
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of single and binary mixtures of collagen amino acids on the structure of synthetic calcium hydroxyapatite as a nanobiomaterial
Sylwester Krukowski * and
Konrad Sztelmach
* and
Konrad Sztelmach
Medical University of Warsaw, Faculty of Pharmacy, Department of Analytical Chemistry, Warsaw, Poland. E-mail: sylwester.krukowski@wum.edu.pl
First published on 22nd August 2022
Abstract
The modifications carried out as part of this study were aimed at examining the effect of the addition of collagen amino acids: glycine, proline and hydroxyproline (used separately and in binary mixtures) on the physicochemical properties of hydroxyapatite obtained in their presence in vitro. The influence of mixtures of amino acids on these properties is an important element of scientific novelty. The obtained samples were tested with the use of instrumental methods: FT-IR, TEM, EDXMA, PXRD and UV spectrophotometry. The results showed the influence of the amino acids used on changes in the relative content of the labile phosphate groups constituting the structure of the hydrated surface layer of crystals. As a consequence, there were differences in some physicochemical properties of the obtained hydroxyapatites (degree of crystallinity, molar Ca/P ratio). It was also determined how the ability of the used amino acids to bind to hydroxyapatite changes.
1. Introduction
Hydroxyapatite, chemically described with the formula Ca10(PO4)6(OH)2, is commonly known as a mineral that occurs naturally in rocks and as an inorganic part of human bones and teeth. With reference to the formula given, the Ca/P molar ratio is approximately 1.67. However, in practice, naturally occurring hydroxyapatites cannot be described by any specific formula and the Ca/P molar ratio does not equal 1.67. This is due to the variability of the composition caused by the ion exchange processes between the hydroxyapatite crystal core and the external environment. This exchange takes place via the hydrated quasi-liquid surface layer.1 In the human body, the hydrated layer on the surface of the crystals plays a key role in the biomineralization process. Type I collagen is also important in the process of bone tissue formation. The main amino acids that build this collagen are glycine (Gly), proline (Pro), and hydroxyproline (HyPro).2From the point of view of the use of hydroxyapatite as a bone-substituting biomaterial or as a drug carrier, modifications with the use of α-amino acids are an interesting and noteworthy direction of research of these minerals. Literature data show a significant influence of amino acid molecules on the physicochemical parameters of nanocrystalline hydroxyapatite powders obtained in laboratory conditions.3 The vast majority of the described tests in this area were carried out using the standard precipitation method. The addition of an amino acid to the solution in which the hydroxyapatite crystals are formed causes the amino acid molecules to interact with them and can affect the size and morphology of the crystals.4 It turns out, therefore, that the amino acids used may have an impact on the process of crystal nucleation and further crystal growth in the solution. Depending on the type of amino acid used, it is known that:
(1) Acidic amino acids (aspartic acid, glutamic acid) as compounds with high affinity for HA, contribute to a significant inhibition of crystal growth. They also reduce the mean crystal size. As a result, the obtained hydroxyapatites are characterized by lower crystallinity compared to the control sample obtained under the same conditions, but without the addition of an amino acid.5–7
(2) Non-polar neutral amino acids (glycine, alanine, phenylalanine, proline) inhibit the crystal growth process, but to a much lesser extent than acidic amino acids. At the same time, they stimulate an increase in the number of formed nuclei of crystallization. They also cause a reduction in the size of the maturing crystals, which results in a reduction in the crystallinity of hydroxyapatite.8–12
(3) Neutral polar amino acids (tyrosine, asparagine) show various effects on the process of formation and growth of apatite crystals. Tyrosine stimulates the growth of crystals, which also results in their larger sizes. Asparagine, on the other hand, like the majority of biogenic amino acids, is an inhibitor of crystal growth and contributes to their reduction and lowering the crystallinity of synthetic hydroxyapatite.11,13
(4) Alkaline amino acids (lysine, arginine) inhibit the growth of crystals and reduce their size. Consequently, they reduce the degree of crystallinity of the hydroxyapatite produced in their presence. However, this effect is relatively lower than that of acidic amino acids.13,14
Moreover, amino acids can also be used to functionalize the surface of hydroxyapatite,15 or to obtain it from other inorganic calcium salts.16
Adsorption is an extremely important phenomenon from which the influence of amino acids directly on the structure of hydroxyapatite results. Considering adsorption at the molecular level, the interactions of amino acids with the surface of the adsorbent (here: hydroxyapatite) have the nature of electrostatic interactions. This is confirmed by the data obtained in theoretical considerations on the affinity of amino acids for the surface of hydroxyapatite crystals carried out with the use of computational molecular modeling methods.9,17 By analyzing the chemical composition and structure of amino acid molecules and the distribution of atoms in the apatite crystal lattice, we can distinguish two types of such interactions: (i) between Ca2+ ions contained in the surface layers of apatite crystals and negatively charged carboxyl groups (–COO−) of amino acid molecules, (ii) between the phosphate (PO43−) ions of HA crystals and positively charged amino groups (–NH3+) of amino acids. Additionally, on the oxygen atoms of apatite's phosphate groups, a negative charge is accumulated, allowing the formation of hydrogen bonds with hydrogen atoms of nearby amino groups of the amino acid. Of course, van der Waals forces must also be considered.8,9,17
The desirability of using biogenic amino acids as simple organic compounds to modify the structure of hydroxyapatite is based on the awareness of the important functions they play in the processes of crystal growth regulation, maturation and resorption in vivo. The results of research on modified apatite biomaterials also suggest that the use of amino acids may lead to an improvement in the biocompatibility of synthetic hydroxyapatite. The results of experiments in which mixtures of amino acids were used to modify the in vitro obtained hydroxyapatite materials have not been published so far. Therefore, the aim of this study is to determine the effect of the main collagen amino acids (glycine, proline and hydroxyproline), used both individually and in binary mixtures, on the structure of hydroxyapatite obtained in their presence. For this purpose, a number of hydroxyapatite samples were obtained in the presence of single amino acids as well as their mixtures: Gly + Pro, Gly + HyPro, HyPro + Pro. All materials had assessed crystallinity and crystal size using powder X-ray diffraction (PXRD), determined morphology using transmission electron microscopy (TEM), and measured chemical composition using energy dispersive X-ray microanalysis (EDXMA). Furthermore, in the employed mid-infrared spectroscopy (FT-IR) to analyze the relative content of labile phosphates and OH groups. Finally, in all materials, the amino acid content was quantified using the UV spectroscopy.
2. Materials and methods
2.1. Material synthesis (preparation)
Ca(NO3)2·4H2O, (NH4)2HPO4, glycine, L-proline, trans-4-hydroxy-L-proline (hereinafter called ‘hydroxyproline’) and ammonia solution were of the analytical grade and were purchased from Merck (Sigma Aldrich). All solutions were prepared with deionized water meeting PN-EN ISO 3696:1999 standards.The reference material was hydroxyapatite synthesized without the addition of amino acids, hereinafter referred to as HA. During its synthesis, a 150 mmol L−1 aqueous solution of (NH4)2HPO4 was added dropwise to an 333 mmol L−1 aqueous solution of Ca(NO3)2, until a final molar ratio Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P of 5:3 was achieved in the reaction mixture. The synthesis was carried out with stirring at 25 °C under pH at 12, adjusted and maintained by means of an aqueous solution of ammonia. The suspension was allowed to maturate for 24 hours. Next, the sediment was centrifuged out and dried at 80 °C for 24 hours. The syntheses of hydroxyapatites in the presence of amino acids were done in the same way as for reference HA, except that an appropriate amount of amino acid (one or two) was dissolved in the Ca(NO3)2 solution before starting the synthesis. Thus, six samples of apatites in the presence of amino acids were obtained: HA-G (molar ratio Ca:P:Gly of 5:3:10), HA-P (molar ratio Ca:P:Pro of 5:3:10), HA-H (molar ratio Ca:P:HyPro of 5:3:10), HA-GH (molar ratio Ca:P:Gly:HyPro of 5:3:10:10), HA-GP (molar ratio Ca:P:Gly:Pro of 5:3:10:10), HA-HP (molar ratio Ca:P:HyPro:Pro of 5:3:10:10). The presented molar ratios were selected on the basis of previously performed preliminary experiments.
:P of 5:3 was achieved in the reaction mixture. The synthesis was carried out with stirring at 25 °C under pH at 12, adjusted and maintained by means of an aqueous solution of ammonia. The suspension was allowed to maturate for 24 hours. Next, the sediment was centrifuged out and dried at 80 °C for 24 hours. The syntheses of hydroxyapatites in the presence of amino acids were done in the same way as for reference HA, except that an appropriate amount of amino acid (one or two) was dissolved in the Ca(NO3)2 solution before starting the synthesis. Thus, six samples of apatites in the presence of amino acids were obtained: HA-G (molar ratio Ca:P:Gly of 5:3:10), HA-P (molar ratio Ca:P:Pro of 5:3:10), HA-H (molar ratio Ca:P:HyPro of 5:3:10), HA-GH (molar ratio Ca:P:Gly:HyPro of 5:3:10:10), HA-GP (molar ratio Ca:P:Gly:Pro of 5:3:10:10), HA-HP (molar ratio Ca:P:HyPro:Pro of 5:3:10:10). The presented molar ratios were selected on the basis of previously performed preliminary experiments.
2.2. Analytical methods
Spectroscopic FT-IR studies were carried out at 25 °C on a PerkinElmer Spectrum 1000 spectrometer. Spectra were acquired in the 400–4000 cm−1 range at a spectral resolution of 2 cm−1 from KBr pellets using a MCT detector and 30 scans. The spectra were processed using GRAMS/AI 8.0 software (Thermo Scientific, 2006). For the 480–720 cm−1 range, this software was used to perform second derivative calculation (identification of component bands), peak fitting (using Gaussian lines) as well as to calculate areas under the component bands.The crystallinity and crystal size of the samples were examined using a PXRD diffractometer (Bruker D8 Discover), having Cu Kα radiation source (λ = 1.54 Å) and TOPAS software (v.3, Bruker). The software uses Rietveld refinement based on analytical profile functions and least squares algorithms to fit a theoretical pattern to a measured PXRD pattern. The degree of crystallinity was calculated using the following formula:
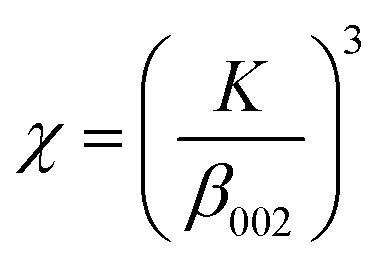
where: d – crystal size [nm], k – constant (0.94 for hydroxyapatite), λ – length of the radiation wave [nm], β – full width at half maximum [rad], θ – the diffraction angle of the corresponding reflex [degrees].18
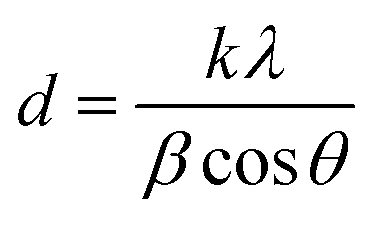
Nanocrystals were observed using a high performance transmission electron microscope JEM 1400 (JEOL Co., Japan, 2008). A drop of the sample suspension in anhydrous ethanol was placed on a nickel grid covered with a formvar film, allowed to dry, and measured under an accelerating voltage of 80 kV. The Ca/P ratio of the synthesized samples was determined using an energy-dispersive full range X-ray microanalysis system (EDS INCA Energy TEM, Oxford Instruments, UK), equipped with a tomographic holder and 11 megapixel TEM camera MORADA G2 (EMSIS GmbH, Germany).
The concentrations of amino acids were determined at 25 °C using a Shimadzu UV 1800 spectrophotometer and quartz cells (l = 1 cm). Absorbance of solutions was measured according to the recently published procedure.19 In order to fully dissolve the obtained hydroxyapatite samples, weights of 400 mg were made and transferred quantitatively to 20 mL volumetric flasks with the use of 1 mol L−1 HCl solution. The solutions prepared in this way were subjected to direct spectrophotometric measurements. The reference to the measurements was a solution of the sample obtained without the addition of amino acids (HA) in 1 mol L−1 HCl. In the case of single-component determinations (mono-amino acid, for: HA-G, HA-P, HA-H), the absorbance was measured at a wavelength of 241 nm. In the case of two-component determinations (di-amino acid, for: HA-GP, HA-GH, HA-HP), the absorbance was measured at 241 and 260 nm, so that on the basis of the Lambert–Beer's law combined with the law of absorbance addictivity it was possible to calculate the content of both amino acids. Amino acid contents were calculated on the basis of appropriate calibration curves obtained after measurements of standard amino acid solutions prepared in 1 mol L−1 HCl (glycine: 37.5–15 mmol L−1; proline: 27.5–11 mmol L−1; hydroxyproline: 25–10 mmol L−1).
The figures and graphs presenting the results were prepared using the KaleidaGraph program (version 3.5 for PC, Synergy Software, 2000) and Microsoft Office Excel.
2.3. Statistical analysis
The differences in mean Ca/P molar ratios, labile/regular phosphate ratios, relative contents of hydroxyl groups, crystallinity indexes, the sizes of the crystals and amino acid contents were statistically analyzed using STATISTICA 13.3 (TIBCO Software Inc.). The following procedures were performed in each case: (i) Shapiro–Wilk test to check the normality of the distribution (p was greater than 0.05 in each case), (ii) Levene's test to check the homogeneity of variance (p was greater than 0.05 in each case), (iii) one-way ANOVA and (iv) post hoc Scheffe test with significance p = 0.05. The statistical significance of the differences for individual parameters was tested in each case for three replications.3. Results and discussion
3.1. Crystal morphology
In the selected TEM photos shown in Fig. 1 it can be seen that both the control hydroxyapatite (HA) crystals and the other samples obtained in the presence of amino acids have nanosize. They take irregular or plate-like shapes with rounded edges and show a high tendency to aggregation, while the aggregates also have free (empty) spaces. Therefore, the main conclusion from the microscopic observations is that the amino acids used in this work, both individually and in mixtures, do not significantly affect either the change of the crystal morphology or the degree of their aggregation. | ||
| Fig. 1 TEM images of all obtained hydroxyapatites. | ||
3.2. Elemental composition
In order to evaluate the deviations from the molar stoichiometry of the synthesized hydroxyapatites modified with various collagen amino acids, X-ray microanalysis (EDXMA) was performed after microscopic observations. For each of the samples, three replications of the measurement analysis were performed, selecting the places where the material appeared to be the thickest under the microscope. The obtained results were used to calculate the mean values of Ca/P molar ratios, the summary of which is presented in the form of a graph in Fig. 2.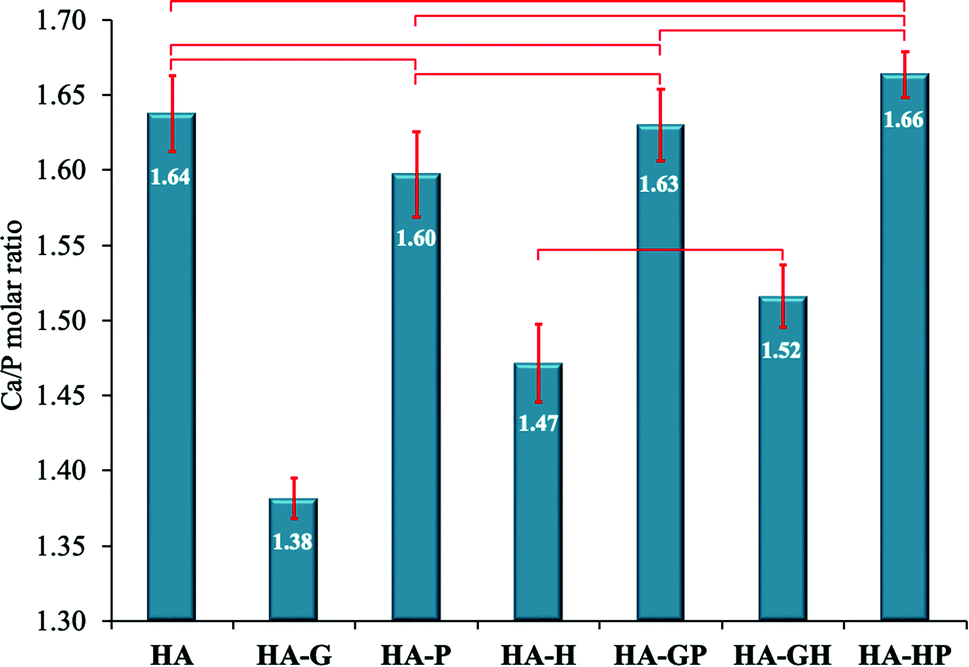 | ||
| Fig. 2 Ca/P molar ratios. Data reported as average ± standard deviation (n = 3). Horizontal bars indicate no statistically significant difference (p > 0.05). | ||
As can be seen, the reference sample (HA) is not a stoichiometric hydroxyapatite, but the mean Ca/P molar ratio calculated for it (1.644 ± 0.025) does not significantly differ from the Ca/P molar ratio of the mixed substrates during the synthesis (1.667) and is within the measurement error.
The relationship regarding the Ca/P molar ratios is clearly visible for all materials obtained in the presence of proline, both alone (HA-P) and in mixtures (HA-GP and HA-HP). Namely, this amino acid did not change the Ca/P molar ratio in a statistically significant manner compared to the reference (HA), with the change in this ratio being the greatest for HA-P. This suggests that the addition of proline during the synthesis of hydroxyapatite materials reduces their deviations from the stoichiometry that could be caused by the presence of other amino acids. This is clearly visible when the samples of HA-G (significant reduction of the Ca/P molar ratio) and HA-GP (the effect of glycine on the reduction of the Ca/P molar ratio were canceled out by proline) were compared. Similar conclusions can be drawn by analyzing the results for HA-H and HA-HP samples.
On the other hand, glycine and, to a lesser extent, hydroxyproline, show a clearly visible ability to reduce the Ca/P molar ratio. HA-G (Ca/P = 1.382 ± 0.013) is characterized by the lowest value of this ratio and only in this sample there are statistically significant differences in relation to all other samples. Interestingly, the effect of the mixture of glycine and hydroxyproline in reducing this ratio is slightly weaker than for the same amino acids used separately.
3.3. Infrared spectroscopy (FT-IR)
The identity of the synthesized materials can be confirmed on the basis of the FT-IR spectra presented in Fig. 3. All the major bands characteristic of hydroxyapatite can be discerned in each spectrum. The most important are the bands corresponding to the stretching vibrations of apatite phosphates: 1092 and 1034 cm−1 (ν3P–O), 962 cm−1 (ν1P–O), 603 and 562 cm−1 (ν4P–O), 473 cm−1 (ν2P–O). Faintly visible, but still observable, are also the bands originating from the stretching vibrations of the structural OH groups of hydroxyapatite: a low intensity band at approx. 3570 cm−1 (masked by an extensive, intense band originating from water molecules) and a band corresponding to libration oscillations at 633 cm−1. There are also visible bands of water molecules, which correspond to bending vibrations in the range of 1630–1650 cm−1 and stretching vibrations of 2900–3500 cm−1 of covalent (O–H) bonds. All the samples obtained are characterized by a very low carbonate content, as evidenced by the residual bands in the 1400–1500 cm−1 range.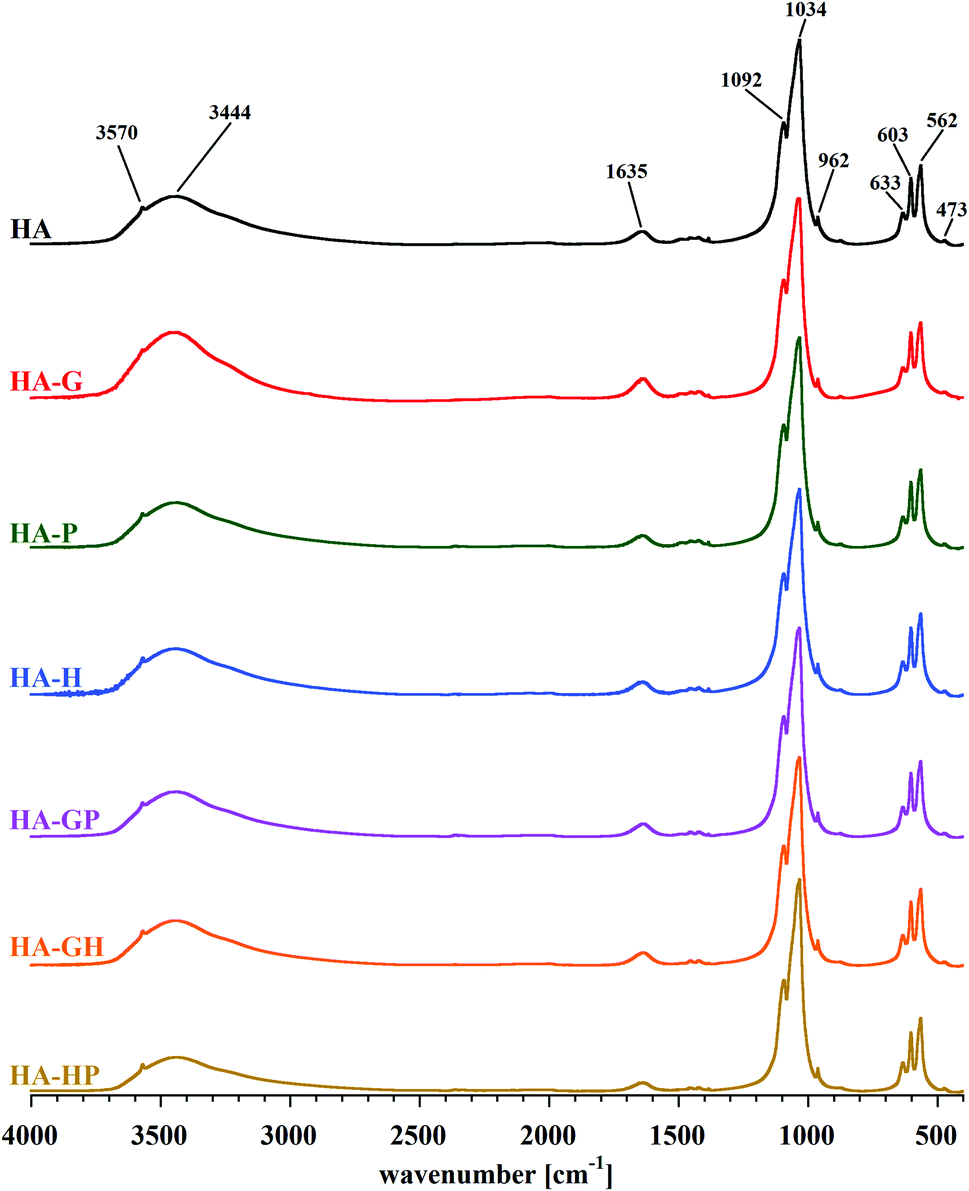 | ||
| Fig. 3 Juxtaposition of FT-IR spectra of all received samples. | ||
On the basis of the FT-IR spectra comparison, it is difficult to determine in the samples the presence of possible residues of amino acids used in the syntheses. The reason is a very low intensity of the bands of the characteristic amino acid functional groups: amine (at approx. 1350 cm−1) and carboxyl (in the range of 1630–1650 cm−1), compared to the very intense bands derived from the mineral itself. An additional difficulty in identifying the bands of carboxyl groups of amino acids is their overlap with the bands occurring in the same range, originating from bending vibrations of water molecules. The spectra presented in Fig. 3 do not differ significantly from each other. Therefore, in order to determine the influence of the amino acids used during the syntheses on the structure of the obtained hydroxyapatites, a detailed analysis of the spectra of each of the samples was carried out in the range limited to 480–720 cm−1. Reducing the wavenumber range made it possible to obtain spectra with higher resolution, which were divided into component bands. A summary of the separated bands for the HA sample is shown in Fig. 4. For the other samples, the component bands were separated in the same way. The values of the coefficients of determination for all peak fittings were higher than 0.998. The interpretation of the identified bands is presented in Table 1.
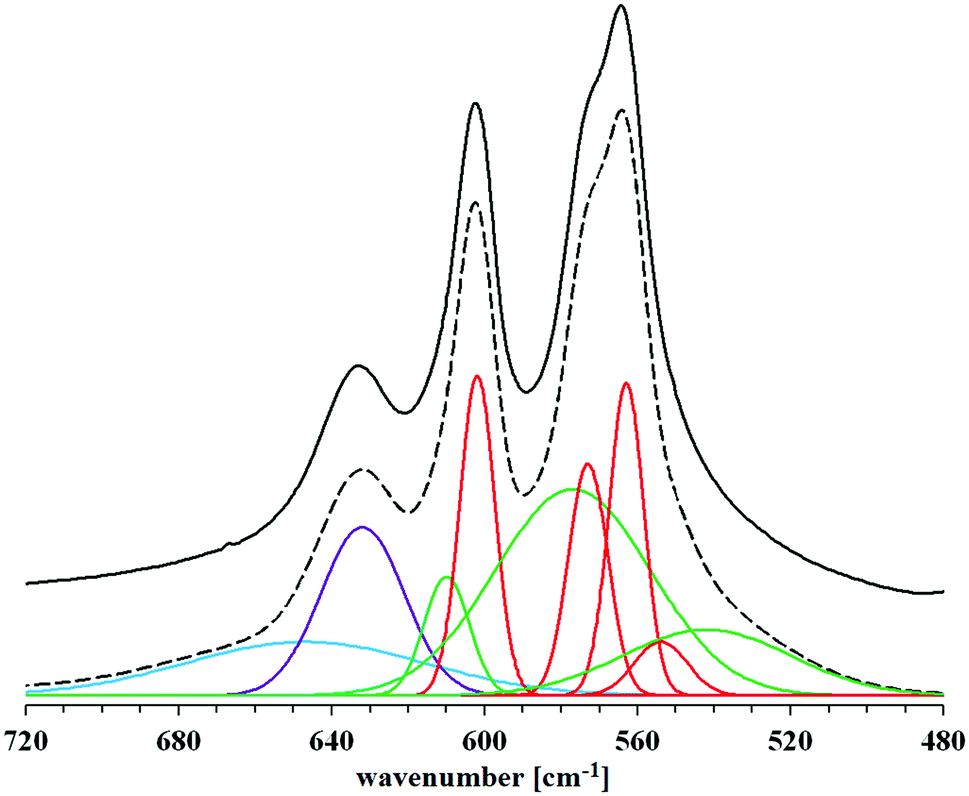 | ||
| Fig. 4 Selected FT-IR spectrum (HA sample) in the range 480–720 cm−1. Bold black line – original spectrum, dashed black line – spectrum obtained after separating and fitting component bands. Color markings: blue – a band corresponding to the vibrations of water molecules; violet – a band corresponding to the vibrations of apatite OH groups; red – bands corresponding to the vibrations of phosphates regularly occurring in the crystal lattice (core); green – bands corresponding to the vibrations of labile phosphates (hydrated surface layer). | ||
| Wavenumber [cm−1] | Assignment | Location in apatite |
|---|---|---|
| 539 | Labile HPO42− | Hydrated surface layer |
| 554 | Regular HPO42− | Crystal core |
| 563 | Regular PO43− | Crystal core |
| 573 | Regular PO43− | Crystal core |
| 577 | Labile PO43− | Hydrated surface layer |
| 602 | Regular PO43− | Crystal core |
| 610 | Labile PO43− | Hydrated surface layer |
| 632 | Apatitic OH− | Crystal core |
| 647 | H2O | Crystal core and/or hydrated surface layer |
As mentioned earlier, the stoichiometric hydroxyapatite has the formula Ca10(PO4)6(OH)2, so it does not contain HPO42− ions. However, detailed analysis of the FT-IR spectra in the range 480–720 cm−1 showed that both the reference sample (HA) and all the others contained these ions. This proves that in each case a more or less non-stoichiometric hydroxyapatite was obtained, which is often represented by the formula Ca10−x(HPO4)x(PO4)6−x(OH)2−x.24 The reason for this is not the use of (NH4)2HPO4 for the synthesis, because the pH was kept at 12 during the precipitation of hydroxyapatite, so that all HPO42− ions could easily convert to PO43−. However, due to the arising disturbances in the crystal lattice (to which hydroxyapatites have a high tendency) and the forced process of intracrystalline partial hydrolysis of PO43− ions, the obtained samples deviate from the stoichiometry. This phenomenon is strongly dependent on the addition of amino acids during the precipitation (see also Fig. 2). HPO42− ions are also formed as a result of the hydrolysis of PO43− ions released into the hydrated surface layer (present on the surface of the crystals even in the case of dry powder), due to the presence of permanently adsorbed water. Thus, the aforementioned hydrated layer contains numerous labile ions (Ca2+, HPO42−, PO43−, OH− and possibly trace amounts of H2PO4−, HCO3− and CO32−).25
Referring to Fig. 4, the two bands corresponding to the lowest wavenumber values, at 539 and 554 cm−1, are assigned to HPO42− ions, respectively: labile and regularly present in the crystal lattice (i.e. in the crystal core). They are characterized by a relatively low intensity compared to the other distinguished bands in the studied wavenumber range. The three highest intensity bands come from phosphates (PO43−) regularly present in the crystal lattice (563, 573 and 602 cm−1). The spectra show two more bands from the labile groups PO43− (577 and 610 cm−1), present in the hydrated surface layer. Among them, a particularly high intensity is characterized by the broad band, the peak of which is located at the lower wavenumber value. The other bands, which appeared in the range above 630 cm−1, are not related to the presence of phosphate groups. The band of greater intensity marked in purple in Fig. 4 (632 cm−1) comes from the vibrations of the structural OH groups of apatite and corresponds to the libration oscillations. The last, relatively wide band indicates the presence of water molecules, which, due to being closed inside the crystal lattice of the tested material and/or firmly bound to its surface, were not removed during the drying process.
All the bands described above appear in the spectrum of each of the samples. Thus, it can be concluded that the crystals of each of the obtained hydroxyapatites contain all types of the mentioned chemical groups. Comparing the blue band on each of the amino acid modified hydroxyapatite spectra, no significant changes in its intensity were observed in relation to the reference hydroxyapatite spectrum. This probably means that the addition of collagen amino acids in the amounts and conditions described in this work does not have a significant effect on the incorporation of water molecules (present in the reaction medium) into the structure of the crystal lattice of the formed hydroxyapatite. Among the bands derived from phosphates regularly occurring in the crystal lattice, visible changes in intensity were observed only for the band at 554 cm−1 associated with the presence of HPO42− groups. They refer to the HA-G and HA-GH samples, but on the basis of the analysis of the other bands belonging to this type, it is difficult to observe clear differences that would explain the changes in intensity. A similar problem was encountered when analyzing the differences in band size at 539 cm−1. It is difficult to associate changes in its intensity with changes taking place in other bands. However, looking at the overall analysis of the presented FT-IR spectra in the range of 480–720 cm−1, it can be concluded that clear differences in intensities occur between the bands at the values of 539 and 554 cm−1, for which the HPO42− groups are responsible. This suggests that the addition of amino acids during the synthesis carried out in this experiment influenced mainly the content of both types of hydrogen phosphates in the obtained materials.21–23,26
In the range of 480–720 cm−1 the percentage of the surface area of each component band was calculated with respect to the total surface area of all the separated bands. Next, the relative labile/regular (L/R) phosphate ratio was calculated by adding up the band surface percentages of both types of phosphates. The ratios obtained are shown in Fig. 5. Using the described methodology allows the L/R ratio to reflect changes in the relative content of labile phosphates, and thus may be a relative measure of the degree of development of the hydrated surface layer.
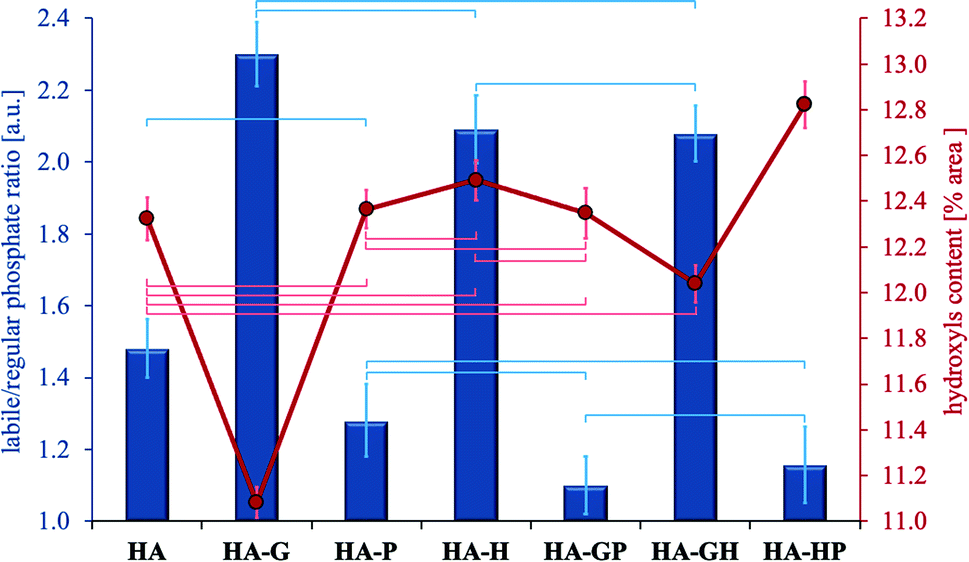 | ||
| Fig. 5 Changes of: the relative labile/regular phosphate ratio (vertical bars, left scale) and the relative content of hydroxyl groups (curve, right scale). According to the peak fitting in the 480–720 cm−1 range of FT-IR spectra. Data reported as average ± standard deviation (n = 3). Horizontal bars of the appropriate color indicate no statistically significant difference (p > 0.05). | ||
The highest L/R ratio is found in hydroxyapatites obtained in the presence of glycine and hydroxyproline (this applies to both single amino acids and their mixtures). Although this ratio is the highest for HA-G, statistical analysis showed no significant differences in comparing this sample to both HA-H and HA-GH. Thus, the glycine and hydroxyproline present in the reaction medium during the synthesis contribute to an increase in the percentage of labile phosphates in the resulting crystals (compared to the reference) in a statistically significant manner. On the other hand, the decrease in the calculated L/R ratio in three types of samples with the addition of proline (HA-P, HA-GP, HA-HP) suggests that its presence during the synthesis of hydroxyapatite promotes the incorporation of phosphate groups into the crystal lattice (samples contain the least labile phosphates). The L/R ratios for the samples HA-P, HA-GP and HA-HP do not differ statistically significantly, but the difference is seen in the comparison of HA-GP and HA-HP to the reference (HA). However, no statistically significant difference was found between HA-P and the reference. It should also be noted that the Ca/P molar ratio values (Fig. 2) are correlated with the L/R ratio values (Fig. 5): samples with the highest labile phosphate values (highest L/R ratio) have the lowest Ca/P molar ratios. Thus, the fact of the greatest influence of glycine and, to a lesser extent, hydroxyproline on the newly formed hydroxyapatite in their presence is again confirmed.
Fig. 5 also shows the relative percentage of hydroxyl groups for all samples as the percentage of the band area at 632 cm−1 in the total area of all bands separated in the range 480–720 cm−1. The difference in the content of the OH groups between the samples with the highest and the lowest content does not exceed 2%. The lowest relative percentage of OH groups is found in hydroxyapatite modified with glycine and only in this sample there are statistically significant differences in relation to all other samples. This is most likely due to the fitting of glycine molecules into channels present in the apatite crystal lattice, originally occupied by hydroxyl groups.8 Other amino acids are not able to integrate into the hydroxyapatite crystal lattice due to the too large size of their molecules.
The other samples did not differ in the content of hydroxyl groups in a statistically significant manner compared to the control hydroxyapatite, except for HA-HP for which the difference is significant. Thus, the presence of proline and hydroxyproline during the synthesis of hydroxyapatite should not be associated with their significant influence on the percentage of hydroxyl groups in the finally obtained materials, except for the use of these amino acids as a mixture.
3.4. Phase purity, crystallinity and crystal size
Diffractometric studies were used to determine the phase purity, crystallinity and crystal size of the obtained hydroxyapatites. The obtained PXRD patterns are summarized in Fig. 6.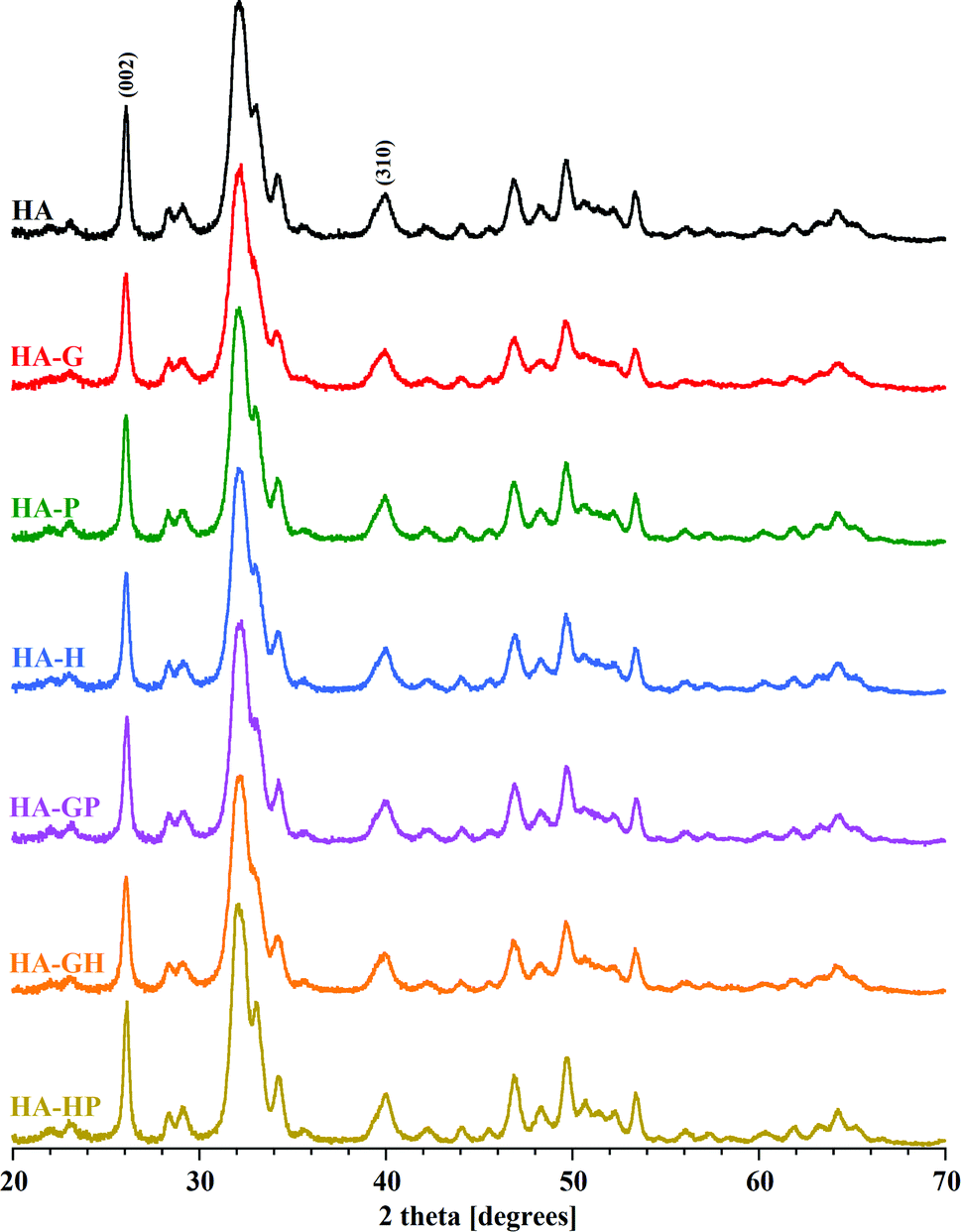 | ||
| Fig. 6 PXRD patterns of all samples. The reflexes (002) and (310), used to calculate the crystallinity and crystal size, are marked. | ||
For all samples, the diffraction lines indicated the presence of a single crystalline phase characteristic of fine crystalline hydroxyapatites. The presence of all the main reflexes coming from the hydroxyapatite lattice planes and the significant similarity of the PXRD patterns of the amino acid modified powders to the reference diffractogram (HA) confirm the purity and phase homogeneity of the products. Looking closely at the diffraction patterns, only a slight distortion and broadening of reflections for the glycine-modified hydroxyapatite sample can be seen. Proline and hydroxyproline seem to have no significant effect on changes in the structure of HA crystals synthesized in their presence. Comparable results of the PXRD studies were obtained in other experiments available in the literature.5,6
It may be difficult to assess the degree of crystallinity of individual samples of hydroxyapatite powders solely on the basis of diffraction patterns. They result from the very high similarity of the obtained diffraction line profiles. It is difficult to see visually very slight changes in the intensity, width and shape of the analyzed reflections between different samples. For these reasons, the degree of crystallinity of all the hydroxyapatites was calculated mathematically. The obtained values of the degree of crystallinity of each of the tested samples are shown in Fig. 7.
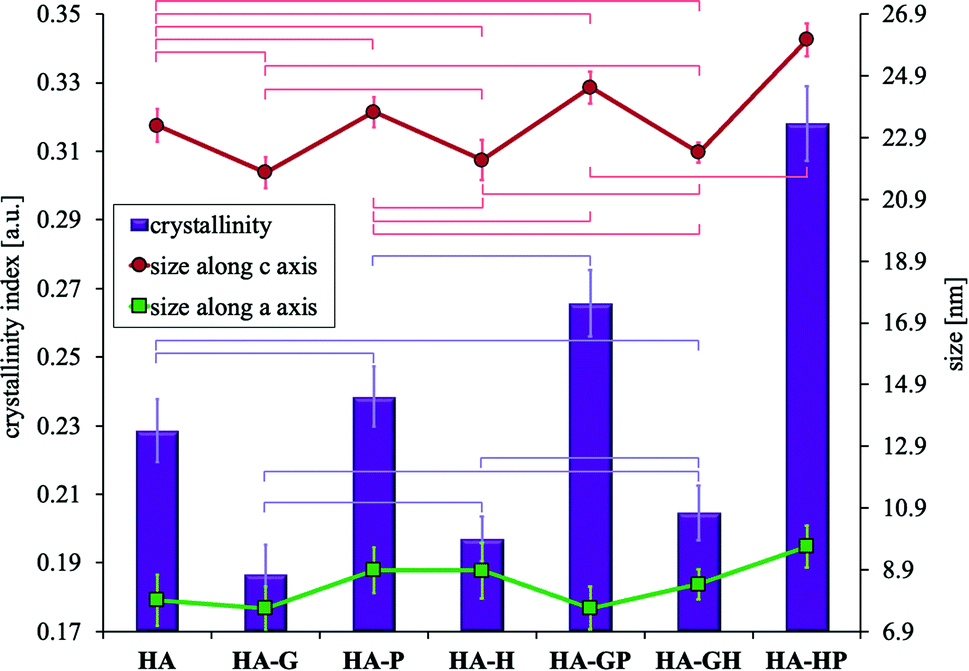 | ||
| Fig. 7 Changes of: the crystallinity (vertical bars, left scale) and the sizes of crystals (right scale) along axes c (red curve with circles) and a (green curve with squares). According to the calculations from PXRD patterns. Data reported as average ± standard deviation (n = 3). Horizontal bars of the appropriate color indicate no statistically significant difference (p > 0.05). For the sizes along the a axis, in each case there are no statistically significant differences (not marked with bars). | ||
All samples modified with amino acids differ statistically significantly in the degree of crystallinity from the reference HA, except for HA-P and HA-GH for which statistically no differences can be demonstrated. Compared to the reference, HA-G (to the greatest extent) and HA-H have significantly reduced crystallinity, while HA-GP and HA-HP (to the greatest extent) have significantly increased crystallinity. Comparing the values of the degree of crystallinity to the results of the Ca/P molar ratio (Fig. 2), consistency between these data was observed for all samples obtained in the presence of amino acids. Therefore, it can be concluded that the discrepancy in the crystallinity of individual samples was mainly influenced by the differences in the content of calcium and phosphate ions, including hydrogen phosphate, which significantly contributed to the reduction of the Ca/P molar ratio.
Due to the strong tendency of nanocrystals of the tested samples to form large aggregates, the visual size assessment is much more difficult. Based on the TEM observations of the tested materials, it can be seen that in each case the sizes of single crystals are below 100 nm. When determining the size of crystals smaller than 100 nm, the analysis of the profile of diffraction lines is often used, consisting in measuring the width of the reflections obtained on the diffraction pattern (Scherrer method). Based on the diffraction patterns, the full widths at half maximum of the reflexes were determined for the two types of lattice planes marked in Fig. 6 as (002) (corresponding to the crystal c axis) and (310) (corresponding to the crystal a axis), to which the diffraction angle 2 theta values were assigned, respectively 26 and 40 degrees. The Scherrer method is one of the indirect methods of determining the size of crystals. It is based on the relationship between the size of the studied crystals and the width of the reflections visible on the diffraction pattern. This dependence connects the broadening of reflections with a decrease in the size of the studied crystals. One of the advantages of this method is that the degree of aggregation of the material does not affect the calculation results. The calculated crystal sizes of the individual samples along the c and a axes are shown in Fig. 7.
The obtained results indicate that all synthetically obtained hydroxyapatite powders are nanocrystalline and can be classified as nanomaterials. In all cases, the size along the c axis is more than twice greater than the size along the a axis. Considering the size along the c axis, the HA-HP sample has the largest crystals (but there was no statistically significant difference with that of the HA-GP sample). For the remaining samples, no statistically significant differences could be demonstrated. On the other hand, when considering the changes in the size of the crystals along the a axis, no statistically significant differences were found in any case. Fig. 7 shows a correlation between the value of the degree of crystallinity and the size of the crystals along the c axis (the greater the crystallinity, the greater the length along the c axis), while the statistical analysis showed no significant relationships.
3.5. Determination of residues of amino acids
Although all samples of hydroxyapatites were thoroughly washed after synthesis, we decided to check whether it would be possible to identify amino acids in them. The determination of the amino acid content was carried out using the method previously described. This method uses spectrophotometric measurements of strong acid solutions, in which the uniform form of the amino acid with a completely undissociated carboxyl group is determined.8,18The analysis of Table 2 clearly shows that the affinity of the used collagen amino acids for hydroxyapatite decreases in the following order: glycine > hydroxyproline > proline. Glycine, as the smallest of the amino acids used, can be located both in the hydroxyapatite crystal lattice and on the crystal surface and is relatively strongly bound to it,8 hence the HA-G sample contains the most amino acid. Hydroxyproline is slightly less bound to hydroxyapatite, but its presence could be found in the HA-H material. Interestingly, the use of glycine and hydroxyproline in the mixture reduces the binding capacity of both these amino acids and, consequently, their content in the HA-GH sample is lower than in HA-G and HA-H. Proline, on the other hand, does not show the ability to bind to hydroxyapatite (it was not detected in the HA-P sample). And most interestingly, proline also prevents the binding of glycine and hydroxyproline with hydroxyapatite, because when used in mixtures in the samples HA-GP and HA-HP, it was impossible to detect the presence of any of the amino acids used.
| Sample code | Gly% (m m−1) ± SD | Pro% (m m−1) ± SD | HyPro% (m m−1) ± SD |
|---|---|---|---|
| HA-G | 24.1 ± 0.3 | N/A | N/A |
| HA-P | N/A | ∼0 | N/A |
| HA-H | N/A | N/A | 8.3 ± 0.2 |
| HA-GP | ∼0 | ∼0 | N/A |
| HA-GH | 18.6 ± 0.4 | N/A | 5.6 ± 0.5 |
| HA-HP | N/A | ∼0 | ∼0 |
The described ability to bind amino acids with hydroxyapatite directly affects most of the parameters described in this study: the Ca/P molar ratio (lowered when amino acids with high affinity for hydroxyapatite are used), the L/R ratio (more labile phosphates, and therefore more expanded hydrated surface layer, have samples for which amino acids with high affinity for hydroxyapatite were used), crystallinity and crystal size along the c axis (decreased in the case of using amino acids with high affinity for hydroxyapatite).
4. Conclusions
(1) All hydroxyapatites obtained in the presence of amino acids are classified as nanomaterials.(2) All collagen amino acids used in this work have an impact on changes in the structure of the quasi-liquid, hydrated surface layer of hydroxyapatites, with it being the most developed in the case of hydroxyapatite obtained in the presence of glycine, followed by hydroxyproline and a mixture of glycine and hydroxyproline, respectively.
(3) The addition of amino acids (glycine, proline, hydroxyproline and their mixtures) during the synthesis of hydroxyapatites did not cause significant changes in the size, morphology and degree of aggregation of the crystals.
(4) Differences were found in the values of the degree of crystallinity and the Ca/P molar ratios for individual hydroxyapatite powders. Changes in both parameters were correlated with each other.
(5) Changes in the Ca/P molar ratios and a detailed analysis of the FT-IR spectra component bands in the range of 480–720 cm−1, with the simultaneous lack of large differences in the crystal sizes of individual samples, indicate that the changes in the crystallinity of the obtained hydroxyapatite materials are caused by different contents labile phosphates included in the hydrated surface layer.
(6) Among the used collagen amino acids, glycine shows the highest affinity for hydroxyapatite. Hydroxyproline binds slightly less, while proline the weakest. The affinity of the amino acid for hydroxyapatite directly affects the Ca/P molar ratio, the degree of expansion of the hydrated surface layer, and the degree of crystallinity of the final product.
(7) Glycine, as the only one of the collagen amino acids used in this work, has a significant impact on the relative content of hydroxide groups in the synthesized materials (it reduces their relative content). However, for the effect to be significant, glycine cannot be used in a mixture with another amino acid.
(8) The influence of amino acid mixtures on the tested parameters is strongly dependent on the composition of the mixture. The dominant effect of proline can be noticed here, which in combination with both glycine and hydroxyproline, to some extent reduces the influence of each of these amino acids on the parameters tested.
5. Possible applications of the results
The obtained nanocrystalline hydroxyapatite powders should be further tested for biological evaluation (cyto-, genotoxicity and effect on osteoblasts). If satisfactory results are obtained, after being given the appropriate form, they could be used as substitutes for hard tissues of the human body. The mean values of the Ca/P molar ratios of the obtained samples are similar to those characteristic for biological hydroxyapatites naturally occurring in the bones and teeth of the human body (1.58–1.73 depending on age27). This great resemblance to bone hydroxyapatite and the content of collagen amino acids in the case of HA-G, HA-P and HA-HP samples may determine the high biocompatibility of the materials synthesized in this study. On the other hand, the fact that the obtained samples contain a homogeneous hydroxyapatite phase without the addition of other calcium phosphates, may (depending on the application) make these materials resistant to the phenomenon of biodegradation.In the case of HA-G, HA-P and HA-HP samples (especially the first of them), amino acids relatively permanently attached to minerals may also affect the ability to bind (adsorb) other organic molecules, including drugs. Therefore, the obtained nanomaterials can potentially also act as carriers of appropriately selected drug molecules.
The research results presented in this paper may be helpful in the development of hydroxyapatite biomaterials with specific, desired physicochemical properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Center (Poland) Miniatura-3 project (number 2019/03/X/NZ7/00783) and Medical University of Warsaw (FW23/N/21).Notes and references
- E. Fiume, G. Magnaterra, A. Rahdar, E. Verne and F. Baino, Ceramics, 2021, 4, 542–563 CrossRef CAS.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- M. Tavafoghi and M. Cerruti, J. R. Soc., Interface, 2016, 13, 20160462 CrossRef PubMed.
- I. Erceg, N. Maltar-Strmečki, D. D. Jurašin, V. Strasser, M. Ćurlin, D. M. Lyons, B. Radatović, N. M. Mlinarić, D. Kralj and M. D. Sikirić, Crystals, 2021, 11, 792 CrossRef CAS.
- T. Matsumoto, M. Okazaki, M. Inoue, Y. Hamada, M. Taira and J. Takahashi, Biomaterials, 2002, 23, 2241–2247 CrossRef CAS PubMed.
- T. Matsumoto, M. Okazaki, M. Inoue, J.-I. Sasaki, Y. Hamada and J. Takahashi, Dent. Mater. J., 2006, 25, 360–364 CrossRef CAS PubMed.
- M. H. Uddin, T. Matsumoto, S. Ishihara, A. Nakahira, M. Okazaki and T. Sohmura, J. Dent. Res., 2010, 5, 488–492 CrossRef PubMed.
- S. Krukowski, N. Lysenko and W. Kolodziejski, J. Solid State Chem., 2018, 264, 59–67 CrossRef CAS.
- N. Almora-Barrios, K. F. Austen and N. H. De Leeuw, Langmuir, 2009, 9, 5018–5025 CrossRef PubMed.
- G. Zhang, J. Chen, S. Yang, Q. Yu, Z. Wang and Q. Zhang, Mater. Lett., 2011, 65, 572–574 CrossRef CAS.
- R. Sharma, R. R. Pandey, A. A. Gupta, S. Kar and M. Dhayal, Mater. Chem. Phys., 2012, 133, 718–725 CrossRef CAS.
- S. Koutsopoulos and E. Dalas, Langmuir, 2000, 16, 6739–6744 CrossRef CAS.
- Z. Chen, Y. Fu, Y. Cai and J. Yao, Mater. Lett., 2012, 68, 361–363 CrossRef CAS.
- S. Koutsopoulos and E. Dalas, J. Colloid Interface Sci., 2000, 231, 207–212 CrossRef CAS PubMed.
- P. Comeau and T. Willet, Sci. Rep., 2018, 8, 12700 CrossRef PubMed.
- S. Yanyan, W. Guangxin, S. Guoqing, W. Yaming, L. Wuhui and A. Osaka, RSC Adv., 2020, 10, 37005–37013 RSC.
- G. Ulian, D. Moro and G. Valdré, Biomolecules, 2021, 11, 728 CrossRef CAS PubMed.
- K. Szurkowska, A. Zgadzaj, M. Kuras and J. Kolmas, Ceram. Int., 2018, 44, 18551–18559 CrossRef CAS.
- S. Krukowski, M. Karasiewicz and W. Kolodziejski, J. Food Drug Anal., 2017, 25, 717–722 CrossRef CAS PubMed.
- D. Eichert, C. Drouet, H. Sfihia, C. Rey and C. Combes, Nanocrystalline Apatite-based Biomaterials, Nova Science Publishers Inc., New York, 2009 Search PubMed.
- A. Kaflak and W. Kolodziejski, J. Mol. Struct., 2011, 990, 263–270 CrossRef CAS.
- J. Kolmas, A. Jaklewicz, A. Zima, M. Bućko, Z. Paszkiewicz, J. Lis, A. Ślósarczyk and W. Kolodziejski, J. Mol. Struct., 2011, 987, 40–50 CrossRef CAS.
- L. Pajchel, V. Kowalska, D. Smolen, A. Kedzierska, E. Pietrzykowska, W. Lojkowski and W. Kolodziejski, Mater. Res. Bull., 2013, 48, 4818–4825 CrossRef CAS.
- F.-H. Lin, C.-J. Liao, K.-S. Chen and J.-S. Sun, Biomaterials, 1998, 19, 1101–1107 CrossRef CAS PubMed.
- C. Drouet, M. Aufray, S. Rollin-Martinet, N. Vandecandelaére, D. Grossin, F. Rossignol, E. Champion, A. Navrotsky and C. Rey, Am. Mineral., 2018, 103, 550–564 CrossRef.
- C. Rey, M. Shimizu, B. Collins and M. J. Glimcher, Calcif. Tissue Int., 1990, 46, 384–394 CrossRef CAS PubMed.
- K. J. Quelch, R. A. Melick, P. J. Bingham and S. M. Mercuri, Arch. Oral Biol., 1983, 8, 665–674 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |