
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorptive separation of para-xylene by nonporous adaptive crystals of phenanthrene[2]arene†
Ying Hou,
Yin-Rong Duan,
Man-Hua Ding*,
Lin-Li Tang and
Fei Zeng *
*
Department of Biology and Chemistry, Hunan University of Science and Engineering, Yongzhou 425199, China. E-mail: zengfei@iccas.ac.cn; 42979930@qq.com
First published on 9th August 2022
Abstract
In this work, we developed a new method for the preparation of phenanthrene[2]arene on a large-scale. Meanwhile, the synthetic phenanthrene[2]arene has been successfully used as nonporous adaptive crystals for the separation of para-xylene (pX) from xylene isomers. The crystal structure revealed that one host molecule can adsorb one pX molecule to form the 1@pX complex, in which pX is located in the cavity of the host.
Xylene is an aromatic hydrocarbon in which two hydrogen atoms on the benzene ring are replaced by two methyl groups, so it has three isomers: ortho-xylene (oX), meta-xylene (mX), and para-xylene (pX). Xylene isomers are important chemical feedstocks in the petrochemical and pharmaceutical industries and used for the production of many polymers, plastics, fibers, solvents, and fuel.1–7 However, most xylenes used in industry are single-component compounds. Therefore, the prepared xylene mixtures need to be individually separated in most cases. The separation of the xylenes was classified as one of the “seven chemical separations to change the world” due to their similar kinetic sizes, close boiling points, and the same molecular weight.8 The boiling point of oX is relatively high (417.55 K) and can be separated by rectification, while the slight difference in boiling points (only 0.8 K) between pX (411.45 K) and mX (412.25 K) makes them almost impossible to separate via a traditional distillation process. However, pX could be isolated by fractional crystallization due to its relatively higher melting point compared with mX.9 However, the energy requirement for fractional crystallization is high because of the need to cool large quantities of material to about −53 °C. Therefore, it is necessary and desirable to develop easy operation and more energy efficient methods to separate pX and mX.
In the past decades, a number of methods have been developed to meet the separation challenge of xylene isomers, including solvent extraction10–16 and adsorption by porous materials.17–21 The method of solvent extraction sometimes has the highly toxic effect on the environment and strong corrosive effect on the equipment impeded its long term applicability. While, the adsorption by porous materials are built using reversible chemistry and have comparatively low moisture or thermal stability, which restrict their further application in industrial. Recently, macrocycle-based nonporous adaptive crystals (NACs) have attracted considerable interest as they display great potentials for practical applications, especially in the adsorption, separation, and storage of hydrocarbons.22–31 Huang and coworkers reported the separation of pX from its structural isomers, mX and oX by nonporous adaptive crystal of pillar[6]arene with a purity of 90%.32 Khashab's group also achieved the separation of pX using a polymorphic azobenzene cage.33 Since the discovery of crown ethers by Pedersen in 1967, supramolecular chemistry has been developed for many years and a series of macrocyclic hosts have been designed and synthesized.34–42 However, they have been studied as xylene isomers selective adsorbents only rarely.43–45
The development of NACs with high pX adsorption capacity and separation purity is still a challenging work. Herein, we report that phenanthrene[2]arene 1 can be used as a host for the encapsulation and separation of pX from the equimolar mixture of pX and mX in the vapor phases through shape selective crystallization under mild and user-friendly conditions. Furthermore, 1 exhibits excellent recyclability, with no significant decrease in performance after seven cycles of adsorption and desorption (Fig. 1).
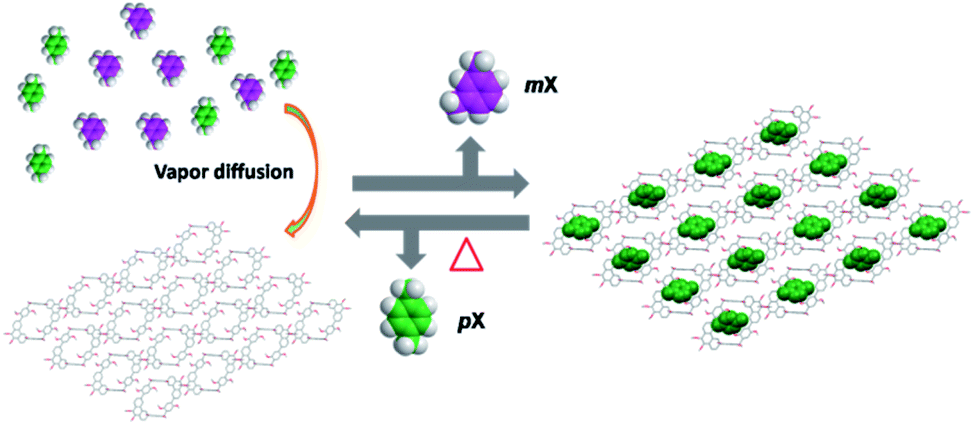 | ||
| Fig. 1 Structural representation of the capture of pX from a pX/mX mixture using phenanthrene[2]arene crystals. | ||
Although, the synthesis of host 1 (ref. 46) have been reported by our group recently. However, we found that the synthetic route can be further optimized to synthesis of the precursor 3, which is beneficial to the subsequent large-scale production. As shown in Scheme 1, starting from the commercially available 3,6-dibromophenanthrene-9,10-dione and 2,5-dimethoxyphenylboronic acid, compound 3 can be quantitatively obtained after work up by simple suction filtration by the Suzuki coupling reaction. Then, the precursor 2 can be quantitatively obtained from compound 3 through sodium dithionite reduction and methylation with dimethyl sulfate.
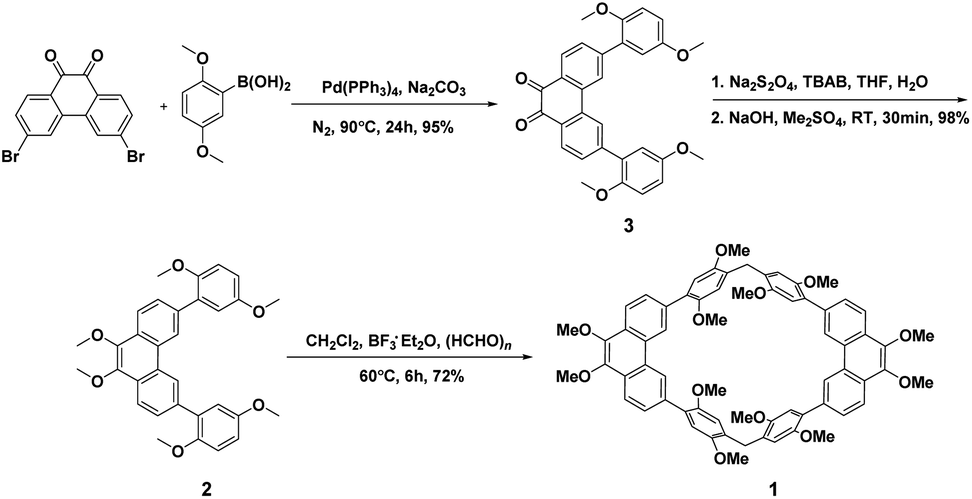 | ||
| Scheme 1 Synthesis of host 1. | ||
Similar to the phenanthrene[2]arene as the nonporous adaptive crystals for benzene adsorption capacity,46 activated host 1 was prepared by recrystallization in the mixture of CH2Cl2/MeOH and dried under vacuum at 150 °C for 6 h. To study the selectivity of pX over mX, single-component solid–vapor sorption experiments were first performed with the activated solid of 1 toward pX and mX. The mole ratio of time-dependent solid–vapor sorption plots were determined by the 1H-NMR analysis. As shown in Fig. 2a, the adsorption capacity of 1 toward pX in the first four hours was negligible, but the adsorption capacity of pX increased significantly with time and it required about 16 hours to reach saturation. The uptake of pX was calculated to be about one pX/1 at saturation. Similar to the pX, activated 1 also showed adsorption capacity toward mX. However, it takes even more time for the adsorption capacity of mX to increase significantly (Fig. 2b). The uptake of mX was calculated to be about 1.2 mX/1 at 20 hours. These results suggest that both pX and mX can be adsorb by the activated solid 1 and the absorption is a relatively slow process. Consistent with the single-component adsorption experiments, TGA analysis further confirmed the absorb number of 1 toward pX and mX. Upon heating the activated 1 that adsorbed pX or mX vapor for 20 hours, a weight loss of 9.2% from 53 to 188 °C and a weight loss of 10.8% from 48 to 145 °C were observed, which corresponded to the release of one pX molecule or 1.2 mX molecules per host 1, respectively (Fig. 2c and d). The higher release temperature of pX than mX revealed that pX could be more steadily stored in 1 crystals than mX.
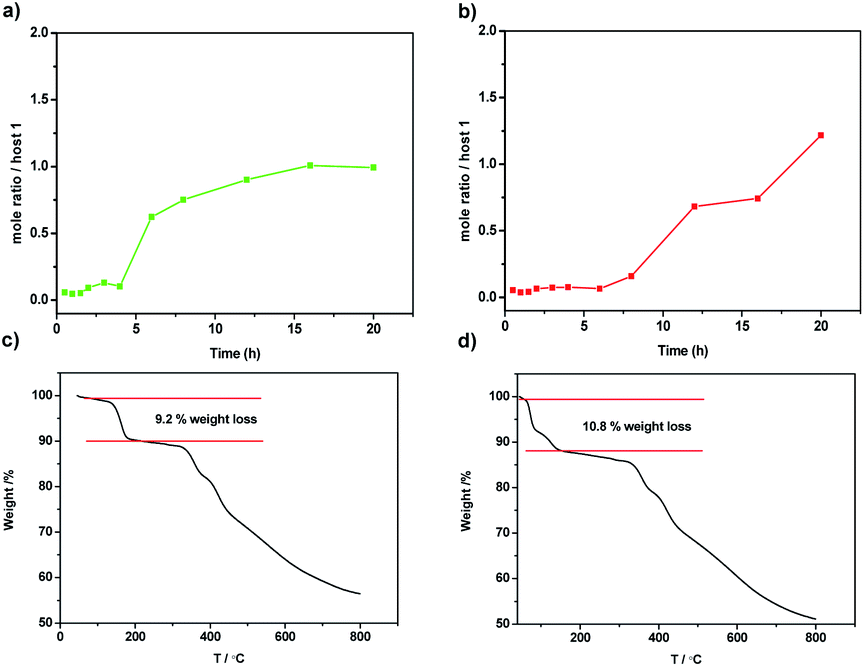 | ||
| Fig. 2 (a and b) Time-dependent solid–vapor sorption plots of 1 for single-component vapour pX and mX respectively; (c and d) thermogravimetric analysis of 1 after sorption of pX or mX adsorbed vapor for 20 h. | ||
To get more information about adsorption capacity of 1 toward pX and mX, We tried to get the single crystals of 1 loaded with pX or mX. Single crystal structures that suitable for X-ray analysis were obtained by slow evaporation of the solution of 1 in pX or mX, providing unambiguous evidence for the formation of 1@pX and 1@2mX. As shown in Fig. 3a, the resulting crystal structure revealed that one host molecule 1 can adsorbs one pX molecule to formation of 1@pX complex, in which pX located in the cavity of host. Interestingly, CH⋯H and CH⋯π interactions between pX and host 1 with the distance of 2.398 Å (a) and 2.865 Å (b) were observed. Because of these multiple noncovalent interactions, the host 1 showed high adsorption capacity for pX. Moreover, it was found that one host molecule 1 can adsorbs two mX molecules to formation of 1@2mX complex (Fig. 3b), in which two mX molecules not located in the cavity of host. Additionally, CH⋯O and CH⋯π intermolecular interactions also exist between the mX and host 1, and their distance are measured to be 2.624 Å (c) and 2.760 Å (d), respectively. From the b-axis direction of 1@pX and 1@2mX complex packing structure (Fig. 3c and d), a large number of pX and mX molecules are absorbed in the crystal structure. These results further support the adsorption capacity of 1 toward pX and mX provides us an opportunity to use host 1 as NACs material for the separation of pX and mX. Moreover, the PXRD pattern of activated solid 1 after absorbing pX showed a significant change in comparison with the original pattern of activated 1, which suggested the structural transformation of activated 1 upon capture of pX vapour (Fig. S8†). The PXRD pattern of activated solid 1 after absorbing mX showed a no change in comparison with the original pattern of activated 1, which suggested no structural transformation of activated 1 upon capture of mX vapour (Fig. S9†). In addition, the PXRD pattern of activated 1 after capturing pX was matched well with the simulated patterns based on single-crystal structures of 1@pX. This indicated that activated 1 transformed into 1@pX after adsorption of pX.
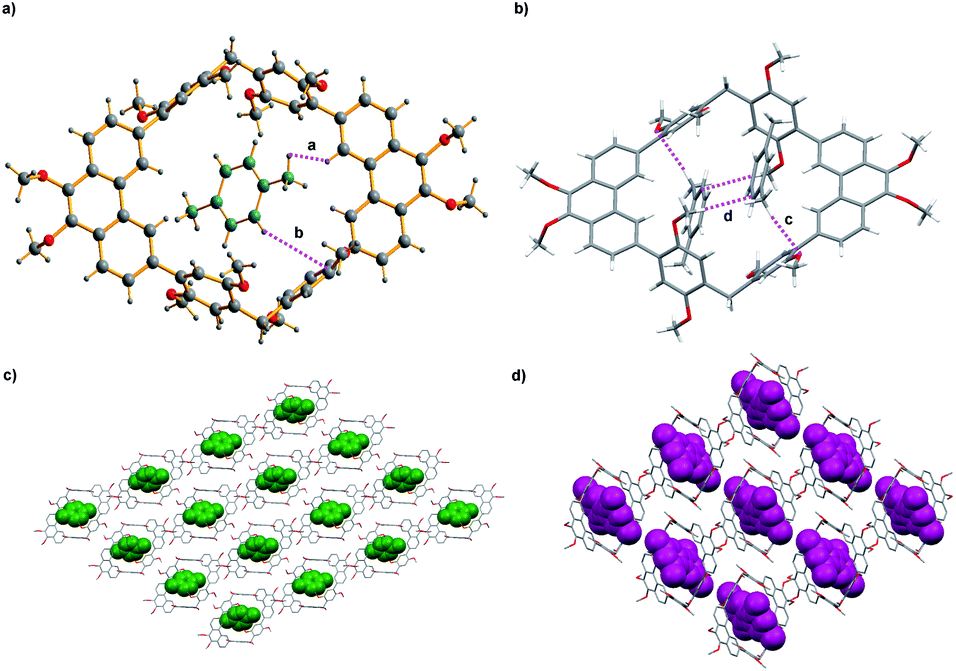 | ||
| Fig. 3 Crystal structure of (a) 1@pX; (b) 1@2mX; (c) and (d) packing of 1@pX and 1@2mX, view along the b axis. | ||
Based on the different sorption ability of activated 1 toward pX and mX, we wondered whether 1 could separate pX/mX mixtures. To confirm this hypothesis, we performed two-component (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) competition sorption experiments. Similar to the single-component solid–vapor sorption experiments, the adsorption capacity of 1 toward pX/mX mixtures in the first three hours was negligible. However, after 3 h, pX could be predominantly adsorbed over mX (Fig. 4a). At saturation, ca. 0.83 pX molecules were adsorbed with 0.18 mX molecules per activated 1 from the 1H-NMR analysis (Fig. S10†). This result indicates that activated 1 has selective adsorption of pX. The Gas chromatography determined the percentage of pX adsorbed by activated 1 to be 81% (Fig. 4c). In addition the powder X-ray diffraction patterns of activated 1 that adsorbed pX/mX mixtures were almost the same with the simulated patterns based on single-crystal structures of 1@pX (Fig. 4b), indicating the similar pX loaded 1 crystals formed. Although, in the crystal structure of 1@2mX, one host molecule can adsorbs two mX molecules, but the host 1 exhibits better pX selectivity over mX, probably due to better matching of pX in the intrinsic cavity of host 1. Considering the further application in real production, recycling capacity is an important criterion for assessing an adsorbent. 1H NMR spectroscopy and TGA analysis revealed that, by heating 1@pX at 150 °C in vacuum for 3 hours, the absorbed pX could be completely released (Fig. S13 and S14†). We also proved that the newly formed crystals were activated 1 as indicated by PXRD (Fig. S15†). Moreover, the re-activated solids can be re-used in next adsorption cycles and no obviously loss of selectivity or capacity were observed after recycling seven times (Fig. 4d).
:1 ratio) competition sorption experiments. Similar to the single-component solid–vapor sorption experiments, the adsorption capacity of 1 toward pX/mX mixtures in the first three hours was negligible. However, after 3 h, pX could be predominantly adsorbed over mX (Fig. 4a). At saturation, ca. 0.83 pX molecules were adsorbed with 0.18 mX molecules per activated 1 from the 1H-NMR analysis (Fig. S10†). This result indicates that activated 1 has selective adsorption of pX. The Gas chromatography determined the percentage of pX adsorbed by activated 1 to be 81% (Fig. 4c). In addition the powder X-ray diffraction patterns of activated 1 that adsorbed pX/mX mixtures were almost the same with the simulated patterns based on single-crystal structures of 1@pX (Fig. 4b), indicating the similar pX loaded 1 crystals formed. Although, in the crystal structure of 1@2mX, one host molecule can adsorbs two mX molecules, but the host 1 exhibits better pX selectivity over mX, probably due to better matching of pX in the intrinsic cavity of host 1. Considering the further application in real production, recycling capacity is an important criterion for assessing an adsorbent. 1H NMR spectroscopy and TGA analysis revealed that, by heating 1@pX at 150 °C in vacuum for 3 hours, the absorbed pX could be completely released (Fig. S13 and S14†). We also proved that the newly formed crystals were activated 1 as indicated by PXRD (Fig. S15†). Moreover, the re-activated solids can be re-used in next adsorption cycles and no obviously loss of selectivity or capacity were observed after recycling seven times (Fig. 4d).
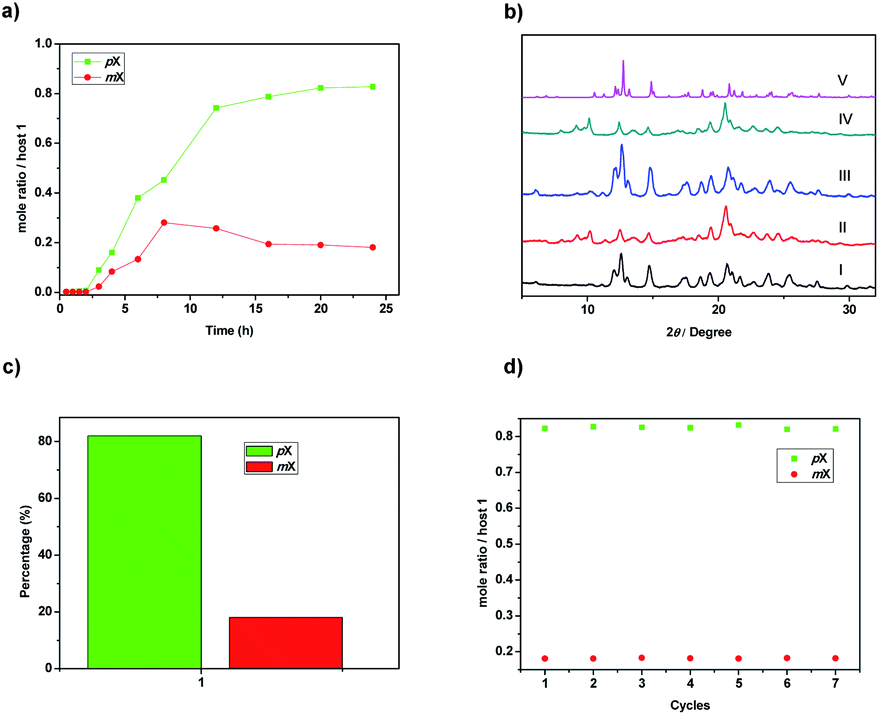 | ||
| Fig. 4 (a) Time-dependent solid–vapor sorption plot of activated 1 for pX and mX equimolar mixture vapor. (b) PXRD patterns of 1: (I) activated 1 adsorption of pX vapour for 20 h; (II) activated 1 adsorption of mX vapour for 20 h; (III) activated 1 adsorption of pX/mX mixture vapour for 20 h; (IV) activated 1 after adsorption of pX/mX mixture vapour for 20 h and then heating at 150 °C under vacuum for 3 h; (V) simulated from single-crystal structure of 1@pX. (c) Relative uptakes of pX and mX adsorbed by activated 1 that adsorbed pX/mX mixtures for 20 h as measured by gas chromatography. (d) Relative uptakes of pX and mX by activated 1 that adsorbed pX/mX mixtures for 20 h after seven recycles. | ||
Conclusions
In summary, we developed a new method for the large-scale preparation of phenanthrene[2]arene 1 and demonstrated that phenanthrene[2]arene can be used for the adsorption and separation of industrially important xylene isomers. The activated crystals of 1 showed higher pX adsorption capacity than mX. One host molecule can at maximum adsorb one pX molecules in its central cavity. Moreover, activated crystals 1 also exhibits excellent recyclability when used as NACs materials to separation of pX and mX. The advantages of simple synthesis, high separation efficiency, and outstanding recycling performance of 1 makes this material possesses enormous potential for applications in the chemical industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China No. 21602055 and 51772091; Natural Science Foundation of Hunan Province No. 2017JJ3094.Notes and references
- Y. Yang, P. Bai and X. Guo, Ind. Eng. Chem. Res., 2017, 56, 14725–14753 CrossRef CAS.
- M. I. Gonzalez, M. T. Kapelewski, E. D. Bloch, P. J. Milner, D. A. Reed, M. R. Hudson, J. A. Mason, G. Barin, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3412–3422 CrossRef CAS PubMed.
- W. J. Cannella, Xylenes and ethylbenzene, Wiley and Sons, New York, 2000 Search PubMed.
- R. Meyers, Handbook of Petroleum Refining Processes, 3th ed., McGraw-Hill, New York, 2003, pp. 2.47–2.53 Search PubMed.
- J. Scheirs and T. E. Long, Industrial Modern Polyesters: chemistry and technology of polyesters and copolyesters, Wiley, Chichester, 2003 Search PubMed.
- J. L. Pellegrino, Energy and Environmental Profile of the Chemicals Industry, U.S. Department of Energy, 2000 Search PubMed.
- N. Sun, S.-Q. Wang, R. Zou, W. G. Cui, A. Zhang, T. Zhang, Q. Li, Z. Z. Zhuang, Y. H. Zhang, J. Xu, M. J. Zaworotko and X. H. Bu, Chem. Sci., 2019, 10, 8850–8854 RSC.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- D. Ruthven, Principles of Adsorption and Adsorption Processes, Wiley, New York, 1984, pp. 401–405 Search PubMed.
- D. A. McCaulay, B. H. Shoemaker and A. P. Lien, Ind. Eng. Chem., 1950, 42, 2103–2107 CrossRef CAS.
- G. Zhang, A.-H. Emwas, U. F. S. Hameed, S. T. Arold, P. Yang, A. Chen, J.-F. Xiang and N. M. Khashab, Chem, 2020, 6, 1082–1096 CAS.
- F. U. Rahman, J.-M. Yang, Y.-H. Wan, H.-B. Zhang, I. D. Petsalakis, G. Theodorakopoulos, J. Rebek and Y. Yu, Chem. Commun., 2020, 56, 6945–6948 RSC.
- G. Zhang, B. Moosa, A. Chen and N. M. Khashab, ChemPlusChem, 2020, 85, 1244–1248 CrossRef CAS PubMed.
- I. Uemasu, J. Inclusion Phenom. Mol. Recognit. Chem., 1992, 13, 1–7 CrossRef CAS.
- I. Uemasu and S. Kushiyama, Fuel Process. Technol., 2004, 85, 1519–1526 CrossRef CAS.
- H. F. Yang and Y. F. Hu, Chem. Eng. Process., 2017, 116, 114–120 CrossRef CAS.
- D. M. Polyukhov, A. S. Poryvaev, S. A. Gromilov and M. V. Fedin, Nano Lett., 2019, 19, 6506–6510 CrossRef CAS PubMed.
- M. du Plessis, V. I. Nikolayenko and L. J. Barbour, J. Am. Chem. Soc., 2020, 142, 4529–4533 CrossRef CAS PubMed.
- M. B. Dewal, M. W. Lufaso, A. D. Hughes, S. A. Samuel, P. Pellechia and L. S. Shimizu, Chem. Mater., 2006, 18, 4855–4864 CrossRef CAS.
- W. Huang, J. Jiang, D. Wu, J. Xu, B. Xue and A. M. Kirillov, Inorg. Chem., 2015, 54, 10524–10526 CrossRef CAS PubMed.
- T. Ogoshi, Y. Shimada, Y. Sakata, S. Akine and T. Yamagishi, J. Am. Chem. Soc., 2017, 139, 5664–5667 CrossRef CAS PubMed.
- J. R. Wu and Y. W. Yang, J. Am. Chem. Soc., 2019, 141, 12280–12287 CrossRef CAS PubMed.
- Y. Zhou, K. Jie, E. Li and F. Huang, Sci. Sin.: Chim., 2019, 49, 832–843 Search PubMed.
- M. Wang, J. Zhou, E. Li, Y. Zhou, R. Zhao, W. Zhu and F. Huang, J. Am. Chem. Soc., 2019, 141, 17102–17106 CrossRef CAS PubMed.
- K. Jie, Y. Zhou, E. Li, R. Zhao, M. Liu and F. Huang, J. Am. Chem. Soc., 2018, 140, 3190–3193 CrossRef CAS PubMed.
- Y. Ding, L. O. Alimi, B. Moosa, C. Maaliki, J. Jacquemin, F. Huang and N. M. Khashab, Chem. Sci., 2021, 12, 5315–5318 RSC.
- H. Yao, Y.-M. Wang, M. Quan, M. U. Farooq, L.-P. Yang and W. Jiang, Angew. Chem., Int. Ed., 2020, 59, 19945–19950 CrossRef CAS PubMed.
- Y. Zhao, H. Xiao, C.-H. Tung, L.-Z. Wu and H. Cong, Chem. Sci., 2021, 12, 15528–15532 RSC.
- J. R. Wu and Y.-W. Yang, Angew. Chem., Int. Ed., 2021, 60, 1690–1701 CrossRef CAS PubMed.
- W. Yang, K. Samanta, X. Wan, T. U. Thikekar, Y. Chao, S. Li, K. Du, J. Xu, Y. Gao, H. Zuilhof and A. C. Sue, Angew. Chem., Int. Ed., 2020, 59, 3994–3999 CrossRef CAS PubMed.
- K. Jie, Y. Zhou, E. Li and F. Huang, Acc. Chem. Res., 2018, 51, 2064–2072 CrossRef CAS PubMed.
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS PubMed.
- B. Moosa, L. O. Alimi, A. Shkurenko, A. Fakim, P. M. Bhatt, G. Zhang, M. Eddaoudi and N. M. Khashab, Angew. Chem., Int. Ed., 2020, 59, 21367–21372 CrossRef CAS PubMed.
- C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC.
- M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- X.-N. Han, Y. Han and C.-F. Chen, J. Am. Chem. Soc., 2020, 142, 8262–8269 CrossRef CAS PubMed.
- H. Chen, J. Fan, X. Hu, J. Ma, S. Wang, J. Li, Y. Yu, X. Jia and C. Li, Chem. Sci., 2015, 6, 197–202 RSC.
- M.-H. Ding, J. Liao, L.-L. Tang and F. Zeng, Chin. Chem. Lett., 2021, 32, 1665–1668 CrossRef CAS.
- Q.-H. Guo, L. Zhao and M.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 8386–8389 CrossRef CAS PubMed.
- Q. Shi, X. Wang, B. Liu, P. Qiao, J. Li and L. Wang, Chem. Commun., 2021, 57, 12379–12405 RSC.
- F. Zeng, L. Cheng, G. C. Ou, L.-L. Tang and M. H. Ding, J. Org. Chem., 2022, 87, 3863–3867 CrossRef PubMed.
- G. Zhang, B. Hua, A. Dey, M. Chosh, B. A. Moosa and N. M. Khashab, Acc. Chem. Res., 2021, 54, 155–168 CrossRef CAS PubMed.
- A. Dey, S. Chand, M. Ghosh, M. Altamimy, B. Maity, P. M. Bhatt, I. A. Bhat, L. Cavallo, M. Eddaoudi and N. M. Khashab, Chem. Commun., 2021, 57, 9124–9127 RSC.
- G. Zhang, Y. Ding, A. Hashem, A. Fakim and N. M. Khashab, Cell Rep. Phys. Sci., 2021, 2, 100470 CrossRef CAS.
- F. Zeng, L. Cheng, L.-L. Tang and X. F. Wang, Org. Chem. Front., 2022, 9, 3307–3311 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 2 and 3; The 1H and 13C NMR spectra of 2 and 3. CCDC 2165484 for 1@pX, 2175792 for 1@mX. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra03773d |
| This journal is © The Royal Society of Chemistry 2022 |