
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePROTACs bearing piperazine-containing linkers: what effect on their protonation state?†
Jenny Desantis‡
 a,
Andrea Mammoli‡b,
Michela Eleuteria,
Alice Coletti§
b,
Federico Crocia,
Antonio Macchiarulob and
Laura Goracci*a
a,
Andrea Mammoli‡b,
Michela Eleuteria,
Alice Coletti§
b,
Federico Crocia,
Antonio Macchiarulob and
Laura Goracci*a
aDepartment of Chemistry, Biology, and Biotechnology, University of Perugia, Via Elce di Sotto 8, 06123 Perugia, Italy. E-mail: laura.goracci@unipg.it
bDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123 Perugia, Italy
First published on 9th August 2022
Abstract
Proteolysis targeting chimeras (PROTACs) represent an emerging class of compounds for innovative therapeutic application. Their bifunctional nature induces the formation of a ternary complex (target protein/PROTAC/E3 ligase) which allows target protein ubiquitination and subsequent proteasomal-dependent degradation. To date, despite great efforts being made to improve their biological efficacy PROTACs rational design still represents a challenging task, above all for the modulation of their physicochemical and pharmacokinetics properties. Considering the pivotal role played by the linker moiety, recently the insertion of a piperazine moiety into the PROTAC linker has been widely used, as this ring can in principle improve rigidity and increase solubility upon protonation. Nevertheless, the pKa of the piperazine ring is significantly affected by the chemical groups located nearby, and slight modifications in the linker could eliminate the desired effect. In the present study, the pKa values of a dataset of synthesized small molecule compounds including PROTACs and their precursors have been evaluated in order to highlight how a fine modulation of piperazine-containing linkers can impact the protonation state of these molecules or similar heterobifunctional ones. Finally, the possibility of predicting the trend through in silico approaches was also evaluated.
Introduction
Proteolysis targeting chimeras (PROTACs) are hetero-bifunctional molecules composed of a warhead capable of binding with a protein of interest (POI), a second ligand for an E3 ubiquitin ligase, and a linker to concatenate the two ligands.1–4 Due to this bifunctionality, PROTACs can induce the formation of a ternary complex composed of the target protein, the PROTAC, and the E3 ligase, allowing the E2 ubiquitin-conjugating enzyme to transfer ubiquitin to the surface of the target protein and activating its successive proteasomal-dependent degradation.1,4 During the design of a PROTAC molecule, besides the two protein-binding ligands, the choice of the linker is crucial.5,6 Indeed, it influences the distance between the POI and E3 ligase and the geometry of the POI–PROTAC–E3 ternary complex, thus affecting the degradation efficacy. Both the length and composition of the linker are critically important for the formation of a productive ternary complex, degradation activity, and target selectivity.7 Moreover, as commonly observed also in other chimeric compounds8 (including antibody–drug conjugates (ADC),9–12 dual-inhibitors, and molecular glues13,14), the linker has a significant impact on the overall physicochemical properties of the PROTAC molecule, which in turn affect its pharmacokinetic (PK) profile.15 Several advantages of PROTAC technology over traditional protein inhibition were highlighted, including the catalytic mechanism (once degradation occurs, the PROTAC molecule can bind to a new target molecule) and the ability to bind targets considered “undruggable” with classical small molecule inhibitors, just to mention a few.16,17 Therefore, PROTACs have opened the way to a new therapeutic modality and nowadays PROTACs rational design represents a novel challenging task. On one hand PROTACs design still requires new in silico methods to predict the ternary complex formation and thus to identify the optimal length and nature of the linker.18,19 On the other hand, it is now generally assumed that classical strategies exploited so far for molecular design and hit-to-lead optimization applied to traditional small molecules to improve bioavailability and absorption, distribution, metabolism, and excretion (ADME) properties cannot be applied tout-court to PROTACs. Indeed, PROTACs are characterized by a high molecular weight (600–1900 Da) and high topological polar surface area (TPSA) (100–400) (data based on PROTAC-DB collection, accessed February 2022),20 making their delivery and bioavailability the most significant hurdles to overcome on the way to the clinic.21 Since PROTACs' chemical space lies beyond the classical Lipinski ‘Rule of 5’ space, efforts have been recently devoted to better characterize this new chemical class not only in terms of their biological activity but also for physicochemical and ADME properties.22–30 To date, although a large collection of PROTACs are available,22,31 their physicochemical properties are generally only calculated values; however, for Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values it has been already demonstrated that in the case of PROTACs the calculated values can be very different from the experimental ones,26 possibly due to the fact that in silico tools have been usually trained with the chemical space of traditional small molecules.30
P values it has been already demonstrated that in the case of PROTACs the calculated values can be very different from the experimental ones,26 possibly due to the fact that in silico tools have been usually trained with the chemical space of traditional small molecules.30
Despite the increasing interest on PROTACs PK properties, we noticed that to date the experimental determination of the ionization constant (pKa) of PROTACs was reported on just one paper and for a small dataset of degraders.30 As known, the protonation state of a molecule is pivotal for pharmaceutical and medicinal research since it influences a large number of properties (pH-dependent solubility, lipophilicity, permeability, and stability) that in turn has a significant impact on ADME.32–34 Despite the hetero-bifunctional nature of PROTACs implies that ionizable centers can be present in each of the three moieties (POI ligand, linker and E3 ligase ligand), we were intrigued by noticing that a general growing trend to improve PROTACs solubility is now to act on the linker by introducing saturated heterocycles bearing a basic center, such as piperidine or piperazine rings, which belong to the classification of rigid linkers used for PROTACs design (Fig. 1).5,6,35 While the insertion of a piperidine or a piperazine in the PROTAC linker has been associated to a potential improved activity due to the increased rigidity,6 another natural effect relies in the insertion of a protonable amino group. Therefore, piperazine containing linkers present the advantage of potentially favoring solubility. However, according to the open-access database collecting PROTACs information (PROTAC-DB) (Fig. 1), these heterocycles are variably connected to the additional part of the linker moiety, and it is well known that the pKa of an acid or a basic center in a molecule is strongly affected by neighboring groups.32,36,37
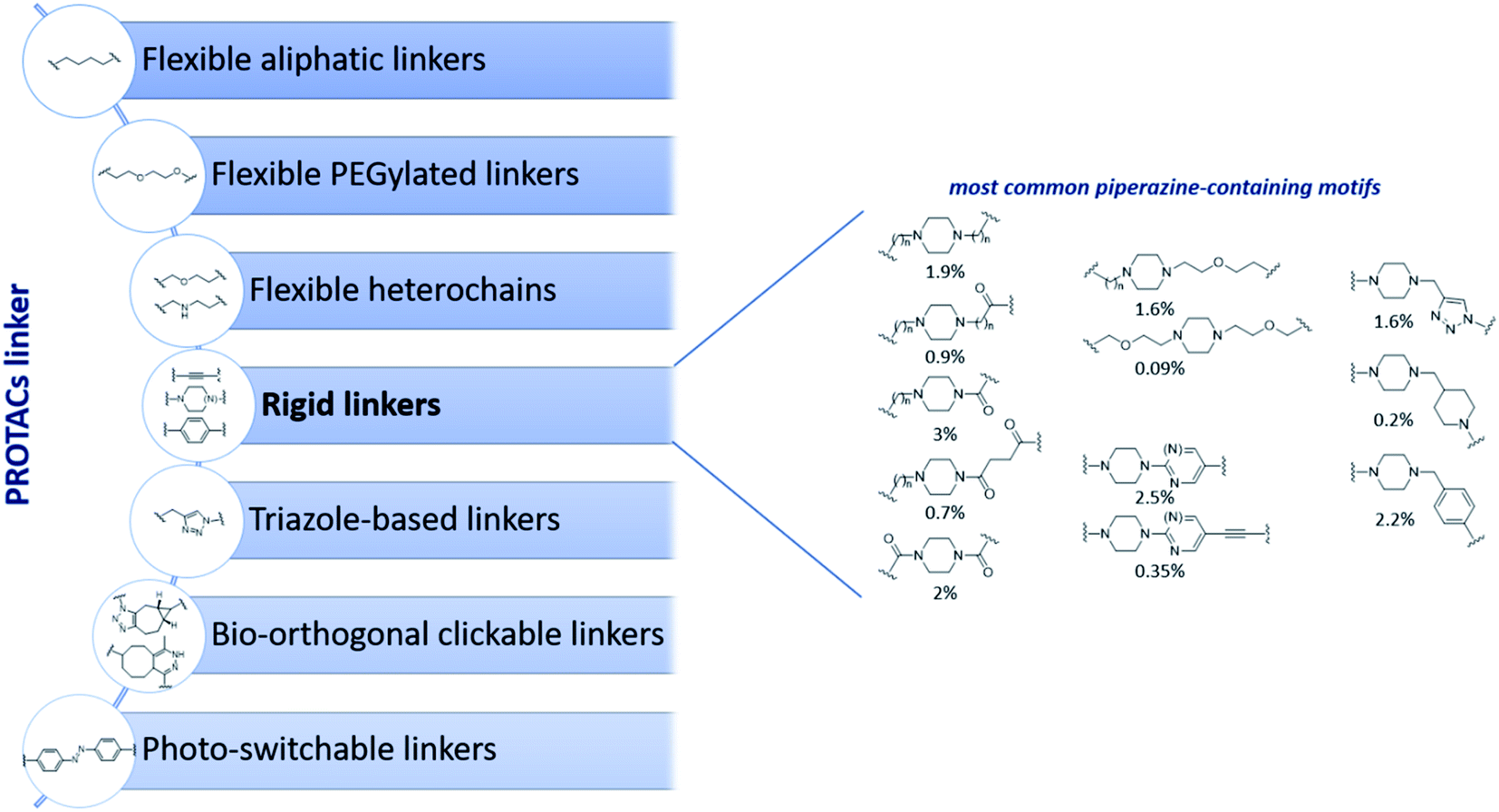 | ||
| Fig. 1 Classification of most common linkers used in PROTAC design based on available structures deposited in PROTAC-DB31 collection accessed on Feb 2022 (left panel) and overview of piperazine-containing motifs used among rigid linkers (right panel). Percentages below piperazine structures indicate their relative abundance on the total on 2258 PROTACs collected in PROTAC-DB database. | ||
In this study, we aimed on shed light on how the protonation state of piperazine moieties used in PROTACs's linkers can be modulated by the structural environment, as Morgenthaler et al. have already provided examples that neighboring groups of nitrogen-containing functional groups can significantly affect the pKa.32 In other words, in this study we desired to answer the following question: to which extent can one modulate the protonation state of a piperazine by modifying the length, the nature, and the anchoring groups in a PROTAC's linker? For this purpose, we designed, collected and synthesized a focused set of piperazine-decorated PROTAC derivatives or precursors, and their pKa were experimentally measured. Our observations led to the definition of general trends that could be easily applied and exploited in PROTACs design and optimization as well as in the development of different heterobifunctional compounds, such as molecular glues, ADC, and dual-inhibitors among others.
Results and discussion
Design of a set of piperazine-containing PROTACs and ligands
In this study, we collected 11 piperazine-containing PROTACs based on four different POI ligands (Fig. 2): the casein kinase 2 (CK2) inhibitor silmitasertib (CX4945),38 the bromodomain and extra-terminal (BET) inhibitor JQ1,39 the FDA-approved poly(ADP-ribose) polymerase (PARP) inhibitor olaparib,40 and the nonsteroidal anti-inflammatory drug indomethacin. Concerning the ligand for E3 ligase, a binder for von Hippel–Lindau (VHL) E3 ligase was selected (Fig. 2) for all PROTACs except for one compound, in which a cereblon (CRBN) ligase binding moiety was used instead. For each POI ligand series, different piperazine-containing linkers among those most commonly used (as summarized in Fig. 1) were investigated and derivative molecules (PROTAC-1/11, Table S1†) were synthesized to evaluate the effect of chemical environment on the pKa. In addition, as proposed/hypothesized by Cantrill et al. assuming that the pKa values of fragments of entire molecules containing the same ionizable moieties remained unchanged,41 precursor molecules constating of POI warheads widely decorated with piperazine-containing motifs were also synthesized (compounds 1–17, Table S2†) and tested for comparative and exploration purposes.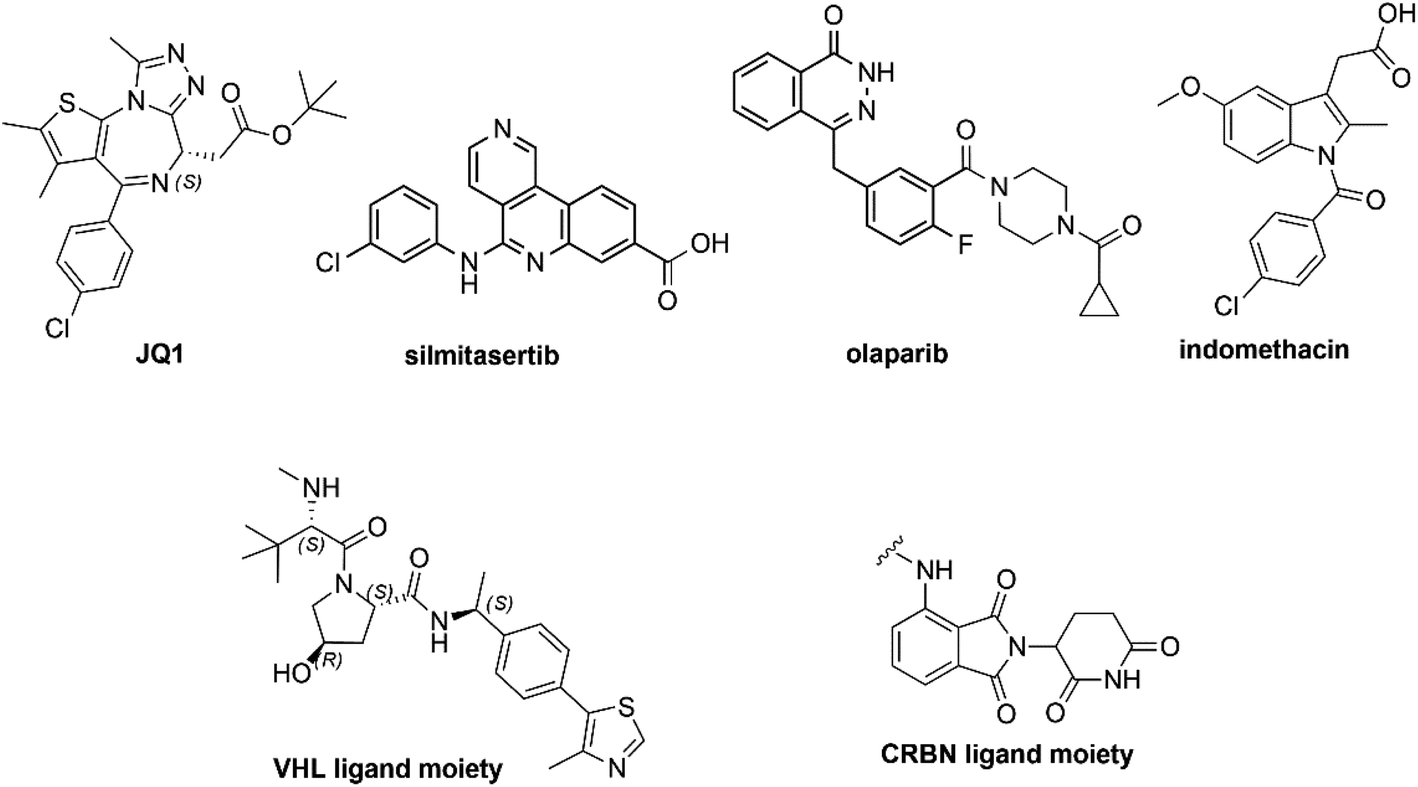 | ||
| Fig. 2 Chemical structures of PROTACs warheads used in this work. | ||
Chemical synthesis
The majority of PROTACs and their precursors used in the present study were synthesized following the general synthetic route showed in Scheme 1. In particular, the four different POI ligands, which have a carboxylic acid group, were coupled by amidation reaction with the appropriated piperazine derivatives or the VHL ligand properly functionalized with piperazine-containing linkers in presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) as coupling reagent and N,N-diisopropylethylamine (DIPEA) at room temperature in dimethylformamide (DMF). For the details of synthetic preparation of all PROTACs (PROTAC-1/11), precursors (compounds 1–17), and the required intermediates see ESI.†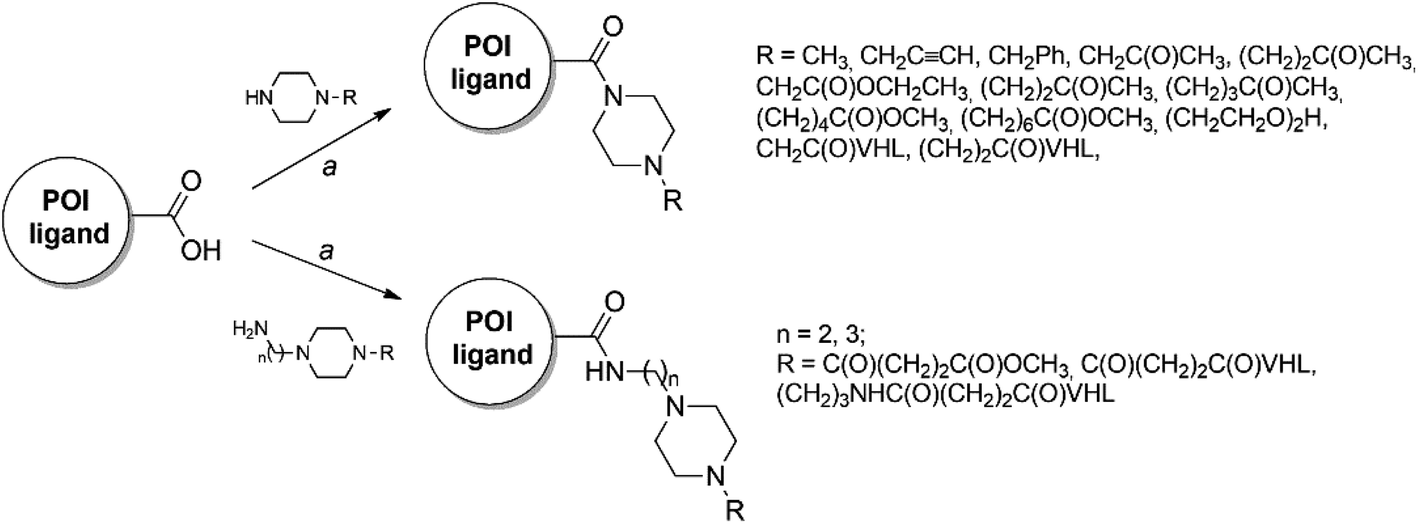 | ||
| Scheme 1 Reagents and conditions: (a) HATU, DIPEA, dry DMF, rt. | ||
Experimental pKa exploration
The pKa values presented herein were all determined using Sirius T3 platform (see Experimental section).First, the pKa values of piperazine and a small set of common substituted piperazines was measured and obtained values displayed a good match with literature ones when available (Table 1). This preliminary analysis represented a starting point to summarize a number of known chemical effects that modulate piperazine basicity. In particular, the methylation of one or both piperazine nitrogens (1-methylpiperazine and 1,4-dimethylpiperazine, respectively; going from secondary to tertiary amino groups) reduces the measured highest pKa value, with ΔpKa of 0.5 or 1.6, respectively, according to our in-house data. Therefore, since piperazines used in PROTACs linkers must be di-alkylated and not monoalkylated by nature, one could consider that the real reference pKa associated to the most basic center for linkers bearing disubstituted piperazines is about 8 (corresponding to 1,4-dimethylpiperazine). Indeed, elongating the aliphatic chain should have limited effect, also supported by the almost identical pKa values for 1-methylpiperazine and 1-ethylpiperazine (Table 1). In terms of percentage of protonated species, the alkylation of the nitrogen atoms in the piperazine ring drops this value from ≈98.5% to ≈78% at pH = 7.5. In addition, as shown in Fig. 1, the piperazine ring in PROTACs linkers is in some cases embedded by one or two amide bonds to the additional linker moiety or to the PROTAC warhead. Recently, we showed as linking a piperazine ring by an amide bond might be also a good strategy to improve metabolic stability, as it prevents N-dealkylation reactions.24 Thus, considering 1-acetyl-4-methylpiperazine as reference, it is noteworthy that the acetylation of one nitrogen atom not only leads to measuring only a single pKa value (associated with the only basic center in the ring) but also reduces the measured pKa value up to 7.06, due to its electron-withdrawing character. As a result, for 1-acetyl-4-methylpiperazine the neutral form results the most abundant species (73.4%) at pH 7.5. Although for the pKa of 1-acetyl-4-methylpiperazine we could not find a confirmation by literature comparison, the measured value well correlates with further data reported in Table 1. In addition, for both pairs 1-methylpiperazine/1,4-dimethylpiperazine and 1-acetylpiperazine/1-acetyl-4-methylpiperazine methylation of the piperazine nitrogen lowered pKa value with the same ΔpKa of 1. This known physicochemical modulation of basic piperazines can be an important task to be considered also in the design and selection of PROTACs linkers.
| Compound | Structure | Experimental pKaa | R2 | Literature pKa | Reference | % protonation state at pH = 7.5b |
|---|---|---|---|---|---|---|
| a Mean ± standard deviation of no. 3 experiments.b P: single protonation state, DP: double protonation state; NI: not-ionized state. | ||||||
| Piperazine |  |
9.67 ± 0.05 | 0.9944 | 9.73 ± 0.02 | Khalili et al.42 | P: 98.48 |
| 5.44 ± 0.03 | 0.9901 | 5.35 ± 0.04 | DP: 0.86 | |||
| NI: 0.67 | ||||||
| 1-Methylpiperazine |  |
9.16 ± 0.00 | 1.0000 | 9.14 ± 0.03 | Khalili et al.42 | P: 97.55 |
| 5.01 ± 0.02 | 0.9995 | 4.63 ± 0.03 | DP: 2.13 | |||
| NI: 0.32 | ||||||
| 1-Ethylpiperazine | 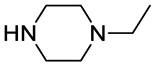 |
9.15 ± 0.02 | 0.9988 | 9.20 ± 0.02 | Khalili et al.42 | P: 97.48 |
| 5.04 ± 0.03 | 0.9983 | 4.76 ± 0.04 | DP: 2.18 | |||
| NI: 0.34 | ||||||
| 1,4-Dimethylpiperazine |  |
8.06 ± 0.01 | 0.9997 | 8.38 ± 0.01 | Khalili et al.42 | P: 78.38 |
| 4.06 ± 0.09 | 0.9846 | 3.81 ± 0.03 | DP: 0.02 | |||
| NI: 21.59 | ||||||
| 1-Acetyl-piperazine |  |
8.05 ± 0.02 | 0.9989 | 7.9 | Morgenthaler et al.32 | P: 78.01 |
| NI: 21.99 | ||||||
| 1-Acetyl-4-methylpiperazine | 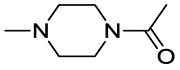 |
7.06 ± 0.00 | 1.0000 | — | — | P: 26.64 |
| NI: 73.36 | ||||||
After having defined and discussed the modulation of pKa in reference substituted piperazines (Table 1), we measured the pKa for a total of 28 compounds, including 17 precursors (Table 2) and 11 PROTACs (Table 3). The reason for testing precursors bearing piperazine-containing linker moieties was to better evaluate the effect of neighbor groups on pKa from the simplest molecules to the final PROTACs. The experimental evaluation of pKa values of the piperazine-containing compounds analyzed in the present work (Tables 2 and 3) led to achieve interesting insights.
|
||||
|---|---|---|---|---|
| Structure | Compound | R1 | Experimental pKa (R2)a | % protonation state at pH = 7.5b |
| a Mean ± standard deviation of no. 3 experiments.b NI: not-ionized state; P: single protonation state, DP: double protonation state. | ||||
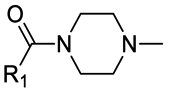 |
1 | A | 2.03 ± 0.18 (0.76) | NI: 74.41 |
| 6.94 ± 0.08 (0.99) | P: 21.59 | |||
| “ | 2 | B | 7.14 ± 0.02 (0.99) | NI: 69.61 |
| P: 30.39 | ||||
| “ | 3 | C | 6.77 ± 0.01 (1.00) | NI: 84.30 |
| P: 15.70 | ||||
| “ | 4 | D | 6.93 ± 0.03 (0.99) | NI: 78.79 |
| P: 21.20 | ||||
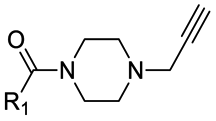 |
5 | B | 4.91 ± 0.14 (0.97) | NI: 99.74 |
| P: 0.26 | ||||
| “ | 6 | C | 4.48 ± 0.06 (1.00) | NI: 99.90 |
| P: 0.01 | ||||
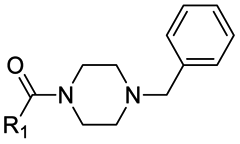 |
7 | C | 5.98 ± 0.01 (1.00) | NI: 97.07 |
| P: 2.93 | ||||
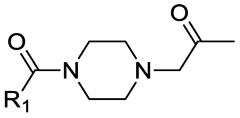 |
8 | A | 2.70 ± 0.19 (0.89) | NI: 99.71 |
| 4.97 ± 0.03 (0.99) | P: 0.29 | |||
| “ | 9 | D | 5.35 ± 0.04 (0.99) | NI: 96.17 |
| P: 3.83 | ||||
 |
10 | D | 4.50 ± 0.06 (0.99) | NI: 99.9 |
| P: 0.01 | ||||
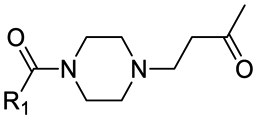 |
11 | B | 6.47 ± 0.02 (0.99) | NI: 91.46 |
| P: 8.54 | ||||
| “ | 12 | D | 5.98 ± 0.05 (0.99) | NI: 97.07 |
| P: 2.93 | ||||
 |
13 | D | 6.75 ± 0.03 (0.99) | NI: 84.90 |
| P: 15.09 | ||||
 |
14 | D | 7.07 ± 0.05 (0.99) | NI: 72.91 |
| P: 27.09 | ||||
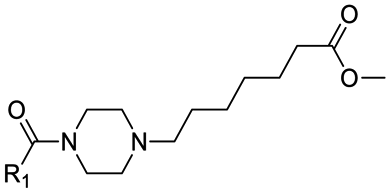 |
15 | D | 7.41 ± 0.08 (0.99) | NI: 55.16 |
| P: 44.84 | ||||
 |
16 | D | 5.61 ± 0.00 (1.00) | NI: 98.73 |
| P: 1.27 | ||||
 |
17 | D | 6.75 ± 0.14 (0.97) | NI: 84.90 |
| P: 15.10 | ||||
|
|||||
|---|---|---|---|---|---|
| Structure | Compound | R1 | R2 | Experimental pKa (R2)a | % protonation state at pH = 7.5b |
| a Mean ± standard deviation of no. 3 experiments.b NI: not-ionized state; P: single protonation state, DP: double protonation state. | |||||
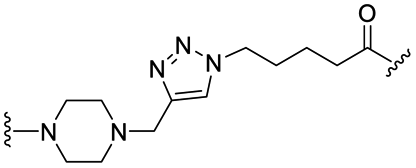 |
PROTAC-1 | B | 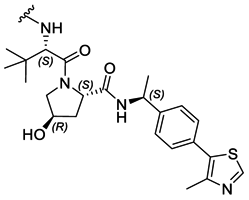 |
5.29 ± 0.03 (0.99) | NI: 99.39 |
| P: 0.62 | |||||
| PROTAC-2 | C | “ | 3.22 ± 0.07 (1.00) | NI: 99.58 | |
| 5.12 ± 0.01 (1.00) | P: 0.41 | ||||
 |
PROTAC-3 | B | “ | 2.32 ± 0.03 (0.98) | NI: 99.89 |
| 4.53 ± 0.08 (0.99) | P: 0.11 | ||||
| PROTAC-4 | D | “ | 5.36 ± 0.19 (0.98) | NI: 99.28 | |
| P: 0.72 | |||||
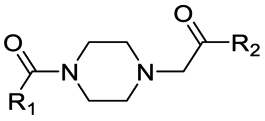
 |
PROTAC-5 | B | “ | 2.51 ± 0.20 (0.80) | NI: 96.17 |
| 6.10 ± 0.06 (0.99) | P: 3.83 | ||||
| PROTAC-6 | D | “ | 2.26 ± 0.02 (0.98) | NI: 98.33 | |
| 5.73 ± 0.04 (0.99) | P: 1.77 | ||||
 |
PROTAC-7 | D | “ | 2.56 ± 0.07 (0.92) | NI: 97.66 |
| 5.88 ± 0.13 (0.97) | P: 2.34 | ||||
 |
PROTAC-8 | D |  |
3.94 ± 0.00 (1.00) | NI: 32.86 |
| 7.81 ± 0.04 (0.99) | P: 67.11 | ||||
| DP: 0.02 | |||||
| PROTAC-9 | B |  |
4.69 ± 0.14 (0.98) | NI: 24.85 | |
| 7.98 ± 0.03 (0.99) | P: 75.04 | ||||
| DP: 0.11 | |||||
 |
PROTAC-10 | D | 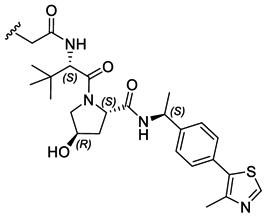 |
2.74 ± 0.12 (0.96) | NI: 94.44 |
| 6.27 ± 0.04 (0.99) | P: 5.56 | ||||
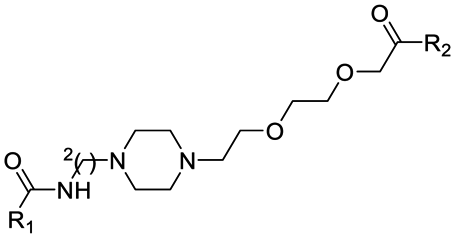 |
PROTAC-11 | D | 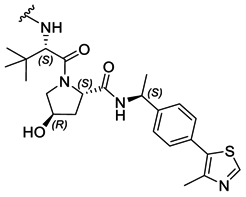 |
3.19 ± 0.11 (0.94) | NI: 51.72 |
| 7.47 ± 0.09 (0.97) | P: 48.27 | ||||
| DP: 0.0023 | |||||
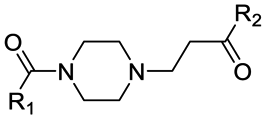
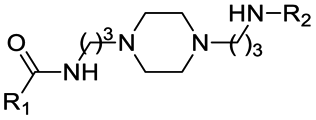
First, to gradually evaluate the increase in structure complexity, compounds containing carbonyl–piperazine–alkyl moiety were first synthesized by linking variably N-substituted piperazine rings to four POI ligands through an amide linkage (compounds 1–7, Table 2). As it could be hypothesized, we confirmed that the POI ligand plays a neglected role on pKa modulation, with measured pKa values (6.77–7.14) very similar to the one of 1-acetyl-4-methylpiperazine (7.06) when the second nitrogen is substituted with a methyl group (compounds 1–4). Among them, compound 3 displayed a slight lower pKa value (6.77), possibly due to the fluorine atom nearby the amide bond. Since we are discussing protonation states at physiological pH of 7.5, even minor changes of pKa induce a significant effect in the % of the protonation state, as shown in Table 2 for compounds 1–4. When the methyl group is replaced by an electron-withdrawing groups as neighbors of the basic nitrogen in the carbonyl–piperazine–alkyl moiety, the pKa significantly decreases, with the alkyne moiety reducing pKa of about 2 units (4.91 and 4.48, compounds 5 and 6, respectively). In the case of a benzyl group instead (compound 7), the pKa results to be slightly higher compared with substitution with the alkyne moiety. The lowering effects observed for compounds 5–7 represented an important point, as when click chemistry is applied on the alkyne moiety to build an aromatic triazole ring next to the piperazine-containing linker, the final triazole ring was expected to hamper the protonation of the piperazine. To confirm the lowering effect on the piperazine's pKa also in PROTAC molecules in the presence of a neighbor triazole ring, PROTAC-1 and PROTAC-2 were tested (Table 3), and a good agreement with compounds 5–7 was found (99.39/99.58% and 97.07–99.90% deprotonation state at pH = 7.5, respectively).
Alternatively to click chemistry, PROTACs synthesis often implies the use of a second amide linkage to connect the linker to the E3 ligase ligand (Fig. 1). Since the linker length is pivotal for the formation of an efficient ternary complex and for modulation of activity, a linear aliphatic chain can be used to tailor the second amide group at variable distance from the piperazine. But how does this distance affect the pKa of piperazine? To answer this question, precursors generated by combining the four POI ligands with piperazines containing a carbonyl group located from 1 to 6 methylene units of distance were synthesized through amidation reaction, and their experimental pKa values were compared with the corresponding PROTAC molecules (compounds 8–15 vs. PROTACs 3/4/5/6 in Tables 2 and 3, respectively). To facilitate the trend analysis, Fig. 3 graphically shows the pKa values for each compound of the series (precursors and whole PROTACs) as well as the average pKa value according to the methylene numbers and the ΔpKa value associated with each methylene insertion. Again, precursors and PROTAC molecules behave similarly in terms of measured pKa (compare compound 9 with and PROTAC-4, compound 11 with and PROTAC-5, compound 12 with and PROTAC-6, Fig. 3a), indicating that in case of low solubility of PROTACs the effect of the linker to the final pKa can be easily estimated by eventually measuring intermediates or smaller analogues. In addition, Fig. 3b indicates that basicity of the piperazine nitrogen reach a maximum when the second carbonyl group is located at least at three methylene units of distance (compound 13). For longer aliphatic chains, the gain of basicity is very low, while the increased hydrophobicity could further reduce the solubility of the compound. These pKa variations are associated with a significant change in the protonation state of the molecules at physiological pH. For example, for compounds 8–15 in Table 2, the calculated percentage of the ionization form at pH 7.5 range from 0% to almost 45%. As an example, by observing the behavior for compounds 10, 14 and 15, which are three small molecules in our dataset in which the linker is attached to the same indomethacin warhead and characterized by the presence of one, four or six methylene units, respectively, the percentage of protonated form at pH = 7.5 goes from 0.01% for compound 10 to 27% and 44.8% for 14 and 15. As expected, when the number of methylene units is kept and the carbonyl group belongs to an inverted amide moiety, the resulting pKa variation is practically identical. In particular, this is observed in compound 16 and PROTAC-7 (5.61 and 5.88, respectively) compared to compound 12 and PROTAC-6 (5.98 and 5.73, respectively).
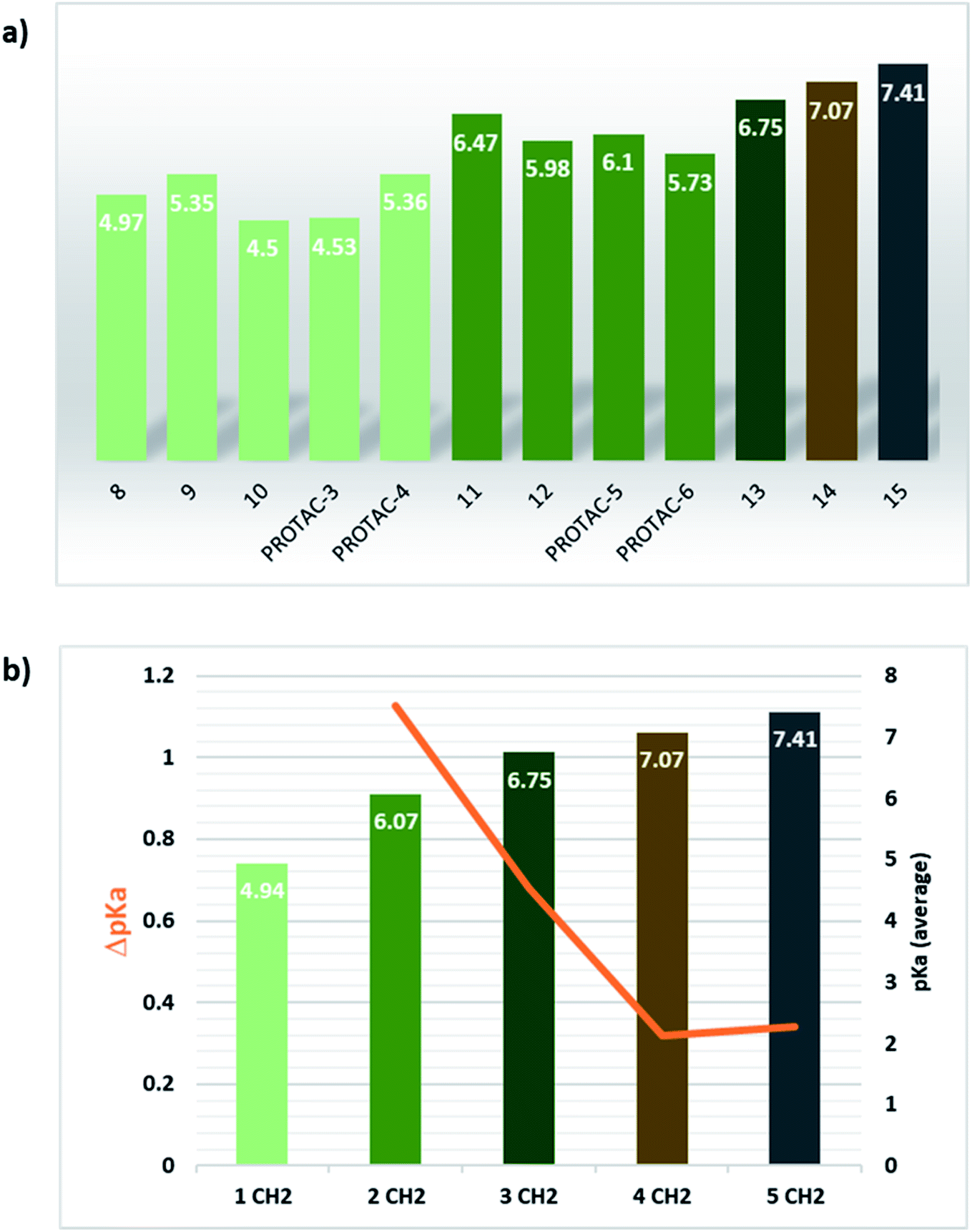 | ||
| Fig. 3 Effect of linear aliphatic chain elongation in the carbonyl–piperazine–(methylene)n–carbonyl moiety series. (a) pKa values for the compounds of this series, with bar colour describing the number of methylene units which separates the piperazine nitrogen from the second carbonyl group (pale green: 1 CH2; green: 2 CH2; dark green: 3 CH2; brown: 4 CH2, grey: 6 CH2). (b) Average pKa bars and line describing the ΔpKa behaviour by increasing the number of methylene units. | ||
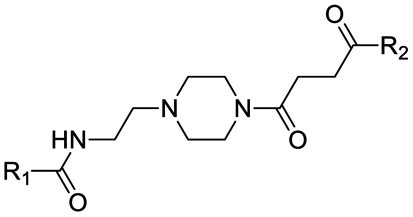
Interestingly, an optimal linker in terms of maximizing the piperazine basicity could be the one used in PROTAC-8 and PROTAC-9 entailing an alkyl–piperazine–alkyl moiety, reaching measured pKa values for the most basic center of 7.81 and 7.98, respectively, and about 70% of protonated species at physiological pH.
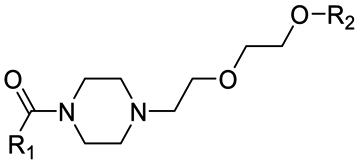
Finally, the use of PEG moieties linked to the piperazine was also analyzed. In particular, compound 17 and PROTAC-10 were characterized by a carbonyl–piperazine–PEG moiety. By comparing the pKa for these compounds (6.75 and 6.27, respectively) with those for compounds 11, 12, PROTAC-5 and PROTAC-6 (in the range 5.62–6.10), it can be highlighted that the oxygen of the ether function, naturally positioned at two methylene units from the basic nitrogen of the piperazine, is less effective than acetyl/amide group in lowering the pKa. To note, similarly to the alkyl–piperazine–alkyl moiety of PROTAC-8 and PROTAC-9, a good option could be to include an alkyl–piperazine–PEG moiety as in PROTAC-11, in which one of the two nitrogen atoms in the piperazine is connected to a PEG linker, while the other is located two methylene units far from an amide group. Indeed, in this case, the latter nitrogen will be the most basic and thus will be protonated; as a result, while in PROTAC-10 the percentage of positively charged species was about 5% at pH = 7.5, in PROTAC-11 it increases up to about 48%.
In silico prediction of pKa values for piperazine derivatives
Experimental pKa determination is routinely performed in drug discovery phase. However, in drug design compounds properties can be only predicted. Empirical, semi-empirical, quantum chemical approaches are currently available,33,43–47 with the first ones being the most used due to their high speed. Since PROTACs design is a critical step in PROTAC technology development, we wanted to evaluate whether the MoKa software could be efficiently used to predict the effect of slight changes in piperazine decorations on pKa. Indeed, MoKa (Molecular Discovery, Ltd., UK)33,46,47 is a quantitative structure/property relationship (QSPR) approach for pKa prediction, which was developed before PROTACs era, and therefore mainly trained with classical small molecules. Observing the plot of predicted vs. experimental pKa values (Fig. 4), Moka similarly estimated pKa values for the most basic centre of piperazine rings both for small molecules or precursors (black points) as well as for PROTACs (red point). The coefficient of determination (R2) for the simple linear regression was 0.88, mostly due to PROTAC-4 that is out of the model. Indeed, excluding this point, R2 becomes 0.93, in line with the general behavior of the most performing pKa prediction software when validated on small molecules. Therefore, despite prior evidences of the unreliable LogP prediction for PROTACs,26 the pKa prediction of piperazine-containing PROTACs with MoKa could find application in PROTAC design.
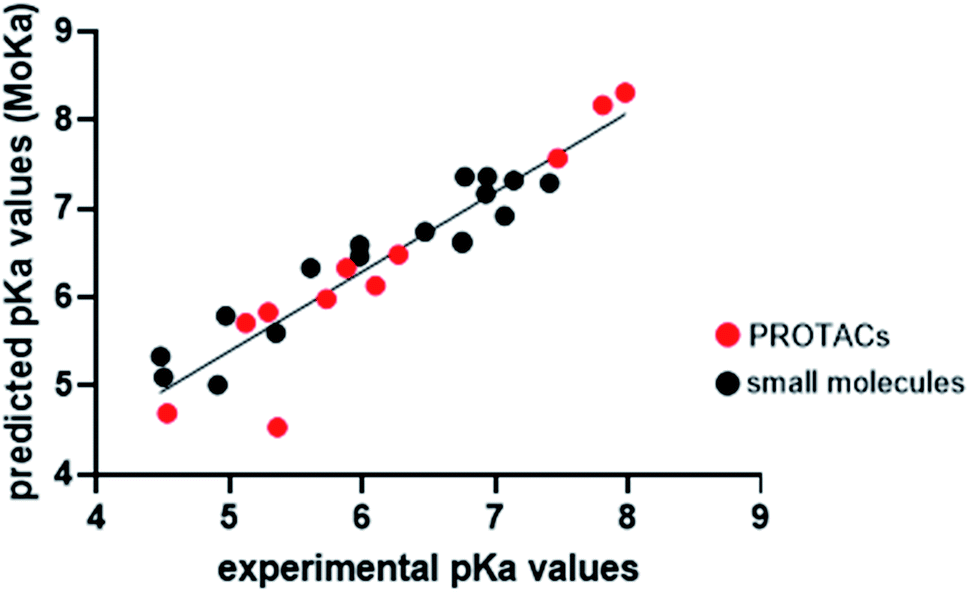 | ||
| Fig. 4 Plot of predicted vs. experimental pKa values. Black points represent small molecules or PROTAC intermediates, while red points represent PROTACs. The simple linear regression was calculated with GraphPad Prism 8.4.3, according to the following equation: y = 0.8968x + 0.9107. | ||
Conclusions
The understanding of PROTACs molecular properties is pivotal for their optimization. Despite pKa is known to be a key physicochemical property with a strong influence on several ADME related properties such as solubility, permeability, and metabolic stability the recent use of piperazine-containing linker has been not explored for its effect on the protonation state of PROTAC molecules. Our study on a dataset of small-molecules, PROTAC intermediates and PROTACs led us to make some considerations to rapidly guide the linker optimization to modulate piperazine basicity. Noteworthy, in this study we intentionally focus on PROTAC molecules and their precursors but many observations related to the effect of piperazine-containing linkers can be beneficial also for the design and optimization of other kind of heterobifunctional compounds e.g., lysosome-targeting chimera LYTAC, autophagy-targeting chimera AUTAC, RIBOTAC,13 molecular glues,14,48 and antibody–drug conjugates,9–12 among others.First, to answer our original question we proved that the basicity of piperazine in PROTAC linker can significantly vary, with pKa values ranging from 4.5 to 8, depending on the linker design. As a result, changes in protonation state highly affected the percentage of protonated species for our dataset, ranging from 0.1 to 75%. Therefore, a large attention should be devoted to modulating the neighbour chemical groups to optimize pKa to the desired value.
The link of piperazine through an amide bond lowers the pKa value of 1 unit compared to 1,4-dimethylpiperazine, while using a click chemistry approach the formed triazole ring decreases pKa of almost 3 pKa units. Therefore, the latter strategy of linker synthesis should be avoided when piperazine is added to increase solubility by protonation.
The elongation of the aliphatic chain bound to the piperazine in compounds 8–15 and PROTACs 3/4/5/6 has highlighted that the detrimental effect on pKa of a carbonyl moiety, usually needed to join the linker to one of the ligands, can be drastically reduced when four or more methylene units separate it from the basic nitrogen of piperazine. Another strategy exploited to increase PROTAC solubility is the use of PEG linkers. We proved that the oxygen of the ether function is less effective than acetyl/amide group in lowering the pKa and a combination of piperazine + PEG linkers could be a good strategy to have a more polar and partially protonated compound. Among all the representative piperazine-containing linker moieties studied in this work, the alkyl–piperazine–alkyl moiety in PROTAC-8 and PROTAC-9 stood out as the best one in maximizing the piperazine basicity, permitting to reach high protonation of compounds at physiological pH. Finally, contrarily to what reported for LogP, whose in silico prediction for PROTACs were considered not reliable, our test with the MoKa software showed that pKa prediction for piperazines embedded in PROTACs structure is efficient, and can be used for drug design purposes. This study centred on the physicochemical properties of PROTACs bearing piperazine-containing linkers could represent the first step for future studies on interpreting PROTACs PK at a molecular level.
Experimental
Chemistry
The experimental procedures and characterization of the precursors and PROTACs tested in this work are reported in the ESI.†General methodology for pKa determination
Calculations of predicted pKa values were done using Marvin (Tables S1 and S2† in ESI†). [Calculator plugins were used for structure property prediction and calculation, Marvin v20.11, 2020, ChemAxon (https://www.chemaxon.com)]. Experimental measurements of pKa values were performed using the SiriusT3 platform (Pion Inc. Ltd., Forest Row, East Sussex, UK), applying a potentiometric acid–base titration.49 All experiments were designed using the Sirius T3 Control software (Sirius T3 v1.1.3.0, Pion Inc. Ltd., Forest Row, East Sussex, UK), setting the volume of the stock solution, molecular weight, number of expected pKa and their predicted values. Considering the basic nature of the compounds, the titration mode was carried out starting from pH 2 to 12. During the dissolution stage, each compound was solubilized in Ionic Strength Adjusted water (ISA Water, KCl 0.15 M). In order to prevent kinetic solubility issue, the molecules were dissolved in 100% DMSO, reaching a final concentration of 30 mM and using 70 μL of organic solutions in each titration. The experiments were carried out in triplicate using an organic cosolvent, methanol, in different ratios (50%, 40%, and 30% of methanol in ISA water). Since the presence of a different solvent modifies the dielectric constant of water, the pKa at 0% of cosolvent is obtained applying the Yasuda–Shedlovsky (YS) method of extrapolation to zero.50 The R2 value was used to evaluate the accuracy of the assay with the cosolvent, and results were considered with acceptable accuracy if R2 value ≥ 0.9. Three compounds (1, 8, and PROTAC-5) showed one experimental pKa close to 2, yielding a R2 < 0.9. The low accuracy of these experiments is ascribed to their closeness of these acidic pKa values to the lower detection limit of the instrument.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The University of Perugia and MIUR are gratefully acknowledged for financial support to the project AMIS, through the program “Dipartimenti di Eccellenza-2018–2022”. J. D. was supported by MIUR-Ministero dell’Istruzione, dell’Università e della Ricerca (Italian Ministry of Education, University and Research), Project “ZODIAC”. We thank Dr Stefano Di Bona (Molecular Horizon srl) for LC-MS analysis.References
- J. L. Gustafson, T. K. Neklesa, C. S. Cox, A. G. Roth, D. L. Buckley, H. S. Tae, T. B. Sundberg, D. B. Stagg, J. Hines, D. P. McDonnell, J. D. Norris and C. M. Crews, Angew. Chem., Int. Ed. Engl., 2015, 54, 9659–9662 CrossRef CAS PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed. Engl., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- M. J. Bond and C. M. Crews, RSC Chem. Biol., 2021, 2, 725–742 RSC.
- S. He, G. Dong, J. Cheng, Y. Wu and C. Sheng, Med. Res. Rev., 2022, 42, 1280–1342 CrossRef PubMed.
- R. I. Troup, C. Fallan and M. G. J. Baud, Exploration of Targeted Anti-tumor Therapy, 2020, 1, 273–312 Search PubMed.
- K. Li and C. M. Crews, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC.
- C. Borsari, D. J. Trader, A. Tait and M. P. Costi, J. Med. Chem., 2020, 63, 1908–1928 CrossRef CAS PubMed.
- P. S. Dragovich, Chem. Soc. Rev., 2022, 51, 3886–3897 RSC.
- Z. Su, D. Xiao, F. Xie, L. Liu, Y. Wang, S. Fan, X. Zhou and S. Li, Acta Pharm. Sin. B, 2021, 11, 3889–3907 CrossRef CAS PubMed.
- R. Sheyi, B. G. de la Torre and F. Albericio, Pharmaceutics, 2022, 14, 396 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- L. Hua, Q. Zhang, X. Zhu, R. Wang, Q. You and L. Wang, J. Med. Chem., 2022, 65, 8091–8112 CrossRef CAS PubMed.
- G. Dong, Y. Ding, S. He and C. Sheng, J. Med. Chem., 2021, 64, 10606–10620 CrossRef CAS PubMed.
- I. Churcher, J. Med. Chem., 2018, 61, 444–452 CrossRef CAS PubMed.
- G. Lottrup, A. Jorgensen, J. E. Nielsen, N. Jorgensen, M. Duno, A. M. Vinggaard, N. E. Skakkebaek and E. Rajpert-De Meyts, J. Clin. Endocrinol. Metab., 2013, 98, 2223–2229 CrossRef CAS PubMed.
- N. Shibata, K. Nagai, Y. Morita, O. Ujikawa, N. Ohoka, T. Hattori, R. Koyama, O. Sano, Y. Imaeda, H. Nara, N. Cho and M. Naito, J. Med. Chem., 2018, 61, 543–575 CrossRef CAS PubMed.
- M. L. Drummond and C. I. Williams, J. Chem. Inf. Model., 2019, 59, 1634–1644 CrossRef CAS PubMed.
- M. L. Drummond, A. Henry, H. Li and C. I. Williams, J. Chem. Inf. Model., 2020, 60, 5234–5254 CrossRef CAS PubMed.
- C. E. Powell, Y. Gao, L. Tan, K. A. Donovan, R. P. Nowak, A. Loehr, M. Bahcall, E. S. Fischer, P. A. Janne, R. E. George and N. S. Gray, J. Med. Chem., 2018, 61, 4249–4255 CrossRef CAS PubMed.
- J. Frost, C. Galdeano, P. Soares, M. S. Gadd, K. M. Grzes, L. Ellis, O. Epemolu, S. Shimamura, M. Bantscheff, P. Grandi, K. D. Read, D. A. Cantrell, S. Rocha and A. Ciulli, Nat. Commun., 2016, 7, 13312 CrossRef CAS PubMed.
- H. J. Maple, N. Clayden, A. Baron, C. Stacey and R. Felix, Medchemcomm, 2019, 10, 1755–1764 RSC.
- G. Ermondi, D. Garcia-Jimenez and G. Caron, Molecules, 2021, 26, 672 CrossRef CAS PubMed.
- L. Goracci, J. Desantis, A. Valeri, B. Castellani, M. Eleuteri and G. Cruciani, J. Med. Chem., 2020, 63, 11615–11638 CrossRef CAS PubMed.
- B. Zhou, J. Hu, F. Xu, Z. Chen, L. Bai, E. Fernandez-Salas, M. Lin, L. Liu, C. Y. Yang, Y. Zhao, D. McEachern, S. Przybranowski, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2018, 61, 462–481 CrossRef CAS PubMed.
- C. Steinebach, I. Sosic, S. Lindner, A. Bricelj, F. Kohl, Y. L. D. Ng, M. Monschke, K. G. Wagner, J. Kronke and M. Gutschow, Medchemcomm, 2019, 10, 1037–1041 RSC.
- C. Steinebach, H. Kehm, S. Lindner, L. P. Vu, S. Kopff, A. Lopez Marmol, C. Weiler, K. G. Wagner, M. Reichenzeller, J. Kronke and M. Gutschow, Chem. Commun., 2019, 55, 1821–1824 RSC.
- C. Steinebach, Y. L. D. Ng, I. Sosic, C. S. Lee, S. Chen, S. Lindner, L. P. Vu, A. Bricelj, R. Haschemi, M. Monschke, E. Steinwarz, K. G. Wagner, G. Bendas, J. Luo, M. Gutschow and J. Kronke, Chem. Sci., 2020, 11, 3474–3486 RSC.
- A. Pike, B. Williamson, S. Harlfinger, S. Martin and D. F. McGinnity, Drug Discovery Today, 2020, 25, 1793–1800 CrossRef CAS PubMed.
- G. Ermondi, M. Vallaro and G. Caron, Drug Discovery Today, 2020, 25, 1585–1591 CrossRef CAS PubMed.
- G. Weng, C. Shen, D. Cao, J. Gao, X. Dong, Q. He, B. Yang, D. Li, J. Wu and T. Hou, Nucleic Acids Res., 2021, 49, D1381–D1387 CrossRef CAS PubMed.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Roder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Muller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS PubMed.
- G. Cruciani, F. Milletti, L. Storchi, G. Sforna and L. Goracci, Chem. Biodiversity, 2009, 6, 1812–1821 CrossRef CAS PubMed.
- C. D. Navo and G. Jimenez-Oses, ACS Med. Chem. Lett., 2021, 12, 1624–1628 CrossRef CAS PubMed.
- X. Han, C. Wang, C. Qin, W. Xiang, E. Fernandez-Salas, C. Y. Yang, M. Wang, L. Zhao, T. Xu, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 941–964 CrossRef CAS PubMed.
- C. Dardonville, B. A. Caine, M. Navarro de la Fuente, G. M. Herranz, B. Corrales Mariblanca and P. L. A. Popelie, New J. Chem., 2017, 41, 11016–11028 RSC.
- K. B. Wiberg, J. Org. Chem., 2002, 67, 1613–1617 CrossRef CAS PubMed.
- F. Pierre, P. C. Chua, S. E. O'Brien, A. Siddiqui-Jain, P. Bourbon, M. Haddach, J. Michaux, J. Nagasawa, M. K. Schwaebe, E. Stefan, A. Vialettes, J. P. Whitten, T. K. Chen, L. Darjania, R. Stansfield, K. Anderes, J. Bliesath, D. Drygin, C. Ho, M. Omori, C. Proffitt, N. Streiner, K. Trent, W. G. Rice and D. M. Ryckman, J. Med. Chem., 2011, 54, 635–654 CrossRef CAS PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
- K. A. Menear, C. Adcock, R. Boulter, X. L. Cockcroft, L. Copsey, A. Cranston, K. J. Dillon, J. Drzewiecki, S. Garman, S. Gomez, H. Javaid, F. Kerrigan, C. Knights, A. Lau, V. M. Loh, Jr., I. T. Matthews, S. Moore, M. J. O'Connor, G. C. Smith and N. M. Martin, J. Med. Chem., 2008, 51, 6581–6591 CrossRef CAS PubMed.
- C. Cantrill, P. Chaturvedi, C. Rynn, J. Petrig Schaffland, I. Walter and M. B. Wittwer, Drug Discovery Today, 2020, 25, 969–982 CrossRef CAS PubMed.
- F. Khalili, A. Henni and A. L. L. East, J. Chem. Eng. Data, 2009, 24, 2914–2917 CrossRef.
- P. Hunt, L. Hosseini-Gerami, T. Chrien, J. Plante, D. J. Ponting and M. Segall, J. Chem. Inf. Model., 2020, 60, 2989–2997 CrossRef CAS PubMed.
- R. Fujiki, T. Matsui, Y. Shigeta, H. Nakano and N. Yoshida, J, 2021, 4, 849–864 CAS.
- E. Selwa, I. M. Kenney, O. Beckstein and B. I. Iorga, J. Comput.-Aided Mol. Des., 2018, 32, 1203–1216 CrossRef CAS PubMed.
- F. Milletti, L. Storchi, G. Sforna and G. Cruciani, J. Chem. Inf. Model., 2007, 47, 2172–2181 CrossRef CAS PubMed.
- F. Milletti, L. Storchi, L. Goracci, S. Bendels, B. Wagner, M. Kansy and G. Cruciani, Eur. J. Med. Chem., 2010, 45, 4270–4279 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- C. Dardonville, Drug Discovery Today: Technol., 2018, 27, 49–58 CrossRef PubMed.
- G. Volgyi, R. Ruiz, K. Box, J. Comer, E. Bosch and K. Takacs-Novak, Anal. Chim. Acta, 2007, 583, 418–428 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemical structures of compounds 1–17 and PROTAC-1/11 are reported in Tables S1 and S2, respectively; synthesis and characterization details, along with 1H and 13C NMR spectra and HRMS spectra of compounds 1–17 and PROTAC-1/11 are reported. See https://doi.org/10.1039/d2ra03761k |
| ‡ Equal contribution. |
| § Present affiliation: Department of Medicine and Surgery, University of Perugia, Polo Unico Sant'Andrea delle Fratte, Perugia, Italy. |
| This journal is © The Royal Society of Chemistry 2022 |