
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards novel tacrine analogues: Pd(dppf)Cl2·CH2Cl2 catalyzed improved synthesis, in silico docking and hepatotoxicity studies†
Aravinda Babua,
Muthipeedika Nibin Joyb,
K. Sunil *a,
Ayyiliath Meleveetil Sajith*a,
Sougata Santrab,
Grigory V. Zyryanovbc,
Olga A. Konovalovab,
Ilya I. Butorinb and
Keesaram Munirajud
*a,
Ayyiliath Meleveetil Sajith*a,
Sougata Santrab,
Grigory V. Zyryanovbc,
Olga A. Konovalovab,
Ilya I. Butorinb and
Keesaram Munirajud
aDepartment of Chemistry, SSIT, Sri Siddhartha Academy of Higher Education, Tumkur, Karnataka, India-572107. E-mail: sunilk999@gmail.com; sajithmeleveetil@gmail.com; Tel: +91-9480146151
bInstitute of Chemical Technology, Ural Federal University, 19 Mira Street, Yekaterinburg, Russia-620002
cI. Ya. Postovskiy Institute of Organic Synthesis, Ural Division of the Russian Academy of Sciences, 22 S. Kovalevskoy Street, Yekaterinburg, Russia-620219
dGovernment Degree College-Puttur (Affiliated to S. V. University, Tirupati), Narayanavanam Road, Puttur, Chittoor (Dt), Andhra Pradesh, India-517583
First published on 11th August 2022
Abstract
A plethora of 6-(hetero)aryl C–C and C–N bonded tacrine analogues has been made accessible by employing palladium mediated (Suzuki–Miyaura, Heck, Sonogashira, Stille and Buchwald) cross-coupling reactions, starting from either halogenated or borylated residues. The successful use of Pd(dppf)Cl2·CH2Cl2 as a common catalytic system in realizing all these otherwise challenging transformations is the highlight of our optimized protocols. The analogues thus synthesized allow the available chemical space around the C-6 of this biologically relevant tacrine core to be explored. The in silico docking studies of the synthesized compounds were carried out against the acetylcholinesterase (AChE) enzyme. The hepatotoxicity studies of these compounds were done against complexes of CYP1A2 and CYP3A4 proteins with known inhibitors like 7,8-benzoflavone and ketoconazole, respectively.
Introduction
Alzheimer's disease (AD) is a chronic and progressive neurodegenerative disease characterized by loss of memory and cognitive capacity.1 Tacrine was the first AChE inhibitor introduced in the market for the treatment of AD. However, the poor selectivity of this drug candidate resulted in side-effects, mainly hepatotoxicity.2 Current AChE inhibitors used in the treatment of AD are donepezil, rivastigmine and glutamine. However, the minor side-effects associated with these drugs further demand the development of new analogues with minimal hepatotoxicity. Efforts have been made by various researchers across the globe to design and synthesize novel tacrine analogues with minimal hepatotoxicity.3 Accordingly, modifications around the tacrine core have been performed by introducing novel functionalities in the ring and by increasing or decreasing the ring sizes.4 Our research was focused on utilizing the bromo group in 6-bromo tacrine as a handle to explore the available chemical space around the tacrine core.Cross-coupling reactions (C–C and C–N) have played a pivotal role in expanding the available chemical space of a lead compound during the drug development process.5 The use of this highly reliable and flexible methodology for developing lead like molecules to the lead optimization stage highlights its significance in medicinal chemistry. The impact of these cross-coupling technologies can be further exemplified by their large-scale applications for accessing various clinical candidates. Recent advancements in this field have further allowed the incorporation of diverse frameworks in a biologically relevant core leading to the generation of complex molecular architectures.6
As a part of our research programme aimed at expanding the available chemical space around the C-6 of the tacrine core,7 we were interested in employing palladium mediated cross-coupling reactions to synthesize novel acetyl cholinesterase inhibitors. The use of 6-bromo tacrine as a handle (for Suzuki–Miyaura, Stille, Heck, Sonogashira and Buchwald cross-coupling) was explored to furnish these novel analogues around this biologically relevant core. Several notable advances in the field of developing novel catalytic systems has led to various benefits such as increased substrate scope, milder reaction conditions, low catalyst loading etc.8 Despite the development of an array of electronically and sterically demanding catalytic systems, the need for different catalysts for various types of cross-coupling reactions still remains as a major hurdle. The use of diverse electronically and sterically demanding catalysts for successful cross-coupling reactions of a common electrophile with distinctive nucleophilic coupling partners is a major challenge prevailing in this field.9 Accordingly, our research was focussed on developing a common catalytic system for the cross-coupling of 6-bromo tacrine with various nucleophilic coupling partners (boronic acids, alkenes, alkynes and amines). In this paper, we herein report the utilization of Pd(dppf)Cl2·CH2Cl2 as a common catalyst in various palladium catalyzed cross-coupling reactions for the synthesis of diverse, pharmacologically relevant 6-substituted tacrine derivatives. The molecular docking and hepatotoxicity studies of these promising compounds were carried out at the later stage.
Results and discussion
Chemistry
The model substrate 6-bromo tacrine 1 was initially synthesized according to the previously reported procedure.7a Our efforts were focussed on the cross-coupling of this model substrate with various nucleophilic coupling partners employing a common catalytic system. The presence of free amine group adds further challenges in developing a common catalytic system for this scaffold.7a We started our synthetic plans by screening the palladium catalyzed borylation reaction of 1 with bis(pinacolonato)diboron 2 (Scheme 1). After the careful screening of various reaction parameters, we obtained the desired 6-borylated tacrine 3 in 73% isolated yield. The detailed optimization studies for developing this methodology have been summarized in Table 1. | ||
| Scheme 1 Synthesis of 6-borylated tacrine derivative. | ||
| Entry | Catalyst | Base | Solvent | Yieldb 3 (%) |
|---|---|---|---|---|
| a Reaction conditions: 6-bromo tacrine (1 mmol), bis(pinacolonato)diboron (1.5 mmol), catalyst (5 mol%), base (2.5 mmol), solvent, 100 °C for 8 h.b Isolated yield. | ||||
| 1 | Pd2(dba)3 | Cs2CO3 | DMF | Traces |
| 2 | Pd2(dba)3 | K2CO3 | DMF | 10 |
| 3 | Pd2(dba)3 | CsF | DMF | Traces |
| 4 | Pd2(dba)3 | Na2CO3 | DMF | 8 |
| 5 | Pd(dppf)Cl2 | K2CO3 | DMF | 30 |
| 6 | PdCl2·(CH3CN)2 | K2CO3 | DMF | Traces |
| 7 | Pd(OAc)2 | K2CO3 | DMF | Traces |
| 8 | Pd(PPh3)4 | K2CO3 | DMF | Traces |
| 9 | PdCl2.(PPh3)2 | K2CO3 | DMF | Traces |
| 10 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | DMF | 50 |
| 11 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane | 60 |
| 12 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | H2O | 40 |
| 13 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
65 |
| 14 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane–H2O (2:1) |
73 |
We screened a variety of Pd catalysts, base and solvent system so as to obtain the 6-borylated tacrine in high yields (Table 1). Among the various catalysts screened, Pd(dppf)Cl2·CH2Cl2 gave better results (Table 1, entries 1–10). The utilization of K2CO3 or Na2CO3 procured superior results when compared to the other bases screened (Table 1, entries 1–4). We also observed that the utilization of dual solvent system plays a pivotal role in improving the yield of the desired product (Table 1, entries 11–14). Finally, the optimized reaction conditions were affixed as detailed in entry 14 of Table 1 since we obtained the desired product in 73% isolated yield (Scheme 1).
After optimizing the suitable reaction conditions for the effective borylation of 6-bromo tacrine, we directed our attention towards utilizing this borylated tacrine 3 as a scaffold for the Suzuki–Miyaura cross-coupling reactions (Scheme 2). Accordingly, this intermediate 3 was treated with three different aryl bromides 4a–c by employing the previously developed protocol of the borylation reaction. The reaction was carried out at 100 °C for 8–10 hours. Gratifyingly, all the aryl bromides reacted well enough to furnish the desired products 7a–c in good yields. The aryl bromides having a chloro substitution at para position gave a slightly lesser yield (73%) of the expected product 7c when compared to aryl bromides possessing m-fluoro and m-difluoro substituents. However, the reaction was found to be compatible for synthesizing a variety of tacrine derivatives by Suzuki–Miyaura cross-coupling conditions.
 | ||
| Scheme 2 Synthesis of 6-arylated tacrine derivatives by Suzuki–Miyaura coupling; reaction conditions: 6-borylated tacrine (1 mmol), aryl bromide (1.2 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), K2CO3 (2.5 mmol), 1,4-dioxane (2 mL), H2O (1 mL), 100 °C for 8–10 h. | ||
The aforementioned results gave us the idea of developing a one-pot protocol for borylation and Suzuki–Miyaura coupling as the developed reaction conditions was identical (Scheme 3). Accordingly, we added 6-bromo tacrine 1, bis(pinacolonato)diboron 2 and aryl bromide 4a along with catalyst, base and solvent and the reaction mixture was heated at 100 °C according to the previously optimized protocol. Unfortunately, the desired product 7a was obtained in 15% yield only whereas the borylated product was procured in 40% isolated yield. Hence, the one-pot protocol was not found to be effective in our optimization studies.
 | ||
| Scheme 3 One-pot protocol for borylation and Suzuki–Miyaura coupling; reaction conditions: 6-bromo tacrine (1 mmol), bis(pinacolonato)diboron (1.5 mmol), aryl bromide (1.2 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), K2CO3 (2.5 mmol), 1,4-dioxane (2 mL), H2O (1 mL), 100 °C for 10 h. | ||
The success of borylation and Suzuki–Miyaura cross-coupling reactions for the synthesis of novel tacrine analogues enlightened us to further explore the 6-bromo tacrine scaffold. We hypothesized that the 6-bromo tacrine 1 can be directly subjected to Suzuki–Miyaura cross-coupling conditions with different boronic acids in our previously optimized protocol. To substantiate this hypothesis, we treated the scaffold 1 with a series of boronic acids 5a–e by employing Pd(dppf)Cl2·CH2Cl2 as the catalyst, K2CO3 as base and 1,4-dioxane/H2O (2:1) as the solvent system at 100 °C for 8–10 hours (Scheme 4). To our delight, we obtained the desired products 7a–e in good to excellent yields (78–87%). The boronic acids bearing electron donating groups afforded the required products (7d, 7e) in good yields whereas the boronic acids possessing electron withdrawing groups yielded the desired products (7a–c) in excellent yields. The Suzuki–Miyaura cross-coupling methodology of 6-bromo tacrine scaffold with boronic acids was found to be slightly better than that of 6-borylated tacrine derivative (with aryl bromides) in terms of isolated yield of the obtained products.
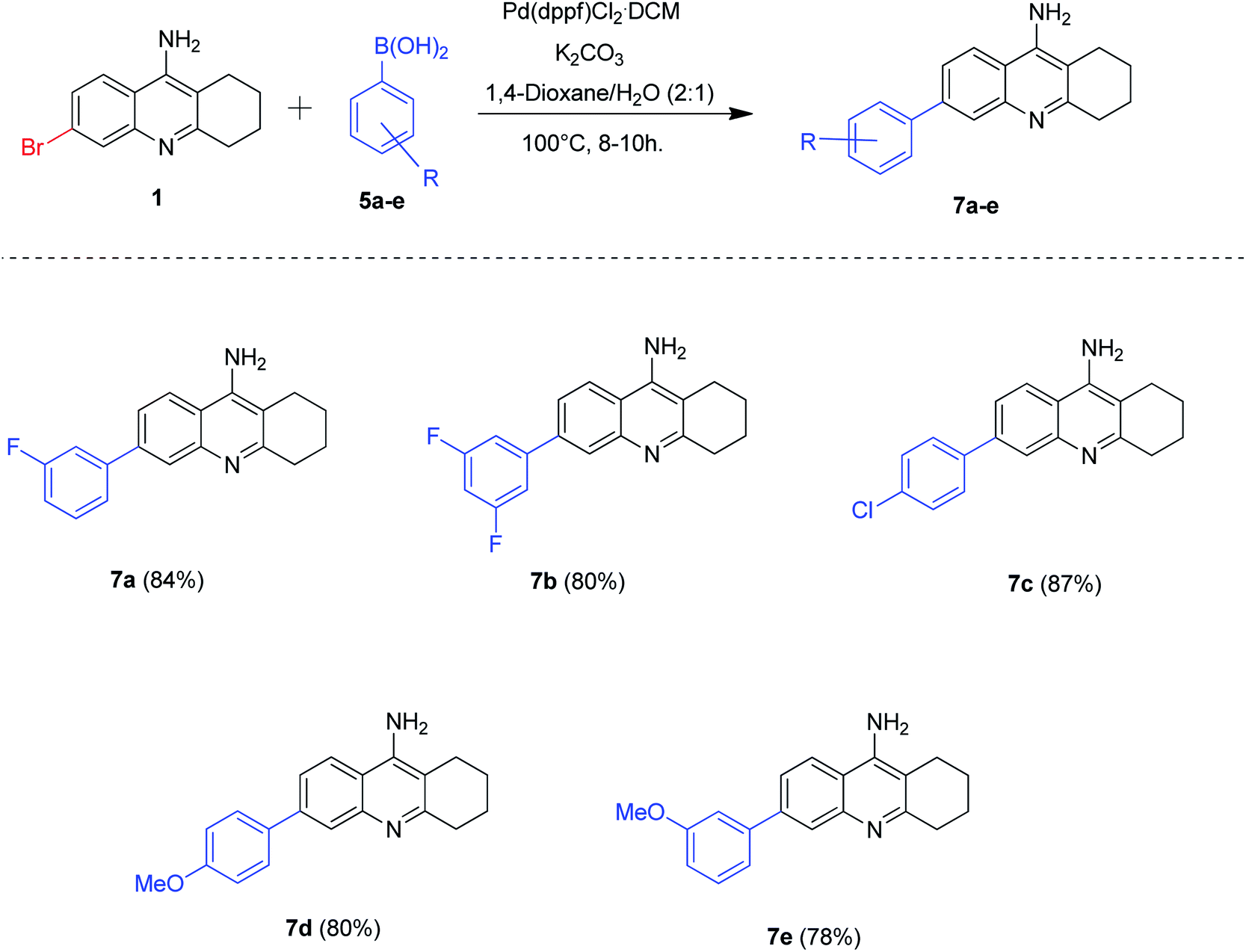 | ||
| Scheme 4 Synthesis of 6-aryl tacrine derivatives by Suzuki–Miyaura coupling; reaction conditions: 6-bromo tacrine (1 mmol), aryl boronic acid (1.2 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), K2CO3 (2.5 mmol), 1,4-dioxane (2 mL), H2O (1 mL), 100 °C for 8–10 h. | ||
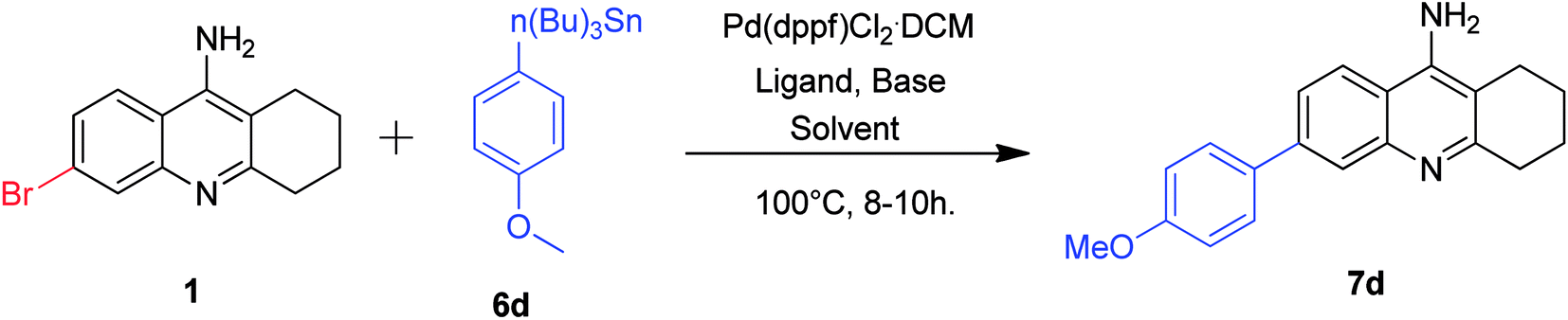
Our next attention was to optimize the developed methodology for Stille cross-coupling conditions. Taking this into consideration, we treated the 6-bromo tacrine 1 with tributyl(4-methoxyphenyl)stannane 6d in our developed protocol. Unfortunately, we obtained the desired product 7d in slightly lesser yield at this time (Table 2, entry 1). We carried out the optimization studies by screening different solvent systems and bases/additives (Table 2). Gratifyingly, we found better yield of the required product when the reaction was carried out in DMF (Table 2, entries 4–8). Finally, we got the expected product in 80% isolated yield when NaCl was employed as an additive (Table 2, entry 8). Even though the actual role of NaCl is unclear in the outcome of this reaction, it is speculated to undergo halide exchange with the oxidative adduct complex before the transmetallation step.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base/additive | Solvent | Yieldb 7d (%) |
| a Reaction conditions: 6-bromo tacrine (1 mmol), tributyl(4-methoxyphenyl)stannane (1 mmol), catalyst (5 mol%), base (1.5 mmol), solvent (2 mL), 100 °C for 8 h.b Isolated yield. | ||||
| 1 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane–H2O (2:1) |
40 |
| 2 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane | 30 |
| 3 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | H2O | Traces |
| 4 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | DMF | 65 |
| 5 | Pd(dppf)Cl2·CH2Cl2 | Na2CO3 | DMF | 60 |
| 6 | Pd(dppf)Cl2·CH2Cl2 | Cs2CO3 | DMF | 50 |
| 7 | Pd(dppf)Cl2·CH2Cl2 | CsF | DMF | 55 |
| 8 | Pd(dppf)Cl2·CH2Cl2 | NaCl | DMF | 80 |
After optimizing the reaction conditions for Stille coupling, we decided to evaluate the substrate scope of this developed protocol (Scheme 5). Accordingly, we synthesized different 6-substituted tacrine analogues 7d–h successfully in good to excellent yields (70–88%). The products 7d and 7e was also obtained in good yields by Suzuki–Miyaura coupling conditions. However, considering the toxicity of the stannanes, it is worth noting that the optimized reaction conditions for Stille coupling could be employed to those substrates (7f–h) which are difficult to obtain via corresponding Suzuki coupling reactions.
 | ||
| Scheme 5 Synthesis of 6-substituted tacrine derivatives by Stille coupling; reaction conditions: 6-bromo tacrine (1 mmol), tributyl(aryl/vinyl)stannane (1 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), NaCl (1.5 mmol), DMF (2 mL), 100 °C for 8–10 h. | ||
Our next attention was to optimize the reaction conditions for Sonogashira cross-coupling reactions of 6-bromo tacrine by employing Pd(dppf)Cl2·CH2Cl2 as the catalyst. As Sonogashira cross-coupling is generally facilitated by copper salts and Hunig-bases,10 we decided to optimize the reaction conditions using copper iodide (CuI) and triethylamine (TEA). Accordingly, we carried out the cross-coupling reaction of 6-bromo tacrine 1 with various terminal acetylenes 8a–d in CuI and TEA at 100 °C for 8–10 hours (Scheme 6). Gratifyingly, we obtained the expected products in good to excellent yields (78–88%). The phenyl acetylenes bearing para substitution (8a, 8c) procured the desired 6-alkynyl tacrines in excellent yields (84–88%) whereas acetylenes containing heteroaryl (pyridine) group yielded the expected product 9d in good yield (78%).
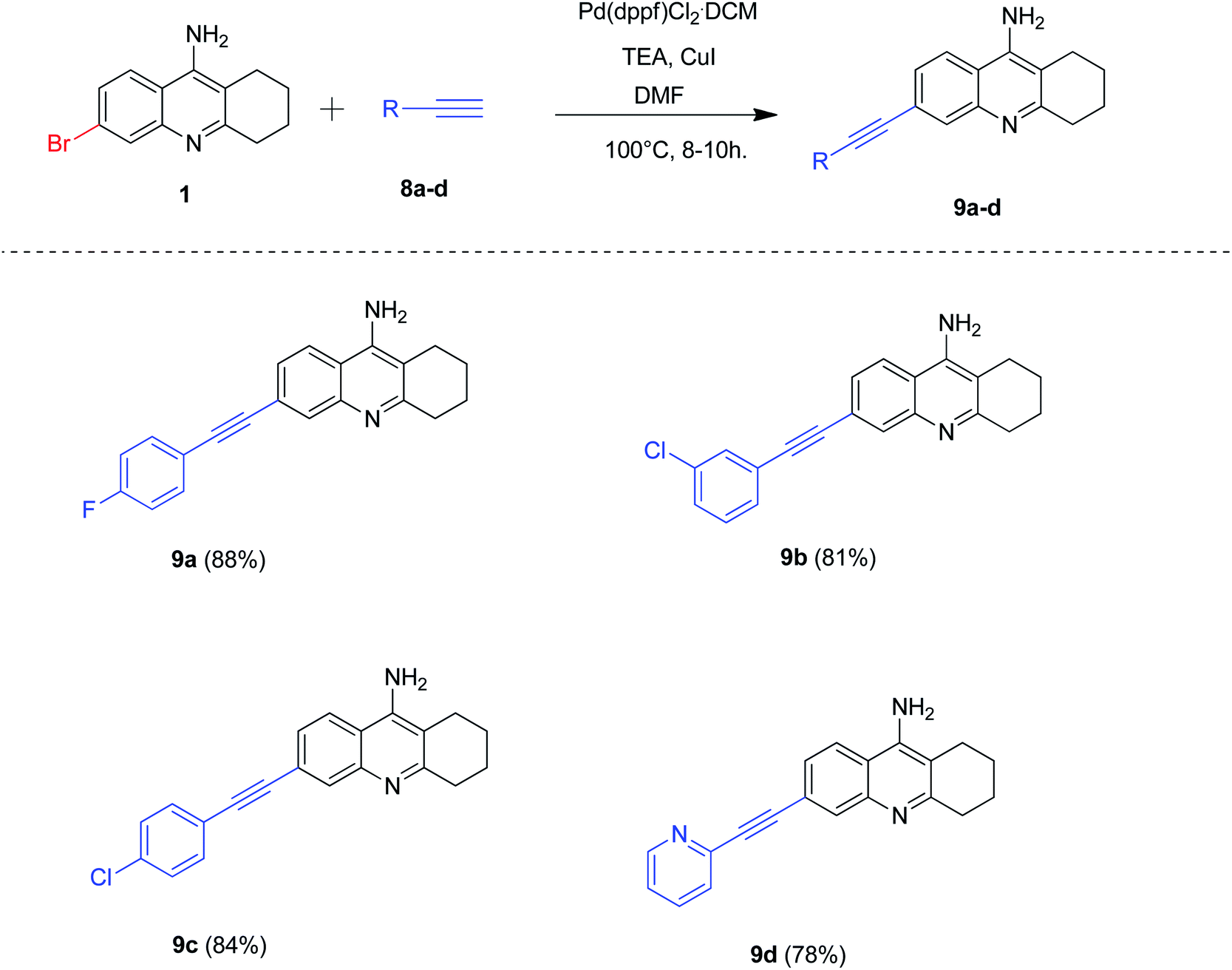 | ||
| Scheme 6 Synthesis of 6-alkynyl tacrine derivatives by Sonogashira coupling; reaction conditions: 6-bromo tacrine (1 mmol), alkyne (1.2 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), CuI (1 mmol), TEA (2 mmol), DMF (2 mL), 100 °C for 8–10 h. | ||
The success of our trials in Suzuki–Miyaura, Sonogashira and Stille cross-couplings enlightened us about the special ability of Pd(dppf)Cl2·CH2Cl2 to act as a common catalyst in palladium catalyzed cross-coupling reactions of 6-bromo tacrine. This observation directed us to investigate the efficiency of this catalyst in Heck and Buchwald cross-coupling conditions.
Accordingly, we treated 6-bromo tacrine 1 with methyl acrylate 10a in order to optimize the suitable conditions for Heck reaction using Pd(dppf)Cl2·CH2Cl2 as the catalyst (Table 3). After careful screening of the reaction parameters, we obtained the desired product 11a in 77% isolated yield when the reaction was carried out in DMF at 100 °C (Table 3, entry 9). It is worth noting that the utilization of an organic base was a key factor for the success of this developed methodology (Table 3, entries 9 and 10).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yieldb 11a (%) |
| a Reaction conditions: 6-bromo tacrine (1 mmol), methyl acrylate (1.2 mmol), catalyst (5 mol%), base (3 mmol), solvent (2 mL), 100 °C for 8 h.b Isolated yield. | ||||
| 1 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane–H2O (2:1) |
30 |
| 2 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | 1,4-Dioxane | 20 |
| 3 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | H2O | Traces |
| 4 | Pd(dppf)Cl2·CH2Cl2 | K2CO3 | DMF | 50 |
| 5 | Pd(dppf)Cl2·CH2Cl2 | NaCl | DMF | 45 |
| 6 | Pd(dppf)Cl2·CH2Cl2 | Na2CO3 | DMF | 50 |
| 7 | Pd(dppf)Cl2·CH2Cl2 | Cs2CO3 | DMF | 40 |
| 8 | Pd(dppf)Cl2·CH2Cl2 | CsF | DMF | 50 |
| 9 | Pd(dppf)Cl2·CH2Cl2 | TEA | DMF | 77 |
| 10 | Pd(dppf)Cl2·CH2Cl2 | DIPEA | DMF | 74 |

After obtaining the suitable reaction conditions in hand, we evaluated the substrate scope of the developed methodology by synthesizing a variety of 6-alkenyl tacrine derivatives (Scheme 7). To our delight, all the alkenes reacted well efficiently to procure the expected products 11a–d in good to excellent yields (75–80%).
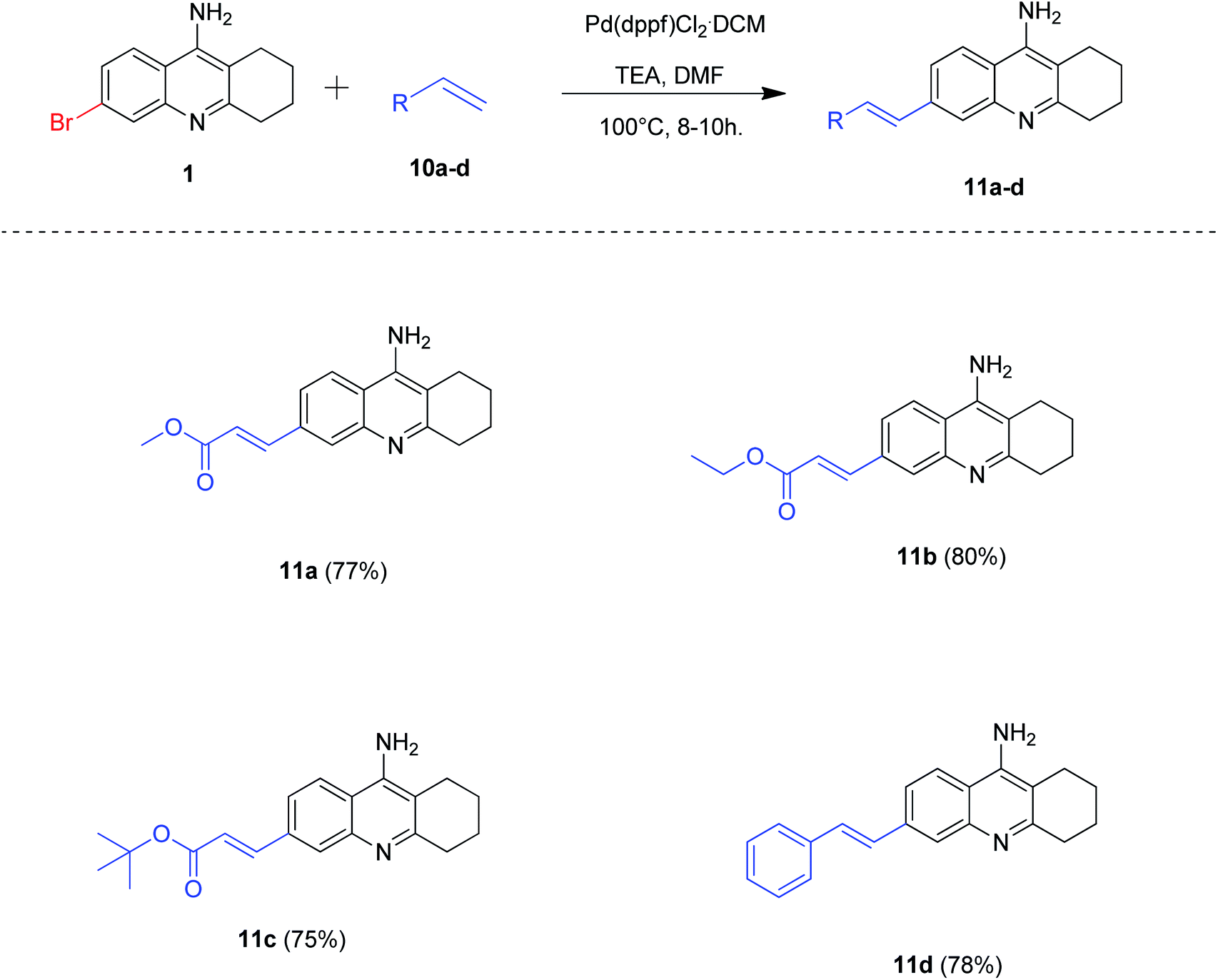 | ||
| Scheme 7 Synthesis of 6-alkenyl tacrine derivatives by Heck coupling; reaction conditions: 6-bromo tacrine (1 mmol), alkene (1.2 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), TEA (3 mmol), DMF (2 mL), 100 °C for 8–10 h. | ||
In order to check the feasibility of our common catalyst in Buchwald coupling, we treated the intermediate 1 with 4-methoxybenzylamine 12a. The detailed optimization studies that we carried out have been summarized in Table 4.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yieldb 13a (%) |
| a Reaction conditions: 6-bromo tacrine (1 mmol), 4-methoxybenzylamine (1.3 mmol), catalyst (5 mol%), ligand (10 mol%), base (3 mmol), solvent (2 mL), 100 °C for 8 h.b Isolated yield.c 4 mmol of base used. | |||||
| 1 | Pd(dppf)Cl2·CH2Cl2 | — | K2CO3 | 1,4-Dioxane–H2O (2:1) |
5 |
| 2 | Pd(dppf)Cl2·CH2Cl2 | — | K2CO3 | 1,4-Dioxane | 20 |
| 3 | Pd(dppf)Cl2·CH2Cl2 | — | K2CO3 | DMF | 15 |
| 4 | Pd(dppf)Cl2·CH2Cl2 | — | Cs2CO3 | 1,4-Dioxane | 10 |
| 5 | Pd(dppf)Cl2·CH2Cl2 | — | TEA | 1,4-Dioxane | Trace |
| 6 | Pd(dppf)Cl2·CH2Cl2 | — | NaOt-Bu | 1,4-Dioxane | 40 |
| 7 | Pd(dppf)Cl2·CH2Cl2 | — | KOt-Bu | 1,4-Dioxane | 65 |
| 8 | Pd(dppf)Cl2·CH2Cl2 | BINAP | KOt-Bu | 1,4-Dioxane | 60 |
| 9 | Pd(dppf)Cl2·CH2Cl2 | Xantphos | KOt-Bu | 1,4-Dioxane | 55 |
| 10 | Pd(dppf)Cl2·CH2Cl2 | PPh3 | KOt-Bu | 1,4-Dioxane | 40 |
| 11c | Pd(dppf)Cl2·CH2Cl2 | — | KOt-Bu | 1,4-Dioxane | 72 |
| 12c | Pd(dppf)Cl2·CH2Cl2 | — | KOt-Bu | DMF | 67 |

After the careful screening of reaction parameters, we could obtain the desired product 13a in 72% isolated yield (Table 4, entry 11). It is noteworthy that the reaction proceeded well when potassium tert-butoxide was employed as a base and no additional ligands were needed for the success of our developed protocol. After the successful optimization of reaction conditions, we evaluated the substrate scope of our methodology for Buchwald coupling (Scheme 8). Gratifyingly, a series of 6-amino tacrine derivatives 13a–g were synthesized in good to excellent yields by our optimized protocol (68–80%).
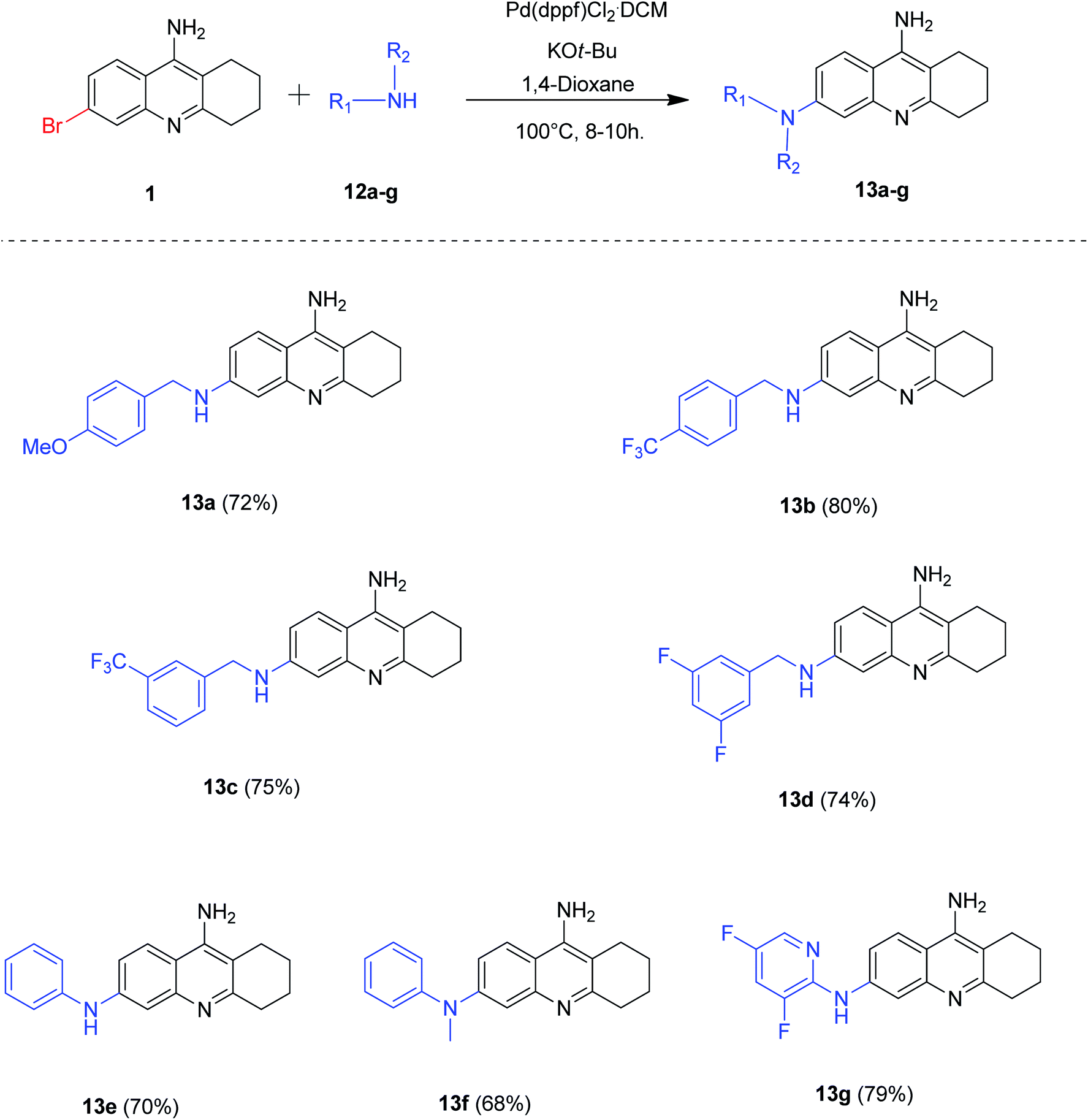 | ||
| Scheme 8 Synthesis of 6-amino tacrine derivatives by Buchwald coupling; reaction conditions: 6-bromo tacrine (1 mmol), amine (1.3 mmol), Pd(dppf)Cl2·CH2Cl2 (5 mol%), KOt-Bu (4 mmol), 1,4-dioxane (2 mL), 100 °C for 8–10 h. | ||
The efficiency of Pd(dppf)Cl2·CH2Cl2 as a common catalyst in all our successful trials encouraged us to rationalize its importance in the cross-coupling methodologies developed.11 It is well-known that most of these cross-coupling reactions between (hetero)aryl halides and nucleophiles involve a series of fundamental steps that occur at the Pd-metal centre (e.g. oxidative addition, coordination-insertion, transmetallation, association of nucleophile, reductive elimination etc.). Once the coordinatively unsaturated true catalytic species is generated, the electronic and steric properties encountered by this catalytic species play a pivotal role in enhancing the reaction rates of various steps involved in the catalytic cycle. Additionally, the steric and electronic properties of the electrophilic and nucleophilic coupling partners are also known to have an impact on the overall outcome of the process. The delicate balance between the steric and electronic parameters of Pd(dppf)Cl2·CH2Cl2 when compared to other phosphine based bidentate catalytic systems is possibly the instrumental factor in achieving successful results in all these cross-coupling reactions employing a wide range of nucleophiles (Scheme 9). The following reasons are rationalized for the ideality of Pd(dppf)Cl2·CH2Cl2 acting as a common catalyst:12 (a) the facile generation of coordinatively unsaturated true catalytic species L2Pd(0); (b) the true catalytic species undergoes rapid oxidative addition; (c) the bidentate ligand attached provides excellent stability to the catalyst; (d) the larger bite angle (99.1 °C), smaller Cl–Pd–Cl angle and forced cis geometry of the groups to be eliminated enhances the reductive elimination step.
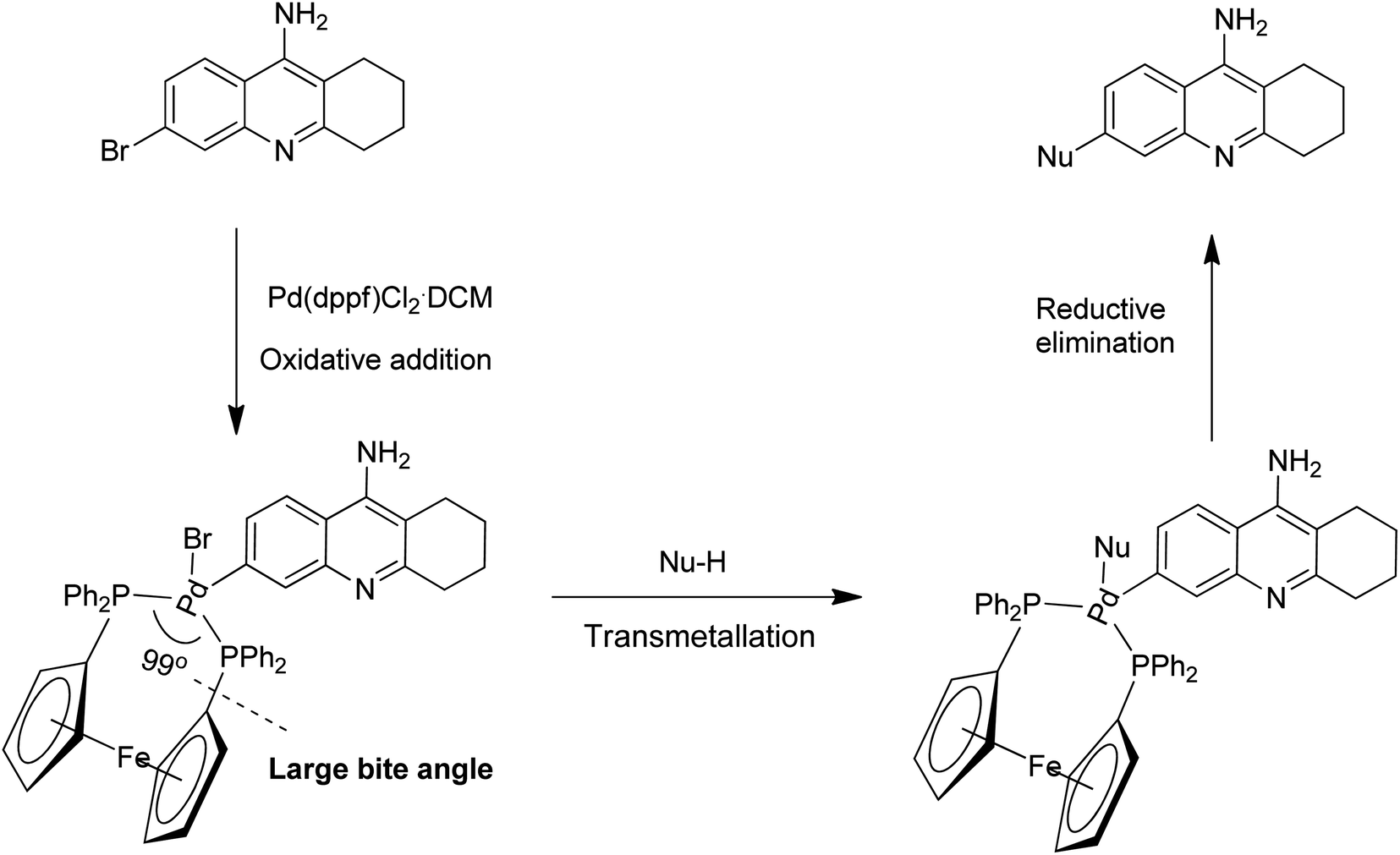 | ||
| Scheme 9 Proposed catalytic steps describing the ideality of Pd(dppf)Cl2·CH2Cl2 catalyst. | ||
In silico studies
All the newly synthesized compounds were then subjected to in silico studies to get an understanding about their drug-likeness and pharmacological activities.Prediction of ADME parameters
The SwissADME service13 was used to determine the key physicochemical and pharmacokinetic parameters of the studied compounds (Table 5). One of the key properties of AChE inhibitors is their ability to pass through the BBB (blood–brain-barrier). Only two compounds 13b and 13c do not meet this criterion. Moreover, all the newly synthesized compounds successfully passed the PAINS filter.| Entry | TPSA | WLOGP | GI absorption | BBB permeant | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Lipinski #violations | PAINS #alerts |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7a | 38,91 | 4, 93 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 7b | 38,91 | 5, 49 | High | Yes | Yes | Yes | No | No | Yes | 1 | 0 |
| 7c | 38,91 | 5, 02 | High | Yes | Yes | Yes | No | No | Yes | 0 | 0 |
| 7d | 48,14 | 4, 38 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 7e | 48, 14 | 4, 38 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 7f | 48, 14 | 3, 71 | High | Yes | Yes | Yes | No | Yes | No | 0 | 0 |
| 7g | 38, 91 | 3, 43 | High | Yes | Yes | Yes | No | Yes | No | 0 | 0 |
| 7h | 51, 8 | 4, 33 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 9a | 38, 91 | 4, 74 | High | Yes | Yes | Yes | Yes | No | No | 1 | 0 |
| 9b | 38, 91 | 4, 84 | High | Yes | Yes | Yes | Yes | No | No | 1 | 0 |
| 9c | 38, 91 | 4, 84 | High | Yes | Yes | Yes | Yes | No | No | 1 | 0 |
| 9d | 51, 8 | 3, 58 | High | Yes | Yes | Yes | Yes | No | Yes | 0 | 0 |
| 11a | 65, 21 | 2, 78 | High | Yes | Yes | Yes | Yes | No | No | 0 | 0 |
| 11b | 65, 21 | 3, 17 | High | Yes | Yes | Yes | Yes | Yes | No | 0 | 0 |
| 11c | 65, 21 | 3, 95 | High | Yes | Yes | Yes | Yes | No | No | 0 | 0 |
| 11d | 38, 91 | 4, 66 | High | Yes | Yes | Yes | No | Yes | No | 0 | 0 |
| 13a | 60, 17 | 3, 98 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13b | 50, 94 | 6, 14 | High | No | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13c | 50, 94 | 6, 14 | High | No | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13d | 50, 94 | 5, 09 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13e | 50, 94 | 4, 45 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13f | 42, 15 | 4, 47 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
| 13g | 63, 83 | 4, 96 | High | Yes | Yes | Yes | No | Yes | Yes | 0 | 0 |
In silico docking studies
Among the five known drugs for the therapy of Alzheimer's disease (AD), four of them (tacrine, donepezil, rivastigmine and galantamine) are inhibitors of acetylcholinesterase (AChE) enzyme.14 In view of this observation, we decided to carry out the molecular docking studies of the newly synthesized tacrine analogs (ligands) at the active site of AChE. The molecular docking studies were done by using ArgusLab and DataWarrior software via ArgusDock algorithm (+empirical evaluation function Ascore). As model compounds, AChE complex with competitive inhibitor tacrine with IC50 = 105 nM (PDB ID 7e3i) and AChE complex with competitive NL1 inhibitor with IC50 = 2.67 nM (PDB ID 7d9o) were used. The validation results were carried out by redocking the native ligand and comparing the in vitro/in silico activity indicators of known AChE inhibitors (Fig. 1). | ||
| Fig. 1 Result of docking studies of known AChE inhibitors. | ||
Molecular docking results
During the docking studies, we observed that almost all the ligands possessed lower values of binding free energy relative to the values for native ligands of various structures (tacrine and NL1). The compounds 7c and 9c have the highest calculated activity (Table 6). However, the compound 7c was found to be the most active one in our investigation.| Compounds | Structures | Binding free energy, kcal mol−1 | Reference | |
|---|---|---|---|---|
| 7d9o | 7e3i | |||
| a Recalculated position, close in position of the scaffold to tacrine. The most active compounds are marked in bold. | ||||
| NL1 |  |
−12.55 (rmsd = 3 Å) | — | IC50 = 2.6 nM |
| Tacrine |  |
— | −10.0, …, −9.0 (rmsd = 2 Å) | IC50 = 105 nM |
| 7a | 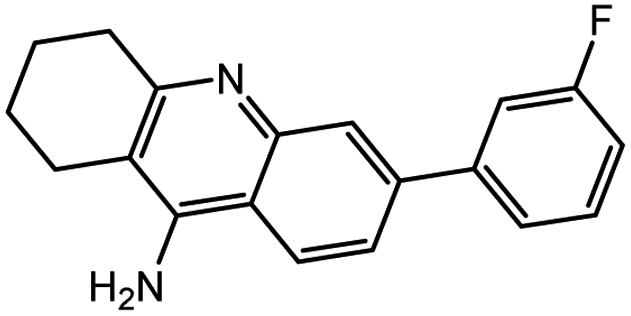 |
−13.46 | −13.54 | |
| 7b |  |
−12.72 | −12.60 | |
| 7c | 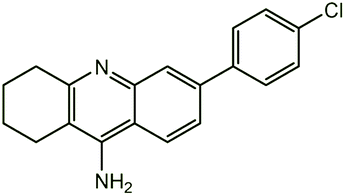 |
−13.92 | −13.67 (−12.41)a | |
| 7d | 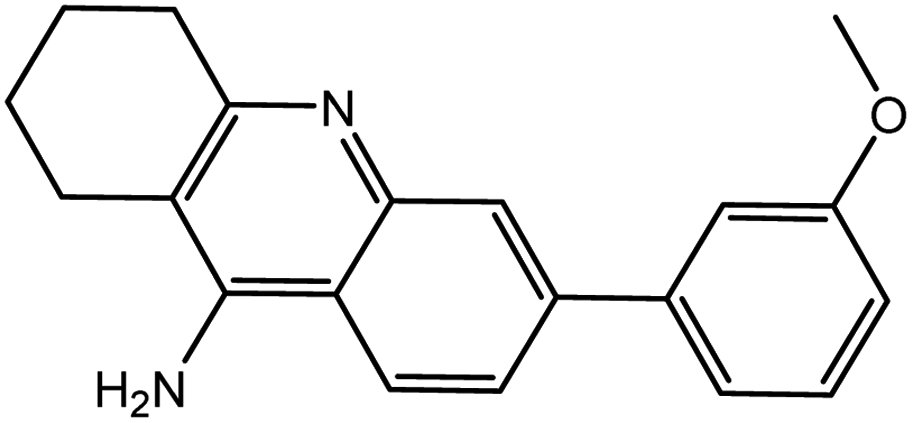 |
−12.66 | −12.77 | |
| 7e | 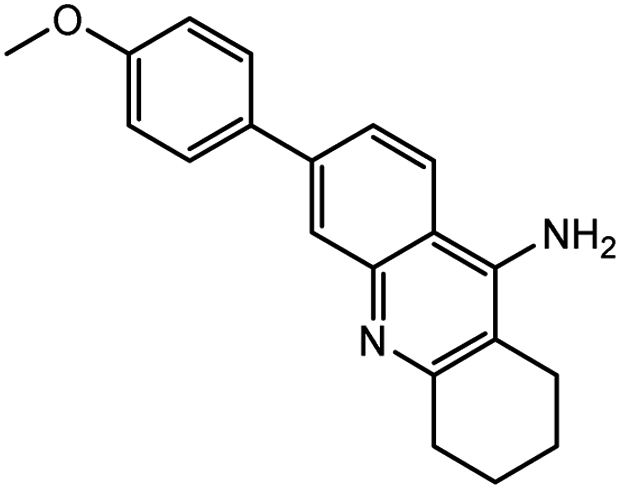 |
−12.83 | −12.95 | |
| 7f |  |
−11.50 | −11.71 | |
| 9a |  |
−13.90 | −11.91 | |
| 9b |  |
−13.38 | −11.83 | |
| 9c |  |
−13.45 | −14.95 | |
| 9d |  |
−11.86 | −11.20 | |
Analysis of non-covalent interactions
The most active compound 7c (Table 6) was recalculated for AChE proteins from complexes “7d9o” and “7e3i”. In the complex with tacrine (PDB 7e3i), ligand 7c was redocked relative to the tacrine position (because they have a common scaffold). An example of redocking for this complex is shown in Fig. 2.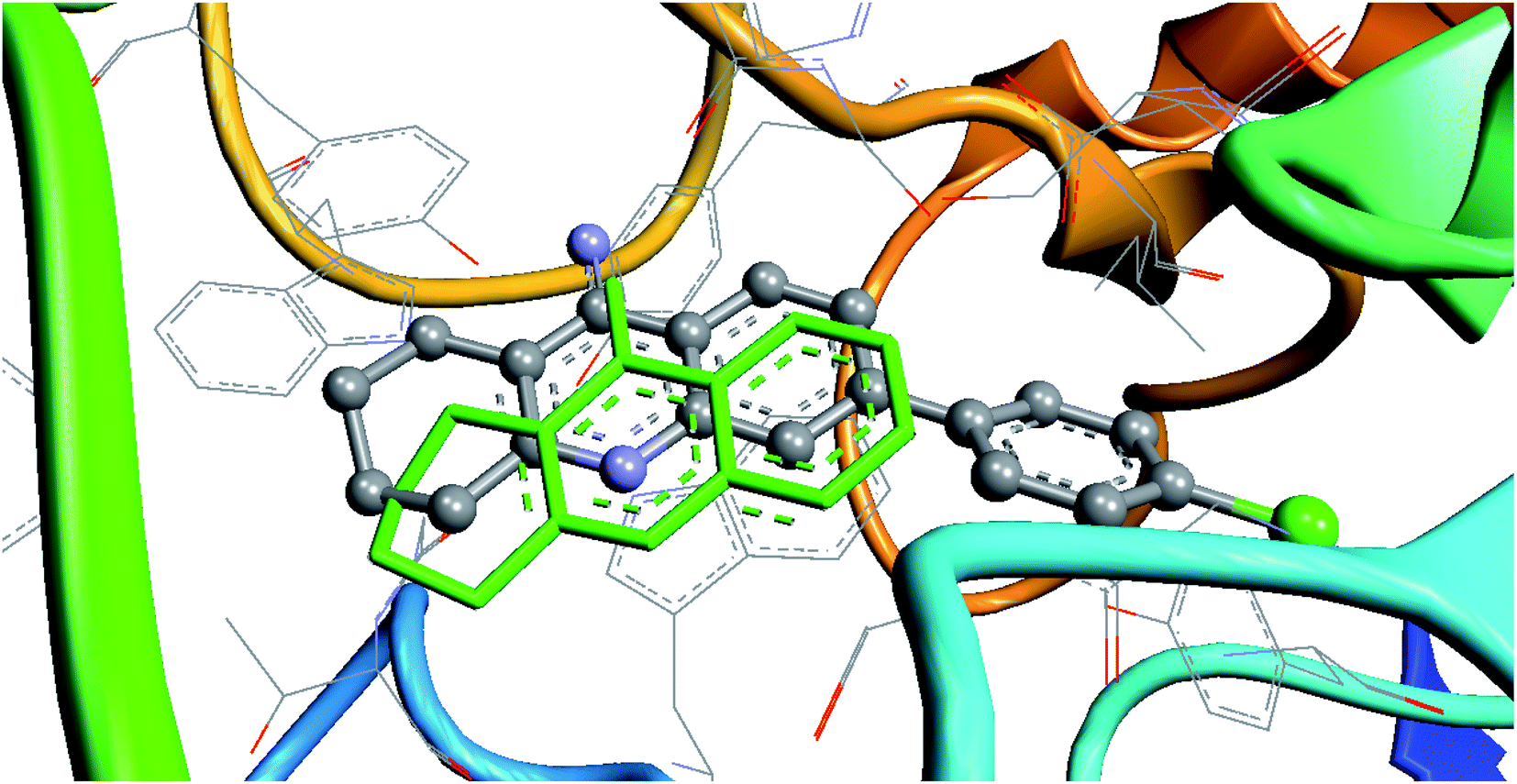 | ||
| Fig. 2 Result of the redocking of compound 7c relative to tacrine (marked in green) in the 7e3i complex. | ||
The maps of non-covalent interactions of native ligands and 7c are presented in Fig. 3. As observed from Fig. 2, 3A and B, the lead compound 7c has a similar non-covalent interaction profile (hydrogen bonding to His447, π–π stacking to Trp86) and a lower binding energy relative to tacrine (probably due to additional van der Waals interactions and π–π stacking with Tyr133). However, there are more energetically favorable states for this ligand in the active site, which are completely inconsistent with the position of tacrine.
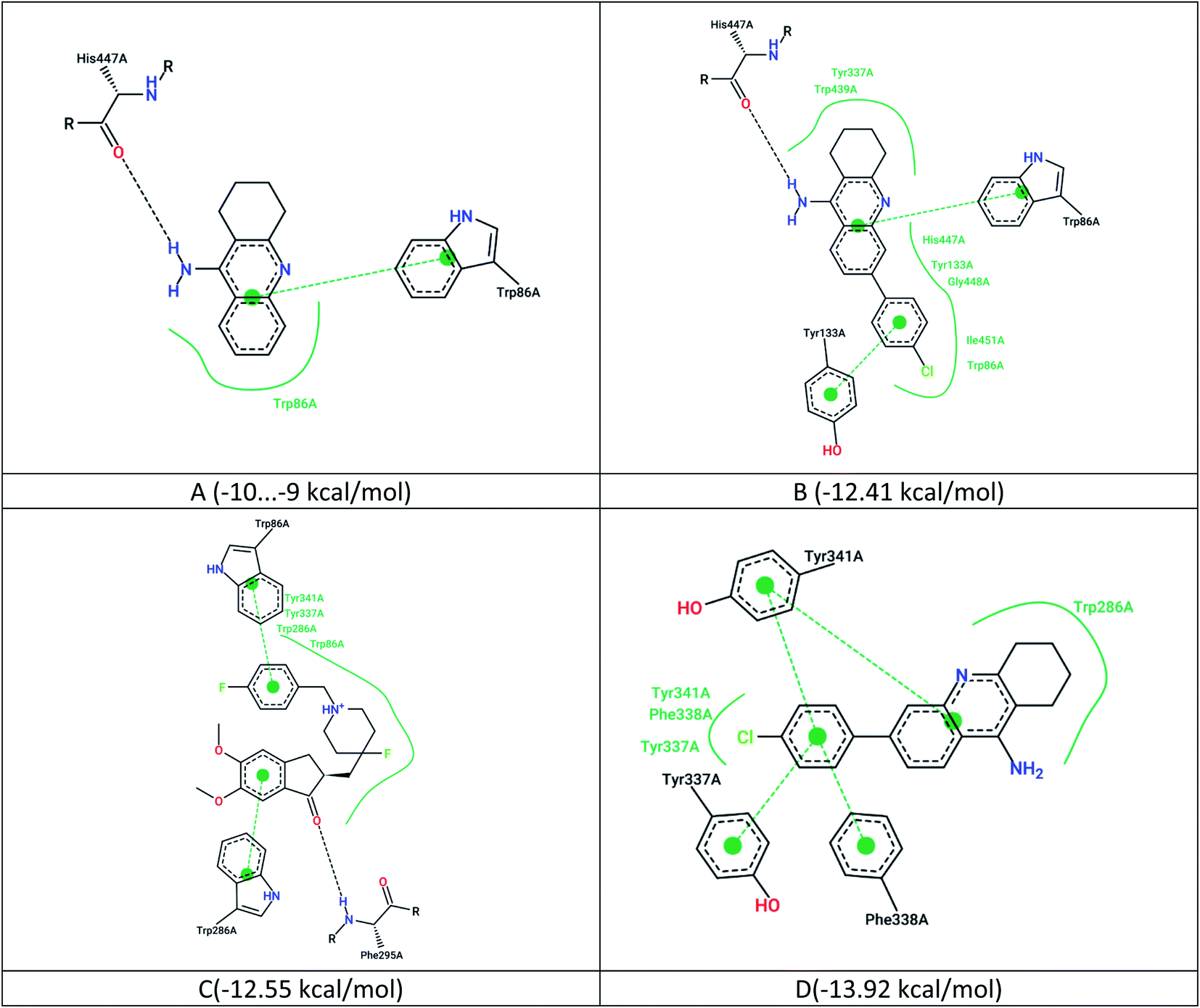 | ||
| Fig. 3 2D maps of non-covalent interactions, (A) native ligand (tacrine) with IC50 = 105 nM; (B) ligand 7c docked relative to tacrine (PDB 7e3i); (C) native nanomolar NL1 inhibitor with IC50 = 2.6 nM; (D) ligand 7c docked to NL1 (PDB 7d9o). | ||
The results of redocking of ligand 7c at the PDB 7d9o complex show a different profile of non-covalent interactions of the ligand relative to the nanomolar NL1 inhibitor (Fig. 3C and D). At the same time, the resulting complex with ligand 7c is more stable due to the many stacking effects.
Toxicity prediction studies
Based on the high toxicity of tacrine, an important issue is the assessment of the acute toxicity of the proposed structural analogues as well as determining the stability of their complexes with CYP3A4 and CYP1A2 proteins. In general, these two proteins play a key role in the formation of hepatotoxic metabolites of tacrine (Fig. 4) and the metabolism of xenobiotics.15 Accordingly, our next attention was to predict the hepatotoxicity of the newly synthesized tacrine derivatives. | ||
| Fig. 4 Hepatotoxic metabolites of tacrine formed with the participation of CYP1A2. | ||
The acute toxicity studies of synthesized tacrine analogues were analyzed using the GUSAR service16 in a model of LD50 prediction in rats administered intravenously. The potential hepatotoxicity of the ligands was assessed by docking against complexes of CYP1A2 (PDB 2hi4) and CYP3A4 (PDB 2v0m) proteins with known inhibitors like 7,8-benzoflavone and ketoconazole respectively. The proteins were prepared in the same way as for AChE. The size of binding sites was calculated relative to native ligands. Docking was carried out in Arguslab using the same algorithm and settings as before. To validate the protocol, redoxing of native ligands was carried out with the following results: 7,8-benzoflavone – RMSD = 1.42 Å, G = 13.47 kcal mol−1, ketoconazole - RMSD = 1.22 Å, ΔG = −10.49 kcal mol−1. The results of docking and calculation of acute toxicity by the GUSAR regression model are shown in Table 7.
| Compounds | Docking score, kcal mol−1 | LD50 (rat, intravenous route of administration//GUSAR model) | |
|---|---|---|---|
| CYP1A2 | CYP3A4 | ||
| a The most active compound is marked in bold; NA – not analyzed. | |||
| 7,8-Benzoflavone (IC50 (1A2) = 340 nM) | −13.47 | NA | NA |
| Ketoconazole IC50 (3A4) = 200 nM | NA | −10.49 | NA |
| Tacrine | −10.9 IC50 = 1.5 μM | −9.8 IC50 = 97 μM | 34.94 (real LD50 = 20 mg kg−1) |
| 7a | −14.42 | −10.27 | 61.38 |
| 7b | −14.24 | −10.65 | 61.39 |
| 7c | −15.05 | −15.36 | 51.65 |
| 7d | −13.54 | −10.74 | 64.52 |
| 7e | −13.64 | −10.19 | 72.60 |
| 7f | −11.38 | −10.90 | 27.57 |
| 7g | −12.51 | −12.05 | 35.07 |
| 7h | −12.33 | −10.70 | 87.19 |
| 9a | −15.40 | −12.0562 | 42.53 |
| 9b | −15.12 | −10.25 | 42.76 |
| 9c | −16.00 | −15.53 | 54.97 |
| 9d | −13.14 | −12.12 | 65.00 |
| 11a | −11.71 | −11.70 | 61.09 |
| 11b | −12.26 | −10.99 | 33.17 |
| 11c | −12.10 | −12.96 | 43.23 |
| 11d | −15.78 | −14.54 | 68.72 |
| 13a | −11.44 | −9.24 | 53.24 |
| 13b | −14.74 | −12.38 | 57.81 |
| 13c | −14.08 | −12.03 | 60.33 |
| 13d | −13.18 | −13.32 | 44.53 |
| 13e | −13.38 | −11.53 | 44.74 |
| 13f | −13.44 | −12.97 | 35.89 |
| 13g | −12.65 | −11.75 | 50.06 |
The results of the GUSAR regression model show that all derivatives, with the exception of 7f, have less acute toxicity (when administered intravenously to rats) compared to the predicted value for tacrine. On average, the lowest toxicity is predicted for 6-aryl-substituted derivatives 7a–h (average LD50 = 62 mg kg−1) in comparison with compounds having alkyne fragment 9a–d (average LD50 = 51 mg kg−1) as linkers. The high predicted acute toxicity of compound 7f appears to be related to the reactivity of the ethylene moiety.
As reported by Rao et al.15 the pathways of tacrine metabolism are directly related to the way it is oriented relative to the heme in CYP1A2. Thus, the proximity of the tetrahydrobenzene fragment of tacrine to heme leads to its enzymatic hydroxylation. Similar positioning of the studied tacrine analogues was established as a result of docking on CYP1A2 and CYP3A4 (Fig. 5).
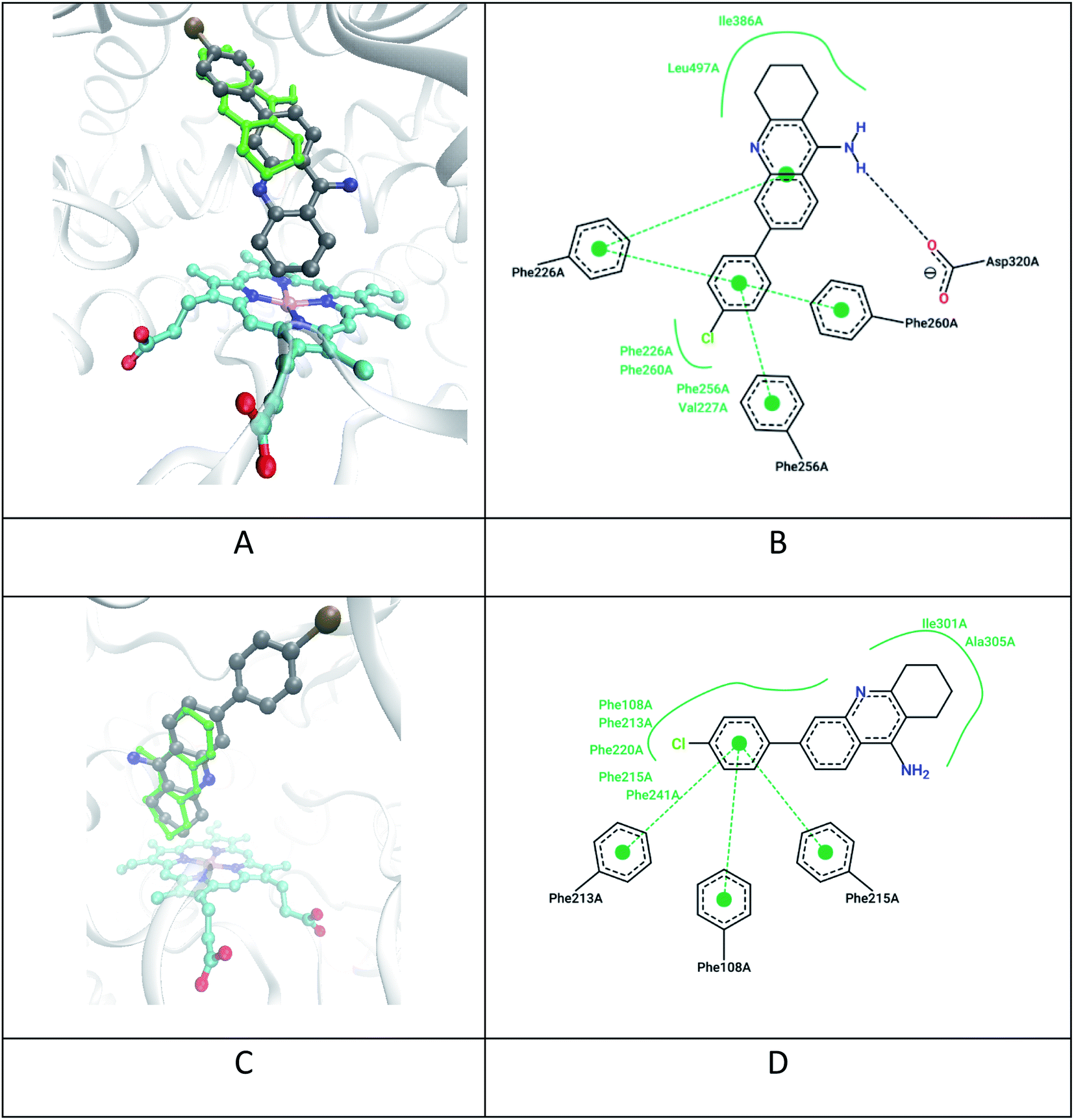 | ||
| Fig. 5 Docking results of the compound 7c and tacrine for CYP1A2 and CYP3A4 subtype proteins: (A) positions 7c (grey), tacrine (green), heme (cyan) in CYP1A2 (PDB 2hi4); (B) 2D map of non-covalent interactions of 7c in CYP1A2; (C) calculated positions 7c (grey), tacrine (green), heme (cyan) in CYP3A4 (PDB 2v0m); (D) 2D map of non-covalent interactions of 7c in CYP3A4. | ||
All ligands show high potential affinity for CYP1A2 (to a greater extent) and CYP3A4 (to a lesser extent). Also, the ligands with highest affinity are characterized by the close location of the tetrahydroacridine fragment of the ligands to heme which contributes to their enzymatic hydroxylation. For example, the distance between the nearest carbon atom of the tetrahydroacridine fragment of ligand 7c and the heme “Fe” ion is about 4.3 Å in CYP1A2 (Fig. 5A) and 5.5 Å in CYP3A4 (Fig. 5C). However, the further metabolism of ligands at the C-7 with the formation of analogues of hepatotoxic metabolites (Fig. 5) can be sterically hampered in the presence of a large substituent at C-6 position of 7c.
Similarly, an aromatic fragment at C-6 can lead to an alternative ligand approach to heme in CYP1A2 and CYP3A4 enzymes. For example, the compound 7e, with the least predicted acute toxicity, orients itself in the CYP1A2 enzyme with the aromatic substituent approaching the heme “Fe” ion at a distance of ∼5.3 Å (Fig. 6) with further metabolism without the participation of the tetrahydroacridine.
 | ||
| Fig. 6 The results of the docking studies for the compound 7e (grey) and tacrine (green) on the CYP1A2 protein with the closest location of the aromatic substituent to heme (cyan). | ||
Thus, as a result of the virtual screening of newly synthesized tacrine compounds by molecular docking, their high inhibitory activity against AChE was established relative to the nanomolar inhibitor – tacrine with a similar profile of non-covalent interactions. Most of the compounds also exhibited 1.5–2 times lower predicted acute toxicity according to the GUSAR model. A high affinity of the compounds for the CYP1A2 enzyme, which plays a key role in tacrine metabolism, has also been established. For the studied derivatives, 2 possible ways of binding to CYP1A2 were established: (1) with the maximum approximation to the heme of the tetrahydroacridine fragment with the possibility of its enzymatic hydroxylation; (2) as close as possible to the heme of the aromatic substituent at the “C-6” of the ligand with its further metabolic transformation, thus preventing the metabolism of the tetrahydroacridine ring to probable hepatotoxic derivatives.
Conclusion
We have successfully demonstrated the utilization of Pd(dppf)Cl2·CH2Cl2 as a common catalyst for the palladium catalyzed cross-coupling reactions of 6-bromo tacrine with various coupling partners. The developed methodologies paved the way for the successful synthesis of diverse array of 6-substituted tacrine analogues of profound pharmacological relevance. The molecular docking and hepatotoxicity prediction studies were carried out for the synthesized compounds. From our studies, we found that most of the synthesized compounds are active as they possessed appreciable binding energy and low toxicity values. The compound 7c showed the highest calculated activity in relative to tacrine when docked at the active site of AchE enzyme. The hepatotoxicity prediction studies revealed the fact that the compound 7e had the least predicted acute toxicity. The synthesis of additional compounds and the evaluation of their biological activities will be carried out in our laboratory in due course.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Aravinda Babu is thankful to Syngene International Ltd. for providing the No Objection Certificate (NOC) to pursue his doctoral work. The authors are thankful to Sri Siddhartha Academy of Higher Education and Karnataka Council for Technological Upgradation (KCTU) for rendering all the facilities to carry out the research work. Grigory V. Zyryanov is thankful to Ministry of Science and Higher Education of the Russian Federation (Grant # 075-15-2020-777) for the financial support. Sougata Santra is thankful to Russian Science Foundation (Grant # 20-73-10205) and Grants Council of the President of the Russian Federation (# NSh-1223.2022.1.3). Muthipeedika Nibin Joy is thankful to Russian Science Foundation (Grants ## 22-23-20189 and 21-13-00304). The detailed experimental procedure and characterization data including spectra are available in the ESI† uploaded along with the manuscript.References
- H. Wang and H. Zhang, ACS Chem. Neurosci., 2019, 10, 852–862 CrossRef CAS.
- X. j. Cheng, J. x. Gu, Y. p. Pang, J. Liu, T. Xu, X. r. Li, Y. Z. Hua, K. A. Newell, X. F. Huang, Y. Yu and Y. Liu, ACS Chem. Neurosci., 2019, 10, 3500–3509 CrossRef CAS PubMed.
- L. Ismaili, B. Refouvelet, M. Benchekroun, S. Brogi, M. Brindisi, S. Gemma, G. Campiani, S. Filipic, D. Agbaba, G. Esteban, M. Unzeta, K. Nikolic, S. Butini and J. M. Contelles, Prog. Neurobiol., 2017, 151, 4–34 CrossRef CAS PubMed.
- (a) L. Gorecki, A. Misiachna, J. Damborsky and R. Dolezal, et al., Eur. J. Med. Chem., 2021, 219, 113434 CrossRef CAS PubMed; (b) W. Liu, L. Wu, D. Li and Y. Huang, et al., Bioorg. Chem., 2022, 126, 105875 CrossRef CAS PubMed; (c) M. Recanatini, A. Cavalli, F. Belluti and L. Piazzi, et al., J. Med. Chem., 2000, 43, 2007–2018 CrossRef CAS PubMed; (d) F. Mao, J. Li, H. Wei, L. Huang and X. Li, J. Enzyme Inhib. Med. Chem., 2015, 30, 995–1001 CrossRef CAS PubMed.
- (a) M. A. Rajendra, K. Sunil, A. M. Sajith, M. N. Joy, V. A. Bakulev and K. R. Haridas, Synlett, 2020, 31, 1629–1633 CrossRef CAS; (b) S. Bhaskaran, M. S. A. Padusha and A. M. Sajith, ChemistrySelect, 2020, 5, 9005–9016 CrossRef CAS.
- (a) R. P. Karuvalam, K. R. Haridas, A. M. Sajith, R. Pakkath, B. Savitha, M. S. A. Padusha, V. A. Bakulev and M. N. Joy, Arkivoc, 2019, 6, 431–445 Search PubMed; (b) B. Savitha, E. K. Reddy, C. S. A. Kumar, R. P. Karuvalam, M. S. A. Padusha, V. A. Bakulev, K. H. Narasimhamurthy, A. M. Sajith and M. N. Joy, Tetrahedron Lett., 2019, 60, 151332 CrossRef CAS.
- (a) E. K. Reddy, C. Remya, K. Mantosh, A. M. Sajith, R. V. Omkumar, C. Sadasivan and S. Anwar, Eur. J. Med. Chem., 2017, 139, 367–377 CrossRef CAS PubMed; (b) C. Remya, K. V. Dileep, E. K. Reddy, K. Mantosh, K. Lakshmi, R. S. Jacob, A. M. Sajith, E. J. Variyar, S. Anwar, K. Y. J. Zhang, C. Sadasivan and R. V. Omkumar, Comput. Struct. Biotechnol. J., 2021, 19, 4517–4537 CrossRef CAS PubMed.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (b) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS.
- (a) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS.
- T. J. Colacot, Platinum Met. Rev., 2001, 45, 22–30 CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- P. Camps, X. Formosa, C. Galdeano, T. Gómez, D. Muñoz-Torrero, L. Ramírez, E. Viayna, E. Gómez, N. Isambert, R. Lavilla, A. Badia, M. V. Clos, M. Bartolini, F. Mancini, V. Andrisano, A. Bidon-Chanal, O. Huertas, T. Dafni and J. Luque, Chem.-Biol. Interact., 2010, 187, 411–415 CrossRef CAS PubMed.
- A. McEneny-King, W. Osman, A. N. Edginton and P. P. N. Rao, Bioorg. Med. Chem. Lett., 2017, 27, 2443–2449 CrossRef CAS.
- A. Lagunin, A. Zakharov, D. Filimonov and V. Poroikov, Mol. Inf., 2011, 30, 241–250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03225b |
| This journal is © The Royal Society of Chemistry 2022 |