
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiezoelectric built-in electric field advancing TiO2 for highly efficient photocatalytic air purification†
Mengmeng Li‡
a,
Qin Cheng‡a,
Cheng Shena,
Bin Hongb,
Yong Jiangc,
Yuxue Weia,
Mengdie Caia,
Jingshuai Chena and
Song Sun *a
*a
aSchool of Chemistry and Chemical Engineering, Anhui University, Hefei, Anhui 230601, China. E-mail: suns@ustc.edu.cn
bHefei Innovation Research Institute, Beihang University, Hefei, Anhui 230013, China
cShanghai Synchrotron Radiation Facility, Zhangjiang National Laboratory, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201204, China
First published on 10th August 2022
Abstract
Photocatalytic air purification is a promising technology; however, it suffers from a limited rate of photocatalytic mineralization (easily inactivated surfactant sites of hydroxyls) and poor kinetics of degradation. Herein, we report a ferroelectric strategy, employing a polyvinylidene fluoride (PVDF) layer embedded with TiO2, where the polarization field of stretched PVDF dramatically enhances and stabilizes active adsorption sites for the promotion of charge separation. The F (−) and H (+) atomic layers with distinct local structures in stretched PVDF increase the electron cloud density around Ti which simultaneously promotes the dissociation of water to form hydroxyl groups which are easier to activate for adsorption of formaldehyde molecules. Besides, the ferroelectric field of stretched PVDF effectively separates the photogenerated charge carriers and facilitates the carriers' transportation of TiO2/PVDF. The optimal stretched TiO2/PVDF exhibits excellent photocatalytic mineralization for formaldehyde with considerable stability. This work may evolve the polarization field as a new method to enhance adsorption and activation of hydroxyls and disclose the mechanism by which hydroxyl radicals mineralize gaseous formaldehyde for photocatalytic air purification.
Introduction
Clear air is vital for human beings but due to current industrial development, air pollution is becoming a serious issue that affects our health and other development.1 Photocatalytic oxidation is an efficient and mild treatment technology and is becoming a hot research topic for air purification due to its room temperature-dependent reaction and direct use of sunlight.2–4 In the photocatalytic degradation of volatile organic compounds (VOCs), VOC molecules are adsorbed on the hydroxyl groups of the catalyst surface, followed by decomposition arising from attack of active radicals that are formed by the photogenerated carriers.5–8 Therefore, effective construction of active and stable surface hydroxyls, as well as promotion of surface reaction kinetics, is extremely important.Recently, ferroelectric polarization for photocatalytic hydrogen evolution has been reported through an all-in-one hybrid photocatalyst.9–11 Polarization field promotion of reactive oxygen species generation can also boost degradation of phenol in the liquid phase. And our group has reported the spontaneous ferroelectric polarization in a single domain for efficient charge separation over a representative semiconductor CdS, for water splitting in absence of any cocatalyst or external voltage.12 Although polarization field has been attempted in photocatalytic water splitting for performance enhancement, the employment of electric- and piezoelectric-induced polarization fields to address the problems of carriers' recombination and improve the properties of active sites for adsorption and radicals attack in photocatalytic degradation of VOCs is still challenging, because of the characteristics of the gas–solid phase catalytic reaction. And the number of isolated hydroxyl groups is not sufficient or stable during the reaction, and is unsatisfactory with the demand of adsorption and degradation of gaseous organic molecules.13–15 On the other hand, polyvinylidene fluoride (PVDF) as a ferroelectricity material can produce a polarization field via the electro-negative difference between F (−) and H (+) atomic layers.16–19 Moreover, the fabrication of flexible polarized PVDF is favorable for embedding photocatalysts particles. So, it is encouraged to develop semiconductor/PVDF hybrid photocatalysts for the improved photocatalytic mineralization VOCs rate.20
Herein, we describe a ferroelectric strategy and fabricate a PVDF layer embedded with TiO2 that can dramatically enhance and stabilize the active adsorption sites and promote charge separation. The reason behind this significant improvement is due to the polarization field of stretched PVDF that can efficiently photocatalytic mineralize formaldehyde, and formaldehyde, one of the typical indoor air pollutants.
Experimental
Polyvinylidene fluoride (PVDF) film was prepared by the solvent evaporation method. First, 1.0 g PVDF powder was dissolved in N,N-dimethylformamide (DMF) and stirred at 60 °C to prepare a solution with a constant mass fraction. Then, the solution was poured into a mold and moved into an oven. The solvent was evaporated at 85 °C to form a PVDF film. To increase the content of β-type PVDF, the film was stretched at a constant speed and temperature using a home-made tensile instrument to obtain the structure with all-trans conformation PVDF film.The synthesis of the hybrid photocatalyst was based on TiO2 (P25). PVDF powder was dissolved in DMF, and stirred at room temperature to prepare a solution with a constant mass fraction, TiO2 was added to the PVDF and DMF mixing solution and continued to stir well, other procedures were described above.
The as-prepared samples were characterized by various analytical techniques. X-ray diffraction (XRD) patterns were recorded on a SmartLab 9 kW X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). The scanning rate of 0.5° S−1 and the scanning step width of 0.02° were applied in the 2θ range of 5–80°. X-ray photoelectron spectroscopy (XPS) was carried out on an X-ray photoelectron spectrometer (Thermo-Fisher Escalab250Xi). Scanning electron microscopy (SEM) was performed on a Zeiss Supra 40 scanning electron microscope at an acceleration voltage of 5 kV. Stress–strain curves using a INSTRON 5967 small tensile machine. Contact angles were measured with in-built DSA30S software with the Drop Shape Analyser DSA 100E instrument. The ferroelectric properties were measured by a ferroelectric tester (aixACCT Systems GmbH, Aachen, Germany). Time-resolved photoluminescence (PL) spectra were measured by Horiba Fluoro max plus (Horiba, Japan). Steady-state PL spectra measured by FLUORMAX-4P (HORIBA JOBIN YUON, France). The electronic structures and partial densities of states (PDOS) of TiO2 with and without polarization field were obtained by density functional theory (DFT).
Results and discussion
In a typical experiment, TiO2/PVDF film was prepared by solvent evaporation method (Fig. S1†).21 PVDF is transformed from α phase (non-piezo phase) to β phase (piezo phase) by hot-stretching of PVDF and TiO2/PVDF film. In this process, the dipole moment increases, and the polarization field is generated (Fig. 1a). X-ray diffraction (XRD) patterns of TiO2/PVDF films before and after hot-stretching are shown in Fig. 1b. 25.2° is the diffraction peak of TiO2 (P25), and the diffraction peak of TiO2 becomes weaker after hot-stretching. The diffraction peaks at approximately 18.2° and 39.0° can be indexed to (020) and (002) of α-phase PVDF,22-respectively. The diffraction peaks at approximately 20.7° and 35.0° can be indexed to (200) and (001) of β-phase PVDF. The sharp diffraction β phase denotes the formation of a large polarization field of stretched-TiO2/PVDF (S-TiO2/PVDF) compared to unstretched-TiO2/PVDF (US-TiO2/PVDF).23,24 It is found that the β phase of stretched-PVDF (S-PVDF) is also stronger than that of PVDF as expected (Fig. S2†). However, the diffraction peak at 20.6° (β phase) of S-PVDF varies less than that of S-TiO2/PVDF. It indicates that hydroxyl of TiO2 promotes the polarization of TiO2/PVDF25-and the addition of TiO2 promotes polarization of PVDF. | ||
| Fig. 1 (a) Schematic illustration of the film stretching process for stablishing polarization filed in TiO2/PVDF. (b) XRD patterns and (c) FTIR of US-TiO2/PVDF and S-TiO2/PVDF. (d) Stress–strain curves of PVDF and TiO2/PVDF films. The inset shows the image of the film stretching process. | ||
The Fourier transform infrared spectroscopy (FTIR) of all these samples was detected as shown in Fig. 1c. The peaks at 1433, 1335, and 845 cm−1 can be assigned to the stretching vibration peak of the C–F bond from PVDF, represent the β crystalline phase in PVDF.17,23,26,27 The peaks at 1335 cm−1 is shifted to 1334 cm−1, and 844 cm−1 is shifted to 849 cm−1 after the hot-stretching, confirming the presence of a polarization field.28 The stress–strain curves further reveal the occurrence of phase transformation over TiO2/PVDF even at smaller tensile stress13 -compared to that of PVDF (Fig. 1d). This suggests that polarization field can be easily formed in TiO2/PVDF.
Scanning electron microscopy (SEM) revealed the morphology of TiO2/PVDF. After hot-stretching, a porous structure formed by spherulites disappears and the oriented fibril structure is formed (Fig. S3†),29,30 and TiO2 nanoparticles are uniformly distributed on the surface of the S-TiO2/PDVF and US-TiO2/PDVF. SEM elemental mapping further confirmed the uniform distribution of TiO2 over the surface of the S-TiO2/PDVF and US-TiO2/PDVF (Fig. S4†). X-ray photoelectron spectroscopy (XPS) (Fig. S5†) shows that S-TiO2/PDVF and US-TiO2/PDVF consist of C 1s, F 1s, Ti 2p, and O 1s. The binding energy of F 1s decreased from 688.2 to 687.7 eV,31,32 -and the binding energy of O 1s shifted toward the high energy direction after hot-stretching of TiO2/PDVF films. It is evident that S-TiO2/PDVF forms a polarization field, and the F (−) atomic layers are efficiently arranged and in contact with the O atoms on TiO2 that consequently enhanced the electron cloud density around Ti, and then the binding energy of Ti 2p increased from 458.4 to 459.2 eV.33
The performance of photocatalytic mineralization of formaldehyde was achieved on the homemade setup (Fig. S6†). Initially, a dark experiment was carried out for 20 min to attain equilibrium of adsorption/desorption on the surface of the photocatalyst followed by UV light irradiation. S-TiO2/PDVF exhibited an adsorption ability of 6%, which was slightly greater than US-TiO2/PDVF (4%) in 20 min. When the adsorption/desorption of formaldehyde equilibrated at around 20 min, then these samples were irradiated by UV light. It is observed that S-TiO2/PDVF has a significantly higher degradation conversion of nearly 100% in 70 min as compare to US-TiO2/PDVF (Fig. 2a). Notably, the mineralization conversion of formaldehyde on S-TiO2/PDVF was also close to 100%, which reflects the complete degradation ability of our photocatalysts for the decomposition of VOC molecules into harmless inorganic species, such as CO2, H2O. These results indicate that the photocatalytic activity over the surface of a polarized electric field (S-TiO2/PDVF) is better than that of non-polarized electric field (US-TiO2/PDVF). The photocatalytic activity of the samples with different degrees of polarization is also considered (Fig. S7†). The efficiency of photocatalytic mineralization of formaldehyde continued to improve along with the increase in the degree of polarization (Fig. S8†). These results demonstrate that the polarization electric field has a positive effect on the photocatalytic mineralization of formaldehyde.
 | ||
| Fig. 2 (a) Photocatalytic performance of samples. Reaction condition: initial concentration of formaldehyde: 100 ppmV; light source: 300 W Xenon lamp; R.H.:70%; temperature: 298 K. (b) Stability test of S-TiO2/PDVF for repeated photocatalytic degradation of with an initial concentration of 100 ppmV. Solid and open symbols represent the concentration of formaldehyde and the concentration of CO2 generated by the degradation of formaldehyde, respectively. (c and d) In situ DRIFT spectra for the absorption of formaldehyde and with UV irradiation after achieving absorption equilibrium on S-TiO2/PDVF. | ||
The S-TiO2/PDVF also exhibits considerable stability after five consecutive reaction cycles during the degradation of formaldehyde (Fig. 2b). This suggests that PVDF serves as a photocatalyst supporter, exhibits good mechanical properties, and great durability, which can enhance the adsorption and activation of hydroxyl groups on the surface of S-TiO2/PDVF and can be remained in the long-term test.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analyses the process of adsorption and photocatalytic degradation of formaldehyde. In the case of S-TiO2/PDVF (Fig. 2c), the band at 3700 cm−1 can be attributed to the isolated hydroxyl group stretching mode ν(O–H), and the peak at 1670 cm−1 corresponds to the typical molecular water bending mode. However, isolated hydroxyl adsorption was not observed over the surface of US-TiO2/PDVF and the adsorption of water molecule at 1670 cm−1 was weaker than S-TiO2/PDVF (Fig. S9†),34 as can be seen from contact angle measurement (Fig. S10†). This demonstrates that S-TiO2/PDVF has better hydrophilic properties and reveals that the polarization field improves the adsorption of hydroxyl at the active site.
With the introduction of formaldehyde (Fig. 2c), new high frequencies bands at 2942, 2906, 2840, and 2775 cm−1 are attributed to C–H stretching vibration ν(C–H) in adsorbed formaldehyde, and low frequencies bands at 1142, 1108, and 1050 cm−1 can be associated with polyoxymethylene, the deformation vibration of ρ(C–H) and molecularly adsorbed formaldehyde were observed. The intensity of bands increased and reached a stable level after 20 min. Meanwhile, the intensities of the bands at 3700 cm−1 are slightly decreased which shows isolated hydroxyl groups. This result demonstrates that formaldehyde is adsorbed on the isolated hydroxyl groups. However, in the case of US-TiO2/PDVF, the bands of formaldehyde were slightly shifted and the intensity is significantly lower than that of S-TiO2/PDVF. This indicates that photocatalytic degradation is not only related to surface hydroxyl groups but also to surface adsorption sites. As we know, hydroxyl groups, particularly isolated hydroxyl groups, serve as adsorption sites for VOC molecules and are the first determining factor in the photocatalytic degradation of VOCs.35
S-TiO2/PDVF was irradiated with UV light after attaining a stable level of formaldehyde adsorption, as can be seen from the characteristic bonds of formaldehyde at 2942, 2906, 2840, and 2759 cm−1 which are decreased and almost disappeared after 3 min of UV irradiation. In addition, the bands at 1142, 1108, and 1051 cm−1 also decreased. Meanwhile, the new bands at 2852, 2746, 1726, 1550, and 1352 cm−1 were observed.
Since PVDF is composed of C, H, and F elements, a large number of C–H vibration peaks (around 3000 cm−1) on PVDF cover up the C–H peaks of formaldehyde adsorbed on TiO2. For a clear observation of the adsorption of formaldehyde and intermediate changes on S-TiO2/PDVF and on US-TiO2/PDVF, FTIR difference spectrum (Fig. S11, and S12†) was used to visually display the conversion of formaldehyde on different samples. Consistent with the above, new bands (1726 cm−1 can be assigned to ν(CO) in HCOO–, 1560 cm−1 can be assigned to ν(OCO)) are observed on S-TiO2/PDVF which are stronger than US-TiO2/PDVF after UV irradiation, but the band at 1279 cm−1 on US-TiO2/PDVF can be assigned to dioxymethylene is stronger than S-TiO2/PDVF. This is a predominate intermediate of photocatalytic degradation on the surface of US-TiO2/PDVF, which means that intermediate species and adsorption forms of S-TiO2/PDVF and US-TiO2/PDVF significantly are different (Fig. S13†). These results indicate that the adsorbed formaldehyde on the TiO2 surface rapidly converts to formate species under UV irradiation, the abundant hydroxyl groups on the surface of S-TiO2/PDVF mean rich active sites for formaldehyde adsorption, favorable for the mineralization during UV light irradiation.
The attribution of adsorption peaks on the film is shown in Tables S1 and S2† (Tables S1, and S2†).36–38 Similarly, S-TiO2/PDVF has also been applied to degrade toluene, which shows an excellent rate of mineralizing (Fig. S14†), further demonstrating the universality of the ferroelectric strategy in air purification.
In order to explore the origin of this significantly promoted photocatalytic activity, caused by ferroelectric polarization, the separation and transportation behaviors of photogenerated charges are examined. The electric hysteresis loop of S-TiO2/PDVF and US-TiO2/PDVF are obtained with the infliction of an alternating voltage (Fig. 3a). The ferroelectric behavior of these TiO2/PDVF films was studied by electric hysteresis loop tests. A broad hysteresis loop was observed for S-TiO2/PDVF, which exhibited a strong spontaneous polarization electric field (Fig. 3b). This hot-stretching provides a strong polarization and forms a positive end (C+) and a negative end (C−), which generates a depolarizing field from C+ toward C−.39 It also induces the upward and downward band bending, respectively, accompanied by the depolarizing field and provides a strong driving force to separate the photogenerated carriers along the reversed polarized locations, rather than that over US-TiO2/PDVF which doesn't show ferroelectric properties (Fig. S15†). And hysteresis loops with different degrees of polarization is also considered (Fig. S16†). Thus, the lifetime of photogenerated carriers is determined from time-resolved photoluminescence (PL) spectra which increased after polarization. This increment demonstrates the substantial contribution of stretching polarization which inhibits the recombination of charge (Fig. 3c). To further investigate the relationship between ferroelectric properties and charge separation, the steady-state PL (Fig. 3d) was used to investigate the charge recombination rate in TiO2/PDVF samples. The fluorescence peak at 396 nm is the emission peak of TiO2, and the peak at 468 nm is the emission peak of PVDF. S-TiO2/PDVF shows the lowest emission intensity at the emission peak position of approximately 396 nm, indicating that hot-stretching treatment can reduce the recombination rate of photogenerated charge carriers. Therefore, the polarization field constructed by stretching TiO2/PDVF not only dramatically enhances the active adsorption sites of hydroxyls but also promotes charge separation.
 | ||
| Fig. 3 (a) Schematic diagram of electric hysteresis loop test of TiO2/PDVF film. (b) Electric hysteresis loop (under 86 kV cm−1) for S-TiO2/PDVF at room temperature. (c) Decay profile of photoluminescence from US-TiO2/PDVF and S-TiO2/PDVF. (d) Steady-state PL spectra of PVDF, US-TiO2/PDVF, and S-TiO2/PDVF. | ||
To further understand the role of polarization field, density functional theory (DFT) calculations are carried out on TiO2 with and without the polarization field. The electronegativity of TiO2 is greatly changed due to the special structure of PVDF after polarization. The electron cloud density around Ti atoms (TiO2) with polarization field is significantly enhanced (Fig. 4a, d), which consequently enhances the adsorption of hydroxyls. In addition, the charge difference of TiO2 with and without polarization field of TiO2 adsorbed hydroxyl models are also calculated, to analyze the charge transfer behavior. It can be clearly seen that the charge accumulation and depletion around TiO2 with polarization field are enhanced, which indicates efficient charge transformation and active surface sites (Fig. 4a–e). Then, the affinities of hydroxyl on TiO2 with and without polarization field are also analyzed. More intense charge transfer is observed between hydroxyl and TiO2 with a polarization field, which is favorable for the hydroxyl adsorption and activation (Fig. 4a–e). This confirms that ferroelectric polarization enhances the electron cloud density around TiO2 and promotes the charge separation on the surface of TiO2, thereby substantially improving the photocatalytic mineralization of formaldehyde. Furthermore, the electronic band composition is analyzed by calculating the densities of states (DOS) of TiO2 with and without polarization field. The top of the valence band (VB) is mainly composed of O 2p orbitals, while the bottom of conduction band (CB) primarily originates from the anti-bonding orbitals of Ti 3d (Fig. S17†). It is concluded that O atom mainly contributes to the bottom of CB and the orbitals of Ti atom are located at the top of VB. The polarization field hardly changes the energy gap of TiO2.
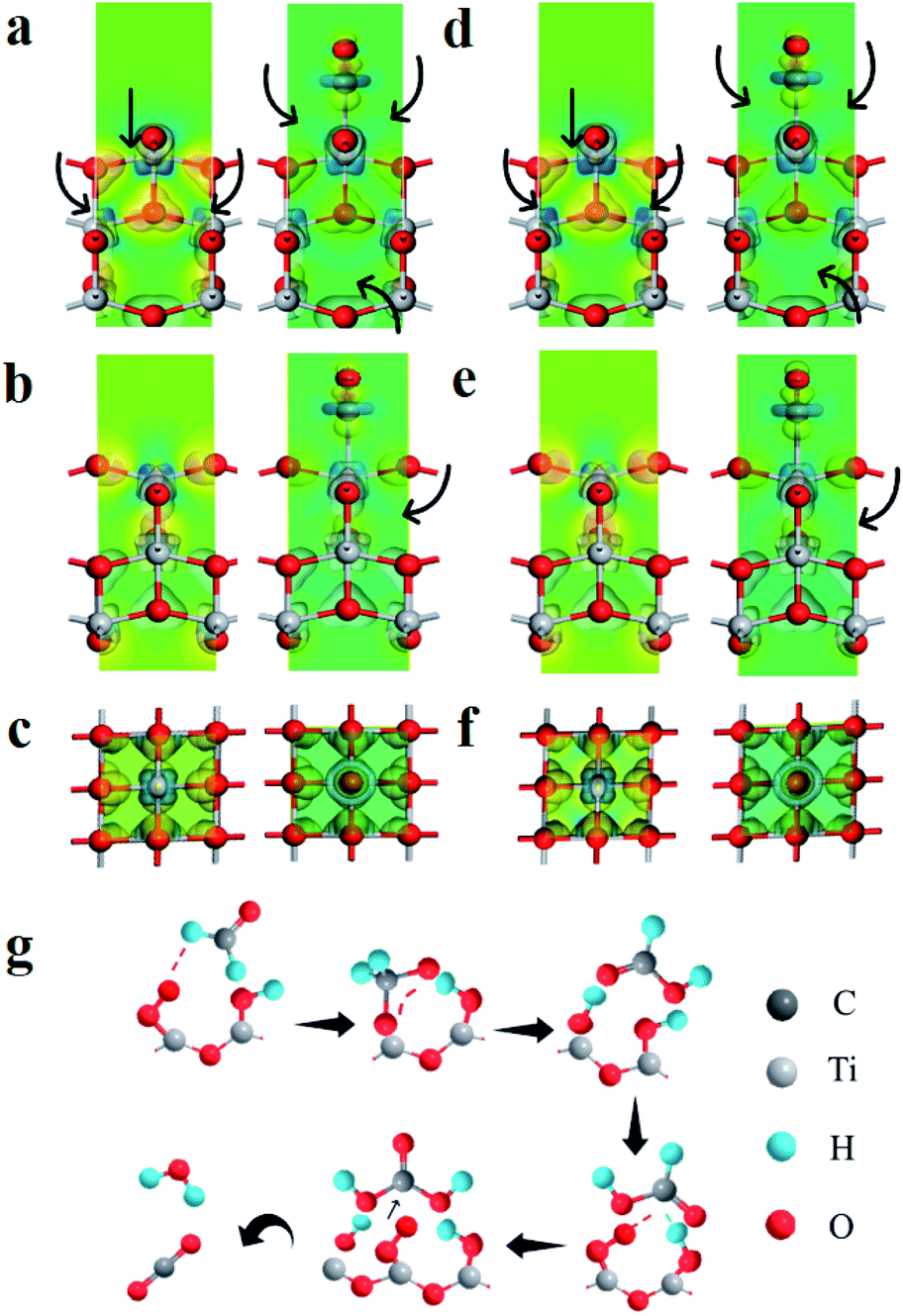 | ||
| Fig. 4 (a–c) Charge difference density of TiO2 without polarization field (left) and hydroxyl absorbed TiO2 (right), (a) main view, (b) side view, (c) top view. (d and e) Charge difference density of TiO2 with polarization field (left) and hydroxyl absorbed TiO2 (right), (d) main view, (e) side view, (f) top view (charge accumulation is depicted in yellow and depletion in blue). (g) Reaction scheme for the photocatalytic mineralized gaseous formaldehyde on the S-TiO2/PDVF. | ||
An electric field is created in the PVDF by hot-stretching. Therefore, this electric field acts as the built-in electric field of the TiO2/PVDF composite catalyst. Under the ultraviolet irradiation, the photo-induced electron and hole pairs are generated in TiO2. The holes are attracted to the negative side of the built-in electric field, while the electrons are repelled in the opposite direction. Photogenerated carriers migrate to the surface of TiO2 to participate in redox reactions in Fig. S18.†
The electron cloud density around Ti is significantly enhanced due to the special structure of PVDF after polarization, which enhances the adsorption of hydroxyl groups. A polarized electric field provides a strong driving force to drive charges migration and activates the hydroxyl groups. The hydroxyl groups attack formaldehyde in time and decompose into CO2 and H2O, the active sites are exposed once again and the hydroxyl groups are adsorbed in time for continuous reaction to achieve spontaneous mineralization.
The degradation mechanism of formaldehyde is proposed as show in Fig. 4g. HCHO is adsorbed on the S-TiO2/PDVF surface OH groups via hydrogen bonding, hydroxyl radical (˙OH) and superoxide radical anion (˙O2−) are generated when exposed to UV light, which then further attacks the C atom of adsorbed HCHO molecule, thus converting HCHO molecule into DOM intermediate, and DOM is further transformed into adsorbed formate species quickly. Subsequently, one active oxygen atom participates in the oxidation of formate species to adsorbed carbonic acid, which quickly decomposes into CO2 and H2O.
Conclusions
In summary, we report a built-in electric field advancing strategy for air purification where the adsorption sites of TiO2/PVDF piezo-photocatalyst are significantly enhanced and stabilized for the photogenerated charge carriers. In situ DRIFTS investigation and theoretical calculations revealed that the F (−) and H (+) atomic layers with distinct local structures in stretched PVDF, increase the electron cloud density around Ti, simultaneously promoting the dissociation of water into hydroxyl groups which are easier to be activated for adsorption of formaldehyde molecules. The built-in electric field induces charge separation, and the optimal stretched TiO2/PVDF also exhibited a formaldehyde mineralization rate of 93% with considerable stability. Besides, this method is also demonstrated for the decomposition of formaldehyde. This work may open a new way for the treatment of air pollution in practical applications, especially to overcome the serious pollution of VOCs.Author contributions
Song Sun conceived the idea and designed the study. Mengmeng Li performed the experiments. Mengmeng Li and Qin Cheng analyzed the results. Yong Jiang guided DRIFTS experiments. Bin Hong guided theoretical calculations. Song Sun, Mengmeng Li and Qin Cheng writing of original draft. All authors participated in the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (U1832165 and 21902001), the Anhui Provincial Natural Science Foundation (2008085QB85), the Key Research and Development Program of Anhui Province (202004a05020015 and 006233172019), and the Higher Education Natural Science Foundation of Anhui Province (K120433040). The authors would also like to acknowledge the staffs from BL01B beamline of National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.Notes and references
- F. He, W. Jeon and W. Choi, Nat. Commun., 2021, 12, 2528 CrossRef CAS PubMed.
- Y. Shi, Z. Li, J. Wang and R. Zhou, Appl. Catal., B, 2021, 286, 119936 CrossRef CAS.
- Q. U. Hassan, A. I. Channa, Q. G. Zhai, G. Zhu, Y. Gao, N. Ali and M. Bilal, Chemosphere, 2022, 288, 132668 CrossRef CAS.
- Q. U. Hassan, D. Yang, J. P. Zhou, Y. X. Lei, J. Z. Wang and S. U. Awan, Inorg. Chem., 2018, 57, 15187–15197 CrossRef CAS.
- Y. Fang, X. Li and X. Wang, ACS Catal., 2018, 8, 8774–8780 CrossRef CAS.
- M. El-Roz, P. Bazin, M. Daturi and F. Thibault-Starzyk, ACS Catal., 2013, 3, 2790–2798 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, e1807660 CrossRef PubMed.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS.
- S. Lin, S. Li, Y. Zhang, T. Ma and H. Huang, J. Mater. Chem. A, 2021, 9, 17936–17944 RSC.
- Z. Zhang, Y. Liang, H. Huang, X. Liu, Q. Li, L. Chen and D. Xu, Angew. Chem., Int. Ed., 2019, 58, 7263–7267 CrossRef CAS PubMed.
- Q. U. Hassan, D. Yang and J. P. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 45531–45545 CrossRef CAS.
- Z. Song, B. Hong, X. Zhu, F. Zhang, S. Li, J. Ding, X. Jiang, J. Bao, C. Gao and S. Sun, Appl. Catal., B, 2018, 238, 248–254 CrossRef CAS.
- Y. Liu, X. Cui, R. Niu, S. Zhang, X. Liao, S. D. Moss, P. Finkel, M. Garbrecht, S. P. Ringer and J. M. Cairney, Nat. Commun., 2022, 13, 335 CrossRef CAS PubMed.
- Y. Chen, D. Li, J. Chen, J. Wang, S. Meng, J. Xian, X. Fu and Y. Shao, Appl. Catal., B, 2013, 129, 403–408 CrossRef CAS.
- H. Li, Z. Ai and L. Zhang, Chem. Commun., 2020, 56, 15282–15296 RSC.
- L. Wan, W. Tian, N. Li, D. Chen, Q. Xu, H. Li, J. He and J. Lu, Nano Energy, 2022, 94, 106930 CrossRef CAS.
- Y. Huang, G. Rui, Q. Li, E. Allahyarov, R. Li, M. Fukuto, G. J. Zhong, J. Z. Xu, Z. M. Li, P. L. Taylor and L. Zhu, Nat. Commun., 2021, 12, 675 CrossRef CAS.
- C. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 16309–16328 CrossRef CAS PubMed.
- R. Tian, Q. Xu, C. Lu, X. Duan and R. G. Xiong, Chem. Commun., 2017, 53, 7933–7936 RSC.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557–564 CrossRef CAS.
- Y. Liu, T. Yang, B. Zhang, T. Williams, Y. T. Lin, L. Li, Y. Zhou, W. Lu, S. H. Kim, L. Q. Chen, J. Bernholc and Q. Wang, Adv. Mater., 2020, 32, e2005431 CrossRef.
- G. Singh, M. Sharma and R. Vaish, ACS Appl. Mater. Interfaces, 2021, 13, 22914–22925 CrossRef CAS.
- S. Han, D. Chen, J. Wang, Z. Liu, F. Liu, Y. Chen, Y. Ji, J. Pang, H. Liu and J. Wang, Nano Energy, 2020, 72, 104688 CrossRef CAS.
- N. Soin, D. Boyer, K. Prashanthi, S. Sharma, A. A. Narasimulu, J. Luo, T. H. Shah, E. Siores and T. Thundat, Chem. Commun., 2015, 51, 8257–8260 RSC.
- D. M. Dhevi, A. A. Prabu and K. J. Kim, Vib. Spectrosc., 2018, 94, 74–82 CrossRef CAS.
- B. Dai, Y. Yu, Y. Chen, H. Huang, C. Lu, J. Kou, Y. Zhao and Z. Xu, Adv. Funct. Mater., 2019, 29, 1807934 CrossRef.
- J. Li, M. Yang, X. Sun, X. Yang, J. Xue, C. Zhu, H. Liu and Y. Xia, Angew. Chem., Int. Ed., 2016, 55, 13828–13832 CrossRef CAS PubMed.
- S. Han, D. Chen, J. Wang, Z. Liu, F. Liu, Y. Chen, Y. Ji, J. Pang, H. Liu and J. Wang, Nano Energy, 2020, 72, 104688 CrossRef CAS.
- B. K. Panigrahi, D. Sitikantha, A. Bhuyan, H. S. Panda and K. Mohanta, Mater. Today, 2021, 41, 335–339 CAS.
- W. Zhang, Y. Zhu, X. Liu, D. Wang, J. Li, L. Jiang and J. Jin, Angew. Chem., Int. Ed., 2014, 53, 856–860 CrossRef CAS.
- L. Yang, H. Chen, Y. Xu, R. Qian, Q. Chen and Y. Fang, Chem. Eng. Sci., 2022, 251, 117435 CrossRef CAS.
- D. Zheng, L. Yang, W. Chen, Y. Fang and X. Wang, ChemSusChem, 2021, 14, 3821–3824 CrossRef CAS PubMed.
- H. Yu, F. Chen, X. Li, H. Huang, Q. Zhang, S. Su, K. Wang, E. Mao, B. Mei, G. Mul, T. Ma and Y. Zhang, Nat. Commun., 2021, 12, 4594 CrossRef CAS.
- Z. Yan, Z. Xu, J. Yu and M. Jaroniec, Appl. Catal., B, 2016, 199, 458–465 CrossRef CAS.
- S. Kato, T. Sato and Y. Kajii, Atmos. Environ., 2011, 45, 5531–5539 CrossRef CAS.
- K. Huang, L. Kong, F. Yuan and C. Xie, Appl. Surf. Sci., 2013, 270, 405–410 CrossRef CAS.
- L. Hao, L. Kang, H. Huang, L. Ye, K. Han, S. Yang, H. Yu, M. Batmunkh, Y. Zhang and T. Ma, Adv. Mater., 2019, 31, e1900546 CrossRef PubMed.
- S. Sun, J. Ding, J. Bao, C. Gao, Z. Qi and C. Li, Catal. Lett., 2010, 137, 239–246 CrossRef CAS.
- S. Liu, S. Xue, W. Zhang, J. Zhai and G. Chen, J. Mater. Chem. A, 2014, 2, 18040–18046 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03751c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |