DOI:
10.1039/D2RA03180A
(Paper)
RSC Adv., 2022,
12, 21111-21121
Ancillary ligand effects on α-olefin polymerization catalyzed by zirconium metallocene: a computational study†
Received
20th May 2022
, Accepted 18th July 2022
First published on 1st August 2022
Abstract
The polymerization of α-olefins catalyzed by zirconium metallocene catalyst was systematically studied through experiments and density functional theory (DFT) calculations. Having achieved an agreement between theory and experiment, it was found that the effect of the catalyst ligand on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C insertion reaction was significantly greater than that on the β-H elimination reaction. Therefore, the molecular weight of polymers can be increased by improving the activity of the CC insertion. In addition, in comparison with propylene, the chain length of α-olefins can directly affect the stereotacticity of polymerization products, owing to steric hindrance between the polymer chain and monomer.
C insertion reaction was significantly greater than that on the β-H elimination reaction. Therefore, the molecular weight of polymers can be increased by improving the activity of the CC insertion. In addition, in comparison with propylene, the chain length of α-olefins can directly affect the stereotacticity of polymerization products, owing to steric hindrance between the polymer chain and monomer.
Introduction
Poly-α-olefins (PAOs) are oligomers of linear α-olefins, which are used as base stocks for synthetic lubricants in automotive and industrial applications. Synthetic base stocks perform better than conventional mineral base stocks in terms of viscosity index, kinematic viscosity, pour point and volatility, thermal and oxidative stability, etc. Synthetic base stocks can be used to prepare all kinds of high-grade lubricating grease products.1–3 PAO synthetic base oil plays a key role in cutting-edge technology fields such as the aerospace and military industries. It is also the key raw material in the production of high-grade lubricating oil used in advanced automobile, wind power, high-speed railway, intelligent manufacturing, and other industries.4 At present, synthesis of PAO base oils is primarily carried out through the oligomerization of pure α-decenes by foreign companies.5
The appropriate selection of catalyst and reaction conditions can aid in the production of PAOs with tailor-made properties. Notably, a catalyst plays a key effect on the polymerization activity, molecular weight, and branching degree.6 The commonly used catalysts for α-olefin polymerization mainly include Lewis acid catalysts, Ziegler Natta catalysts, metallocene catalysts, ionic liquid catalysts, etc.7–16 Among them, Ziegler Natta catalysts are traditional and the most common catalyst for olefin polymerization.17–19 However, because Ziegler Natta catalysts have multiple active sites, olefins have different growth rates at different sites of catalyst, and the various properties of polymerization cannot be effectively controlled.20 In comparison to Ziegler–Natta, metallocene catalysts can control various important parameters, such as co-monomer distribution, molecular weight, molecular weight distribution, molecular architecture, stereo-specificity, degree of linearity, and branching of the polymer, due to the single active sites available on the metallocene catalysts.20,21 Thus, metallocene catalysts (with group IV metal) have become a research hotspot in the recent years and have been applied in industrial production owing to their single reaction active center and high catalytic activity.22,23 There are four types of metallocene catalysts commonly used: (a) monometallocene catalyst,24–27 (b) constrained geometry catalysts (CGC),28–30 (c) unbridged metallocene catalysts,31,32 (d) bridged metallocene catalysts33–35 (Fig. 1). Compared with other types of metallocene catalysts, rigid bridging groups of bridged metallocene catalysts can produce relatively stable spatial-specific stereoscopic effects on chain propagation and coordination monomers, affording synthesis polymers with high molecular weight and regular structure.31,36 Moreover, the bridged rigid structure makes the connection between the metallocene ring and the transition metal more stable, and the metallocene ring does not slip at high temperatures.37 Therefore, the bridged metallocene catalyst also shows high catalytic activity at high temperatures.38,39 As has been reported in the literature,40–43 different electronic or stereoscopic effects of catalyst ligands can significantly affect the polymerization performance, but the relationship between the ligand of group IV bridged metallocene catalysts and activity or the molecular weight of α-olefins (e.g. 1-decene), polymerization at the molecular level remains poorly understood.44–50
 |
| | Fig. 1 Four common metallocene catalyst structures: (a) Monometallocene catalyst (b) Constrained Geometry Catalysts (CGC) (c) unbridged metallocene catalyst (d) bridged metallocene catalyst. | |
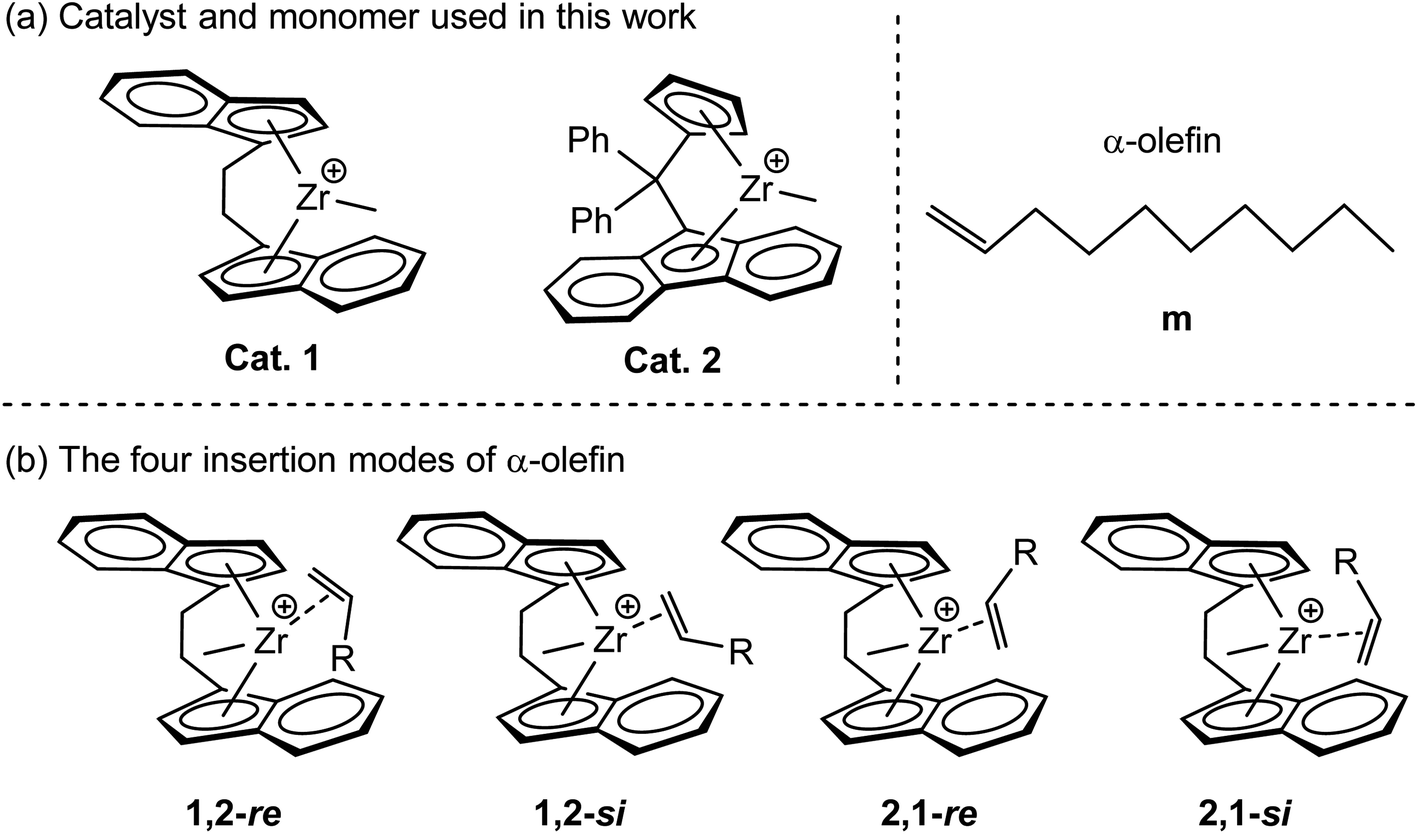
In general, the occurrence of polymerization requires MAO to activate the metallocene catalyst precursor (Fig. 1) to obtain active species with methyl groups, but the overall mechanisms of metallocene catalyst activation are not precisely understood. All computational studies dealing with the MAO activator suffer from its elusive structure, thus the use of DFT model systems is required (Fig. 2a). The starting point of this work was the activation of the dimethylated metallocene complexes, omitting the preceding catalyst alkylation step. This study mainly focuses on the systematic experimental comparison and theoretical calculation of the polymerization of 1-decene catalyzed by bridged metallocene complexes 1 (ref. 51) and 2 (ref. 52) (Fig. 2a) in order to explore the factors affecting both catalyst activity and polymer molecular weight. The research results will provide us with a theoretical basis for the design of high-efficiency catalysts. Four possible modes of olefin coordination insertion are shown in Fig. 2b. Therefore, the four different insertion modes were calculated and the results were compared, and the most favorable reaction path was selected for further analysis.
 |
| | Fig. 2 (a) The structures of catalysts and monomers used for DFT calculation in this work (b) four possible modes of olefin coordination insertion. | |
Computational details
All calculations were performed using the Gaussian 16 program.53 The B3PW91 hybrid exchange-correlation functional was utilized for geometry optimization.54–56 Each optimized structure was subsequently analyzed by harmonic vibration frequencies for the characterization of a minimum (Nimag = 0) or a transition state (Nimag = 1), providing thermodynamic data. The transition state structures were used to connect the reactant and product on either side via intrinsic reaction coordinate (IRC) tracking. The 6-31G* basis set was considered for C, H atoms. The Zr atoms were treated by the Stuttgart/Dresden effective core potential (ECP) and the associated basis sets.57 This basis set was denoted as “BSI”. Such a computational strategy has been widely used for the study of transition-metal containing systems.58 To obtain more reliable relative energies, the single-point calculations of optimized structures were carried out at the level of M06 (ref. 59)/BSII, taking into account a solvation effect of toluene with the SMD60 solvation model. In the BSII, the 6-311G** basis set was used for the nonmetal atoms, whereas the basis sets together with associated pseudopotentials for Zr atoms were the same as that in geometry optimization. Therefore, unless otherwise mentioned, the free energy (ΔG, 298.15 K, 1 atm) in solution, which was used for the description of energy profiles, was obtained from solvation single-point calculation and gas-phase Gibbs free energy correction. The 3D molecular structures displayed herein were created using CYLview.61
Results and discussion
Catalyst 1 and catalyst 2 have been synthesized according to the procedure reported in the literature51,52 and are applied for 1-decene (m) polymerization. The experimental results are summarized in Table 1. It can be seen from the experimental results that the activity and molecular weight of catalyst 2 for m are significantly higher than that of catalyst 1. These differences in activity and molecular weight suggest that the ligand of the catalyst significantly influences the polymerization behavior of 1-decene. To systematically explore the factors affecting polymerization performance, density functional theory (DFT) calculations provided additional insight into the polymerization of m with catalysts 1 and 2.
Table 1 Polymerization of 1-decene by catalyst 1 and 2a
| Cat. (μmol) |
[Al]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Zr] :[Zr] |
Act.b |
Mnc |
| Conditions: 1-decene, 200 mL; [B]:[Zr] = 1.5:1; 90 °C, 1 h. 106 g (mol Cat.)−1 M−1 h−1. kg mol−1. |
| 1 (10) |
50:1 |
6.3 |
6875 |
| 2 (10) |
50:1 |
11.1 |
14802 |
Effects of catalyst ligands on polymerization activity molecular weight
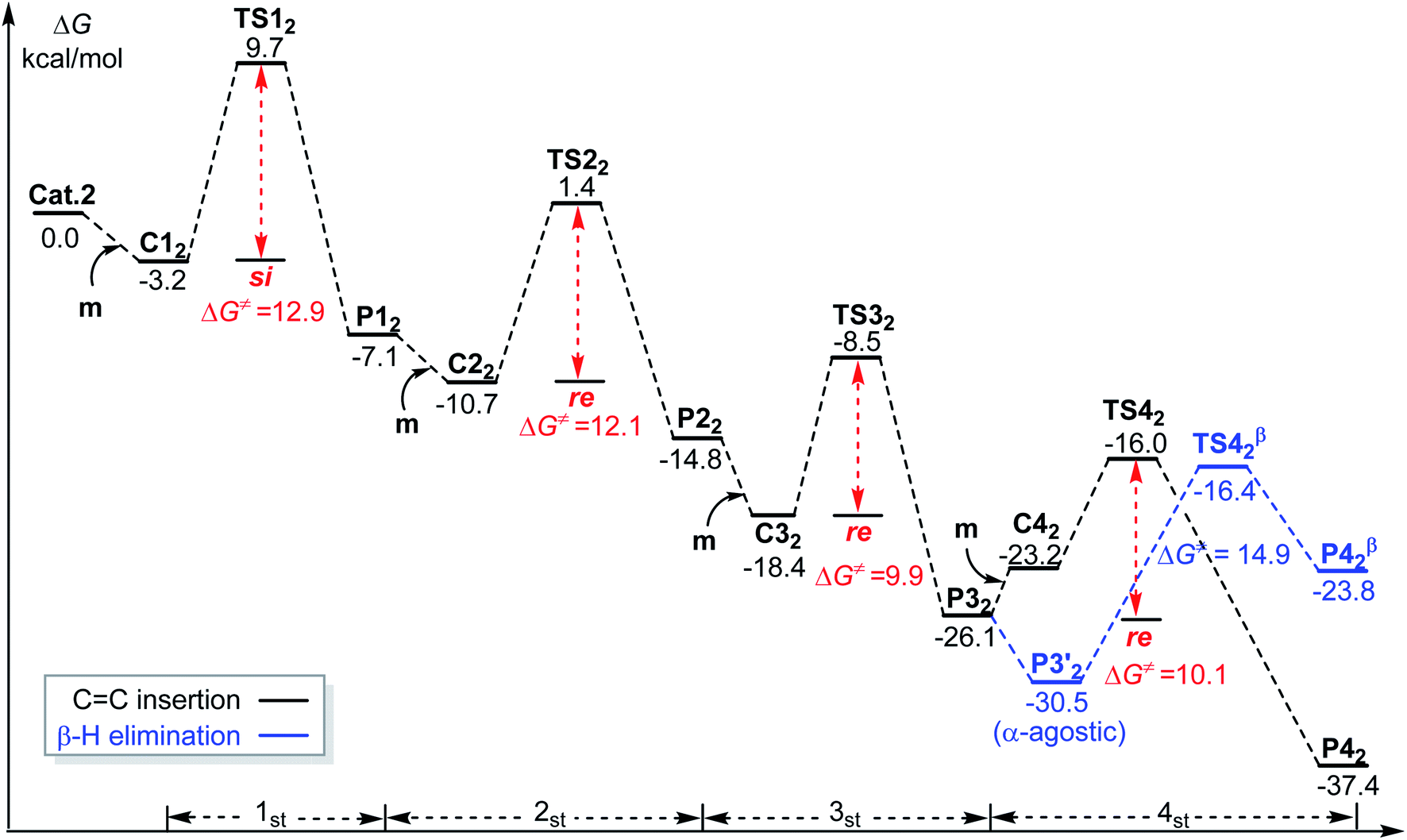
Firstly, four possible modes of m coordination insertion in Cat.1 and Cat.2 systems were calculated (Tables S1 and S2†). The computed results indicate that 1,2-insertion is both kinetically and thermodynamically more favorable than 2,1-insertion in the chain initiation stage. Based on this results, only 1,2-insertion mode was considered here for the chain propagation stage. Next, all kinetically favorable paths of chain initiation and chain growth stages are summarized in Fig. 3 and Fig. 4. As shown in Fig. 3, the average insertion barrier of Cat.1 in the chain propagation (second to fourth monomer insertion) was 11.6 kcal mol−1, which is 0.9 kcal mol−1 higher than that of Cat.2 (10.7 kcal mol−1, Fig. 4). These results are consistent with the experimental results shown above. In addition, For the rate-limiting step (first insertion), the energy barrier of Cat.1 (12.1 kcal mol−1) is slightly lower than that of Cat.2 (12.9 kcal mol−1), but the coordination complex of Cat.1 (−0.7 kcal mol−1) is significantly higher in energy than that of Cat.2 (−3.2 kcal mol−1). If taking Cat.1 and Cat. 2 as one system, one can roughly estimate the probability ratio of the occurring of the two rate-limiting steps via Boltzmann statistics62 on the basis of the energies of the complexes and transition states. The result indicates that the insertion probability ratio of the first monomer by Cat.1 and Cat.2, respectively, was calculated to be 5.4/94.6, suggesting the higher activity of Cat.2 compared to Cat.1. Besides, the calculation results show that the isotactic polymers (si-re-re-re) can be obtained by Cat.1 and Cat.2 (Tables S1 and S2†). Moreover, the calculation results show that dispersion correction does not affect the energetic significantly in this work (Fig. 3 vs. S1†).
 |
| | Fig. 3 Computed energy profiles for Cat.1 mediated chain initiation and chain propagation of m. The CC insertion and β-H elimination pathways are indicated in black and blue, respectively. | |
 |
| | Fig. 4 Computed energy profiles for Cat.2 mediated chain initiation and chain propagation of m. The CC insertion and β-H elimination pathways are in black and blue, respectively. | |
Moreover, it can been seen that isomerization of the third molecule insertion product is always easier to occur than next CC coordination (Fig. 3 and 4). It is worth noting that the formation of the P3’1 intermediate (−29.7 kcal mol−1) is more favorable than the m coordination (C41, -16.5 kcal mol−1) by more than 13 kcal mol−1 (Fig. 3), thus potential wells (P3’1) may be formed to reduce the polymerization activity. However, for Cat. 2, it can be seen from Fig. 4 that the thermodynamic energy difference between P3’2 (−30.5 kcal mol−1) and C42 (−23.2 kcal mol−1) is only 7.3 kcal mol−1. At the same time, the β-H elimination is an obvious endothermic process, which makes the β-H elimination is less favorable than CC insertion. Therefore, it can be further proved that the polymerization activity of Cat. 2 is higher than that of Cat. 1, which can partially explain the experimental results.
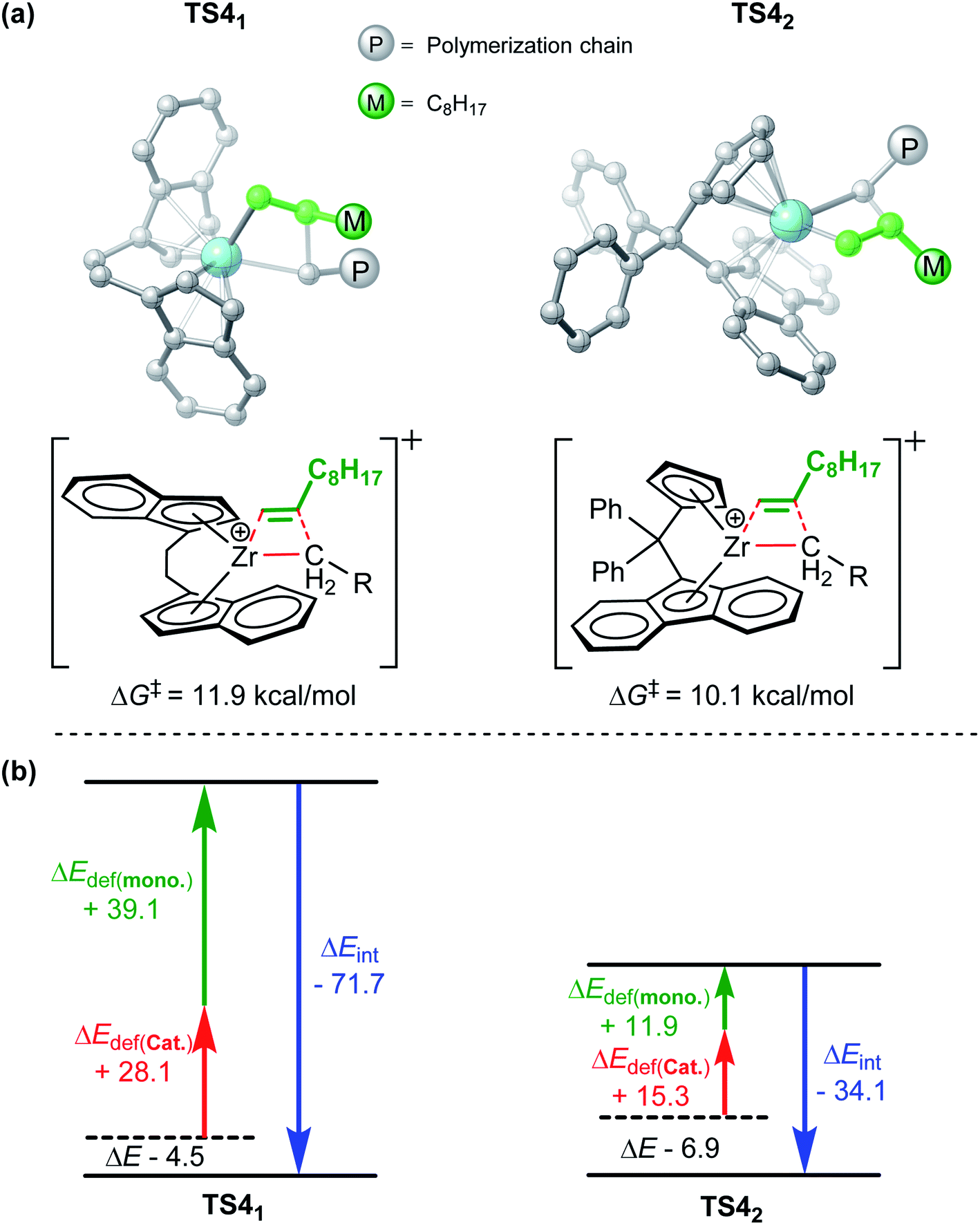
To obtain a deeper insight into the activity difference in chain propagation, activation strain analysis63 was performed for the corresponding two transition states (TS41 and TS42, Fig. 5). For this purpose, the transition state (TS) structures were divided into two fragments, viz., the catalyst part (cat.) and monomer moiety (mono). The energies of the fragments in the TS geometries were evaluated via single-point energy calculations. Such single-point energies of the fragments and the energy (corrected by BSSE) of the TS were used to estimate the interaction energy ΔEint between the two fragments. The single-point energies, together with the energies of the respective fragments in their optimal geometries, allowed for the estimation of the distortion energies of the two fragments, ΔEstrain(cat.) and ΔEstrain(mono.). As the energy of TS, ΔETS, is evaluated with respect to the energy of the two separated fragments, the relation ΔETS = ΔEint + ΔEstrain(cat.) + ΔEstrain(mono.) holds. As shown in Fig. 5, in the case of TS41, the interaction energy ΔEint was significantly stronger than that in TS42 (−71.7 kcal mol−1 vs. −34.1 kcal mol−1), but the total distortion energy of TS41 was larger (67.2 kcal mol−1). Therefore, TS41 needed to overcome the higher energy barrier 11.9 kcal mol−1. Therefore, the larger steric repulsion between the metal center and monomer moiety in TS41 accounted for its lower stability in comparison with TS42.
 |
| | Fig. 5 Geometric structures (a) and activation strain analysis (b) of TS41 and TS42. | |
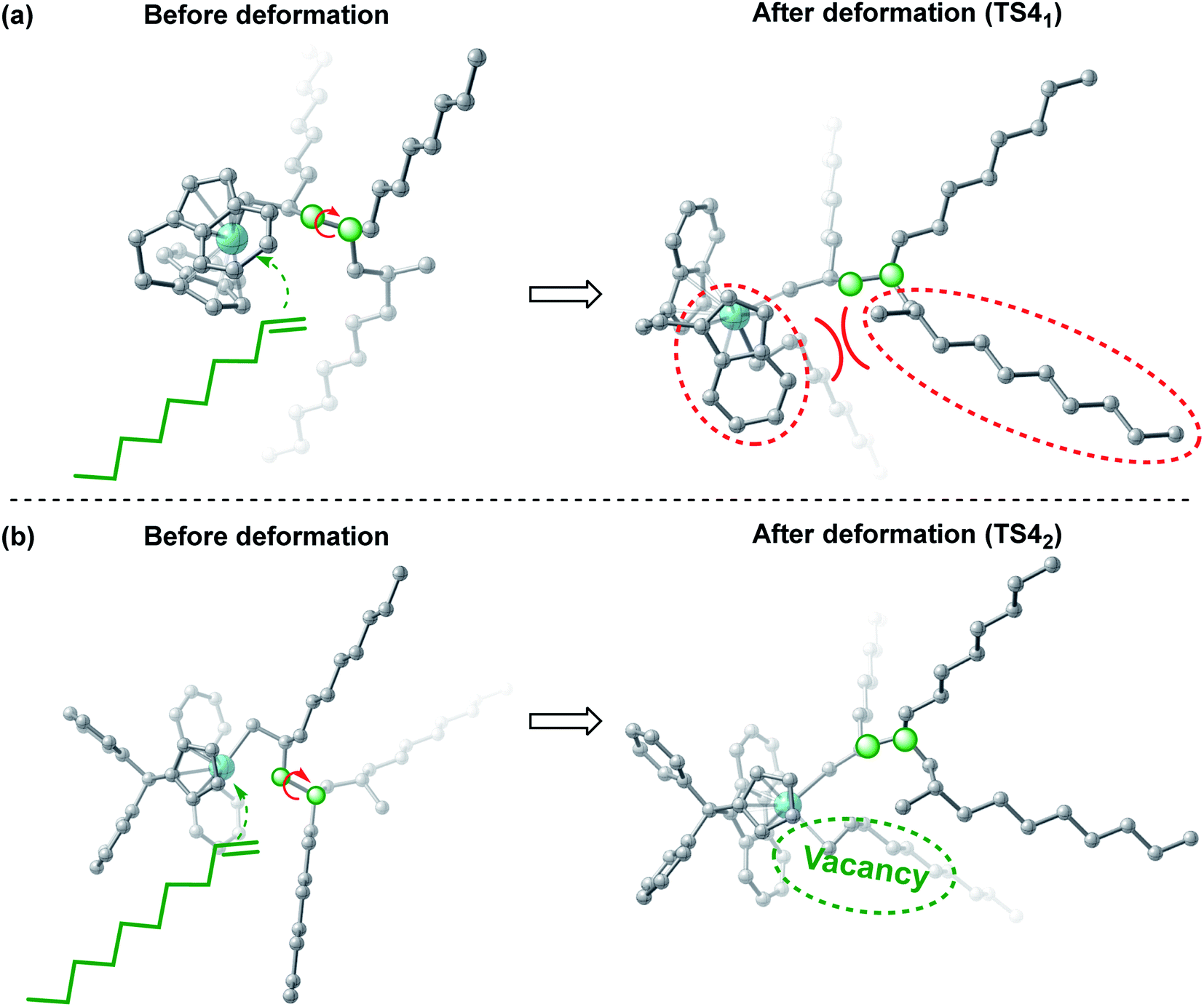
When considering the polymerization behavior, it is necessary to clarify the nature of the monomer and active species. First, the frontier molecular orbital energy gap between m and two active species (Cat.1 and Cat.2) was calculated. Generally, the reactivity is strongly influenced by the frontier molecular orbital energy gap of the two reactants: the smaller the energy gap between the two reactants, the higher the reaction activity. As shown in Fig. 6, the energy gap between m and Cat.1 is significantly smaller than that between m and Cat.2; therefore, there is a relatively strong interaction between m and Cat.1 in TS41 (−71.7 kcal mol−1). The difference in the reactivity between the two catalysts originates mainly from the steric hindrance effect. However, the difference in activity is not manifested in the topographical steric map analyses,64 because buried volume %VBur of Cat. 1 and 2 are close to each other (81.7 vs. 82.3, Fig. S2†). Therefore, in the subsequent sections, we will discuss the structures of the two transition states TS41 and TS42 in detail (Fig. 7). In TS41, the polymer chain has to be twisted to a certain extent for coordination and insertion of the next monomer, but the steric hindrance of the fused-ring on the ligand of Cat.1 is large, resulting in a large repulsion between the polymer chain and the fused-ring. As such, TS41 needs to overcome the higher energy barrier. However, one of the metallocene rings of Cat.2 is cyclopentadienyl with small steric hindrance. The above-mentioned repulsion does not exist in this system, and thus, the energy barrier is relatively low. Consequently, the polymerization activity can be improved by reducing the repulsion between the ligands and polymer chains.
 |
| | Fig. 6 Comparison of the frontier molecular orbital energy gap between m and Cat.1(a)/Cat.2(b). | |
 |
| | Fig. 7 Geometric deformation of the transition states TS41 and TS42. | |
Next, the effects of different catalyst ligands on the molecular weight of polymers will be discussed. In general, the larger the energy barrier difference of the CC insertion and β-H elimination, the higher the molecular weight of the polymerization products. In this study, the insertion of the fourth molecular monomer and β-H elimination based on the third insertion product mediated by Cat.1 and Cat.2 were investigated (Fig. 3 and 4). The results showed that when the catalyst was changed from Cat.1 to Cat.2, the CC insertion barrier decreased by 1.8 kcal mol−1 and the β-H elimination energy barrier decreased by 1.5 kcal mol−1. As a result, the energy barrier difference between the CC insertion and β-H elimination mediated by Cat.2 was 0.3 kcal mol−1 higher than that by Cat.1, indicating that CC insertion is relatively easier when using Cat.2. These results are consistent with the above-mentioned experimental results (Table 1). In addition, the effect of changing the ligand on CC insertion is greater than that on β-H elimination. In other words, the molecular weight of the polymerization product can be increased by reducing the CC insertion energy barrier.
Effect of monomer chain length on stereoselectivity
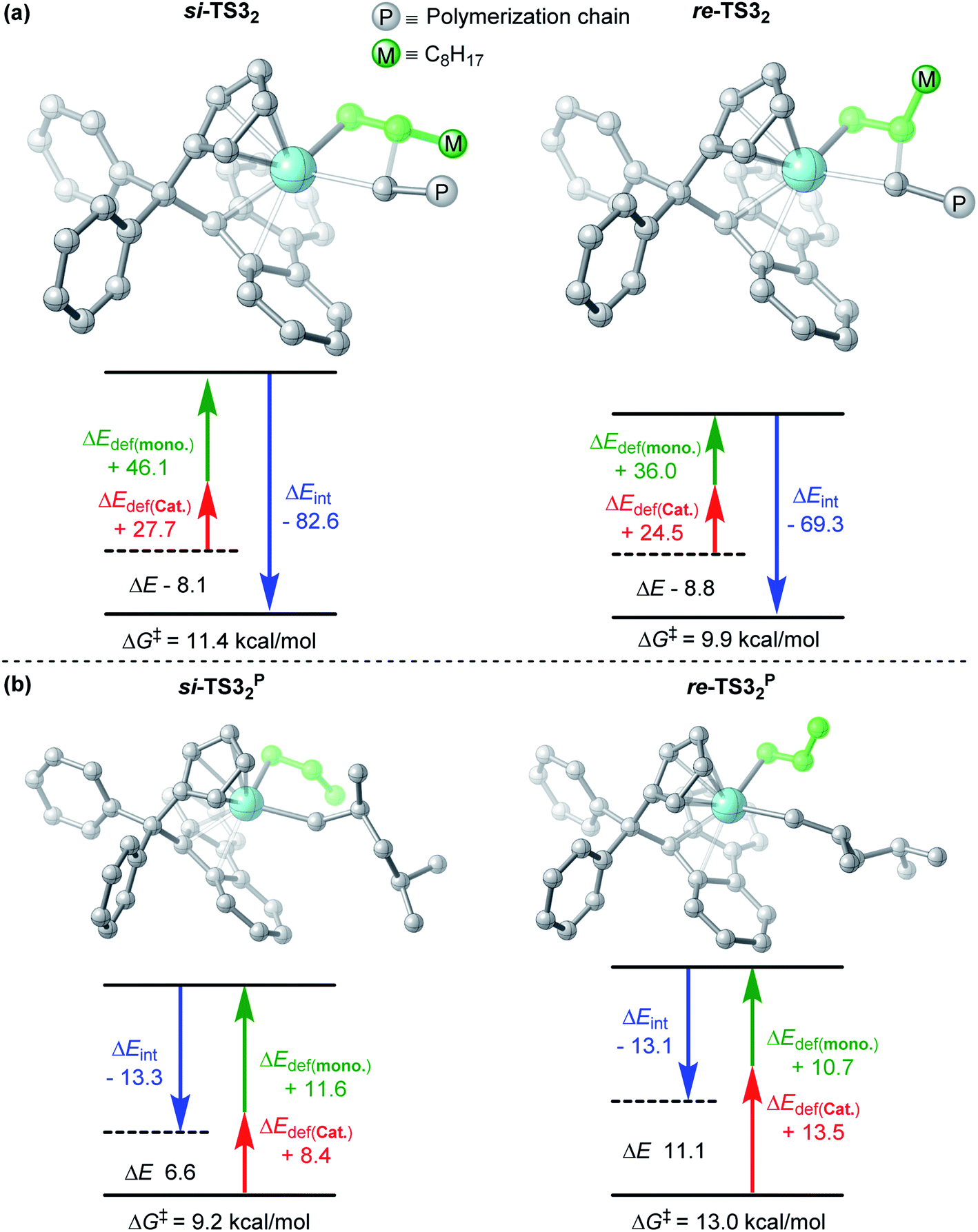
It has been reported in the literature52 that a syndiotactic polymerization product can obtained through the polymerization of propylene catalyzed by Cat.2, which in contrast to what is mentioned above, that isotactic products can be obtained through the polymerization of m catalyzed using the same catalyst. To further understand the effect of the monomer chain length on stereoselectivity, propylene polymerization with Cat.2 was conducted and the results are depicted in Fig. 8. The computational results are consistent with those reported in the literature,52 and syndiotactic polymerization products (si-re-si-re) were obtained. Further, m and propylene showed different stereoselectivities at the third molecule insertion (si-re-re-re vs. si-re-si-re). Therefore, the activation strain analysis for the TSs of the third insertion (both m and propylene) was performed. As shown in Fig. 9a, in the case of re-TS32, the total distortion energy ΔEstrain is 60.5 kcal mol−1, which could be partly balanced out by its ΔEint (−69.3 kcal mol−1) leading to a ΔETS of −8.8 kcal mol−1. By contrast, a larger total distortion energy (ΔEstrain = 73.8 kcal mol−1) in the si-TS32 is difficult to be compensated by the interaction energy of −82.6 kcal mol−1, thus producing a slightly higher ΔETS (si-TS32, −8.1 kcal mol−1). Therefore, the larger steric repulsion between the metal center and monomer moiety in si-TS32 could account for its lower stability in comparison with re-TS32. In addition, as displayed in Fig. 9b, the interaction energy between the monomer and catalyst in si-TS32P and re-TS32P are almost the same, and the large total deformation energy of the monomer and catalyst in re-TS32P is the primary reason for the higher energy barrier (13.0 kcal mol−1).
 |
| | Fig. 8 Computed energy profiles for 2 mediated chain initiation and chain propagation of propylene. | |
 |
| | Fig. 9 Activation strain analysis of si-TS32, re-TS32, si-TS32P and re-TS32P. | |
Based on the above-mentioned results, it was observed that the different types of stereoselectivity in the polymerization of m and propylene can be explained based on the steric hindrance between the catalyst and monomer. Therefore, by analyzing the geometric structure of the key transition states, the main causes of different deformations can be explored. As can be seen in Fig. 10, in si-TS33, due to the long monomer (m) and polymer chain, it is easy to produce a large repulsion between the monomer and polymer chain, resulting in a relatively high energy barrier (11.4 kcal mol−1). However, in re-TS33, the relative orientation of monomer insertion is changed from si surface to re surface, effectively avoiding a large repulsion and lowering the energy barrier (9.9 kcal mol−1). However, such a steric repulsion does not exist in propylene polymerization because the molecular chain of propylene is relatively short. Therefore, syndiotactic products can be obtained by the polymerization of propylene.
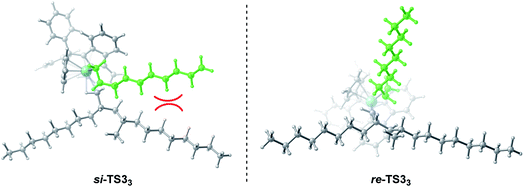 |
| | Fig. 10 Geometric structures of si-TS33 and re-TS33. | |
Conclusion
In summary, the origin of activity and molecular weight metallocene difference in the polymerization of 1-decene, catalyzed by various zirconium catalysts, have been computationally elucidated. The obtained results indicate that the effect of different catalyst ligands on β−H elimination is less than that on CC insertion. Therefore, these systems can be used to indirectly regulate the molecular weight of polymerization products, by changing the energy barrier of the CC insertion. The easier the CC insertion, the higher the molecular weight of the polymerization product. Moreover, improving the activity of catalytic polymerization can be achieved by reducing the repulsion between the catalyst ligand and the polymer growth chain, such as reducing the steric hindrance of one aromatic ring of the catalyst. In addition, through the analysis of the transition state structure, the chain length of α-olefins will directly affect the stereoselectivity of polymerization products. When the length of the monomer is short, syndiotactic products are obtained, otherwise isotactic products are obtained. It is worth noting that, in practice, a polymerization reaction system is more complex. When the steric hindrance is regulated, the electronic factors may also change accordingly. Therefore, specific analysis must be carried out for specific systems.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was partly supported by the NSFC (22101041, 22071015, U1862115) and China Postdoctoral Science Foundation (2021M700664). The authors also thank the RICC (RIKEN Integrated Cluster of Clusters) and the Network and Information Center of the Dalian University of Technology for part of the computational resources.
References
- S. Ray, P. Rao and N. Choudary, Poly-α-Olefin-Based Synthetic Lubricants: A Short Review on Various Synthetic Routes, Lubr. Sci., 2012, 24, 23–44 CrossRef CAS
 .
. - G. Yadav and N. Doshi, Development of a Green Process for Poly-α-Olefin Based Lubricants, Green Chem., 2002, 4, 528–540 RSC .
- J. Hogg, A. Ferrer-Ugalde, F. Coleman and M. Swadźba-Kwaśny, Borenium Ionic Liquids as Alternative to BF3 in Polyalphaolefins (PAOs) Synthesis, ACS Sustainable Chem. Eng., 2019, 7, 15044–15052 CrossRef CAS .
- M. Lahtela and T. Pakkanen, Molecular Modeling of Poly-α-olefin Synthetic Oils, J. Phys. Chem., 1995, 99, 10267–10271 CrossRef CAS .
- R. Leslie and L. Ronald Synthetic Lubricants and High-Performance Functional Fluids[M]. Translated by Li P Q, Guan Z J, Geng Y J., China Petroleum Industrial Press, Beijing, 2006 Search PubMed .
- R. Shubkinand and M. Kerkemeyer, Tailor-making Polyalphaolefins, Lubr. Sci., 2010, 8, 115–134 CrossRef .
- J. Baek, Y. Hyun, H. Lee, J. Lee, S. Bae, Y. Seo, D. Lee and B. Lee, Selective Trimerization of α-Olefins with Immobilized Chromium Catalyst for Lubricant Base Oils, Catalysts, 2020, 10, 990–1012 CrossRef CAS .
- Q. Huang, L. Chen, L. Ma, Z. Fu and W. Yang, Synthesis and Characterization of Oligomer from 1-Decene Catalyzed by Supported Ziegler–Natta Catalyst, Eur. Polym. J., 2005, 41, 2909–2915 CrossRef CAS .
- Q. Huang, L. Chen, Y. Sheng, L. Ma, Z. Fu and W. Yang, Synthesis and Characterization of Oligomer from 1-Decene Catalyzed by AlCl3/TiCl4/SiO2/Et2AlCl, J. Appl. Polym. Sci., 2006, 101, 584–590 CrossRef CAS .
- H. Shao, X. Gu, R. Wang, X. Wang, T. Jiang and X. Guo, Preparation of Lubricant Base Stocks with High Viscosity Index through 1-Decene Oligomerization Catalyzed by Alkylaluminum Chloride Promoted by Metal Chloride, Energy Fuels, 2020, 34, 2214–2220 CrossRef CAS .
- C. Zhou, W. Li, M. Qiu, W. Li, H. Liu, H. Liu, K. Zhang and X. Chen, Precisely Regulating the Acidity of Mesoporous Silica on the Catalytic Performance of 1-Decene Oligomerization, New J. Chem., 2021, 45, 9109–9117 RSC .
- R. Brüll, H. Pasch, H. Raubenheimer, R. Anderson and U. Wahner, Polymerization of Higher Linear α-Olefins with (CH3)2Si(2- methylbenz[e]indenyl)2ZrCl2, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2333–2339 CrossRef .
- I. Nifanťev and P. Ivchenko, Fair Look at Coordination Oligomerization of Higher α-Olefins, Polymers, 2020, 12, 1082–1113 CrossRef PubMed .
- A. Jalali, M. Nekoomanesh, S. Dehghani and N. Bahri-Laleh, Effect of Metal Type on the Metallocene-Catalyzed Oligomerization of 1-Hexene and 1-Octene to Produce Poly α-Olefin-Based Synthetic Lubricants, Appl. Organomet. Chem., 2019, e5338 Search PubMed .
- S. Dong, P. Mi, S. Xu, J. Zhang and R. Zhao, Preparation and Characterization of Single-Component Poly-α-olefin Oil Base Stocks, Energy Fuels, 2019, 33, 9796–9804 CrossRef CAS .
- H. Shao, H. Li, J. Lin, T. Jiang, X. Guo and J. Li, Metallocene-Catalyzed Oligomerizations of 1-Butene and α-Olefins: Toward Synthetic Lubricants, Eur. Polym. J., 2014, 59, 208–217 CrossRef CAS .
- M. Masoori, M. Nekoomanesh, S. Posada-Pérez, R. Rashedi and N. Bahri-Laleh, Exploring Cocatalyst Type Effect on the Ziegler-Natta Catalyzed Ethylene Polymerizations: Experimental and DFT Studies, J. Polym. Res., 2022, 29, 197–208 CrossRef CAS .
- A. Piovano, M. D'Amore, K. Thushara and E. Groppo, Spectroscopic
Evidences for TiCl4/Donor Complexes on the Surface of MgCl2-Supported Ziegler-Natta Catalysts, J. Phys. Chem. C, 2018, 122, 5615–5626 CrossRef CAS .
- J. Kumawat and V. Gupta, Fundamental Aspects of Heterogeneous Ziegler-Natta Olefin Polymerization Catalysis: an Experimental and Computational Overview, Polym. Chem., 2020, 11, 6107–6128 RSC .
- A. Shamiri, M. Chakrabarti, S. Jahan, M. Hussain, W. Kaminsky, P. Aravind and W. Yehye, The Influence of Ziegler-Natta and Metallocene Catalysts on Polyolefin Structure, Properties, and Processing Ability, Materials, 2014, 7, 5069–5108 CrossRef PubMed .
- S. Rahmatiyan, N. Bahri-Laleh, A. Hanifpour and M. Nekoomanesh-Haghighi, Different Behaviors of Metallocene and Ziegler-Natta Catalysts in Ethylene/1,5-Hexadiene Copolymerization, Polym. Int., 2019, 68, 94–101 CrossRef CAS .
- W. Kaminsky, Highly Active Metallocene Catalysts for Olefin Polymerization, J. Chem. Soc., Dalton Trans., 1998, 1413–1418 RSC .
- R. Stockland, S. Foley and R. Jordan, Reaction of Vinyl Chloride with Group 4 Metal Olefin Polymerization Catalysts, J. Am. Chem. Soc., 2003, 125, 796–809 CrossRef CAS PubMed .
- J. Huang, T. Wu and Y. Qian, Ethylene trimerization with a half-sandwich titanium complex bearing a pendant thienyl group, Chem. Commun., 2003, 2816–2817 RSC .
- K. Endo, T. Nomaguchi and Y. Tsuchiya, Control of Molecular Weight in Polymerization of Vinyl Chloride with Cp*Ti(OPh)3/MAO Catalyst, J. Polym. Sci., Part A: Polym. Chem., 2010, 45, 3872–3876 CrossRef .
- K. Endo and M. Saitoh, Polymerization of Vinyl Chloride with Cp*Ti(OCH3)3 -Methylaluminoxane Catalyst, Polym. J., 2000, 32, 300–302 CrossRef CAS .
- S. Gharajedaghi, Z. Mohamadnia, E. Ahmadi, M. Marefat, G. Pareras, S. Simon, A. Poater and N. Bahri-Laleh, Experimental and DFT Study on Titanium-based Half-Sandwich Metallocene Catalysts and their Application for Production of 1-Hexene from Ethylene, Mol. Catal., 2021, 509, 111636–111648 CrossRef CAS .
- J. McInnis, M. Delferro and T. Marks, Multinuclear Group 4 Catalysis: Olefin Polymerization Pathways Modified by Strong Metal–Metal Cooperative Effects, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed .
- A. McKnight and R. Waymouth, Group 4 ansa-Cyclopentadienyl-Amido Catalysts for Olefin Polymerization, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS PubMed .
- U. Siemeling, Chelate Complexes of Cyclopentadienyl Ligands Bearing Pendant O-Donors, Chem. Rev., 2000, 100, 1495–1526 CrossRef CAS PubMed .
- H. Alt and A. Köpp, Effect of the Nature of Metallocene Complexes of Group IV Metals on Their Performance in Catalytic Ethylene and Propylene Polymerization, Chem. Rev., 2000, 100, 1205–1221 CrossRef CAS PubMed .
- A. Zhou, Y. Zhang, Y. Shia and Q. Xu, Integrated Synthesis of Metallocene@support Catalysts Based on Glyphosate and its Zirconium Derivatives, RSC Adv., 2017, 7, 55866–55873 RSC .
- W. Kaminsky, K. Kiilper, H. Brintzinger and F. Wild, Polymerization of Propene and Butene with a Chid Zirconocene and Methylalumoxane as Cocatalyst, Angew. Chem., Int. Ed., 1985, 24, 507–508 CrossRef .
- J. Eisch, F. Owuor and P. Otieno, Metalations with Group 4 Alkylmetal(IV) Halides: Expeditious Route to Metallocene and Nonmetallocene Procatalysts, Organometallics, 2001, 20, 4132–4134 CrossRef CAS .
- J. Buffet, T. Arnold, Z. Turner, P. Angpanitcharoen and D. O'Hare, Synthesis and Characterisation of Permethylindenyl Zirconium Complexes and Their Use in Ethylene Polymerization, RSC Adv., 2015, 5, 87456–87464 RSC .
- F. Wild, L. Zsolnai, G. Huttner and H. Brintzinger, Synthesis and Molecular Structures of Chiral ansa-Titanocene Derivatives with Bridged Tetrahydroindenyl Ligands, J. Organomet. Chem., 1982, 232, 233–247 CAS .
- F. Wild, M. Wasiucionek, G. Huttner and H. Brintzinger, Synthesis and Crystal Structure of a Chiral ansa-Zirconocene Derivative with Ethylene-Bridged Tetrahydroindenyl Ligands, J. Organomet. Chem., 1985, 288, 63–67 CrossRef CAS .
- C. Zuccaccia, N. Stahl, A. Macchioni, M. Chen, J. Roberts and T. Marks, NOE and PGSE NMR Spectroscopic Studies of Solution Structure and Aggregation in Metallocenium Ion-Pairs, J. Am. Chem. Soc., 2004, 126, 1448–1464 CrossRef CAS PubMed .
- F. Song, S. Lancaster, R. Cannon, M. Schormann, S. Humphrey, C. Zuccaccia, A. Macchioni and M. Bochmann, Synthesis, Ion Aggregation, Alkyl Bonding Modes, and Dynamics of 14-Electron Metallocenium Ion Pairs [(SBI)MCH2SiMe3+⋯, X−] (M = Zr, Hf): Inner-Sphere (X = MeB(C6F5)3) versus Outer-Sphere (X = B(C6F5)4) Structures and the Implications for “Continuous” or “Intermittent” Alkene Polymerization Mechanisms, Organometallics, 2005, 24, 1315–1328 CrossRef CAS .
- Y. Zhao, H. Lu, G. Luo, X. Kang, Z. Hou and Y. Luo, Origin of Stereoselectivity and Multidimensional Quantitative Analysis of Ligand Effects on Yttrium Catalysed Polymerization of 2-Vinylpyridine, Catal. Sci. Technol., 2019, 9, 6227–6233 RSC .
- C. Wang, G. Luo, M. Nishiura, G. Song, A. Yamamoto, Y. Luo and Z. Hou, Heteroatom-Assisted Olefin Polymerization by Rare-Earth Metal Catalysts, Sci. Adv., 2017, 3, e1701011 CrossRef PubMed .
- X. Wang, F. Lin, J. Qu, Z. Hou and Y. Luo, DFT Studies on Styrene Polymerization Catalyzed by Cationic Rare-Earth-Metal Complexes: Origin of Ligand-Dependent Activities, Organometallics, 2016, 35, 3205–3214 CrossRef CAS .
- A. Kronast, D. Reiter, P. Altenbuchner, S. Vagin and B. Rieger, 2-Methoxyethylamino-bis(phenolate) yttrium Catalysts for the Synthesis of Highly Isotactic Poly(2-vinylpyridine) by Rare-Earth Metal-Mediated Group Transfer Polymerization, Macromolecules, 2016, 49, 6260–6267 CrossRef CAS .
- A. Laine, B. Coussens, J. Hirvi, A. Berthoud, N. Friederichs, J. Severn and M. Linnolahti, Effect of Ligand Structure on Olefin Polymerization by a Metallocene/Borate Catalyst: A Computational Study, Organometallics, 2015, 34, 2415–2421 CrossRef CAS .
- S. Karimia, N. Bahri-Laleha, G. Parerasb, S. Sadjadic, M. Nekoomanesh-Haghighia and A. Poater, Pd on Nitrogen Rich Polymer-Halloysite Nanocomposite as an Environmentally Benign and Sustainable Catalyst for Hydrogenation of Polyalfaolefin based Lubricants, J. Ind. Eng. Chem., 2021, 97, 441–451 CrossRef .
- M. D'Alterio, C. Rosa and G. Talarico, Stereoselective Lactide Polymerization: the Challenge of Chiral Catalyst Recognition, ACS Catal., 2020, 10, 2221–2225 CrossRef .
- A. Hanifpour, N. Bahri-Laleh, M. Nekoomanesh-Haghighi and A. Poater, Coordinative Chain Transfer Polymerization of 1-Decene in the Presence of A Ti-Based Diamine Bis(phenolate) Catalyst: A Sustainable Approach to Produce Low Viscosity PAOs, Green Chem., 2020, 22, 4617–4626 RSC .
- M. Tabrizi, S. Sadjadi, G. Pareras, M. Nekoomanesh-Haghighi, N. Bahri-Laleh and A. Poater, Efficient Hydro-Finishing of Polyalfaolefin Based Lubricants under Mild Reaction Condition using Pd on Ligands Decorated Halloysite, J. Colloid Interface Sci., 2021, 581, 939–953 CrossRef CAS PubMed .
- A. Hanifpoura, N. Bahri-Laleha, M. Nekoomanesh-Haghighia and A. Poater, Group IV Diamine Bis(Phenolate) Catalysts for 1-Decene Oligomerization, Mol. Catal., 2020, 493, 111047 CrossRef .
- A. Hanifpour, N. Bahri-Laleh, M. Nekoomanesh-Haghighi and A. Poater, 1-Decene Oligomerization by New Complexes Bearing Diamine-Diphenolates Ligands: Effect of Ligand Structure, Appl. Organomet. Chem., 2021, 35, e6227 CrossRef CAS .
- I. Lee, W. Gauthier, J. Ball, B. Iyengar and S. Collins, Electronic Effects in Ziegler-Natta Polymerization of Propylene and Ethylene Using Soluble Metallocene Catalysts, Organometallics, 1992, 11, 2115–2122 CrossRef CAS .
- A. Razavi and J. Atwood, Preparation and Crystal Structures of the Complexes (η5-C5H4CPh2-η5-C13H8)MCl2, (M = Zr, Hf) and the Catalytic Formation of High Molecular Weight
High Tacticity Syndiotactic Polypropylene, J. Organomet. Chem., 1993, 459, 117–123 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed .
- A. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5653 CrossRef CAS .
- C. Lee, W. Yang and R. Parr, Development of the Colic-Salvetti Correlation-Energy Formula into A Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed .
- J. Perdew, K. Burke and Y. Wang, Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System, Phys. Rev. B, 1996, 54, 16533–16539 CrossRef CAS PubMed .
-
(a) P. Hay and W. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS ;
(b) P. Hay and W. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K To Au Including the Outermost Core Orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS ;
(c) W. Wadt and P. Hay, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS .
-
(a) Y. Zhao, G. Luo, X. Wang, X. Kang, D. Cui, Z. Hou and Y. Luo, DFT Studies on the Polymerization of Functionalized Styrenes Catalyzed by Rare-Earth-Metal Complexes: Factors Affecting C-H Activation Relevant to Step-Growth Polymerization, Organometallics, 2018, 37, 3210–3218 CrossRef CAS ;
(b) Y. Zhao, G. Luo, X. Kang, F. Guo, X. Zhu, R. Zheng, Z. Hou and Y. Luo, “C–H…π Interaction” Regulates the Stereoselectivity in Olefin Polymerization, Chem. Commun., 2019, 55, 6689–6692 RSC ;
(c) L. Perrin, F. Bonnet, M. Visseaux and L. Maron, A DFT Study of Conjugated Dienes Polymerization Catalyzed by [Cp*ScR]+: Insights into the Propensity for cis-1,4 Insertion, Chem. Commun., 2010, 46, 2965–2967 RSC ;
(d) L. Perrin, F. Bonnet, T. Chenal, M. Visseaux and L. Maron, A Joint Experimental/Theoretical Investigation of the Statistical Olefin/Conjugated Diene Copolymerization Catalyzed by a Hemi-Lanthanidocene [(Cp*)(BH4)LnR], Chem.–Eur. J., 2010, 16, 11376–11385 CrossRef CAS PubMed .
-
(a) Y. Zhao and D. Truhlar, Benchmark Energetic Data in a Model System for Grubbs II Metathesis Catalysis and their Use for the Development, Assessment, and Validation of Electronic Structure Methods, J. Chem. Theory Comput., 2009, 5, 324–333 CrossRef CAS PubMed ;
(b) Y. Zhao and D. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed .
- A. Marenich, C. Cramer and D. Truhlar, Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed .
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed .
- Y. Luo, Y. Luo, J. Qu and Z. Hou, QM/MM Studies on Scandium-Catalyzed Syndiospecific Copolymerization of Styrene and Ethylene, Organometallics, 2011, 30, 2908–2919 CrossRef CAS .
-
(a) F. Bickelhaupt and K. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed ;
(b) I. Fernández and F. Bickelhaupt, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC ;
(c) F. Liu, Y. Liang and K. Houk, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed ;
(d) K. Kitaura and K. Morokuma, A New Energy Decomposition Scheme for Molecular Interactions within the Hartree-Fock Approximation, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS .
-
(a) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps, Organometallics, 2016, 35, 2286–2293 CrossRef CAS ;
(b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the Online Computer-Aided Design of Catalytic Pockets, Nat. Chem., 2019, 11, 872–880 CrossRef CAS PubMed , see https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html..
Footnote |
| † Electronic supplementary information (ESI) available: Optimized cartesian coordinates of all stationary points together with their single-point energies (a.u.) in solution and the imaginary frequencies (cm−1) of transition states (XYZ file). See https://doi.org/10.1039/d2ra03180a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xianming Xuc,
Yulong Wangc,
Tong Liuc,
Hongpeng Lic,
Yongjun Zhangc,
Libo Wangc,
Xiuhui Wangc,
Simeng Zhaoc and
Yi Luo
a,
Xianming Xuc,
Yulong Wangc,
Tong Liuc,
Hongpeng Lic,
Yongjun Zhangc,
Libo Wangc,
Xiuhui Wangc,
Simeng Zhaoc and
Yi Luo