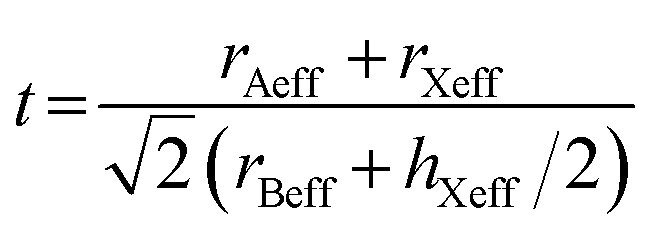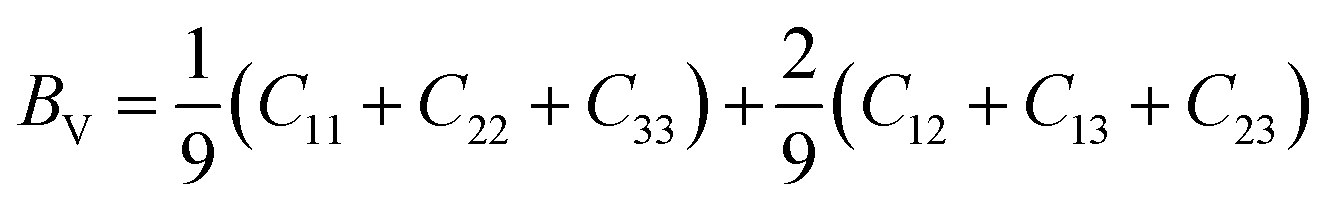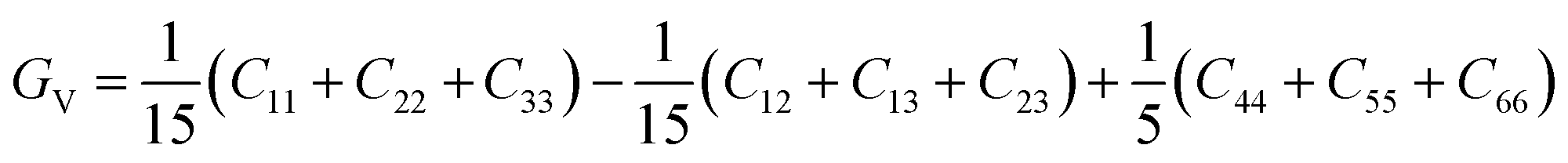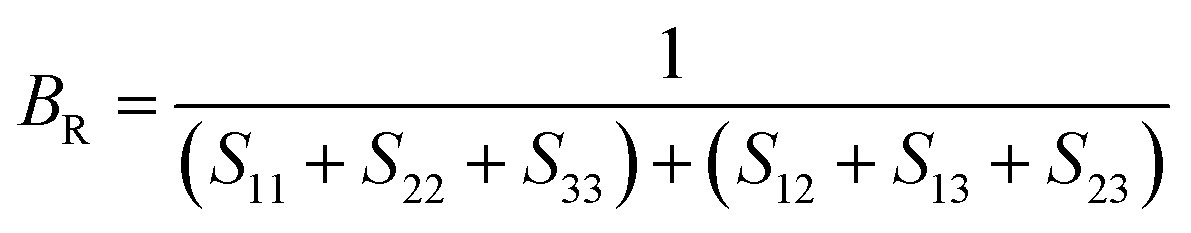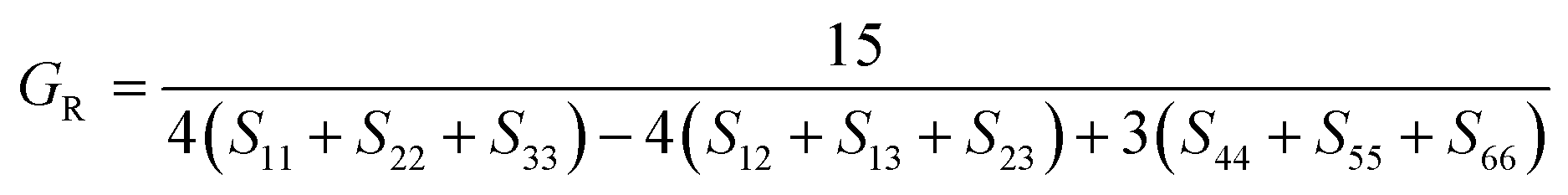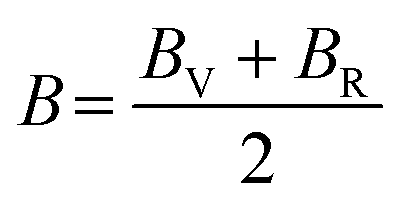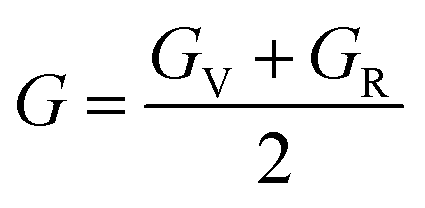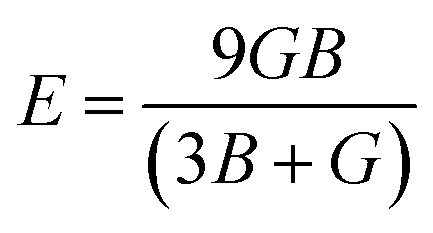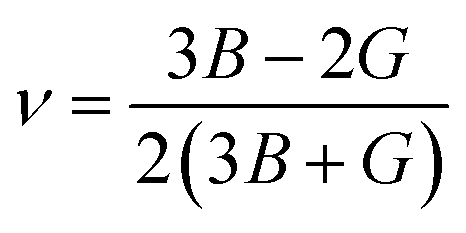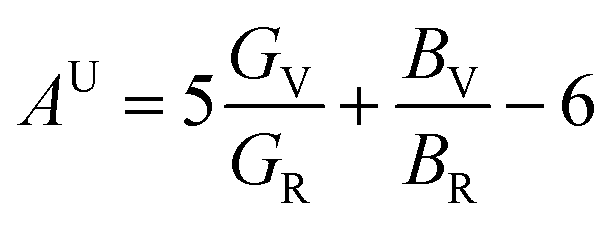DOI:
10.1039/D2RA03407G
(Paper)
RSC Adv., 2022,
12, 24647-24653
First-principles study on the mechanical and electronic properties of energetic molecular perovskites AM(ClO4)3 (A = C6H14N22+, C4H12N22+, C6H14N2O2+; M = Na+, K+)†
Received
1st June 2022
, Accepted 20th July 2022
First published on 31st August 2022
Abstract
Density functional theory (DFT) simulations were conducted to study the crystal structures, and mechanical and electronic properties of a series of new energetic molecular perovskites, including (C6H14N2)[Na(ClO4)3], (C6H14N2)[K(ClO4)3], (C4H12N2)[Na(ClO4)3] and (C6H14N2O)[K(ClO4)3], abbreviated as DAP-1, DAP-2, PAP-1, and DAP-O2, respectively. By calculating the elastic constants, moduli (Young's modulus E, bulk modulus B, and shear modulus G), Poisson ratio ν and Pugh's ratio B/G, we found that the four energetic molecular perovskites not only possessed good mechanical stability but excellent mechanical flexibility and ductility. In addition, DFT calculations were used to investigate the electronic properties of all of the perovskite compounds. The band gaps of DAP-1 and DAP-2 were comparable, and the band gap of PAP-1 was the smallest and that of DAP-O2 was the largest. A comprehensive analysis of the density of states and the M–O bonding characteristics provided a good explanation for the band gap characteristics. Besides, we found that the modulus properties of these molecular perovskite energetic compounds are also tightly bound to the strength of M–O bonding.
Introduction
Molecular perovskites are a type of compound with an inorganic perovskite structure but at least one component is a molecular unit.1 Owing to their unique structures and excellent properties,2 molecular perovskites have played important roles in solar cells,3,4 thermoelectricity,5 ferroelectrics,6 piezoelectricity,7 lasers,8 etc. Recently, by using a molecular assembly strategy, a series of ABX3-type high-energy-density molecular perovskites (C6H14N2)[M(ClO4)3] (M = Na+, K+, Rb+ and NH4+) have been synthesized by Chen's team.9 The synthesis of these energetic molecular perovskites created a precedent for molecular perovskites in the field of energetic materials. Different from traditional energetic compounds, these energetic molecular perovskites consist of organic cations like C6H14N22+ (as the fuel part) and special anions like ClO4− (as the oxidizing part). Furthermore, an increasing number of analogues have been produced by exploiting the compositionally adjustable and modifiable nature of the A, B and X components, such as (C4H12N2)[Na(ClO4)3], (C6H14N2O)[K(ClO4)3], (C6H14N2)[NH4(ClO4)3], (C6H14N2)(NH2NH3)(ClO4)3, etc.10–12 Best of all, these energetic molecular perovskites are characterized by excellent detonation performance, good thermal stability and low cost.9–12 For example, energetic molecular perovskite (C6H14N2)[Na(ClO4)3] with a density of 2.02 g cm−3 exhibits high detonation performance (detonation velocity D = 8.781 km s−1, detonation pressure P = 36.5 GPa) and good thermal stability (thermal decomposition temperature Td = 344 °C), which is superior to that of cyclotrimethylene trinitramine (RDX, D = 8.634 km s−1, P = 33.3 GPa, Td = 210 °C).12 Similarly, (C6H14N2)[K(ClO4)3] with a density of 2.04 g cm−3 also has high detonation performance (D = 8.591 km s−1, P = 35.2 GPa) and good thermal stability (Td = 364 °C).11 The emergence of energetic molecular perovskites has greatly promoted the development of energetic materials.
Energetic molecular perovskite as a unique energetic material has stood at the forefront of material science and attracted extensive attention of researchers. Mechanical properties are one of the vital properties related to the preparation, storage, transportation and usage of energetic materials.13 Electronic structure is not only tightly bound to the basic physicochemical properties of energetic materials, but also an internal factor determining the sensitivity of energetic materials.14–16 The lack of knowledge about fundamental mechanical properties and electronic properties of energetic molecular perovskites is not conducive to the further development of these promising energetic materials. However, the studies on energetic molecular perovskites mainly focus on the synthesis of new structures and thermal decomposition properties,17–23 very few studies have been conducted on the mechanical and electronic properties on energetic molecular perovskites.24 Therefore, it is a great interest to research the mechanical and electronic properties of molecular perovskite high-energetic materials.
To attract more attention on energetic molecular perovskite, more efforts are required to dedicated to this important area. In this paper, we focused our attentions on a family of metal-based energetic molecular perovskite AM(ClO4)3 (A = C6H14N22+, C4H12N22+, C6H14N2O2+; M = Na+, K+). Due to ion size mismatch, the molecular perovskites (C4H12N2)[K(ClO4)3] and (C6H14N2O)[Na(ClO4)3] could not be formed.11 Hence, we studied four energetic molecular perovskites including (C6H14N2)[Na(ClO4)3], (C6H14N2)[K(ClO4)3], (C4H12N2)[Na(ClO4)3] and (C6H14N2O)[K(ClO4)3], called DAP-1, DAP-2, PAP-1, DAP-O2, respectively. And the mechanical properties and electronic properties of them were studied and compared comprehensively and systematically. The research results could not only obtain the key properties which hard to be acquired by experiment, but also provide important theoretical guidance for designing more advanced energetic materials.
Computational methods
All calculations of this paper based on density functional theory (DFT) were performed by using the Vienna Ab initio Simulation Package (VASP) code.25,26 The projector augmented wave (PAW) method was applied to describe the interaction between core and valence electrons.27 And the Perdew, Burke, and Ernzerhof (PBE)28 functional revised for solids (PBEsol)29 was used as the exchange-correlation functional in this work. The H(1s1), C(2s22p2), N(2s22p3), O(2s22p4), Cl(3s23p5), Na(2p63s1) and K(3s23p64s1) were considered as valence electrons.
The crystal structures of DAP-1 and DAP-2 crystallized in cubic lattice with Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group,9 while the crystal structures of PAP-1 and DAP-O2 crystallized in monoclinic lattice with P21/c space group.11 The crystal structures of the four molecular perovskites were displayed in Fig. 1. The experimental crystal structures of AM(ClO4)3 were used as input structures and conducted full geometry optimizations which allowing atomic positions and lattice parameters fully relaxed. The geometry optimizations were completed when the total energy is less than 10−5 eV and the Hellmann–Feynman force on each atom is less than 0.03 eV Å−1. The cutoff energy of plane waves was set to 450 eV. The Gamma-centered Monkhorst–Pack k-point mesh employed were set as 2 × 2 × 2, 2 × 2 × 2, 3 × 3 × 2 and 2 × 3 × 3 for DAP-1, DAP-2, PAP-1, and DAP-O2 in geometry optimizations, respectively. The k-point mesh was the same as that of geometric optimization during the calculation of mechanical properties but increased twice when calculating density of states. The finite difference method by VASP30 was used in calculating mechanical properties.
space group,9 while the crystal structures of PAP-1 and DAP-O2 crystallized in monoclinic lattice with P21/c space group.11 The crystal structures of the four molecular perovskites were displayed in Fig. 1. The experimental crystal structures of AM(ClO4)3 were used as input structures and conducted full geometry optimizations which allowing atomic positions and lattice parameters fully relaxed. The geometry optimizations were completed when the total energy is less than 10−5 eV and the Hellmann–Feynman force on each atom is less than 0.03 eV Å−1. The cutoff energy of plane waves was set to 450 eV. The Gamma-centered Monkhorst–Pack k-point mesh employed were set as 2 × 2 × 2, 2 × 2 × 2, 3 × 3 × 2 and 2 × 3 × 3 for DAP-1, DAP-2, PAP-1, and DAP-O2 in geometry optimizations, respectively. The k-point mesh was the same as that of geometric optimization during the calculation of mechanical properties but increased twice when calculating density of states. The finite difference method by VASP30 was used in calculating mechanical properties.
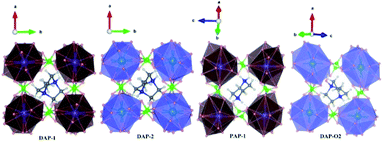 |
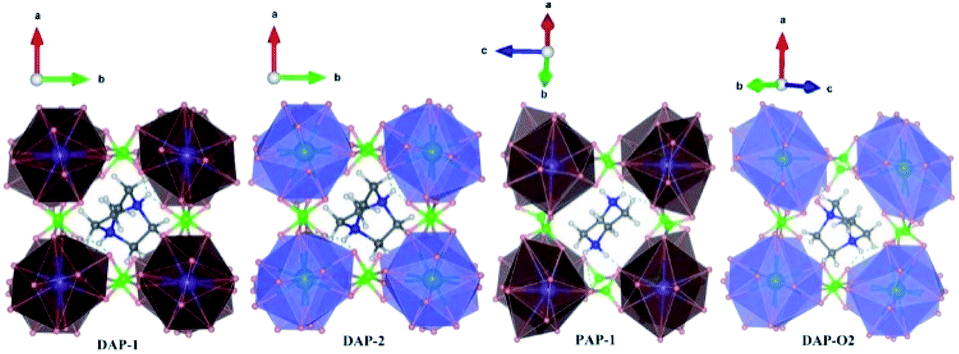
| | Fig. 1 Cage structures of AM(ClO4)3. H atom in light grey, C atom in dark gray, N atom in blue, O atom in pink, Cl atom in green, Na atom in purple, K atom in dark green. Hydrogen bonds are represented by dashed lines. | |
Results and discussion
Crystal structures and geometric stability
Energetic molecular perovskite AM(ClO4)3 (A = C6H14N22+, C4H12N22+, C6H14N2O2+; M = Na+, K+) belonged to ABX3-type perovskite compounds, where organic cations occupy the A site, metal cations M+ occupy the B site and ClO4− act as the X-site bridging ligand. Each M+ ion was coordinated by twelve oxygen atoms from six ClO4− to form MO12 polyhedron. And each polyhedron was connected by bridging ligand ClO4− to form a three-dimensional anionic inorganic framework [M(ClO4)3]2−. The A-site cation resided in the framework cavity and interacted with the X linkers through hydrogen bonding.31 In addition to hydrogen and coordination bonds, there are also Coulomb interactions between A, B, and X, which are beneficial for the geometric stability of unique perovskites.9 The unit cell parameters of AM(ClO4)3 by experiment and by DFT relaxation are tabulated in Table 1. The results showed that these crystal structures relaxed by PBEsol are in good accordance with the experimental structure. And the optimized crystal structures were shown in Fig. S1 and S2.†
Table 1 The lattice parameters of molecular perovskites AM(ClO4)3 by calculation and experiment
| Materials |
Methods |
a (Å) |
b (Å) |
c (Å) |
β (deg) |
V (Å3) |
| DAP-1 |
Expt9 |
14.154 |
14.154 |
14.154 |
90.00 |
2835.371 |
| Cal. |
14.116 |
14.116 |
14.116 |
90.00 |
2812.635 |
| |
(−0.34%) |
(−0.34%) |
(−0.34%) |
(0.00%) |
(−0.80%) |
| DAP-2 |
Expt9 |
14.291 |
14.291 |
14.291 |
90.00 |
2918.689 |
| Cal. |
14.288 |
14.288 |
14.288 |
90.00 |
2917.954 |
| |
(−0.02%) |
(−0.02%) |
(−0.02%) |
(0.00%) |
(-0.03%) |
| PAP-1 |
Expt11 |
10.152 |
9.720 |
13.162 |
91.90 |
1298.034 |
| Cal. |
10.146 |
9.770 |
12.943 |
91.78 |
1282.320 |
| |
(−0.06%) |
(+0.51%) |
(−1.66%) |
(−0.13%) |
(−1.21%) |
| DAP-O2 |
Expt11 |
14.692 |
10.453 |
10.045 |
91.17 |
1542.264 |
| Cal. |
14.674 |
10.414 |
10.085 |
91.89 |
1540.450 |
| |
(−0.12%) |
(−0.37%) |
(+0.40%) |
(+0.79%) |
(−0.12%) |
Notably, the geometric stability of the perovskite structure can be predicted by using the Goldschmidt tolerance factor (t).32 Due to the existence of molecular ions at the A and X sites, we adopted the extended tolerance factor (t)33–36 to evaluate the t value of molecular perovskites. According to the method reported by Kieslich and Cheetham,33,34 the t value can be calculated through eqn (1) and (2).33,34,36
| |
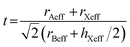 | (2) |
where
rmass is defined as the distance between the center of mass of the A-site cation and the atom with the largest distance to the center of mass (excluding hydrogen atoms), while
rion refers to the ionic radius of the atom mentioned above. The sum of
rmass and
rion is equal to the effective ionic radius of A-site cation
rAXeff.
33 rB is defined as the ionic radius of the B-site metal ions,
rXeff and
hXeff refer to the effective radius and height of the X-site molecular ions, respectively.
33,34,36 It was worth mentioning that the atom with the largest distance to the center of mass of C
6H
14N
22+ and C
4H
12N
22+ is N atom, while the atom with the largest distance to the center of mass of C
6H
14N
2O
2+ is O atom. Here, the ionic radii for N, O, Na and K atom adopted Shannon crystal radius.
37 The calculated Goldschmidt tolerance factors are shown in
Table 2.
Table 2 The calculated tolerance factor t for molecular perovskites AM(ClO4)3
| |
A |
B |
X |
t |
| rmass (pm) |
r(N/O) (pm) |
rAeff (pm) |
r (Na/K) (pm) |
rXeff (pm) |
hXeff (pm) |
| (C6H14N2)[Na(ClO4)3] |
125 |
132 |
257 |
153 |
240 |
410 |
0.982 |
| (C6H14N2)[K(ClO4)3] |
125 |
132 |
257 |
178 |
240 |
410 |
0.918 |
| (C4H12N2)[Na(ClO4)3] |
145 |
132 |
277 |
153 |
240 |
410 |
1.021 |
| (C6H14N2O)[K(ClO4)3] |
232 |
121 |
353 |
178 |
240 |
410 |
1.095 |
It has been reported that the t values of structurally stable perovskites generally lie in the range between 0.76 and 1.13.38 In ideal case, the perovskite crystal would have a perfectly packed structure when the value of t reaches 1. And any deviation from t = 1 demonstrates a structural distortion in the crystal thus leading to a lower symmetry.32,39 According to the calculated t values, 0.982 for DAP-1, 0.918 for DAP-2, 1.021 for PAP-1, 1.095 for DAP-O2, it could be preliminarily judged that these perovskite structures have structural stability. In addition, according to the degree of deviation of the t value from 1, we could find that the t values of Na-based molecular perovskites (DAP-1 and PAP-1) are closer to 1 than those of K-based molecular perovskites (DAP-2 and DAP-O2). Hence, we inferred that the geometric stability of Na-based molecular perovskites is better than that of K-based molecular perovskites. From this point of view, we could further speculate that the strength of Na–O bonds is stronger than that of K–O bonds, where the strength is directly related to the stability of the framework structure of molecular perovskites.
Mechanical properties
The elastic constants (Cij) of the four molecular perovskites were obtained from first-principles calculations, as given in Table 3. To the best of our knowledge, there were no experimental result on the elastic constant Cij about AM(ClO4)3 but one theoretical research about cubic phase DAP-2 that can be used as a comparison with the present results.24 The calculated elastic constants C11, C12 and C44 (40.9 GPa, 16.3 GPa and 14.6 GPa) about DAP-2 by Feng's group were higher than those of our calculated results (about 29.4 GPa, 13.9 GPa and 7.7 GPa). However, it was mentioning that our calculated results were comparable to those of cubic phase of molecular perovskite CH3NH3PbI3 whose calculated elastic constants C11, C12 and C44 were 27.1 GPa, 11.1 GPa and 9.2 GPa, respectively.40
Table 3 Elastic constants of energetic molecular perovskite AM(ClO4)3. The unit is GPaa
| Materials |
DAP-1 |
DAP-2 |
PAP-1 |
DAP-O2 |
| The elastic constant value corresponding to the ellipsis is zero. |
| C11 |
34.402 |
29.452 |
36.838 |
29.607 |
| C22 |
… |
… |
30.171 |
30.211 |
| C33 |
… |
… |
32.246 |
24.855 |
| C44 |
9.589 |
7.719 |
10.559 |
6.945 |
| C55 |
… |
… |
5.145 |
6.552 |
| C66 |
… |
… |
9.562 |
5.599 |
| C12 |
12.938 |
13.900 |
11.955 |
10.030 |
| C13 |
… |
… |
13.010 |
6.853 |
| C23 |
… |
… |
13.859 |
8.560 |
| C15 |
… |
… |
1.559 |
1.292 |
| C25 |
… |
… |
0.069 |
−1.497 |
| C35 |
… |
… |
3.955 |
−3.346 |
| C46 |
… |
… |
0.517 |
1.170 |
To investigate the mechanical stability of the four molecular perovskites, we adopted the Born stability conditions41,42 to theoretically judge the mechanical stability. For a cubic crystal, its mechanical stability required that its three independent elastic constants satisfy the following relations:
| C11 − C12 > 0, C11 + 2C12 > 0, C44 > 0 |
For the crystal structures with monoclinic symmetry, there were thirteen independent elastic constants to be obtained, and the following conditions were required for their mechanical stability,
| C11 > 0, C22 > 0, C33 > 0, C44 > 0, C55 > 0, C66 > 0, |
| [C11 + C22 + C33 + 2(C12 + C13 + C23)] > 0, |
| C33 × C55 − C35^2 > 0, C44C66 − C46^2 > 0, C22 + C33 − 2C23 > 0, |
| C22(C33C55 − C35^2) + 2C23C25C35 − (C23^2)C55 − (C25^2)C33 > 0, |
| 2[C15C25(C33C12 − C13C23) + C15C35(C22C13 − C12C23) + C25C35(C11C23 − C12C13)] − [C15C15(C22C33 − C23^2) + C25C25(C11C33 − C13^2) + C35C35(C11C22 − C12^2)] + C55g > 0 |
| (g = C11C22C33 − C11(C23^2) − C22(C13^2) − C33(C12^2) + 2C12C13C23) |
From Table 3, it could be deduced that our calculated elastic constants for the four perovskite compounds satisfy these conditions, suggesting that all structures are mechanically stable.
After obtaining the elastic constants, the most important mechanical parameters of a compound such as the bulk modulus B, shear modulus G, Young's modulus E, Pugh's ratio B/G,43 Poisson's ratio ν and universal anisotropy index AU (ref. 44) of these perovskite compounds were calculated using the Voigt–Reuss–Hill approximation.45 The relationships were given as
| |
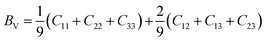 | (3) |
| |
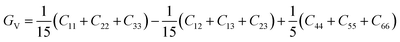 | (4) |
| |
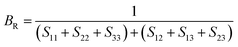 | (5) |
| |
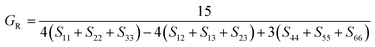 | (6) |
| |
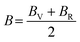 | (7) |
| |
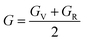 | (8) |
| |
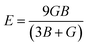 | (9) |
| |
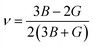 | (10) |
| |
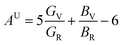 | (11) |
where
Sij is the elastic compliance matrix;
BV and
GV are the Voigt average bulk and shear modulus, respectively. And
BR and
GR are the Reuss average bulk and shear modulus, respectively. The calculated results are presented in
Table 4.
Table 4 Calculated mechanical parameters of AM(ClO4)3, Bulk modulus B, Shear modulus G, Young's modulus E, Pugh's ratio B/G, Poisson's ratio ν and Universal anisotropy index AU
| Materials |
DAP-1 |
DAP-2 |
PAP-1 |
DAP-O2 |
| B (GPa) |
20.09 |
19.08 |
19.28 |
14.79 |
| G (GPa) |
10.03 |
7.74 |
8.49 |
7.36 |
| E (GPa) |
25.8 |
20.46 |
22.21 |
18.95 |
| B/G |
2.00 |
2.46 |
2.27 |
2.01 |
| ν |
0.29 |
0.32 |
0.31 |
0.29 |
| AU |
0.02 |
0 |
0.79 |
0.62 |
As can be seen from Table 4, each kind modulus in the four molecular perovskites in descending order were DAP-1 > PAP-1 > DAP-2 > DAP-O2. And their bulk moduli ranged from 14 to 21 GPa, the shear moduli ranged from 7 to 11 GPa and the Young's moduli ranged from 18 to 26 GPa. The low Young's moduli revealed the excellent mechanical flexibility of the four molecular perovskites. Furthermore, it was evident that the shear moduli are relatively lower than bulk moduli and Young's moduli for all structures. Therefore, we can conclude that the ability of the four molecular perovskites to resist shear strain is relatively weak, and the shear deformation is the main reason for the instability of mechanical properties. This conclusion echoed the characteristics about the mechanical sensitivity of energetic molecular perovskites reported by experiment, in which friction sensitivity was higher than other types of sensitivity.9,11
The ductility of materials has been extensively indexed by the critical value 1.75 for Pugh’s ratio.43 In other words, the material will be ductile (brittle) if Pugh's ratio is larger (smaller) than 1.75.46 The calculated B/G of molecular perovskites ranged from 2.00 to 2.46, indicating that all molecular perovskites exhibit good ductility. The Poisson's ratio ν was another useful indicator for judging the ductility and brittleness of materials. Generally, the solid with a large ν (ν > 0.26) usually exhibits ductility. And the larger the Poisson’s ratio, the higher the ductility.47 The calculated ν values of all the samples ranged from 0.29 to 0.32 thus further implying the ductile nature of all the compounds.
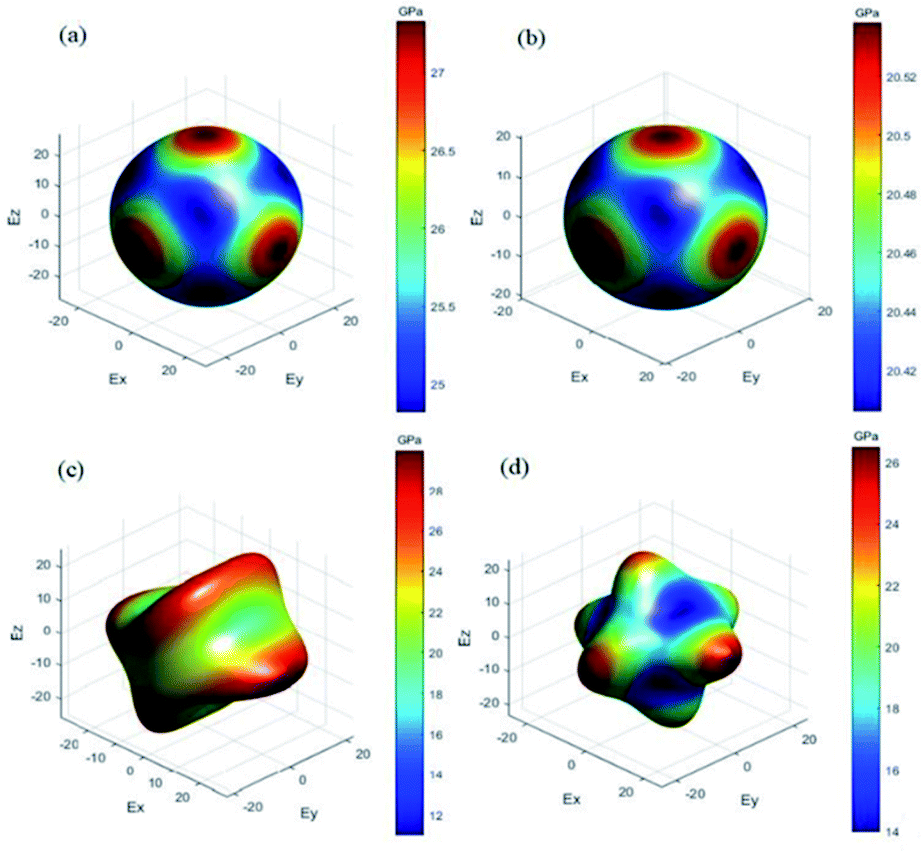
The anisotropy of elastic properties was an important topic from the view of applications. The anisotropy in elastic properties might be estimated using various anisotropy indexes and factors.45,48 Among them, the universal anisotropy index (AU)49 was utilized to quantify the single crystalline elastic anisotropy. AU showed the degree of anisotropy of materials, and the crystal with a zero value of AU was isotropic. The deviation from zero meant anisotropy.50 What's more, the three-dimensional (3D) surface contour of Young's modulus can express the elastic anisotropy more intuitively. For a perfectly isotropic medium, this surface would be a sphere, while the deviation of this surface from the spherical shape indicated the presence of a certain degree of elastic anisotropy. The 3D surface contour of Young's modulus for each perovskite compounds are depicted in Fig. 2.
 |
| | Fig. 2 3D surface contour of Young's modulus for (a) DAP-1, (b) DAP-2, (c) PAP-1, (d) DAP-O2. | |
In general, the elastic response of a single-crystal is seldom isotropic; almost all the known crystals exhibit anisotropic elastic properties. Surprisingly, for the compound DAP-2, the AU with a value of zero as well as the spherical three-dimensional (3D) surface contour of Young's modulus all implied the isotropy of mechanical properties. The AU value of DAP-1 was 0.02, which suggested that DAP-1 has a low anisotropy in elastic properties. Moreover, the nearly spherical 3D surface contour of Young's modulus of DAP-1 further indicated the low anisotropy in elastic properties of DAP-1. The AU value of PAP-1 and DAP-O2 were 0.79 and 0.62, respectively. Hence, PAP-1 and DAP-O2 showed obvious anisotropic characteristics in elastic properties. Furthermore, the 3D surface contour of Young's modulus of both PAP-1 and DAP-O2 had a large deviation from the spherical shape, which confirmed the strong anisotropy from another angle. In addition, the universal anisotropy indexes for the two cubic structures DAP-1 and DAP-2 were far less than those of monoclinic structures PAP-1 and DAP-O2, which meant that the universal anisotropy index of these energetic molecular perovskites decrease with the rising structure symmetry. And the reason may be that the higher crystallographic symmetry gives more constraints on elastic tensors and intrinsically favors isotropy.51
Electronic properties
For energetic compounds, the band gap was tightly correlated with the impact sensitivity. This may be result from the fact that there is accompanied by the breaking of chemical bonds requiring population of anti-bonding state when the energetic material was initiated. Therefore, the electronic excitation usually led to a weakness of the chemical bonds. As a consequence, the smaller the band gap was, the easier the electronic was to reach antibonding orbital, and the easier the chemical bond was to break.52,53 According to the first-principles band gap criterion,14 for a series of energetic crystal with similar structure or with similar thermal decomposition mechanism, a smaller band gap meant a higher impact sensitivity.15,54
The calculated band gaps for these title compounds are given in Table 5. As can be seen from Table 5, the four compounds exhibited large band gaps, thus revealing that the four energetic compounds have low impact sensitivities. By comparation, we found that the band gaps of DAP-1 and DAP-2 were comparable, and the band gap of PAP-1 was the smallest and that of DAP-O2 was the largest. The band gaps of the four crystals in ascending order were PAP-1 < DAP-1 < DAP-2 < DAP-O2. According to the first-principles band gap criterion, it can be predicted that the impact sensitivities of the four molecular perovskites in descending order are PAP-1 > DAP-1 > DAP-2 > DAP-O2.
Table 5 The calculated band gaps of molecular perovskite AM(ClO4)3
| Materials |
DAP-1 |
DAP-2 |
PAP-1 |
DAP-O2 |
| Band gap (eV) |
5.62 |
5.66 |
5.55 |
6.54 |
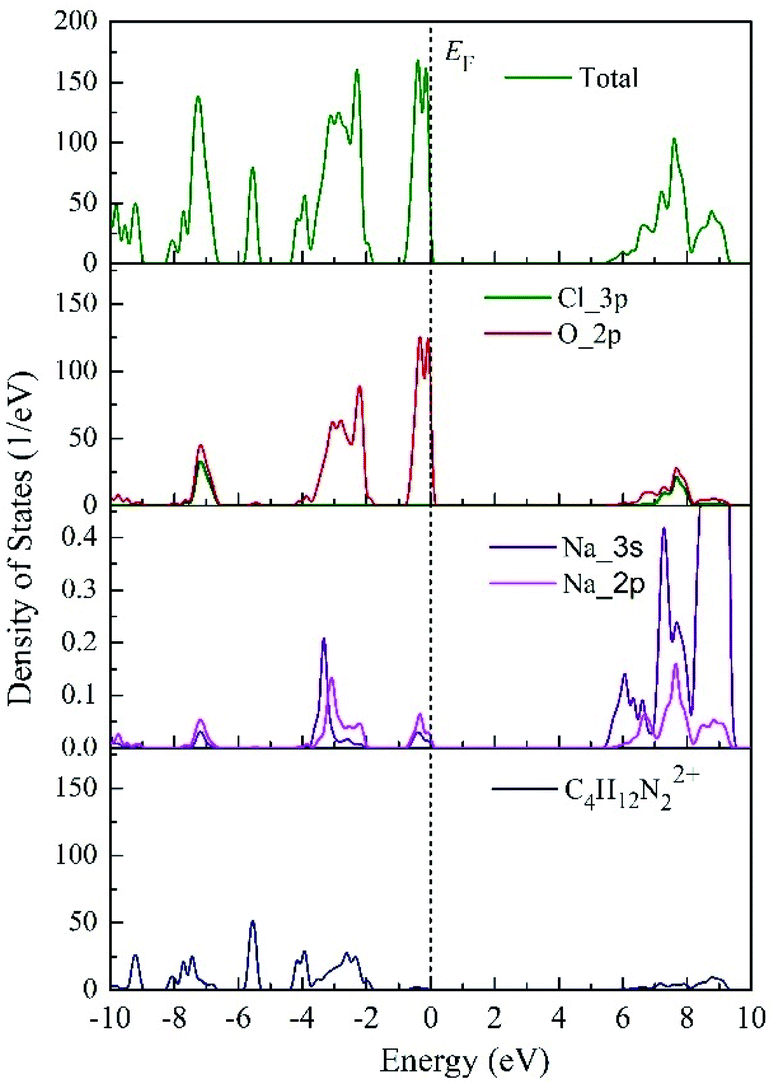
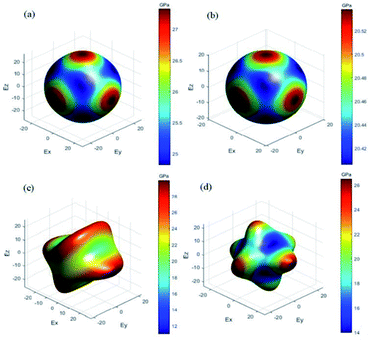
To obtain more information about the electronic properties of the four titled compounds, the density of states of each compound was calculated. According to our research, all compounds had similar electronic properties. Here, PAP-1 was taken as an example to analyze the characteristics of density of states of them. Fig. 3 plots the calculated partial density of states (PDOS) of (C4H12N2)[Na(ClO4)3] (PAP-1). It could be seen that the valence bands maximum (VBM) and the conduction bands minimum (CBM) are mainly dominated by the O 2p and Na 3s (2p) orbital, while the organic cations (C4H12N22+) have no contribution to the both band edges. In addition, some strong peaks occurred at the same energy in the PDOS of Na and O atom, it may be inferred that the two atoms are bonded strongly. Hence, we can speculate that the band gap of these compounds will be closely related to the M–O bonding. However, both DAP-1 and PAP-1 exhibited different band gap though having the same kind of M–O bonds. The similar argument applied to DAP-2 and DAP-O2. Therefore, the band gaps of these energetic molecular perovskites can also be influenced by organic cations.
 |
| | Fig. 3 PDOS of (C4H12N2)[Na(ClO4)3] (PAP-1). | |
Analysis of M–O bonds
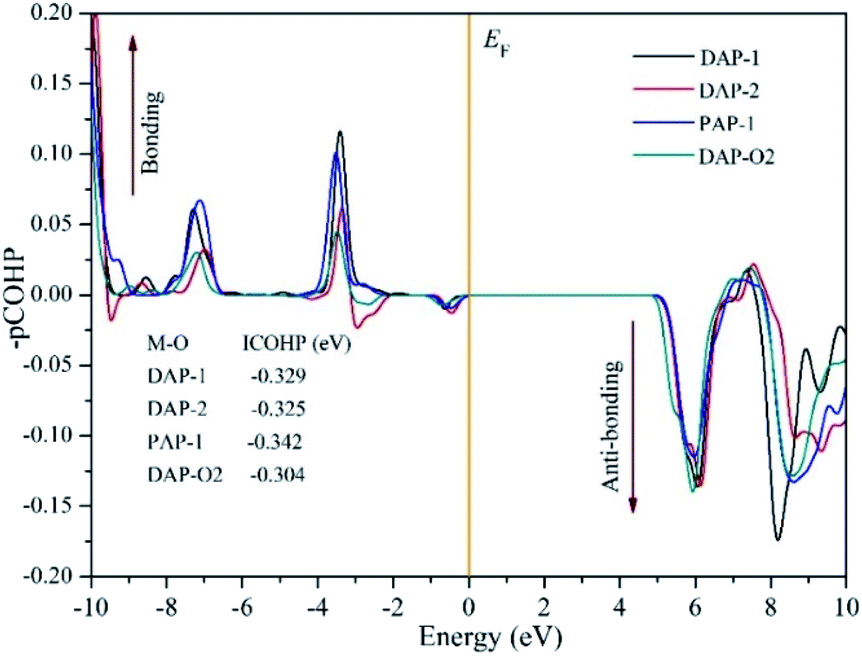
To explore the relationship between the band gap and M–O bonds in AMX3 perovskites, we calculated the M–O bond interactions using the projected crystal orbital Hamilton population (pCOHP) method.55,56 The criterion to judge the bonding states depended on the positive and negative of the pCOHP value. Positive pCOHP values corresponded to anti-bonding states, while negative pCOHP values corresponded to bonding states. The integrated COHP (ICOHP) can reflect the bonding strength, and the bonding strength increased with the absolute value of ICOHP.57
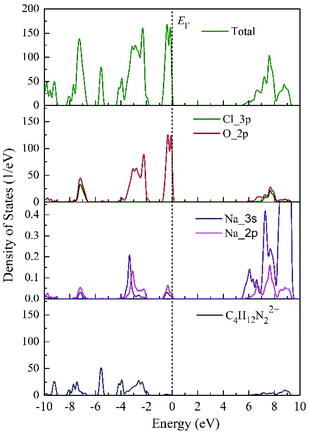
The average value of pCOHP and ICOHP over the M–O atom pairs of each AMX3 compound are depicted in Fig. 4. The value of ICOHP of Na–O bond in DAP-1 and PAP-1 were −0.329 eV and −0.342 eV, respectively. And the value of ICOHP of K–O bonding in DAP-2 and DAP-O2 were −0.325 eV and −0.304 eV, respectively. Therefore, we found that the same kind of M–O bonds in the molecular perovskites exhibit different strength due to the influence of the A-site cations. And the strength of M–O bonds in the four molecular perovskites were arranged in descending order as PAP-1 > DAP-1 > DAP-2 > DAP-O2. It also could be seen that the VBM in all perovskites is an anti-bonding state of the M–O hybridization. The VBM would shift down in energy when the bonding strength decreases and result in the increase of band gap, thus providing a good explanation for the tendency of band gap mentioned above. Furthermore, we could find that the tendency of bonding strength is roughly consistent with the sequence of the elastic moduli in the four perovskites discussed in the preceding part. Thus, we speculated that magnitude of the elastic modulus is also closely related to the strength of the M–O bonds. And the stronger the M–O bond, the bigger the elastic modulus. In addition, the ICOHP values of Na-based molecular perovskites (DAP-1 and PAP-1) were more negative than those of K-based molecular perovskites (DAP-2 and DAP-O2), which meant that the strength of Na–O bonds is stronger than that of K–O bonds, which also provided a good explanation for the conclusion about geometric stability discussed above.
 |
| | Fig. 4 COHP and ICOHP analysis of AM(ClO4)3. | |
Conclusions
In conclusion, the crystal structures, mechanical properties and electronic properties of energetic molecular perovskites AM(ClO4)3 (A = C6H14N22+, C4H12N22+, C6H14N2O2+; M = Na+, K+) were investigated systematically by performing the first-principles calculations based on DFT. The analysis of the elastic modulus demonstrated that the four energetic molecular perovskites possessed excellent mechanical flexibility, and shear deformation was the main reason for the instability of mechanical properties. In addition, the calculated Poisson ratio ν and Pugh's ratio B/G all revealed the ductility characteristic. The calculated universal anisotropy index AU as well as the three-dimensional (3D) surface contours of Young's modulus indicated that the structures with lower symmetry generally show stronger anisotropic properties. According to the calculated band gap values, we can infer that the sensitivity of DAP-1 and DAP-2 was comparable, while the sensitivity of PAP-1 was the highest and that of DAP-O2 was the lowest. A comprehensive analysis of the density of states and the M–O bonding characteristics revealed that the band gap values of these compounds were strongly related to the strength of M–O bonds and the A-site organic cations affected the band gap by the way of influencing the strength M–O bonds. Besides, it could be found that the elastic moduli were positively correlated with the strength of M–O bonds. To summarize, this study demonstrated the important role of the type and strength of M–O bonds in the mechanical and electronic properties of energetic molecular perovskite AM(ClO4)3. Therefore, researchers can rationally choose the B-site cations to modulate the mechanical and electronic properties of molecular perovskite high-energetic materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the grant from the National Natural Science Foundation of China (Grant No. 11572160).
Notes and references
- G. Kieslich and A. L. Goodwin, Mater. Horiz., 2017, 4, 362–366 RSC.
- X. Qi, Y. P. Zhang, Q. D. Ou, S. T. Ha, C. W. Qiu, H. Zhang, Y. B. Cheng, Q. H. Xiong and Q. L. Bao, Small, 2018, 14, 1800682 CrossRef PubMed.
- B. Kshirsagar, N. Jaykhedkar, K. Jain, S. Kishor, V. Shah, L. M. Ramaniah and S. Tiwari, J. Phys. Chem. C, 2021, 125, 2592–2606 CrossRef CAS.
- M. N. Xue, H. Zhou, Y. Xu, J. Mei, L. Yang, C. Ye, J. Zhang and H. Wang, Sci. China Mater., 2017, 60, 407–414 CrossRef CAS.
- Y. P. He and G. Galli, Chem. Mater., 2014, 26, 5394–5400 CrossRef CAS.
- H. Y. Ye, Y. Y. Tang, P. F. Li, W. Q. Liao, J. X. Gao, X. N. Hua, H. Cai, P. P. Shi, Y. M. You and R. G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS PubMed.
- W. Q. Liao, D. W. Zhao, Y. Y. Tang, Y. Zhang, P. F. Li, P. P. Shi, X. G. Chen, Y. M. You and R. G. Xiong, Science, 2019, 363, 1206–1210 CrossRef CAS PubMed.
- G. C. Xing, N. Mathews, S. S. Lim, N. Yantara, X. F. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- S. L. Chen, Z. R. Yang, B. J. Wang, Y. Shang, L. Y. Sun, C. T. He, H. L. Zhou, W. X. Zhang and X. M. Chen, Sci. China Mater., 2018, 61, 1123–1128 CrossRef CAS.
- Y. Shang, R. K. Huang, S. L. Chen, C. T. He, Z. H. Yu, Z. M. Ye, W. X. Zhang and X. M. Chen, Cryst. Growth Des., 2020, 20, 1891–1897 CrossRef CAS.
- S. L. Chen, Y. Shang, C. T. He, L. Y. Sun, Z. M. Ye, W. X. Zhang and X. M. Chen, CrystEngComm, 2018, 20, 7458–7463 RSC.
- Y. Shang, Z. H. Yu, R. K. Huang, S. L. Chen, D. X. Liu, X. X. Chen, W. X. Zhang and X. M. Chen, Engineering, 2020, 6, 1013–1018 CrossRef CAS.
- T. Sun, J. J. Xiao, Q. Liu, F. Zhao and H. M. Xiao, J. Mater. Chem. A, 2014, 2, 13898–13904 RSC.
- W. H. Zhu and H. M. Xiao, Struct. Chem., 2010, 21, 657–665 CrossRef CAS.
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Phys. Chem. C, 2013, 117, 16830–16839 CrossRef CAS.
- Q. Wu, W. H. Zhu and H. M. Xiao, RSC Adv., 2014, 4, 15995–16004 RSC.
- P. Deng, H. Ren and Q. J. Jiao, Ionics, 2020, 26, 1039–1044 CrossRef CAS.
- P. Deng, H. Ren and Q. J. Jiao, Vacuum, 2019, 169, 108882 CrossRef CAS.
- Q. Jia, P. Deng, X. X. Li, L. S. Hu and X. Cao, Vacuum, 2020, 175, 109257 CrossRef CAS.
- X. X. Li, S. Q. Hu, X. Cao, L. S. Hu, P. Deng and Z. B. Xie, J. Energ. Mater., 2020, 38, 162–169 CrossRef CAS.
- Y. Liu, L. s. Hu, S. D. Gong, C. Y. Guang, L. Q. Li, S. Q. Hu and P. Deng, Cent. Eur. J. Energ. Mater., 2020, 17, 451–469 CrossRef CAS.
- S. D. Zhu, X. Cao, X. Q. Cao, Y. Q. Feng, X. B. Lin, K. H. Han, X. X. Li and P. Deng, Mater. Des., 2021, 199, 109426 CrossRef CAS.
- J. Zhou, L. Ding, F. Q. Zhao, B. Z. Wang and J. L. Zhang, Chin. Chem. Lett., 2020, 31, 554–558 CrossRef CAS.
- G. Q. Feng, X. X. Jiang, W. J. Wei, P. F. Gong, L. Kang, Z. H. Li, Y. C. Li, X. D. Li, X. Wu and Z. S. Lin, Dalton Trans., 2016, 45, 4303–4308 RSC.
- W. Kohn and L. J. Sham, Phys. Rev. Lett., 1965, 140, A1133–A1138 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- Y. Le Page and P. Saxe, Phys. Rev. B, 2002, 65, 104104 CrossRef.
- Y. L. Sun, X. B. Han and W. Zhang, Chem.–Eur. J., 2017, 23, 11126–11132 CrossRef CAS PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- G. Kieslich, S. J. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- D. B. Mitzi, Dalton Trans., 2001, 1–12 RSC.
- W. Li, Z. M. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 1–18 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Hagenmuller, Inorganic Solid Fluorides: Chemistry and Physics, Academic Press, London, 1985 Search PubMed.
- K. Wang, D. Yang, C. Wu, M. Sanghadasa and S. Priya, Prog. Mater. Sci., 2019, 106, 100580 CrossRef CAS.
- J. Feng, APL Mater., 2014, 2, 081801 CrossRef.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B, 2014, 90, 224104 CrossRef.
- Z. J. Wu, E. J. Zhao, H. P. Xiang, X. F. Hao, X. J. Liu and J. Meng, Phys. Rev. B, 2007, 76, 054115 CrossRef.
- S. Pugh, Philos. Mag., 1954, 45, 823–843 CAS.
- S. I. Ranganathan and M. Ostoja-Starzewski, Phys. Rev. Lett., 2008, 101, 055504 CrossRef PubMed.
- J. Feng, APL Mater., 2014, 2, 081801 CrossRef.
- L. Guo, G. Tang and J. W. Hong, Chin. Phys. Lett., 2019, 36, 056201 CrossRef CAS.
- M. Z. Rahaman, M. L. Ali and M. A. Rahman, Chin. J. Physiol., 2018, 56, 231–237 CrossRef CAS.
- A. Benahmed, A. Bouhemadou, B. Alqarni, N. Guechi, Y. Al-Douri, R. Khenata and S. Bin-Omran, Philos. Mag., 2018, 98, 1217–1240 CrossRef CAS.
- Z. Q. Lv, Z. F. Zhang, Q. Zhang, Z. H. Wang, S. H. Sun and W. T. Fu, Solid State Sci., 2016, 56, 16–22 CrossRef CAS.
- S. Yalameha, P. Saeidi, Z. Nourbakhsh, A. Vaez and A. Ramazani, J. Appl. Phys., 2020, 127, 085102 CrossRef CAS.
- A. B. Cairns and A. L. Goodwin, Phys. Chem. Chem. Phys., 2015, 17, 20449–20465 RSC.
- A. A. Michalchuk, S. Rudić, C. R. Pulham and C. A. Morrison, Phys. Chem. Chem. Phys., 2018, 20, 29061–29069 RSC.
- A. A. Michalchuk, M. Trestman, S. Rudić, P. Portius, P. T. Fincham, C. R. Pulham and C. A. Morrison, J. Mater. Chem. A, 2019, 7, 19539–19553 RSC.
- G. N. Pallewela and R. P. Bettens, Comput. Theor. Chem., 2021, 1201, 113267 CrossRef CAS.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Gratzel and F. D. Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS PubMed.
- M. Kato, T. Fujiseki, T. Miyadera, T. Sugita, S. Fujimoto, M. Tamakoshi, M. Chikamatsu and H. Fujiwara, J. Appl. Phys., 2017, 121, 115501 CrossRef.
- P. F. Shen, K. Q. Nie, X. H. Sun, L. J. Liu and J. A. McLeod, Phys. Status Solidi RRL, 2016, 10, 677–681 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Shenshen Li and
Jijun Xiao*
,
Shenshen Li and
Jijun Xiao*