and B were isolated from the Nocardia sp. Acta 3026 and the structural characterization of the compounds was performed by mass spectrometry and NMR spectroscopy. The chemical structures of these siderophores are closely related to mycobactin siderophores. In contrast, comparison of nocardichelins A and B to brasilibactin A reveals that only the 4,5-dihydro-2-(2-hydroxyphenyl)-4-oxazolecarboxylic acid moiety is identical.139
Nocardimicins A, B, C, D, E and F members of the family of siderophores isolated from Nocardia sp. TP-A0674 (ref. 140) bear an undecyl chain and oxazole moiety. Nocardimicins are the first example of siderophores that have demonstrated inhibition activity to the muscarinic M3 receptor.140 Nocardimicins G, H and I were isolated from Nocardia nova JCM 6044 and their chemical structures were determined by spectroscopic analysis using NMR and MS.141
First preparations of mycobactin type siderophores were described by Miller and coworkers.142,143 They prepared mycobactin S2, an analog of mycobactin S with a long alkyl chain of the mycobactic acid residue replaced by a methyl group.142 However, the modification resulted in lowered lipid solubility which was found essential for the siderophore activity in mycobacteria.143 A total synthesis of mycobactin S was thus designed using a similar strategy: coupling of cobactin T and mycobactic acid derivative, i.e. two compounds formed in the saponification process of mycobactins.143 The first component was prepared from two building blocks, one 148 containing 2-aryloxazoline residue which was constructed from methyl salicylate 146 and L-serine benzyl ester, using Burgess' reagent to induce cyclization (Scheme 28). It was connected with L-lysine derivative 150 containing hydroxamic acid moiety and a pentadecane chain (Schemes 29 and 30). Cobactin T component was also based on the blocked L-lysine as a staring material, converted to the corresponding hydroxylamine 151 which underwent cyclization to yield a seven-membered ring (caprolactam derivative) with a preserved configuration of a stereogenic centre 152. Coupling with (R)-3-hydroxybutanoic acid and deprotection gave cobactin T (153, Scheme 30). Its reaction with mycobactic acid followed by treatment with TFA afforded mycobactin S 155 (27% yield of the last stages). As it could be seen, L-amino acids and 3-hydroxybutanoic acid were used as sources of chirality.
 | ||
| Scheme 28 Preparation of oxazoline derivative 148 as a part of synthesis of mycobactin S.143 | ||
 | ||
| Scheme 29 The use of L-lysine for the preparation of two building blocks 150, and cobactin T 153 in mycobactin S synthesis.143 | ||
 | ||
| Scheme 30 Assembling of mycobactic acid and its conversion to mycobactin S 155.143 Note that the configuration of a stereogenic centre bearing the methyl group was apparently wrong in the original paper. | ||
Eleven years later, Miller's group accomplished the synthesis of amamistatin B, one of siderophores isolated form a strain of Nocardia.127 Using methods previously developed in Miller's laboratory,136 they prepared 2-aryl-oxazole and D-lysine-derived hydroxamic acid; a third component, (S)-3-hydroxy-2,2-dimethyldecanoic acid was also obtained following the procedure introduced by Yokokawa et al. (vide infra).144 The last building block, a cyclic hydroxamic acid 157, was synthesized by a modified route from Cbz-L-lysine 149 in 19% overall yield (Scheme 31); its configuration was preserved in the caprolactam derivative formed in the cyclization step. This component was then reacted with hydroxydecanoic acid 156, and the product esterified using a linear hydroxamate 160. Two enantiomers of the latter were used (one is shown in Scheme 31), which allowed preparation (after deprotection and reaction with oxazole part 163) of amamistatin B 164 and its epimer. A similar protocol was applied in the synthesis of an analog lacking two hydroxyl groups, however, only the original siderophore was (moderately) active against M. tuberculosis.
 | ||
| Scheme 31 Preparation of amamistatin B 164 by Miller and co-workers.127 | ||
A similar synthetic approach was used earlier by Yokokawa, Shioiri, and co-workers who prepared amamistatin A, which differs only by the presence of a methoxy substituent from B form.144 In the paper describing the synthesis, a stereoselective preparation of (S)-3-hydroxy-2,2-dimethyldecanoic acid 158 was presented utilizing aldol reaction of octanal 166 and methyl trimethylsilyl dimethylketene acetal 165 in the presence of stoichiometric amounts of a chiral oxazaborolidinone 167 obtained from D-valine; hydrolysis of the resulting ester 168 afforded the desired acid 158 in 50% overall yield (Scheme 32). The oxazole component 171 was prepared starting from 5-methoxysalicylic acid 169, and ring closure of 170 was achieved through a Wipf's variant of Robinson–Gabriel synthesis (oxidation with Dess–Martin reagent followed by dehydration of the intermediate ketoamides, Scheme 32). Amamistatin A 175 was constructed by a stepwise addition of subsequent building blocks to the caprolactam derivative 152 (Scheme 33; this compound 152 and a linear hydroxamate 173 were prepared by previously described methods143), and the overall yield of the seven steps was ca. 7.5% (part of the unreacted intermediates, however, could be recovered).
 | ||
| Scheme 32 Preparation of two components 158 and 171 of amamistatin A 175 by Yokokawa et al.144 | ||
 | ||
| Scheme 33 Synthesis of amamistatin A 175.144 | ||
Among other compounds from the mycobactin family, a total synthesis of J form 191 was described by Kapur and co-workers in 2018.145 The specific component of this compound, a long-chain carboxylic acid 178 with a double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with (Z) configuration, was prepared in 48% overall yield via oxidation of 1-tetradecanol 176 to tetradecanal, its Corey–Fuchs conversion to alkyne, its esterification with chloroformate, and hydrolysis (Scheme 34). Thus obtained acid 177 with a carbon–carbon triple bond was stereospecifically hydrogenated over Lindlar catalyst to give (Z)-pentadec-2-enoic acid 178. It was attached to a mycobactic acid fragment which in turn was constructed from salicylic acid 179, L-threonine methyl ester, and a fragment derived from Cbz-L-lysine 184; oxazoline 181 formation was achieved by molybdate-mediated cyclization with a retention of configuration (Schemes 35 and 36). The second synthon also required L-lysine which was converted to a cyclic amide using HATU; coupling with (2S,3R)-3-hydroxy-2-methylpentanoic acid 189 (a source of two stereogenic centres in the final molecule) resulted in the cobactin fragment 190. However, in contrast to Miller's preparations,142,143 the attempts of esterification to yield mycobactin J were unsuccessful. Instead, a chiral pentanoic acid derivative 189 was first connected to mycobactic acid 188, and a cyclic hydroxamic acid 186 was added in a subsequent step yielding the desired final product 191 (21% yield over three steps).
C bond with (Z) configuration, was prepared in 48% overall yield via oxidation of 1-tetradecanol 176 to tetradecanal, its Corey–Fuchs conversion to alkyne, its esterification with chloroformate, and hydrolysis (Scheme 34). Thus obtained acid 177 with a carbon–carbon triple bond was stereospecifically hydrogenated over Lindlar catalyst to give (Z)-pentadec-2-enoic acid 178. It was attached to a mycobactic acid fragment which in turn was constructed from salicylic acid 179, L-threonine methyl ester, and a fragment derived from Cbz-L-lysine 184; oxazoline 181 formation was achieved by molybdate-mediated cyclization with a retention of configuration (Schemes 35 and 36). The second synthon also required L-lysine which was converted to a cyclic amide using HATU; coupling with (2S,3R)-3-hydroxy-2-methylpentanoic acid 189 (a source of two stereogenic centres in the final molecule) resulted in the cobactin fragment 190. However, in contrast to Miller's preparations,142,143 the attempts of esterification to yield mycobactin J were unsuccessful. Instead, a chiral pentanoic acid derivative 189 was first connected to mycobactic acid 188, and a cyclic hydroxamic acid 186 was added in a subsequent step yielding the desired final product 191 (21% yield over three steps).
 | ||
| Scheme 34 Synthesis of unsaturated acid component 178 of mycobactin J 191.145 | ||
 | ||
| Scheme 35 Synthesis of three synthons 181, 184 and 186 of mycobactin J 191.145 | ||
 | ||
| Scheme 36 Assembling of four components into mycobactin J 191.145 | ||
Carboxymycobactin T 203 and its three analogs were prepared by Slomczynska and co-workers using a solid-phase approach.146 L-Norleucine 192 derivative was reacted with nosyl-activated hydroxylamine bound to Wang resin 194; PyAOP-induced cyclization afforded an immobilized caprolactam derivative 197 (Scheme 37) to which subsequent fragments were connected (Scheme 38): (S)-3-hydroxybutyric acid 198, another L-norleucine-derived component 200, and 2-(2-hydroxyphenyl)oxazoline-4-carboxylic acid 181 which preparation was described by Miller's group.143 To the obtained skeleton, a monomethyl ester of heptane-1,7-dioic acid 202 was attached as a side chain and the carboxymycobactin T 203 was removed from the resin. Its epimer with the opposite configuration of carbon atom bearing methyl group was prepared by the same route with (R)-3-hydroxybutyric acid ent-198 as one of components; in case of two analogs bearing amide instead of ester linkage, separation of diastereomeric mixture appeared necessary due to unexpected epimerization.
 | ||
| Scheme 37 Synthesis of synthons used for preparation of carboxymycobactin T.146 | ||
 | ||
| Scheme 38 Solid-phase synthesis of carboxymycobactin T 203.146 | ||
In 2007, two research groups reported on a synthesis of brasilibactin A. In a paper of Mitchell and Shaw, preparation of two diastereomers differing in the configuration of the β-hydroxyacid component allowed the assignment of absolute configuration of the naturally occurring form.147 This fragment was prepared using the titanium-mediated aldol reaction of chiral thiazolidinethione auxiliary 204 with hexadecanal which led to syn products 205 and dia-205 with the absolute configuration dependent on the amount of (−)-sparteine added (45% yield and diasteromeric ratio >95:5 were noted in both cases, Scheme 39). Coupling with caprolactam-based amine 186 released the auxiliary and resulted in the respective diastereomeric products 206 and dia-206. Their subsequent reactions with the known building blocks derived from D-lysine 173 and, finally, oxazoline fragment 181 prepared from D-serine followed the previously reported procedures and led to brasilibactin A 208 (found to be a 17R,18S isomer; Scheme 40) and its (17S,18R)-analog. Independently, Ying and Hong prepared four diastereomers differing by configurations of these two stereocentres.138 The four hydroxyacid components 213, ent-213, dia-213 and dia′-213 were prepared by diastereoselective aldol reactions from N-propanoyloxazilidinones (syn isomers) and O-propanoylnorephedrines (anti selectivity, Scheme 41). The overall synthetic strategy was similar to the one of Mitchell and Shaw, and the stereochemical assignment of natural brasilibactin A 208 was identical.
 | ||
| Scheme 39 Preparation of two diastereomers of hydroxyacid-caparolactam part of brasilibactin A 208.147 | ||
 | ||
| Scheme 40 Synthesis of brasilibactin 208 by Mitchell and Show.147 | ||
 | ||
| Scheme 41 Preparation of four diastereomers of hydroxyacid part of brasilibactin A by Ying and Hong.138 | ||
Banks and Moody prepared nocardimicin B 233 from four usual components, though they modified the synthesis of particular building blocks.148 The 2-aryloxazole part 219 was obtained by rhodium-catalyzed reaction of 2-benzoyloxybenzonitrile 216 with dimethyl diazomalonate 217; removal of the methoxy group and hydrolysis gave the 2-aryloxazole-4-carboxylic acid 219 in ca. 67% yield (Scheme 42). The linear hydroxamic acid component 231 was prepared by a previously established method, while for the β-hydroxycarboxylic acid, an anti-selective aldol reaction was applied between a lactate-derived (R)-ketone and dodecanal under Patterson conditions.149 Standard four-step manipulation provided the desired hydroxyacid 224 in 25% overall yield. The cyclic hydroxamic acid 229 was prepared using ring-closing metathesis (RCM) as a key step. O-Benzyl-N-Boc-hydroxylamine 50 was converted into N-allyl derivative 226 which in turn reacted with (R)-Boc-allylglycine 227 to give the respective amide 228. RCM employing Grubbs' catalyst yielded a seven-membered caprolactam derivative with a double bond 229 (59% yield from hydroxylamine derivative, Scheme 42). Starting from this building block 229, all fragments were connected subsequently, and the final step involved a hydrogenative deprotection, but also reduction of the CC bond, and yielded the expected siderophore 233 (Scheme 43).
 | ||
| Scheme 42 Preparation of three components 219, 224 and 229 of nocoardimicin B.148 | ||
 | ||
| Scheme 43 Synthesis of nocardimicin B 233.148 | ||
Concluding this part, sidorophores containing an oxazole or oxazoline ring exhibit a significant similarity of four main chiral synthons which can be appropriately assembled. Part of these fragments inherit chirality of the starting amino acid residues, however, others are prepared in stereoselective reactions (for example, 3-hydroxycarboxylic acid in aldol reaction). Though a total synthesis of all known members of the family has not been accomplished yet (as exemplified by other mycobactins, nocobactin, and formobactin), the general procedure for their preparation should not differ much from their analogs obtained in the laboratories.
3 Conclusions
In this comprehensive review we presented the total synthesis of naturally occurring siderophores bearing arylthiazoline and aryloxazoline subunits. During the years a development of synthetic strategies has been observed to find the optimal approach for synthons used for many siderophores. The progress of characterization techniques allowed to determine configurations of all stereogenic centres and to correctly describe all stereoisomers. The yield of the total synthesis and enantiomeric purity of prepared compounds have been greatly improved which is important from the application point of view. Amounts of siderophores required in biomedical studies are typically in the range of hundred milligrams, two orders of magnitude more than typically isolated from natural sources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Polish National Science Centre (UMO-2017/26/A/ST5/00363) is gratefully acknowledged.References
- K. D. Krewulak and H. J. Vogel, Structural biology of bacterial iron uptake, Biochim. Biophys. Acta, Biomembr., 2008, 1778(9), 1781–1804, DOI:10.1016/j.bbamem.2007.07.026.
- (a) C. Ratledge and L. G. Dover, Iron metabolism in pathogenic bacteria, Annu. Rev. Microbiol., 2000, 54, 881–941, DOI:10.1146/annurev.micro.54.1.881; (b) M. L. Guerinot, Microbial iron transport, Annu. Rev. Microbiol., 1994, 48, 743–772, DOI:10.1146/annurev.mi.48.100194.003523; (c) L. Johnson, Iron and siderophores in fungal-host interactions, Mycol. Res., 2008, 112, 170–183, DOI:10.1016/j.mycres.2007.11.012; (d) M. Miethke, Molecular strategies of microbial iron assimilation: from high-affinity complexes to cofactor assembly systems, Metallomics, 2013, 5(1), 15–28, 10.1039/c2mt20193c.
- M. Miethke and M. A. Marahiel, Siderophore-based iron acquisition and pathogen control, Microbiol. Mol. Biol. Rev., 2007, 71(3), 413–451, DOI:10.1128/mmbr.00012-07.
- C. E. Lankford and B. R. Byers, Bacterial Assimilation of iron, Crit. Rev. Microbiol., 1973, 2(3), 273–311, DOI:10.3109/10408417309108388.
- U. Mollmann, L. Heinisch, A. Bauernfeind, T. Kohler and D. Ankel-Fuchs, Siderophores as drug delivery agents: application of the “Trojan Horse” strategy, BioMetals, 2009, 22(4), 615–624, DOI:10.1007/s10534-009-9219-2.
- (a) M. G. P. Page, Siderophore conjugates, Antimicrob. Therapeut., 2013, 1277, pp. 115–126, DOI:10.1111/nyas.12024; (b) R. C. Hider and X. Kong, Chemistry and biology of siderophores, Nat. Prod. Rep., 2010, 27(5), 637–657, 10.1039/b906679a.
- F. Garzon-Posse, Y. Quevedo-Acosta, C. Mahecha-Mahecha and P. Acosta-Guzman, Recent Progress in the Synthesis of Naturally Occurring Siderophores, Eur. J. Org. Chem., 2019, 2019(48), 7747–7769, DOI:10.1002/ejoc.201901257.
- Y. N. Ho, H. J. Lee, C. T. Hsieh, C. C. Peng and Y. L. Yang, Chemistry and Biology of Salicyl-Capped Siderophores, Stud. Nat. Prod. Chem., 2019, 59, 431–490, DOI:10.1016/b978-0-444-64179-3.00013-x.
- S. M. Barry and G. L. Challis, Recent advances in siderophore biosynthesis, Curr. Opin. Chem. Biol., 2009, 13(2), 205–215, DOI:10.1016/j.cbpa.2009.03.008.
- (a) A. L. Crumbliss and J. M. Harrington, Iron sequestration by small molecules: thermodynamic and kinetic studies of natural siderophores and synthetic model compounds, Adv. Inorg. Chem., 2009, 61, 179–250, DOI:10.1016/s0898-8838(09)00204-9; (b) M. Sandy and A. Butler, Microbial Iron Acquisition: Marine and Terrestrial Siderophores, Chem. Rev., 2009, 109(10), 4580–4595, DOI:10.1021/cr9002787.
- Y. N. Ho, H. J. Lee, C. T. Hsieh, C. C. Peng and Y. L. Yang, Chemistry and Biology of Salicyl-Capped Siderophores, Stud. Nat. Prod. Chem., 2019, 59, 431–490, DOI:10.1016/b978-0-444-64179-3.00013-x.
- (a) J. Kramer, O. Oezkaya and R. Kuemmerli, Bacterial siderophores in community and host interactions, Nat. Rev. Microbiol., 2020, 18(3), 152–163, DOI:10.1038/s41579-019-0284-4; (b) B. R. Wilson, A. R. Bogdan, M. Miyazawa, K. Hashimoto and Y. Tsuji, Siderophores in Iron Metabolism: From Mechanism to Therapy Potential, Trends Mol. Med., 2016, 22(12), 1077–1090, DOI:10.1016/j.molmed.2016.10.005; (c) V. I. Holden and M. A. Bachman, Diverging roles of bacterial siderophores during infection, Metallomics, 2015, 7(6), 986–995, 10.1039/c4mt00333k; (d) A. Szebesczyk, E. Olshvang, A. Shanzer, P. L. Carver and E. Gumienna-Kontecka, Harnessing the power of fungal siderophores for the imaging and treatment of human diseases, Coord. Chem. Rev., 2016, 327, 84–109, DOI:10.1016/j.ccr.2016.05.001; (e) D. J. Raines, T. J. Sanderson, E. J. Wilde and A. K. Duhme-Klair, Siderophores, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2015 Search PubMed.
- E. Sansinenea and A. Ortiz, Bacterial Siderophores Containing a Thiazoline Ring, Mini-Rev. Org. Chem., 2009, 6(2), 120–127, DOI:10.2174/157019309788167701.
- P. V. Liu and F. Shokrani, Biological activities of pyochelins: iron-chelating agents of Pseudomonas aeruginosa, Infect. Immun., 1978, 22(3), 878–890 CrossRef CAS.
- C. D. Cox and R. Graham, Isolation of an iron-binding compound from pseudomonas-aeruginosa, J. Bacteriol., 1979, 137(1), 357–364, DOI:10.1128/jb.137.1.357-364.1979.
- R. G. Ankenbauer, T. Toyokuni, A. Staley, K. L. Rinehart and C. D. Cox, Synthesis and biological-activity of pyochelin, a siderophore of pseudomonas-aeruginosa, J. Bacteriol., 1988, 170(11), 5344–5351, DOI:10.1128/jb.170.11.5344-5351.1988.
- C. D. Cox, K. L. Rinehart, M. L. Moore and J. C. Cook, Pyochelin: novel structure of an iron-chelating growth promoter for Pseudomonas aeruginosa, Proc. Natl. Acad. Sci. U. S. A., 1981, 78(7), 4256–4260, DOI:10.1073/pnas.78.7.4256.
- (a) L. Serino, C. Reimmann, P. Visca, M. Beyeler, V. D. Chiesa and D. Haas, Biosynthesis of pyochelin and dihydroaeruginoic acid requires the iron-regulated pchDCBA operon in Pseudomonas aeruginosa, J. Bacteriol., 1997, 179(1), 248–257, DOI:10.1128/jb.179.1.248-257.1997; (b) C. Reimmann, L. Serino, M. Beyeler and D. Haa, Dihydroaeruginoic acid synthetase and pyochelin synthetase, products of the pchEF, are induced by extracellular pyochelin in Pseudornonas aeruginosa, Microbiology, 1998, 144(11), 3135–3148, DOI:10.1099/00221287-144-11-3135.
- P. Darling, M. Chan, A. D. Cox and P. A. Sokol, Siderophore Production by Cystic Fibrosis Isolates of Burkholderia cepacia, Infect. Immun., 1998, 66(2), 874–877, DOI:10.1128/iai.66.2.874-877.1998.
- (a) H. Hayen and D. A. Volmer, Different iron-chelating properties of pyochelin diastereoisomers revealed by LC/MS, Anal. Bioanal. Chem., 2006, 385(3), 606–611, DOI:10.1007/s00216-006-0443-0; (b) J. Brandel, N. Humbert, M. Elhabiri, I. J. Schalk, G. L. A. Mislin and A. M. Albrecht-Gary, Pyochelin, a siderophore of Pseudomonas aeruginosa: Physicochemical characterization of the iron(III), copper(II) and zinc(II) complexes, Dalton Trans., 2012, 41(9), 2820–2834, 10.1039/c1dt11804h.
- R. F. Seipke, L. Song, J. Bicz, P. Laskaris, A. M. Yaxley, G. L. Challis and R. Loria, The plant pathogen Streptomyces scabies 87–22 has a functional pyochelin biosynthetic pathway that is regulated by TetR- and AfsR-family proteins, Microbiology, 2011, 157(9), 2681–2693, DOI:10.1099/mic.0.047977-0.
- K. Schlegel, J. Lex, K. Taraz and H. Budzikiewicz, The X-ray structure of the pyochelin Fe3+ complex, Z. Phys. Chem., 2006, 61(3–4), 263–266, DOI:10.1515/znc-2006-3-418.
- G. L. A. Mislin, F. Hoegy, D. Cobessi, K. Poole, D. Rognan and I. J. Schalk, Binding related properties of pyochelin and structurally molecules to FptA of Pseudomonas aeruginosa, J. Mol. Biol., 2006, 357(5), 1437–1448, DOI:10.1016/j.jmb.2006.01.080.
- K. B. Mathur, R. N. Iyer and M. L. Dhar, Studies in Potential Antimycobacterial Agents 14. Synthesis of Some 2-(2′-Hydroxyphenyl)-2-Thiazoline-4-N-Substituted Carboxyamides, J. Sci. Ind. Res., Sect. B, 1962, 21(1), 34–37 Search PubMed.
- K. L. Rinehart, A. L. Staley, S. R. Wilson, R. G. Ankenbauer and C. D. Cox, Stereochemical Assignment of the Pyochelins, J. Org. Chem., 1995, 60(9), 2786–2791, DOI:10.1021/Jo00114a029.
- A. Zamri and M. A. Abdallah, An improved stereocontrolled synthesis of pyochelin, siderophore of Pseudomonas aeruginosa and Burkholderia cepacia, Tetrahedron, 2000, 56(2), 249–256, DOI:10.1016/S0040-4020(99)00946-1.
- A. Zamri and M. A. Abdallah, An improved stereocontrolled synthesis of pyochelin, siderophore of Pseudomonas aeruginosa and Burkholderia cepacia (vol. 56, p.249, 2000), Tetrahedron, 2000, 56(47), 9397, DOI:10.1016/S0040-4020(00)00875-9.
- R. J. Bergeron, J. Wiegand, J. B. Dionis, M. Eglikarmakka, J. Frei, A. Huxleytencer and H. H. Peter, Evaluation of Desferrithiocin and Its Synthetic Analogs as Orally Effective Iron Chelators, J. Med. Chem., 1991, 34(7), 2072–2078, DOI:10.1021/Jm00111a023.
- R. G. Ankenbauer, A. L. Staley, K. L. Rinehart and C. D. Cox, Mutasynthesis of siderophore analogues by Pseudomonas aeruginosa, Proc. Natl. Acad. Sci. U. S. A., 1991, 88(5), 1878–1882, DOI:10.1073/pnas.88.5.1878.
- A. Zamri, I. J. Schalk, F. Pattus and M. A. Abdallah, Bacterial siderophores: synthesis and biological activities of novel pyochelin analogues, Bioorg. Med. Chem. Lett., 2003, 13(6), 1147–1150, DOI:10.1016/s0960-894x(03)00010-6.
- (a) G. L. Mislin, F. Hoegy, D. Cobessi, K. Poole, D. Rognan and I. J. Schalk, Binding properties of pyochelin and structurally related molecules to FptA of Pseudomonas aeruginosa, J. Mol. Biol., 2006, 357(5), 1437–1448, DOI:10.1016/j.jmb.2006.01.080; (b) S. Noel, F. Hoegy, F. Rivault, D. Rognan, I. J. Schalk and G. L. Mislin, Synthesis and biological properties of thiazole-analogues of pyochelin, a siderophore of Pseudomonas aeruginosa, Bioorg. Med. Chem. Lett., 2014, 24(1), 132–135, DOI:10.1016/j.bmcl.2013.11.054.
- G. L. Mislin, A. Burger and M. A. Abdallah, Synthesis of new thiazole analogues of pyochelin, a siderophore of Pseudomonas aeruginosa and Burkholderia cepacia. A new conversion of thiazolines into thiazoles, Tetrahedron, 2004, 60(52), 12139–12145, DOI:10.1016/j.tet.2004.10.030.
- J. Brandel, N. Humbert, M. Elhabiri, I. J. Schalk, G. L. Mislin and A. M. Albrecht-Gary, Pyochelin, a siderophore of Pseudomonas aeruginosa: physicochemical characterization of the iron(III), copper(II) and zinc(II) complexes, Dalton Trans., 2012, 41(9), 2820–2834, 10.1039/c1dt11804h.
- S. Kobayashi, Y. Ikenishi, Y. Takinami, M. Takema, W. Y. Sun, A. Ino and Y. Hayase, Preparation and antimicrobial activity of micacocidin, J. Antibiot., 2000, 53(5), 532–539, DOI:10.7164/antibiotics.53.532.
- S. Kobayashi, S. Hidaka, Y. Kawamura, M. Ozaki and Y. Hayase, Micacocidin A, B and C, novel antimycoplasma agents from Pseudomonas sp. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities, J. Antibiot., 1998, 51(3), 323–327, DOI:10.7164/antibiotics.51.323.
- S. Kobayashi, H. Nakai, Y. Ikenishi, W. Y. Sun, M. Ozaki, Y. Hayase and R. Takeda, Micacocidin A, B and C, novel antimycoplasma agents from Pseudomonas sp. II. Structure elucidation, J. Antibiot., 1998, 51(3), 328–332, DOI:10.7164/antibiotics.51.328.
- M. F. Kreutzer, H. Kage, J. Herrmann, J. Pauly, R. Hermenau, R. Muller, D. Hoffmeister and M. Nett, Precursor-directed biosynthesis of micacocidin derivatives with activity against Mycoplasma pneumoniae, Org. Biomol. Chem., 2014, 12(1), 113–118, 10.1039/c3ob41839a.
- M. F. Kreutzer, H. Kage, P. Gebhardt, B. Wackler, H. P. Saluz, D. Hoffmeister and M. Nett, Biosynthesis of a Complex Yersiniabactin-Like Natural Product via the mic Locus in Phytopathogen Ralstonia solanacearum, Appl. Environ. Microbiol., 2011, 77(17), 6117–6124, DOI:10.1128/aem.05198-11.
- A. Ino, Y. Hasegawa and A. Murabayashi, Total synthesis of the antimycoplasma antibiotic micacocidin, Tetrahedron Lett., 1998, 39(21), 3509–3512, DOI:10.1016/s0040-4039(98)00518-8.
- A. Ino and A. Murabayashi, Synthetic studies of thiazoline and thiazolidine-containing natural products—1. Phosphorus pentachloride-mediated thiazoline construction reaction, Tetrahedron, 1999, 55(34), 10271–10282, DOI:10.1016/S0040-4020(99)00582-7.
- A. Ino, Y. Hasegawa and A. Murabayashi, Synthetic studies of thiazoline and thiazolidine-containing natural products—2. Total synthesis of the antimycoplasma antibiotic micacocidin, Tetrahedron, 1999, 55(34), 10283–10294, DOI:10.1016/S0040-4020(99)00583-9.
- I. Kubo, M. Kim, K. Naya, S. Komatsu, Y. Yamagiwa, K. Ohashi, Y. Sakamoto, S. Hirakawa and T. Kamikawa, Prostaglandin Synthetase Inhibitors from the African Medicinal PlantOzoroa mucronata, Chem. Lett., 1987, 16(6), 1101–1104, DOI:10.1246/cl.1987.1101.
- D. S. Kemp and R. I. Carey, Boc-L-Dmt-OH as a Fully N,S-Blocked Cysteine Derivative for Peptide Synthesis by Prior Thiol Capture. Facile Conversion of N-Terminal Boc-L-Dmt-Peptides to H-Cys(Scm)-Peptides, J. Org. Chem., 1989, 54(15), 3640–3646, DOI:10.1021/jo00276a026.
- A. Ino, S. Kobayashi, K. Ueda, S. Hidaka and Y. Hayase, Structure-activity relationship of the antimycoplasma antibiotic micacocidin – a preliminary study, J. Antibiot., 2001, 54(9), 753–756, DOI:10.7164/antibiotics.54.753.
- M. F. Kreutzer, H. Kage, J. Herrmann, J. Pauly, R. Hermenau, R. Muller, D. Hoffmeister and M. Nett, Precursor-directed biosynthesis of micacocidin derivatives with activity against Mycoplasma pneumoniae, Org. Biomol. Chem., 2014, 12(1), 113–118, 10.1039/c3ob41839a.
- (a) H. Haag, K. Hantke, H. Drechsel, I. Stojiljkovic, G. Jung and H. Zahner, Purification of yersiniabactin – a siderophore and possible virulence factor of yersinia-enterocolitica, J. Gen. Microbiol., 1993, 139, 2159–2165, DOI:10.1099/00221287-139-9-2159; (b) C. E. Chambers and P. A. Sokol, Comparison of siderophore production and utilization in pathogenic and environmental isolates of yersinia-enterocolitica, J. Clin. Microbiol., 1994, 32(1), 32–39, DOI:10.1128/jcm.32.1.32-39.1994.
- R. D. Perry, P. B. Balbo, H. A. Jones, J. D. Fetherston and E. DeMoll, Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation, Microbiology, 1999, 145, 1181–1190, DOI:10.1099/13500872-145-5-1181.
- S. R. Petermann, J. S. Sherwood and C. M. Logue, The Yersinia high pathogenicity island is present in Salmonella enterica Subspecies I isolated from turkeys, Microb. Pathog., 2008, 45(2), 110–114, DOI:10.1016/j.micpath.2008.04.001.
- B. Lesic and E. Carniel, The high pathogenicity island: a broad-host-range pathogenicity island, in Yersinia molecular and cellular biology, ed. Carniel, E. and Hinnebusch, B. J., Horizon Bioscience, 2004, pp. 285–306 Search PubMed.
- J. P. Henderson, J. R. Crowley, J. S. Pinkner, J. N. Walker, P. Tsukayama, W. E. Stamm, T. M. Hooton and S. J. Hultgren, Quantitative Metabolomics Reveals an Epigenetic Blueprint for Iron Acquisition in Uropathogenic Escherichia coli, PLoS Pathog., 2009, 5(2), e1000305, DOI:10.1371/journal.ppat.1000305.
- A. Ino and A. Murabayashi, Synthetic studies of thiazoline and thiazolidine-containing natural products. Part 3: total synthesis and absolute configuration of the siderophore yersiniabactin, Tetrahedron, 2001, 57(10), 1897–1902, DOI:10.1016/S0040-4020(01)00012-6.
- G. Ageno, L. Banfi, G. Cascio, G. Guanti, E. Manghisi, R. Riva and V. Rocca, Enantiospecific and Diastereoselective Synthesis of 4,4-Disubstituted-3-Amino-2-Azetidinones, Starting from D-Serine, Tetrahedron, 1995, 51(29), 8121–8134, DOI:10.1016/0040-4020(95)00427-A.
- A. Souto, M. A. Montaos, A. J. Rivas, M. Balado, C. R. Osorio, J. Rodríguez, M. L. Lemos and C. Jiménez, Structure and Biosynthetic Assembly of Piscibactin, a Siderophore from Photobacterium damselae subsp. piscicida, Predicted from Genome Analysis, Eur. J. Org. Chem., 2012, 2012(29), 5693–5700, DOI:10.1002/ejoc.201200818.
- M. C. de la Fuente, Y. Segade, K. Valderrama, J. Rodriguez and C. Jimenez, Convergent Total Synthesis of the Siderophore Piscibactin as Its Ga3+ Complex, Org. Lett., 2021, 23(2), 340–345, DOI:10.1021/acs.orglett.0c03850.
- Y. Segade, M. A. Montaos, J. Rodriguez and C. Jimenez, A Short Stereoselective Synthesis of Prepiscibactin Using a Sml2-Mediated Reformatsky Reaction and Zn2+-Induced Asymmetric Thiazolidine Formation, Org. Lett., 2014, 16(21), 5820–5823, DOI:10.1021/ol502958u.
- R. O. Duthaler and B. Wyss, Conversion of L-Cysteine into D-alpha-Amino Acids and Related Transformations, Eur. J. Org. Chem., 2011, 2011(24), 4667–4680, DOI:10.1002/ejoc.201100247.
- (a) P. B. Alper, S. C. Hung and C. H. Wong, Metal catalyzed diazo transfer for the synthesis of azides from amines, Tetrahedron Lett., 1996, 37(34), 6029–6032, DOI:10.1016/0040-4039(96)01307-x; (b) R. B. Yan, F. Yang, Y. F. Wu, L. H. Zhang and X. S. Ye, An efficient and improved procedure for preparation of triflyl azide and application in catalytic diazotransfer reaction, Tetrahedron Lett., 2005, 46(52), 8993–8995, DOI:10.1016/j.tetlet.2005.10.103.
- L. A. Actis, W. Fish, J. H. Crosa, K. Kellerman, S. R. Ellenberger, F. M. Hauser and J. Sandersloehr, characterization of anguibactin, a novel siderophore from vibrio-anguillarum 775(PJM1), J. Bacteriol., 1986, 167(1), 57–65, DOI:10.1128/jb.167.1.57-65.1986.
- I. Frans, C. W. Michiels, P. Bossier, K. A. Willems, B. Lievens and H. Rediers, Vibrio anguillarum as a fish pathogen: virulence factors, diagnosis and prevention, J. Fish Dis., 2011, 34(9), 643–661, DOI:10.1111/j.1365-2761.2011.01279.x.
- H. Lee, W. Y. Song, M. Kim, M. W. Lee, S. Kim, Y. S. Park, K. Kwak, M. H. Oh and H. J. Kim, Synthesis and Characterization of Anguibactin to Reveal Its Competence to Function as a Thermally Stable Surrogate Siderophore for a Gram-Negative Pathogen, Acinetobacter baumannii, Org. Lett., 2018, 20(20), 6476–6479, DOI:10.1021/acs.orglett.8b02789.
- M. A. F. Jalal, M. B. Hossain, D. Vanderhelm, J. Sandersloehr, L. A. Actis and J. H. Crosa, Structure of anguibactin, a unique plasmid-related bacterial siderophore from the fish pathogen vibrio-anguillarum, J. Am. Chem. Soc., 1989, 111(1), 292–296, DOI:10.1021/ja00183a044.
- Y. Takeuchi, S. Ozaki, M. Satoh, K. Mimura, S. Hara, H. Abe, H. Nishioka and T. Harayama, Synthesis of Acinetobactin, Chem. Pharm. Bull., 2010, 58(11), 1552–1553, DOI:10.1248/cpb.58.1552.
- R. Grigg, Z. Rankovic and M. Thoroughgood, Acyclic oxyiminium ions. Mannich reactions and addition of Grignard reagents, Tetrahedron, 2000, 56(40), 8025–8032, DOI:10.1016/s0040-4020(00)00721-3.
- R. Sulsky and J. P. Demers, Alkylation of N-benzyloxyureas and carbamates, Tetrahedron Lett., 1989, 30(1), 31–34, DOI:10.1016/s0040-4039(01)80314-2.
- J. Kim, J. E. Lee, H. Ree and H. J. Kim, Total Synthesis of Acinetobactin, Bull. Korean Chem. Soc., 2015, 36(1), 439–441, DOI:10.1002/bkcs.10102.
- S. Sakaguchi, D. Kikuchi and Y. Ishii, Oxidation of diols and ethers by NaBrO3/NaHSO3 reagent, Bull. Chem. Soc. Jpn., 1997, 70(10), 2561–2566, DOI:10.1246/bcsj.70.2561.
- Y. Igarashi, D. Asano, M. Sawamura, Y. In, T. Ishida and M. Imoto, Ulbactins F and G, Polycyclic Thiazoline Derivatives with Tumor Cell Migration Inhibitory Activity from Brevibacillus sp, Org. Lett., 2016, 18(7), 1658–1661, DOI:10.1021/acs.orglett.6b00531.
- J. A. Shapiro, K. R. Morrison, S. S. Chodisetty, D. G. Musaev and W. M. Wuest, Biologically Inspired Total Synthesis of Ulbactin F, an Iron-Binding Natural Product, Org. Lett., 2018, 20(18), 5922–5926, DOI:10.1021/acs.orglett.8b02599.
- J. A. Frump, Oxazolines - their preparation, reactions, and applications, Chem. Rev., 1971, 71(5), 483–505, DOI:10.1021/cr60273a003.
- J. W. Zhang, W. J. Zhang, S. P. Wei and W. J. Wu, A new dihydrooxazole antibiotic from the fermentation broth of streptomyces djakartensis, Heterocycles, 2014, 89(7), 1656–1661, DOI:10.3987/com-14-13001.
- E. Djurendic, S. D. Vujaskovic, M. Sakac, J. Ajdukovic, A. Gakovic, V. Kojic, G. Bogdanovic, O. Klisuric and K. P. Gasi, Synthesis and biological evaluation of some new 2-oxazoline and salicylic acid derivatives, Arkivoc, 2011, 83–102, DOI:10.3998/ark.5550190.0012.207.
- P. Fu, P. P. Liu, H. J. Qu, Y. Wang, D. F. Chen, H. Wang, J. Li and W. M. Zhu, alpha-Pyrones and Diketopiperazine Derivatives from the Marine-Derived Actinomycete Nocardiopsis dassonvillei HR10-5, J. Nat. Prod., 2011, 74(10), 2219–2223, DOI:10.1021/np200597m.
- P. Garg, S. Chaudhary and M. D. Milton, Synthesis of 2-Aryl/Heteroaryloxazolines from Nitriles under Metal- and Catalyst-Free Conditions and Evaluation of Their Antioxidant Activities, J. Org. Chem., 2014, 79(18), 8668–8677, DOI:10.1021/jo501430p.
- V. K. Yadav and K. G. Babu, A remarkably efficient Markovnikov hydrochlorination of olefins and transformation of nitriles into imidates by use of AcCl and an alcohol, Eur. J. Org. Chem., 2005, 2005(2), 452–456 CrossRef.
- (a) P. J. Reider, R. S. E. Conn, P. Davis, V. J. Grenda, A. J. Zambito and E. J. J. Grabowski, Synthesis of (R)-serine-2-d and its conversion to the broad-spectrum antibiotic fludalanine, J. Org. Chem., 1987, 52(15), 3326–3334, DOI:10.1021/jo00391a029; (b) H. Ait-Haddou, O. Hoarau, D. Cramailere, F. Pezet, J. C. Daran and G. G. A. Balavoine, New dihydroxy bis(oxazoline) ligands for the palladium-catalyzed asymmetric allylic alkylation: experimental investigations of the origin of the reversal of the enantioselectivity, Chem.–Eur. J., 2004, 10(3), 699–707, DOI:10.1002/chem.200204649.
- L. B. Li, W. J. Dan, F. F. Tan, L. H. Cui, Z. P. Yuan, W. J. Wu and J. W. Zhang, Synthesis and Antibacterial Activities of Yanglingmycin Analogues, Chem. Pharm. Bull., 2015, 63(1), 33–37, DOI:10.1248/cpb.c14-00578.
- W. J. Dan, H. L. Geng, J. W. Qiao, R. Guo, S. P. Wei, L. B. Li, W. J. Wu and J. W. Zhang, Efficient Synthesis and Antibacterial Evaluation of (+/−)-Yanglingmycin and Its Analogues, Molecules, 2016, 21(1), 96, DOI:10.3390/molecules21010096.
- H. Huang, W. Yang, Z. L. Chen, Z. W. Lai and J. W. Sun, A mild catalytic synthesis of 2-oxazolines via oxetane ring-opening: rapid access to a diverse family of natural products, Chem. Sci., 2019, 10(41), 9586–9590, 10.1039/c9sc03843d.
- S. Yamamoto, N. Okujo and Y. Sakakibara, Isolation and structure elucidation of acinetobactin, a novel siderophore from acinetobacter-baumannii, Arch. Microbiol., 1994, 162(4), 249–254, DOI:10.1007/bf00301846.
- M. Najimi, M. L. Lemos and C. R. Osorio, Identification of siderophore biosynthesis genes essential for growth of Aeromonas salmonicida under iron limitation conditions, Appl. Environ. Microbiol., 2008, 74(8), 2341–2348, DOI:10.1128/aem.02728-07.
- M. Balado, A. Souto, A. Vences, V. P. Careaga, K. Valderrama, Y. Segade, J. Rodriguez, C. R. Osorio, C. Jimenez and M. L. Lemos, Two Catechol Siderophores, Acinetobactin and Amonabactin, Are Simultaneously Produced by Aeromonas salmonicida subsp salmonicida Sharing Part of the Biosynthetic Pathway, ACS Chem. Biol., 2015, 10(12), 2850–2860, DOI:10.1021/acschembio.5b00624.
- E. S. Sattely and C. T. Walsh, A latent oxazoline electrophile for N–O–C bond formation in pseudomonine biosynthesis, J. Am. Chem. Soc., 2008, 130(37), 12282–12284, DOI:10.1021/ja804499r.
- W. M. Wuest, E. S. Sattely and C. T. Walsh, Three Siderophores from One Bacterial Enzymatic Assembly Line, J. Am. Chem. Soc., 2009, 131(14), 5056–5057, DOI:10.1021/ja900815w.
- U. Anthoni, C. Christophersen, P. H. Nielsen, L. Gram and B. O. Petersen, Pseudomonine, an isoxazolidone with siderophoric activity from Pseudomonas fluorescens AH2 isolated from Lake Victorian Nile Perch, J. Nat. Prod., 1995, 58(11), 1786–1789, DOI:10.1021/np50125a026.
- J. A. Shapiro and T. A. Wencewicz, Acinetobactin Isomerization Enables Adaptive Iron Acquisition in Acinetobacter baumannii through pH-Triggered Siderophore Swapping, ACS Infect. Dis., 2016, 2(2), 157–168, DOI:10.1021/acsinfecdis.5b00145.
- W. Y. Song, D. Jeong, J. Kim, M. W. Lee, M. H. Oh and H. J. Kim, Key Structural Elements for Cellular Uptake of Acinetobactin, a Major Siderophore of Acinetobacter baumannii, Org. Lett., 2017, 19(3), 500–503, DOI:10.1021/acs.orglett.6b03671.
- W. F. Penwell, N. DeGrace, S. Tentarelli, L. Gauthier, C. M. Gilbert, B. A. Arivett, A. A. Miller, T. F. Durand-Reville, C. Joubran and L. A. Actis, Discovery and Characterization of New Hydroxamate Siderophores, Baumannoferrin A and B, produced by Acinetobacter baumannii, Chembiochem, 2015, 16(13), 1896–1904, DOI:10.1002/cbic.201500147.
- T. J. Bohac, L. T. Fang, D. E. Giblin and T. A. Wencewicz, Fimsbactin and Acinetobactin Compete for the Periplasmic Siderophore Binding Protein BauB in Pathogenic Acinetobacter baumannii, ACS Chem. Biol., 2019, 14(4), 674–687, DOI:10.1021/acschembio.8b01051.
- M. Balado, Y. Segade, D. Rey, C. R. Osorio, J. Rodriguez, M. L. Lemos and C. Jimenez, Identification of the Ferric-Acinetobactin Outer Membrane Receptor in Aeromonas salmonicida subsp salmonicida and Structure-Activity Relationships of Synthetic Acinetobactin Analogues, ACS Chem. Biol., 2017, 12(2), 479–493, DOI:10.1021/acschembio.6b00805.
- A. Sakakura, S. Umemura and K. Ishihara, Convergent total syntheses of fluvibactin and vibriobactin using molybdenum(VI) oxide-catalyzed dehydrative cyclization as a key step, Chem. Commun., 2008,(30), 3561–3563, 10.1039/b805880f.
- T. J. Bohac, J. A. Shapiro and T. A. Wencewicz, Rigid Oxazole Acinetobactin Analog Blocks Siderophore Cycling in Acinetobacter baumannii, ACS Infect. Dis., 2017, 3(11), 802–806, DOI:10.1021/acsinfecdis.7b00146.
- H. Ree, J. Kim, W. Y. Song, J. E. Lee and H. J. Kim, Total Syntheses and Evaluation of the Siderophore Functions of Fimsbactin B and Its Analogs, Bull. Korean Chem. Soc., 2015, 36(5), 1520–1523, DOI:10.1002/bkcs.10265.
- S. Kim, H. Lee, W. Y. Song and H. J. Kim, Total Syntheses of Fimsbactin A and B and Their Stereoisomers to Probe the Stereoselectivity of the Fimsbactin Uptake Machinery in Acinetobacter baumannii, Org. Lett., 2020, 22(7), 2806–2810, DOI:10.1021/acs.orglett.0c00790.
- A. Sakakura, R. Kondo and K. Ishihara, Molybdenum oxides as highly effective dehydrative cyclization catalysts for the synthesis of oxazolines and thiazolines, Org. Lett., 2005, 7(10), 1971–1974, DOI:10.1021/ol050543j.
- E. D. Laganis and B. L. Chenard, Metal silanolates - organic soluble equivalents for O-2, Tetrahedron Lett., 1984, 25(51), 5831–5834, DOI:10.1016/s0040-4039(01)81697-x.
- W. Tang and S. Y. Fang, Mono-acylation of symmetric diamines in the presence of water, Tetrahedron Lett., 2008, 49(41), 6003–6006, DOI:10.1016/j.tetlet.2008.07.174.
- G. H. Tait, Identification and biosynthesis of siderochromes formed by micrococcus-denitrificans, Biochem. J., 1975, 146(1), 191–204, DOI:10.1042/bj1460191.
- T. Peterson and J. B. Neilands, Revised structure of a catecholamide spermidine siderophore, Tetrahedron Lett., 1979, 50, 4805–4808 CrossRef.
- T. Peterson, K. E. Falk, S. A. Leong, M. P. Klein and J. B. Neilands, Structure and behavior of spermidine siderophores, J. Am. Chem. Soc., 1980, 102(26), 7715–7718, DOI:10.1021/ja00546a013.
- S. A. Ong, T. Peterson and J. B. Neilands, Agrobactin, a siderophore from agrobacterium-tumefaciens, J. Biol. Chem., 1979, 254(6), 1860–1865 CrossRef CAS PubMed.
- B. E. Allred, C. Correnti, M. C. Clifton, R. K. Strong and K. N. Raymond, Siderocalin Outwits the Coordination Chemistry of Vibriobactin, a Siderophore of Vibrio cholerae, ACS Chem. Biol., 2013, 8(9), 1882–1887, DOI:10.1021/cb4002552.
- M. Mansson, L. Gram and T. O. Larsen, Production of Bioactive Secondary Metabolites by Marine Vibrionaceae, Mar. Drugs, 2011, 9(9), 1440–1468, DOI:10.3390/md9091440.
- G. L. Griffiths, S. P. Sigel, S. M. Payne and J. B. Neilands, Vibriobactin, a siderophore from vibrio-cholerae, J. Biol. Chem., 1984, 259(1), 383–385, DOI:10.1016/S0021-9258(17)43671-4.
- D. P. Henderson and S. M. Payne, Vibrio-cholerae iron transport-systems - roles of heme and siderophore iron transport in virulence and identification of a gene associated with multiple iron transport-systems, Infect. Immun., 1994, 62(11), 5120–5125, DOI:10.1128/iai.62.11.5120-5125.1994.
- N. Okujo, M. Saito, S. Yamamoto, T. Yoshida, S. Miyoshi and S. Shinoda, Structure of vulnibactin, a new polyamine-containing siderophore from vibrio-vulnificus, BioMetals, 1994, 7(2), 109–116, DOI:10.1007/BF00140480.
- W. Tan, V. Verma, K. Jeong, S. Y. Kim, C.-H. Jung, S. E. Lee and J. H. Rhee, Molecular characterization of vulnibactin biosynthesis in Vibrio vulnificus indicates the existence of an alternative siderophore, Front. Microbiol., 2014, 5, 1, DOI:10.3389/fmicb.2014.00001.
- G. Ehlert, K. Taraz and H. Budzikiewicz, Bacterial constituents 59. Serratiochelin, a new catecholate siderophore from serratia-marcescens, Z. Phys. Chem., 1994, 49(1–2), 11–17 CAS.
- Y. Schneider, M. Jenssen, J. Isaksson, K. O. Hansen, J. H. Andersen and E. H. Hansen, Bioactivity of Serratiochelin A, a Siderophore Isolated from a Co-Culture of Serratia sp. and Shewanella sp., Microorganisms, 2020, 8(7), 1042, DOI:10.3390/microorganisms8071042.
- M. R. Seyedsayamdost, S. Cleto, G. Carr, H. Vlamakis, M. J. Vieira, R. Kolter and J. Clardy, Mixing and Matching Siderophore Clusters: Structure and Biosynthesis of Serratiochelins from Serratia sp. V4, J. Am. Chem. Soc., 2012, 134(33), 13550–13553, DOI:10.1021/ja304941d.
- R. J. Bergeron and S. J. Kline, Short synthesis of parabactin, J. Am. Chem. Soc., 1982, 104(16), 4489–4492, DOI:10.1021/ja00380a032.
- R. J. Bergeron, S. J. Kline, N. J. Stolowich, K. A. McGovern and P. S. Burton, Flexible synthesis of polyamine catecholamides, J. Org. Chem., 1981, 46(22), 4524–4529, DOI:10.1021/jo00335a041.
- A. Tawson and F. Pyman, Amidines of pharmacological interest, J. Chem. Soc., 1931, 2991–3001, 10.1039/JR9310002991.
- Y. Nagao, T. Miyasaka, Y. Hagiwara and E. Fujita, Utilization of sulfur-containing leaving group .3. total synthesis of parabactin, a spermidine siderophore, J. Chem. Soc., Perkin Trans. 1, 1984,(2), 183–187, 10.1039/p19840000183.
- Y. Nagao, T. Miyasaka, K. Seno, M. Yagi and E. Fujita, Monitored aminolysis of 3-acyl-1,3-thiazolidine-2-thione with amino-acid and its derivative – peptide-bond formation, chemoselective acylation, and bridging reaction, Chem. Lett., 1981,(4), 463–466, DOI:10.1246/cl.1981.463.
- R. J. Bergeron, J. S. McManis, J. B. Dionis and J. R. Garlich, An efficient total synthesis of agrobactin and its gallium(III) chelate, J. Org. Chem., 1985, 50(15), 2780–2782, DOI:10.1021/jo00215a037.
- W. H. Rastetter, T. J. Erickson and M. C. Venuti, Synthesis of iron chelators – enterobactin, enantioenterobactin, and a chiral analog, J. Org. Chem., 1981, 46(18), 3579–3590, DOI:10.1021/jo00331a001.
- R. J. Bergeron, J. R. Garlich and J. S. McManis, Total synthesis of vibriobactin, Tetrahedron, 1985, 41(3), 507–510, DOI:10.1016/s0040-4020(01)96492-0.
- R. J. Bergeron, M. G. Xin, W. R. Weimar, R. E. Smith and J. Wiegand, Significance of asymmetric sites in choosing siderophores as deferration agents, J. Med. Chem., 2001, 44(15), 2469–2478, DOI:10.1021/jm010019s.
- R. J. Bergeron, N. Bharti and S. Singh, Total synthesis of vulnibactin: A natural product iron chelator, Synthesis, 2007,(7), 1033–1037, DOI:10.1055/s-2007-965960.
- R. J. Bergeron, J. R. Garlich and N. J. Stolowich, Reagents for the stepwise functionalization of spermidine, homospermidine, and bis(3-aminopropyl)amine, J. Org. Chem., 1984, 49(16), 2997–3001, DOI:10.1021/jo00190a028.
- A. Sakakura, R. Kondo, S. Umemura and K. Ishihara, Catalytic synthesis of peptide-derived thiazolines and oxazolines using Bis(quinolinolato)dioxomolybdenum(VI) complexes, Adv. Synth. Catal., 2007, 349(10), 1641–1646, DOI:10.1002/adsc.200700068.
- K. Ishihara, Y. Kuroki, N. Hanaki, S. Ohara and H. Yamamoto, Antimony-templated macrolactamization of tetraamino esters. Facile synthesis of macrocyclic spermine alkaloids, (+/−)-buchnerine, (+/−)-verbacine, (+/−)-verbaskine, and (+/−)-verbascenine, J. Am. Chem. Soc., 1996, 118(6), 1569–1570, DOI:10.1021/ja953541a.
- T. Ueda and K. Ishizaki, Syntheses of 3-aminopropylguanidine derivatives, Chem. Pharm. Bull., 1967, 15(2), 228–232 CrossRef CAS.
- G. M. Buckley, G. Pattenden and D. A. Whiting, New synthetic probes of the iron transport-system of paracoccus-denitrificans, Tetrahedron, 1994, 50(40), 11781–11792, DOI:10.1016/s0040-4020(01)85669-6.
- (a) R. J. Bergeron, J. B. Dionis and M. J. Ingeno, Synthesis of a parabactin photoaffinity label, J. Org. Chem., 1987, 52(1), 144–149, DOI:10.1021/jo00377a026; (b) T. Miyasaka, Y. Nagao, E. Fujita, H. Sakurai and K. Ishizu, Synthesis of parabactin analogs and formation of transition-metal complexes of parabactin and related-compounds, J. Chem. Soc., Perkin Trans. 2, 1987,(11), 1543–1549, 10.1039/p29870001543; (c) S. Yoganathan, C. S. Sit and J. C. Vederas, Chemical synthesis and biological evaluation of gallidermin-siderophore conjugates, Org. Biomol. Chem., 2011, 9(7), 2133–2141, 10.1039/c0ob00846j.
- (a) G. A. Snow and A. J. White, Chemical and biological properties of mycobactins isolated from various mycobacteria, Biochem. J., 1969, 115(5), 1031–1045, DOI:10.1042/bj1151031; (b) G. A. Snow, Mycobactins – iron-chelating growth factors from mycobacteria, Bacteriol. Rev., 1970, 34(2), 99–125, DOI:10.1128/mmbr.34.2.99-125.1970.
- K. A. Fennell, U. Mollmann and M. J. Miller, Syntheses and biological activity of amamistatin B and analogs, J. Org. Chem., 2008, 73(3), 1018–1024, DOI:10.1021/jo7020532.
- J. Francis, J. Madinaveitia, H. M. Macturk and G. A. Snow, Isolation from acid-fast bacteria of a growth-factor for mycobacterium-johnei and of a precursor of phthiocol, Nature, 1949, 163(4140), 365–366, DOI:10.1038/163365b0.
- C. F. McQueen and J. T. Groves, A reevaluation of iron binding by Mycobactin J, J. Biol. Inorg. Chem., 2018, 23(7), 995–1007, DOI:10.1007/s00775-018-1592-2.
- B. D. Schwartz and J. J. De Voss, Structure and absolute configuration of mycobactin J, Tetrahedron Lett., 2001, 42(21), 3653–3655, DOI:10.1016/s0040-4039(01)00531-7.
- S. J. Lane, P. S. Marshall, R. J. Upton, C. Ratledge and M. Ewing, Novel extracellular mycobactins, the carboxymycobactins from mycobacterium-avium, Tetrahedron Lett., 1995, 36(23), 4129–4132, DOI:10.1016/0040-4039(95)00676-4.
- J. Gobin, C. H. Moore, J. R. Reeve, D. K. Wong, B. W. Gibson and M. A. Horwitz, Iron acquisition by mycobacterium-tuberculosis – isolation and characterization of a family of iron-binding exochelins, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(11), 5189–5193, DOI:10.1073/pnas.92.11.5189.
- J. Gobin, D. K. Wong, B. W. Gibson and M. A. Horwitz, Characterization of exochelins of the Mycobacterium bovis type strain and BCG substrains, Infect. Immun., 1999, 67(4), 2035–2039 CrossRef CAS PubMed.
- C. Ratledge and G. A. Snow, Isolation and structure of nocobactin na, a lipid-soluble iron-binding compound from nocardia-asteroides, Biochem. J., 1974, 139(2), 407–413, DOI:10.1042/bj1390407.
- S. Kokubo, K. Suenaga, C. Shinohara, T. Tsuji and D. Uemura, Structures of amamistatins A and B, novel growth inhibitors of human tumor cell lines from Nocardia asteroides, Tetrahedron, 2000, 56(35), 6435–6440, DOI:10.1016/s0040-4020(00)00591-3.
- K. A. Fennell and M. J. Miller, Syntheses of amamistatin fragments and determination of their HDAC and antitumor activity, Org. Lett., 2007, 9(9), 1683–1685, DOI:10.1021/ol070382e.
- M. Tsuda, M. Yamakawa, S. Oka, Y. Tanaka, Y. Hoshino, Y. Mikami, A. Sato, H. Fujiwara, Y. Ohizumi and J. Kobayashi, Brasilibactin A, a cytotoxic compound from actinomycete Nocardia brasiliensis, J. Nat. Prod., 2005, 68(3), 462–464, DOI:10.1021/np0496385.
- Y. Ying and J. Hong, Synthesis of brasilibactin A and confirmation of absolute configuration of beta-hydroxy acid fragment, Tetrahedron Lett., 2007, 48(46), 8104–8107, DOI:10.1016/j.tetlet.2007.09.112.
- K. Schneider, I. Rose, S. Vikineswary, A. L. Jones, M. Goodfellow, G. Nicholson, W. Beil, R. D. Suessmuth and H.-P. Fiedler, Biosynthetic capacities of actinomycetes. 39. Nocardichelins A and B, siderophores from Nocardia strain Acta 3026, J. Nat. Prod., 2007, 70(6), 932–935, DOI:10.1021/np060612i.
- Y. Ikeda, H. Nonaka, T. Furumai, H. Onaka and Y. Igarashi, Nocardimicins A, B, C, D, E, and F, siderophores with muscarinic M3 receptor inhibiting activity from Nocardia sp TP-A0674, J. Nat. Prod., 2005, 68(7), 1061–1065, DOI:10.1021/np050091j.
- Y. Ikeda, T. Furumai and Y. Igarashi, Nocardimicins G, H and I, siderophores with muscarinic M3 receptor binding inhibitory activity from Nocardia nova JCM 6044, J. Antibiot., 2005, 58(9), 566–572, DOI:10.1038/ja.2005.77.
- P. J. Maurer and M. J. Miller, Total synthesis of a mycobactin – mycobactin-S2, J. Am. Chem. Soc., 1983, 105(2), 240–245, DOI:10.1021/ja00340a017.
- J. D. Hu and M. J. Miller, Total synthesis of a mycobactin S, a siderophore and growth promoter of Mycobacterium smegmatis, and determination of its growth inhibitory activity against Mycobacterium tuberculosis, J. Am. Chem. Soc., 1997, 119(15), 3462–3468, DOI:10.1021/ja963968x.
- F. Yokokawa, K. Izumi, J. Omata and T. Shioiri, Total synthesis of amamistatin A, an antiproliferative linear peptide from an actinomycete, Tetrahedron, 2000, 56(19), 3027–3034, DOI:10.1016/s0040-4020(00)00170-8.
- C. Ghosh, S. Pal, A. Patel and M. Kapur, Total Synthesis of the Proposed Structure of Mycobactin J, Org. Lett., 2018, 20(20), 6511–6515, DOI:10.1021/acs.orglett.8b02832.
- A. R. Poreddy, O. F. Schall, G. R. Marshall, C. Ratledge and U. Slomczynska, Solid-phase synthesis of methyl carboxymycobactin T 7 and analogues as potential antimycobacterial agents, Bioorg. Med. Chem. Lett., 2003, 13(15), 2553–2556, DOI:10.1016/s0960-894x(03)00473-6.
- J. M. Mitchell and J. T. Shaw, Synthesis and stereochemical assignment of brasilibactin A, Org. Lett., 2007, 9(9), 1679–1681, DOI:10.1021/ol070355o.
- J. C. Banks and C. J. Moody, Synthesis of the reported structure of the naturally occurring siderophore nocardimicin B, Tetrahedron Lett., 2009, 50(26), 3371–3373, DOI:10.1016/j.tetlet.2009.02.124.
- I. Paterson, D. J. Wallace and C. J. Cowden, Polyketide synthesis using the boron-mediated, anti-aldol reactions of lactate-derived ketones: total synthesis of (-)-ACRL, toxin IIIB, Synthesis, 1998, 639–652, DOI:10.1055/s-1998-5929.
| This journal is © The Royal Society of Chemistry 2022 |