
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilization of a plasticized PVC optical sensor for the selective and efficient detection of cobalt(II) in environmental samples
Hesham H. El-Feky ,
Alaa S. Amin* and
Eslam M. I. Moustafa
,
Alaa S. Amin* and
Eslam M. I. Moustafa
Chemistry Department, Faculty of Science, Benha University, Benha, Egypt. E-mail: asamin2005@hotmail.com
First published on 22nd June 2022
Abstract
A novel sensitive, selective, and reversible cobalt(II) ion optical sensor was prepared by the incorporation of 5-[o-carboxyphenylazo]2,4-dihydroxybenzoic acid [CPDB] and sodium tetraphenylborate (NaTPB) in a plasticized polyvinyl chloride (PVC) membrane containing dioctyl adipate (DOA) as a plasticizer. The influence of several parameters such as pH, base matrix, solvent mediator and reagent concentration was optimized. A comparison of the obtained results with those of previously reported sensors revealed that the proposed method, in addition to being fast and simple, provided a good linear range (0.05–45.20 μM) and low detection limit (0.015 μM). Low detection and quantification limits and excellent selectivity in the presence of interfering ions such as Fe3+, Cu2+, Ni2+, Ag+, Au3+, Cr3+, Cd2+, Zn2+, Hg2+, and SO42− make it feasible to monitor Co2+ ion content accurately and repeatedly in environmental samples with complicated matrices. The optode was regenerated successfully using 0.3 M nitric acid (HNO3) solution while its response was reversible with a relative standard deviation (RSD) lower than 1.9% for seven replicate determinations of 20 μM Co2+ in various membranes. The optode was stable and was stored for at least 15 days without observing any change in its sensitivity.
Introduction
Environmental pollutants have attracted attention due to their ability to gradually accumulate in the human body through the food chain and cause diseases and additional complications. In recent years, all over the world, heavy metal pollution has become a serious concern due to the increase in urban development and industry. As a constituent of vitamin B12, cobalt is a critical metal in the environment, having important roles in the body. Depending on its concentration, it can be an essential metal but can also be toxic for many living beings.1 In the medicine and toxicology fields, the assessment of trace levels of Co2+ ions plays a significant role in environmental sample evaluation.2 Owing to its inexpensive procedures, colorimetry is a frequently used analytical technique for assessing ultra-trace metals in biological samples. Preliminary preconcentration and sample clean-up procedures are required to determine trace metals because of their low abundance and high complexity.3,4A variety of sensitive and selective analytical methodologies such as electroanalysis,5 atomic absorption spectrometry (AAS),6 flame atomic absorption spectrometry (FAAS),2,7–12 electrothermal atomic absorption spectrometry (ET-AAS),13,14 chemiluminescence,15 X-ray fluorescence,16 laser-induced thermal lens spectrometry,17 neutron activation analysis (NAA),18 inductively coupled plasma optical emission spectroscopy (ICP-OES),19,20 inductively coupled plasma mass spectrometry (ICP-MS),21 and spectrophotometry22–30 have been demonstrated for the assessment of cobalt in various samples. However, many of these methods involve the risk of sample contamination and analyte loss due to the sample preparation and preconcentration steps. Among these techniques, spectrophotometry is the most convenient one due to its simplicity, availability, specificity, and low cost.
Many types of samples in their natural state cannot be analyzed due to the presence of analytes at ultra-trace levels. Furthermore, due to a great demand for analytical techniques in the environmental field, the established sample number has also been increased by these labs. The methodologies of optical sensor optodes have been prescribed to separate and/or preconcentrate the analyte, thereby defining the detection limits of current instrumentation. To achieve high sensitivity and productivity, coupling between optical sensor optodes and colorimetry has been shown to be an effective strategy.
To ensure operator safety and environmental preservation, green analytical chemistry studies various analytical procedures that minimize toxic substance consumption, reagent use, waste generation and waste decontamination.31 Recently, optical sensors have been of great interest in analytical chemistry.32–35 In clinical and environmental monitoring, optodes have played a significant role owing to their miniaturization potential, good flexibility, electrical interference freedom, low cost and remote sensing potential. Moreover, it is possible to develop optical sensors for species that cannot be sensed in other ways.
These sensors usually consist of diverse reagents that are immobilized by applying diverse procedures within appropriate membranes, wherein analyte mass transport into the membrane changes its optical characteristics. The ideal sensor must possess a rapid response time, long lifetime and reversibility, which are affected by the immobilization technique used.36–39
Lately, many optodes have been synthesized with diverse membranes. In most of these sensors, the reagent is immobilized by simple adsorption onto the membranes, covalent bonding or entrapment.40–43 Transparent triacetyl-cellulose has been extensively applied for chemical reagent immobilization as a polymeric membrane. Many spectrophotometry-based optodes have several drawbacks such as high detection limits, vast interfering effects of ions, and narrow ranges of assessment. To exhibit good sensitivity and selectivity toward Co2+ ions, the 5-[o-carboxyphenylazo]2,4-dihydroxybenzoic acid (CPDB)44 ionophore has been applied, which well enhances the detection limits, decreases the effect of interference, and widens the concentration range. Previously reported optical sensors depending on a one-at-a-time approach have various limitations such as high experiment number (which is expensive due to costly reagent consumption) and possible errors in the optimum value attainment of variables and their consequences.
In our laboratory, CPDB was synthesized and used to determine cobalt in synthetic mixtures and in pharmaceutical formulations.44 The goal of the present study is to successfully incorporate this reagent in plasticized PVC film to construct a novel optical sensor for Co2+ determination. The sensor response time and pH value were optimized separately. The influence of several parameters such as pH, base matrix, solvent mediator, and reagent concentration were optimized.
Experimental
Chemicals
All the chemicals were of analytical grade. Low molecular weight polyvinyl chloride (LPVC, mol wt ≈ 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), tetraphenylborate (NaTPB), dioctyl adipate (DOA), sodium dibutylphthalate (DBP), tributylphosphate (TBP), dioctylphthalate (DOP), o-nitrophenyloctyl ether (o-NPOE) and tetrahydrofuran (THF) were used without purification.
000 g mol−1), tetraphenylborate (NaTPB), dioctyl adipate (DOA), sodium dibutylphthalate (DBP), tributylphosphate (TBP), dioctylphthalate (DOP), o-nitrophenyloctyl ether (o-NPOE) and tetrahydrofuran (THF) were used without purification.
5-[o-Carboxyphenylazo]-2,4-dihydroxybenzoic acid (CPDB) was prepared as described in our previous work.44 Universal, thiel, phosphate, borate and acetate buffer solutions at different pH values of 2.0–12 were prepared as described previously.45 0.01 M stock solutions of interfering ions were prepared by dissolving appropriate amounts of suitable salts in double-distilled water.
Stock solutions of Co2+ (1.7 × 10−3 M) were prepared by dissolving 0.4947 g of the respective pure nitrate salt Co(NO3)2·6H2O (Merck, Darmstadt, Germany) in the minimum amount of deionized water, completed to the mark of a 100 mL measuring flask with water, and standardized volumetrically against EDTA. Working standard solutions were obtained daily by the suitable stepwise dilution of the stock solutions with deionized water and shaking them just before use.
Instrumentation
An Orion research model 601 A/digital ionalyzer pH meter was used to check the pH of the solutions. A Shimadzu model 670 atomic absorption spectrometer with flame atomization was used. The operating parameters were set as recommended by the manufacturer. Atomic absorption measurements were carried out in a nitrous oxide-acetylene flame. A UV-vis spectrophotometer model V-670 from JASCO (Tokyo, Japan) was used to record the spectra and the absorbance measurements. The absorbance measurements were obtained by mounting optical membrane sensor samples (1 × 9 × 50 mm3) inside a quartz cuvette. The absorbance measurements of the optical membrane sensor samples were obtained with respect to air as well as the blank optode sample. The film thicknesses of the sensing slides were measured using a digital microscope (Ray Vision Y 103) that was coupled to a video camera (JVC TK-C 751 EG) and a digital micrometer (Mitutoy, Japan) with an accuracy of ±0.001 mm.Membrane preparation
The transparent glass slide membranes were purchased from microscope slides in sizes of 1 × 9 × 50 mm3 and were kept in a HNO3:H2O2 (3:1) volume ratio solution. The optical sensor was washed with THF and double-distilled water before use. The sensing mixture was prepared by dissolving 5.0 mg NaTPB, 8.0 mg CPDB, 30 mg PVC and 75 mg DOA in 2.0 mL THF. The mixture was stirred with a magnetic stirrer to obtain a homogeneous solution. Glass plates were placed in the spin-on device and spin-coated onto a glass plate at 1000 rpm for 30 s. The thickness of the obtained membrane was estimated to be about 0.005 mm. The membrane was placed in ambient air and allowed to dry in air for 5.0 min, and the optode was maintained in a buffer solution of pH 7.5.
General procedure
The sensing membrane (optode) was placed in a beaker filled with 25 mL of the test solutions containing different levels of Co2+ (0.05–45.20 μM) and 5.0 mL of pH 7.5 buffer. After 5.0 min, the optode was mounted into the spectrophotometer directly and its net absorbance was assessed at λmax = 595 nm against a blank membrane prepared in the same manner except for cobalt ions.Procedure for pharmaceutical formulations
A suitable weight of vitamin B12 powder or 5.0 mL of the injectable was placed in a 25 mL crucible and digested with a few drops of HNO3 at 500 °C. The residue was heated in a water bath after dissolving in 3.0–5.0 mL HNO3 for 2.0 min. Then, it was diluted with 5.0–10.0 mL double-distilled water and filtered if necessary. Three 5.0 mL portions of double-distilled water were used to wash the crucible and the filter paper. The cobalt content of the prepared solution was assessed as stated according to the previous procedure.Water samples
Water samples were directly used after filtration with a filter paper (Whatman No. 1). The analyses of the alloy samples were conducted by weighing about 1.8 g of the sample to the nearest 0.1 mg and transferring the sample into a 250 mL Erlenmeyer flask with 10–12 mL HNO3:HCl (1:3) and a few drops of concentrated HF. The mixture was heated until dissolution was complete. The solution was heated to dryness. The residue was dissolved in water and diluted to 100 mL in a 100 mL volumetric flask. The solution was used for analysis.
Biological samples
Urine and saliva samples were gathered from male and female volunteers aged 25 to 35 years, living in Benha (Egypt), without eating breakfast. In order to minimize the possibility of contamination with cigarette or food debris and airborne particles, the subjects were asked to thoroughly rinse their mouths three times with ultra-pure water. Human saliva samples were gathered between 8 and 9 h to minimize possible circadian contributions in Co-free polystyrene test tubes.46 The samples (7.0 mL) were acidified with HNO3 to pH 2.0 and placed in a graduated centrifuge tube and centrifuged for 20 min at 1500 rpm (377.2 g). Five milliliters of the supernatant were diluted to 25 mL with double-distilled water. Co2+ was assessed by the proposed method. Reasonable dilution for analysis is practical as the subsequent collection of great volumes may be tedious for the donors. Blanks were prepared with the same reagents without the samples, and underwent an identical procedure.Urine samples were digested by UV-photolysis, as described previously.3 Briefly, 5.0 mL of the sample was transferred in a decomposition glass beaker. 200 μL 30% (w/w) H2O2 was added. The mixture was irradiated for 45 min. Another 200 μL aliquot of 30% (w/w) H2O2 was added, and the irradiation process was continued for 45 min. Finally, 10 mL water was added, and the irradiation process was repeated for another 120 min. The digested sample volume was set to 25 mL after the completion of the irradiation method.
Food samples
Fifteen grams of flour and dried vegetable samples (obtained from Benha, Egypt) were first carbonized at a low temperature. The samples were burned in a muffle furnace for about 3.0 h at 600 °C. They were extracted after cooling at room temperature by adding 2.0 mL 9.0 M H2SO4 with heating. The mixture was filtered. The filtrate pH was adjusted to ≈7.5 with NaOH. A reasonable amount of EDTA for masking was added, and the sample was taken for analysis by the suggested technique.Results and discussion
Preliminary investigations
5-[o-Carboxyphenylazo]-2,4-dihydroxybenzoic acid (CPDB) is described as a spectrophotometric reagent,44 and it exhibits a pink color when it chelates with Co2+. Owing to CPDB being an organic reagent, it is immobilized efficiently in the hydrophobic part of the membrane without prior lyophilization.47 It was shown that by optimized fabrication, a sensor made by the incorporation of CPDB in a plasticized PVC membrane, including DOA, could be colorimetrically practical for the assessment of cobalt. When Co2+ ions diffuse into the membrane, it forms a complex with CPDB; thus, the membrane color varied from orange to pink. The absorption spectra of the CPDB control (λmax = 444 nm) and cobalt-based optodes upon contact with 20 μM Co2+ at pH 7.5 (λmax = 595 nm) are shown in Fig. 1. | ||
| Fig. 1 Absorption spectra of a control sensor (PVC; DOA; NaTPB; CPDB) and cobalt based sensor upon contact with 20 mmol L−1 Co2+ at pH 7.5. | ||
Membrane composition
The response features and working concentration range of an optical sensor depend on certain ingredients such as the base matrix, solvent mediator, ionophore and additive applied in the membrane structure. Consequently, the sensor matrix must be optimized through the comparison of various polymers; low molecular weight PVC was found to be the best choice for the membrane base due to various factors such as its reasonable transmittance, appropriate immobilization of CPDB without any leakage, good mechanical stability and reliable permeability to Co2+ ions.The plasticizer is essential and must be physically compatible with the polymer. To obtain a homogenous organic phase, plasticizers such as DBP, TBP, DOP, o-NPOE and DOA were screened. As shown in Fig. 2, it was established that the membranes including DBP, TBP, DOP and o-NPOE did not obtain a suitable band due to either improper physical features with LPVC or the leakage of CPDB from the membrane. It was established that DOA was the optimum choice due to its better physical characteristics as well as its maximum sensitivity and minimum leakage.
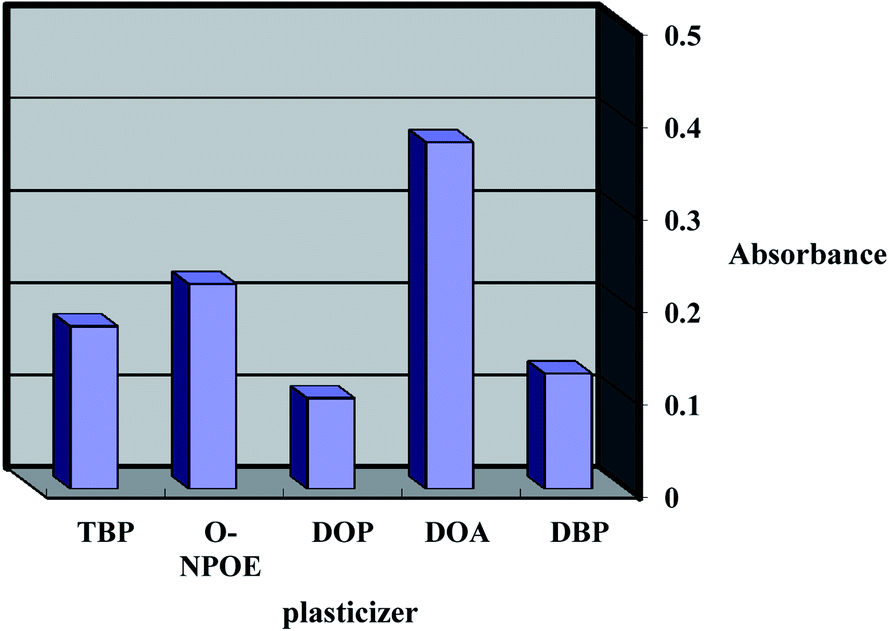 | ||
| Fig. 2 Effect of Plasticizer types on the sensor formed to complexed with 20 mmol L−1 Co2+ at pH 7.5. | ||
As listed in Table 1 (membrane no. 1–4), the sensors with a weight ratio of DOA to LPVC of 2.5 give the highest absorbance, and the results are highly concordant. Thus, 30 mg PVC and 75 mg DOA were selected as the optimum levels. The decrease in the Co2+ uptake efficiency at values lower than 75 mg is attributed to the improper solidity of the optode, which led to the low diffusion of analyte cations into the membrane. At quantities more than 75 mg, the flexibility of the optode increased, which led to ionophore leakage into the test solution.
| Sensor | LPVC (mg) | DOA (mg) | CPDB (mg) | NaTPB (mg) | Response time (min) | Absorbancea (595 nm) |
|---|---|---|---|---|---|---|
| a Mean absorbance ± SD (n = 3) of each parameter is recorded from three solutions of 20 μM Co2+ at pH 7.5. | ||||||
| 1 | 30 | 45 | 8 | 5 | 5.0 | 0.177 ± 0.021 |
| 2 | 30 | 60 | 8 | 5 | 5.0 | 0.268 ± 0.012 |
| 3 | 30 | 75 | 8 | 5 | 5.0 | 0.375 ± 0.005 |
| 4 | 30 | 90 | 8 | 5 | 5.0 | 0.323 ± 0.009 |
| 5 | 30 | 75 | 4 | 5 | 5.0 | 0.201 ± 0.017 |
| 6 | 30 | 75 | 6 | 5 | 5.0 | 0.327 ± 0.011 |
| 7 | 30 | 75 | 8 | 5 | 5.0 | 0.375 ± 0.005 |
| 8 | 30 | 75 | 10 | 5 | 5.0 | 0.337 ± 0.008 |
| 9 | 30 | 75 | 8 | 2 | 5.0 | 0.219 ± 0.024 |
| 10 | 30 | 75 | 8 | 4 | 5.0 | 0.326 ± 0.011 |
| 11 | 30 | 75 | 8 | 5 | 5.0 | 0.375 ± 0.006 |
| 12 | 30 | 75 | 8 | 7 | 5.0 | 0.292 ± 0.032 |
| 13 | 30 | 75 | 8 | 5 | 2.0 | 0.132 ± 0.023 |
| 14 | 30 | 75 | 8 | 5 | 4.0 | 0.279 ± 0.011 |
| 15 | 30 | 75 | 8 | 5 | 5.0 | 0.375 ± 0.004 |
| 16 | 30 | 75 | 8 | 5 | 10 | 0.366 ± 0.006 |
The effect of varying amounts of CPDB on the response of the membrane is shown in Table 1. As shown by membrane no. 5–8, the absorbance increased with increasing amounts of CPDB up to 8.0 mg, while the absorbance decreased at greater levels owing to membrane leakage. Thus, 8.0 mg CPDB was the optimal value.
The presence of an anionic additive such as NaTPB leads to ion-exchange equilibrium due to the decrease in the response time and the complete mass transfer of Co2+ ions into the membrane.48 The influence of NaTPB was demonstrated in the range of 2.0–7.0 mg (Table 1 membrane no. 9–12). It is found that the maximum absorbance is achieved by applying 5.0 mg NaTPB. Therefore, at this optimum concentration of NaTPB, the complete transfer of the Co2+ ion occurs only to the optode, in addition to the low response time and the complete equilibrium of ion-exchange.
Response time of the optode
The response time of the optodes is defined as the diffusion time of the metal ions from the solution into the membrane (slowest step in the complexation process).49 The influence of this factor on the optode response was determined (Table 1 membrane no. 13–16). The response time is affected by various factors, including Co2+ ion concentration, which controls its diffusion into the membrane; the ionophore loading technique;50 and the membrane thickness. As observed, a time interval of at least 5.0 min is required for quantitative Co2+ uptake at 25 ± 2.0 °C. It was noted that the optode response remained constant for more than 15 days.Effects of pH
Briefly, the ionophore structure indicates the presence of more reactive atoms such as the hydroxyl group and nitrogen atoms, which are used for binding Co2+ ions at high pH based on its complexation tendency. Consequently, the response of the optodes depends on the incorporation of the ionophore, which is significantly affected by the pH of the buffer. The influence of pH on the optode response (extent of complexation) in the pH range of 2.5–12 in buffer and non-buffer solutions was demonstrated, and the results are shown in Fig. 3. As shown, the optode absorbance increased at pH 7.5 and then decreased. At pH < 6.5, the protonation of the reagent prevents its reaction with Co2+ ions, while at pH > 9.5, the decrease in the response could be because of the hydrolysis of Co2+ ions, which is produced by the incomplete diffusion of Co2+ cations into the optode. Hence, a buffer with pH 7.5 was used in all further investigations. In comparison, the optimum pH for the Co–CPDB complex in aqueous solution has been reported as 9.5.44 | ||
| Fig. 3 Effect of pH value on the sensor response for 20 mmol L−1 of Co2+ at the optimum conditions. | ||
Lifetime
The lifetime of the optode film was assessed by adding the buffer solution (pH 7.5) to the prepared membrane. The absorbance was measured at a wavelength of 595 nm over a period of time (about 10 h). During this time, no significant loss of the carriers occurred. A stable absorbance versus time plot was achieved through the exposure of radiation to the membrane. This may be attributed to the fixed composition of the membrane (the absence of significant carrier bleeding from the membrane to the bulk aqueous solution). No drift in the absorbance occurred. Nevertheless, the applied membrane was stored under water when not in use to protect it from drying out.Membrane properties
The characteristics of the optode membrane were obtained by assessing the change in the absorbance at 595 nm from individual solutions of 0.8, 1.6 and 3.2 μM Co2+. In all of the three cases, the optodes reached 98% absorbance after 5.0 min. The membrane stability was studied for 10 h and during this period, the mean difference of absorbance for the tested solutions was ±0.009. Moreover, the membrane response was stable in air for 15 days.The effect of the salting-out phenomenon on the response of the optode was examined by adding different concentrations of sodium nitrate. The results demonstrate that this factor had no influence on the response of the membrane up to 0.05 M NaNO3 and it significantly decreased above this concentration due to the reduction in the activity of Co2+ ions at higher concentrations of the electrolyte, which reduces the interaction of Co2+ cations with the CPDB in the membrane.
Repeatability and regeneration
The repeatability of the membrane response at 595 nm was checked by performing various replicate measurements for a 20 μM solution. The RSD for these determinations was found to be less than 2.0%. It is essential that the sensor membrane be regenerated by an appropriate solution to prepare it for subsequent measurements. To select the best regeneration solution, mineral acids such as HNO3, H2SO4, HCl, and H3PO4 as well as solutions of EDTA and KCl (all 0.3 M) were checked. To do this, the optode was put in 20 μM cobalt solution and after attaining equilibrium, it was taken out and put in a regenerating solution until the absorption of the membrane was stabilized. Nitric acid (0.3 M) showed the highest percentage of regeneration of the sensor.Great regeneration was achieved for a Co2+ ion concentration of 20 μM. The corresponding RSD value was found to be ±1.95%. The short-term stability of the optodes was determined by their absorbance difference measurements in contact with a 20 μM Co2+ ion solution at pH 7.5 over a period of 10 h. From the absorbance readings taken every 15 min (n = 40), it was established that the response is almost complete with only a 1.7% increase in the absorbance after 10 h of monitoring. The lifetime of the membrane was checked over a period of two weeks, during which four prepared membranes were kept in 5.0% (v/v) ethanol at 4 °C. The mean absorbance differences of the sensors were found to be 0.043 (±0.002) and 0.055 (±0.003) before and after this period, respectively.
Sensor selectivity
One of the most necessary parameters of a proposed sensor, which dictates its applicability for the analysis of real samples, is the selectivity coefficient. The influence of various cations and anions on the reactivity with the proposed optode at the optimum conditions was examined, revealing that there is no response to any of the examined ions and thereby indicating that no reaction occurs or there is no change in the color of the CPDB optode. The interference of various inorganic cations towards the proposed optical sensor was assessed using a 20 μM Co2+ solution in the presence of various concentrations of the interfering cations in a pH 7.5 buffer. The tolerance ratio was defined as the ratio of the interfering ion concentration to the Co2+ ion concentration, which causes a relative error of ±5.0%. The resulting tolerance ratios ([Mn+]/[Co2+]) for the various interfering ions (Mn+) listed in Table 2 indicate that the Co2+ ion content can be distinguished selectively with high accuracy using the proposed optical sensor in the presence of excess levels of the investigated potential interferents. This moderate selectivity demonstrates the applicability of the proposed sensor for evaluating and monitoring the cobalt content of various real samples with complicated matrices in the presence of an excess of many other coexisting cationic species. The high selectivity in the presence of interfering ions such as Fe2+, Cu2+, Ni2+, Ag+, Au3+, Cr3+, Cd2+, Zn2+, Hg2+, and SO42− make it possible to precisely and repeatedly monitor Co2+ ion content in environmental samples with complicated matrices.| Foreign ion | Tolerance limit (μM) | Foreign ion | Tolerance limit (μM) |
|---|---|---|---|
| K+, CH3COO− | 20000 |
Cr3+, CO32− | 3500 |
| Na+, PO43− | 17500 |
W6+, Cl− | 3000 |
| Ba2+, NO3−, N3− | 15000 |
Cr6+, SO42− | 2500 |
| Al3+, BrO3− | 13000 |
Fe3+, I−, Br−, F− | 2000 |
| Ca2+, Mg2+, SO42− | 11000 |
Ti3+, HCO3− | 1500 |
| Ag+, Au3+, citrate | 10000 |
Mo6+, IO4− | 1250 |
| Fe2+, Zn2+, NO2− | 8500 | La3+, Y3+, Sc3+ | 1000 |
| Ni2+, Cu2+, NH4+ | 7000 | V5+, Be2+ | 800 |
| Zn2+, Hg2+, oxalate | 5500 | Th4+, UO22+ | 600 |
| Cd2+, Be2+, S2O32− | 4500 | Pb2+, Mn2 | 500 |
Analytical characteristics
The optode response, in the form of a change in absorbance at 595 nm, towards Co2+ ion concentration was obtained within 0.05–45.20 μM (Table 3). The blank absorbance at 595 nm was assessed after equilibrating the optode sample with a blank solution at pH 7.5. The absorbance changes linearly as a function of Co2+ ion concentration in the range of 0.05–45.20 μM. The minimum concentration of Co2+ ions required in the 25 mL equilibrating solution to establish a distinct color change of the optode was found to be 20 μM. However, the detection limit of Co2+ ion concentration can be further improved using a larger volume of the aqueous sample. The detection and quantification limits,51 defined as CL = 3SB/m and CQ = 10SB/m (where CL, CQ, SB, and m are the detection limit, quantification limit, standard deviation of the blank and slope of the calibration graph, respectively), were 0.015 and 0.048 μM, respectively.| Parameters | Proposed sensor |
|---|---|
| a Relative standard deviation. | |
| pH | 7.5 |
| λmax (nm) | 595 |
| Beer's range (μM) | 0.05–42.5 |
| Ringbom range (μM) | 0.2–40.5 |
| Molar absorptivity (L mol−1 cm−1) | 8.82 × 107 |
| Sandell sensitivity (ng cm−2) | 0.007 |
| Detection limit (μM) | 0.015 |
| Quantification limit (μM) | 0.048 |
| Regression equation | |
| Slope (μg mL−1) | 24.7 |
| Intercept | 0.09 |
| Correlation coefficient (r) | 0.999 |
| RSDa (%) | 1.9 |
Tables 4 and 5 demonstrate a comparison between the prepared optode and other previously reported sensors3,35,40,42,43 or spectrophotometric52–62 procedures for the determination of cobalt. It is clear that the achieved results are comparable with those of current sensors. The proposed optode provides a greater linear range and detection limit in some cases.
| Sensing material | Type of sensor (membrane) | Dynamic range (M) | Response time (min) | Detection limit (M) | Ref. |
|---|---|---|---|---|---|
| Methyltrioctylammonium chloride | Triacetyl cellulose | 8.5 × 10−6 to 1.3 × 10−4 | 7.0 | 5.9 × 10−6 | 43 |
| m-(Mercapto acetamido) phenol | Triacetyl cellulose | — | 0.5–3.0 | 5.8 × 10−6 | 2 |
| Pyrogallol red | Triacetyl cellulose | 1.7 × 10−6 to 1.52 × 10−4 | 2.0 | 3.6 × 10−7 | 40 |
| 1-(2-Pyridylazo)-2-naphthol | Cellulose acetate + poly(vinyl acetate) | 0.02–0.5 mg L−1 | 10 | 0.07 mg L−1 | 42 |
| Potassium thiocyanate | Polyvinyl chloride | 1.0 × 10−6 to 1.0 × 10−3 | 10 | 6.10 × 10−7 | 35 |
| 5-[o-Carboxyphenylazo]2,4-dihydroxybenzoic acid | Polyvinyl chloride | 5.0 × 10−8 to 4.52 × 10−5 | 5.0 | 1.5 × 10−8 | This work |
| Reagent | Medium/solvent | λmax (nm) | ε (L mol−1 cm−1) × 104 | Linear range (μg mL−1) | Ref. |
|---|---|---|---|---|---|
| a CTAB: hexadecyltrimethylammonium bromide.b Units μg.c ADS: ammoniumdodecyl sulphate.d Units mg.e Triton X-100.f Detection limit. | |||||
| Disodium 1-nitroso-2-naphthol-3,6-disulphonate | DMF/CHCl3 | — | 1.33 | 0.84–1.44 | 52 |
| Sodium diethyldithiocarbamate | Aqueous (CTABa) | 324 | 2.17 | 0.0377 ± 0.0082b | 53 |
| N-Hydroxy-N,N-diphenyl benzamidine | Toluene | 405 | 0.70 | 0.10–12 | 54 |
| 1-Hydroxy-2-carboxyanthraquinone | Ethanol–water | 494.5 | — | 0.75–4.5 | 55 |
| Diethyl thiocarbamate | Aqueous (ADSc) | 322 | 2.22 | 0.0493 ± 0.0018b | 56 |
| 3-Hydroxy-2-methyl-1,4-napthoquinone 4-oxime | Naphthalene/DMF | 430 | 2.09 | 0.12–1.8 | 57 |
| Bis(2,4,4-trimethylpentyl)phosphinic acid | Benzene | 635 | 0.03 | 0.295–2.36d | 58 |
| 1-Nitroso-2-naphthol | Aqueous (TX-100e) | 420 | 3.18 | 0.0056–3.00 (1.68 × 10−3)f | 27 |
| 2-(2-Benzothiazolyazo)-2-p-cresol | Aqueous (TX-100e) | 615 | 1.62 | 0.08–1.06 (10)f | 59 |
| 2-Nitroso-1-naphthol-4-sulphonic acid | DMF | 620 | — | 0.2–12 | 60 |
| Phenanthraquinone monothiosemicarbazone | Water–methanol | 550 | 1.24 | 0.8–4.0 | 61 |
| 3-(4-Phenyl-2-pyridinyl)-5-phenyl-1,2,4-triazine + picric acid | 1,2-Dichloroethane | — | — | 0.0072–0.50 | 62 |
| 5-[o-Carboxyphenylazo]2,4-dihydroxybenzoic acid | PVC NaTPB | 595 | This work | ||
Analytical applications
The proposed sensor was applied to assess cyanocobalamin phosphate (vitamin B12) in various pharmaceutical, serum, saliva and urine samples. Using the recommended method, the results of the analysis of capsules, syrups and ampoules showed a good correlation with those accomplished using the BP technique.63 The relative standard deviation of the suggested procedure is established to be better than 1.9% (six determinations). The procedure performance was judged through the calculation of student's t- and F-values at a 95% confidence limit,64 and the results show that the calculated values did not exceed the theoretical values (Table 6).| Sample | Vitamin B12 content (mg) | Founda | t-Valueb | F-Value | |
|---|---|---|---|---|---|
| Optode ± SD | Official ± SD | ||||
| a Average of six determinations.b Theoretical values of t- and F- are 2.57 and 5.05, respectively, for five degrees of freedom and a 95% confidence limit.c The Nile Company for Pharmaceutical and Chemical Industries, Egypt.d Egyptian International Pharmaceutical Industries Company, Egypt.e The Memphis Chemical Company, Cairo, Egypt.f Misr Company for Pharmaceutical Industries, Cairo, Egypt.g The Arab Drug Company for Pharm. & Chem. Industries, Egypt.h Chemical Industries Development Company, Egypt.i Not detected. | |||||
| Tablets | |||||
| Tri-Bc | 0.125 | 0.126 ± 0.07 | 0.122 ± 0.22 | 1.37 | 2.09 |
| Mineravitd | 1.000 | 0.997 ± 0.05 | 0.990 ± 0.18 | 1.56 | 2.35 |
| Trivarole | 0.125 | 0.124 ± 0.10 | 0.129 ± 0.26 | 1.14 | 1.88 |
| Beco fortef | 12.00 | 11.98 ± 0.08 | 12.10 ± 0.17 | 1.22 | 2.03 |
|
|||||
| Ampoules | |||||
| B12 Depotg | 0.5 mg mL−1 | 0.502 ± 0.12 | 0.494 ± 0.26 | 1.10 | 1.71 |
| Trivarole | 1 mg per amp | 1.008 ± 0.06 | 1.025 ± 0.20 | ||
| Tri-vitacidh | 1 mg per amp | 0.994 ± 0.11 | 1.028 ± 0.28 | ||
| Tri-Bc | 1 mg per amp | 0.991 ± 0.14 | 1.031 ± 0.19 | 0.99 | 1.65 |
| Serum ng mL−1 | — | 75.0 ± 0.08 | 73.50 ± 0.23 | ||
| 25 | 103.6 ± 0.05 | 106.60 ± 0.14 | 1.43 | ||
| 50 | 127.2 ± 0.07 | 102.20 ± 0.13 | 2.26 | ||
| Urine ng mL−1 | 22.0 ± 0.10 | 23.00 ± 0.17 | |||
| 40 | 63.8 ± 0.07 | 61.20 ± 0.12 | 1.21 | ||
| 80 | 100.5 ± 0.09 | 106.40 ± 0.17 | 2.00 | ||
| Saliva ng mL−1 | 0 | n.d.i | |||
| 35 | 37.1 ± 0.10 | 36.70 ± 0.21 | 1.65 | ||
| 70 | 72.5 ± 0.12 | 68.40 ± 0.27 | 3.17 | ||
Cobalt is usually applied in dental cast alloys, orthodontic wires and implantable orthopedic instruments, releasing it into human tissue due to corrosion.65 Saliva is a simple and low-cost sample to collect, and is used successfully to screen great populations;66 it can therefore be applied to monitor Co2+ released from orthopedic devices. However, a major challenge for the assessment of chemical contaminants in saliva is that concentrations are often 1 or 2 orders of magnitude lower than in blood.67 Consequently, blood and urine have been suggested as biomarkers of recent exposure to soluble Co2+ species.1 Nevertheless, urine is favored for the monitoring of heavy metals because it allows for non-invasive sampling and simple collection.66 To the best of our knowledge, there have been no reports on the optical sensor viability for metal preconcentration from non-invasive biological samples such as serum, saliva and urine. Consequently, after urine and saliva analysis, the results obtained are listed in Table 6. Moreover, analyte recovery in the presence of a biological matrix was examined. The applied procedure was used for six portions of both saliva and urine matrices, and the average concentrations of Co2+ were taken as the base values. Then, 1.0 μg L−1 Co2+ was added to the samples and the same method was employed. The results achieved with the proposed technique were in great agreement with those previously described for urine samples,68 while the Co2+ recoveries were highly satisfactory for all the cases.
The final test of the effectiveness of the sensor as an environmental monitoring device is to test real water samples and alloy samples since the system has been optimized with laboratory prepared samples. The recovery of spiked Co2+ in Nile River water (Benha) was established with the suggested optode. The results are given in Table 7. The standard deviations of the analysis and the recoveries of the added Co2+ to the samples indicate that the recommended procedure is capable of real sample analysis.
| Sample | Added (μg L−1) | Found (μg L−1) | Recoverya (%) | |
|---|---|---|---|---|
| Sensor ± SD | FAAS ± SD | |||
| a Nile River water (Benha City).b Certified reference standard materials with cobalt contents of 0.020% for SRM-12 and 0.110% for SRM-589. | ||||
| River water | — | 0.37 ± 0.03 | 0.40 ± 0.14 | — |
| 1.00 | 1.40 ± 0.08 | 1.35 ± 0.21 | 102.19 | |
| 2 0 | 2.32 ± 0.02 | 2.55 ± 0.17 | 97.89 | |
| Tap water | — | 0.25 ± 0.02 | 0.30 ± 0.14 | — |
| 2.5 | 2.80 ± 0.07 | 2.70 ± 0.09 | 101.82 | |
| 5.0 | 5.15 ± 0.03 | 5.45 ± 0.18 | 98.10 | |
| Wastewater | — | 1.05 ± 0.09 | 1.00 ± 0.25 | — |
| 4.0 | 4.95 ± 0.11 | 5.15 ± 0.32 | 98.02 | |
| 8.0 | 9.15 ± 0.04 | 8.80 ± 0.27 | 101.10 | |
| SRM-12b | — | 0.019 ± 0.02 | 0.020 ± 0.24 | — |
| 0.2 | 2.02 ± 0.09 | 2.03 ± 0.16 | 99.06 | |
| 0.4 | 0.415 ± 0.13 | 4.41 ± 0.23 | 103.26 | |
| SRM-589b | — | 0.112 ± 0.03 | 0.110 ± 0.17 | — |
| 0.1 | 0.215 ± 0.09 | 0.220 ± 0.22 | 101.42 | |
| 0.2 | 0.310 ± 0.14 | 0.320 ± 0.28 | 99.36 | |
The suggested procedure was used for the assessment of Co2+ in tap, waste and river water samples (Table 7). The recovery of Co2+ was between 98.02% and 103.26%. The Co levels were 0.37 μg L−1 in the river water samples, 0.25 μg L−1 in tap water and 1.05 μg L−1 in wastewater. The results did not significantly vary compared to those previously described for river, tap and wastewater samples.69
Furthermore, the accuracy of the proposed methodology was estimated by analyzing the certified reference materials (CRMs) of SRM-12 and SRM-589 with Co2+ contents of 0.020% and 0.110%, respectively. These CRMs include many ions frequently present in such samples. Subsequently, the certified concentration values in the CRMs were greater than the upper limit of the linear range obtained by the proposed sensor; a dilution by a factor of 25 had to be employed for analysis. Applying the developed methodology, the Co contents present in the CRMs were 0.019% and 0.112%, respectively (95% confidence interval, n = 6).
The proposed optical sensor was applied for the assessment of cobalt in flour and vegetable samples after standard addition, under the described procedure. The results recorded in Table 8 indicate that the concentrations of Co2+ ions assessed using the optical sensor are in good agreement with those obtained by the FAAS procedure.
| Samples | Added (μg g−1) | Found (μg g−1) | Recovery (%) | |
|---|---|---|---|---|
| Optodea | FAAS | |||
| a Mean ± standard deviation (n = 6).b Not detected. | ||||
| Tomato | — | 1.45 ± 0.03 | 1.51 ± 0.20 | — |
| 5.00 | 6.30 ± 0.05 | 6.30 ± 0.13 | 97.67 | |
| 10.0 | 11.60 ± 0.09 | 11.80 ± 0.21 | 101.31 | |
| Soybean meal | — | 312.10 ± 0.12 | 314.20 ± 0.27 | — |
| 25.0 | 335.80 ± 0.08 | 340.90 ± 0.32 | 99.61 | |
| 50.0 | 385.6 0.15 | 383.30 ± 0.24 | 99.94 | |
| Tea | — | 101.60 ± 0.11 | 105.40 ± 0.19 | — |
| 40.0 | 142.50 ± 0.07 | 147.70 ± 0.36 | 100.64 | |
| 80.0 | 180.30 ± 0.10 | 182.90 ± 0.29 | 99.28 | |
| Spinach | — | 0.83 ± 0.03 | 0.91 ± 0.24 | — |
| 4.00 | 4.65 ± 0.06 | 4.72 ± 0.34 | 96.27 | |
| 8.00 | 8.95 ± 0.08 | 9.20 ± 0.19 | 101.36 | |
| Mint | — | 13.50 ± 0.11 | 14.20 ± 0.32 | — |
| 10.0 | 22.90 ± 0.09 | 25.00 ± 0.27 | 97.45 | |
| 20.0 | 34.50 ± 0.12 | 33.60 ± 0.38 | 102.99 | |
| Cabbage | — | N.D. | N.D. | — |
| 15.0 | 14.91 ± 0.04 | 15.25 ± 0.33 | 99.40 | |
| 30.0 | 30.45 ± 0.12 | 29.60 ± 0.35 | 101.50 | |
| Flour | — | N.D.b | N.D.b | — |
| 12.5 | 12.55 ± 0.03 | 12.40 ± 0.23 | 100.40 | |
| 25.0 | 24.70 ± 0.07 | 25.25 ± 0.34 | 98.80 | |
Conclusion
The proposed optode is a precise, accurate, low-cost, sensitive and selective device for cobalt assessment based on a PVC membrane. Additionally, the suggested procedure is rapid and easy and provides a broader dynamic range, reliable reproducibility and better detection and quantification limits. A comparison with previously reported sensors demonstrates that the proposed method provides a wider linear range and lower detection and quantification limits in some cases. Finally, the fabricated sensor can be applied successfully for the monitoring of cobalt in various environmental samples. According to the best of our knowledge, no manufactured optode has been described in the literature for the determination of cobalt using the studied reagent.Ethics approval and consent to participate
All biological studies were carried out in strict accordance with the animal welfare guidelines of the World Organization for Animal Health. All biological experiments were performed using protocols approved by the Laboratory Animal Ethics Committee of “Egypt” and the Commission on the Ethics of Scientific Research, Faculty of Medicine, Benha University. In all cases, informed written consent was obtained from each participant.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the Department of Chemistry, Faculty of Science, Benha University, as well as for providing instrumental facilities.References
- D. Lison, in Handbook on the Toxicology of Metals, ed. G. F. Nordberg, B. A. Fowler, M. Nordberg and L. T. Friberg, Academic Press Inc, 3rd edn, 2007, pp. 511–528 Search PubMed.
- D. Afzali and A. Mostafavi, Anal. Sci., 2008, 24, 1135–1139 CrossRef CAS PubMed.
- L. Husakova, A. Bobrowski, J. Sramkova, A. Krlicka and K. Vytras, Talanta, 2005, 66, 999–1004 CrossRef CAS PubMed.
- S. J. Hill, T. A. Arowolo, O. T. Butler, J. M. Cook, M. S. Cresser, C. Harrington and D. L. Miles, J. Anal. At. Spectrom., 2003, 18, 170–202 RSC.
- A. Mohadesi, E. Teimoori, M. A. Taher and H. Beitollah, Int. J. Electrochem. Sci., 2011, 6, 301–308 CAS.
- H. A. Zadeh and E. Ebrahimzadeh, Cent. Eur. J. Chem., 2010, 8, 617–625 Search PubMed.
- A. Ghasemi, M. R. Jamali and Z. Eshaghi, Anal. Lett., 2021, 54, 378–393 CrossRef CAS.
- F. G. Almeida, M. P. Ferreira, M. G. Segatelli, A. Beal, W. A. Spinosa, F. A. S. Cajamarca and C. R. T. Tarley, React. Funct. Polym., 2021, 164, 104934 CrossRef.
- M. Shirani, F. Salari, S. Habibollahi and A. Akbari, Microchem. J., 2020, 152, 104340 CrossRef CAS.
- Z. Tekin, T. Unutkan, F. Erulaş, E. G. Bakırdere and S. Bakırdere, Food Chem., 2020, 310, 125825 CrossRef CAS PubMed.
- E. Yazici, M. Firat, D. S. Chormey, E. G. Bakirdere and S. Bakirdere, Food Chem., 2020, 302, 125336 CrossRef CAS PubMed.
- C. Arpa and I. Arıdaşır, Food Chem., 2019, 284, 16–22 CrossRef CAS PubMed.
- P. Berton and R. G. Wuilloud, Anal. Chim. Acta, 2010, 662, 155–162 CrossRef CAS PubMed.
- A. AlKinani, M. Eftekhari and M. Gheibi, Int. J. Environ. Anal. Chem., 2021, 101, 17–34 CrossRef CAS.
- H. Li, J. Wang and J. Du, Talanta, 2021, 223, 121712 CrossRef CAS PubMed.
- C. Roldan, J. Coll, J. L. Ferrero and D. Juanes, X-Ray Spectrom., 2004, 33, 28–32 CrossRef CAS.
- F. Shemirani and N. Shokoufi, Anal. Chim. Acta, 2006, 577, 238–243 CrossRef CAS PubMed.
- U. Repinc, L. Benedik and B. Pihlar, Microchim. Acta, 2008, 162, 141–146 CrossRef CAS.
- X. Kong, Q. Jia and W. Zhou, Microchem. J., 2007, 87, 132–138 CrossRef CAS.
- Y. Xu, J. Zhou, G. Wang, J. Zhou and G. Tao, Anal. Chim. Acta, 2007, 584, 204–209 CrossRef CAS PubMed.
- H. L. Xie, X. D. Nie and Y. G. Tang, Chin. Chem. Lett., 2006, 17, 1077–1093 CAS.
- M. Bahram and S. Khezri, Anal. Methods, 2012, 4, 384–393 RSC.
- R. Elsheikh, A. A. Gouda, H. A. Elsayed and E. M. Alamin, Int. J. Pharm. Pharm. Sci., 2015, 7, 213–221 CAS.
- K. Mahmood, F. H. Wattoo, M. H. S. Wattoo, M. Imran, M. J. Asad, S. A. Tirmizi and A. Wadood, Saudi J. Biol. Sci., 2012, 19, 247–250 CrossRef CAS PubMed.
- N. V. Scheglova, T. V. Popova, A. V. Druzhinina and T. V. Smotrina, J. Mol. Liq., 2019, 286, 110909 CrossRef CAS.
- M. Shaimaa and H. W. Hindawi, Nano Biomed. Eng., 2020, 12, 160–166 Search PubMed.
- H. B. Singh, N. K. Agnihotri and V. K. Singh, Talanta, 1999, 48, 623–631 CrossRef CAS PubMed.
- M. Ezati, S. Moinfar, S. Mohammadi and G. Khayatian, J. Anal. Chem., 2021, 76, 172–179 CrossRef.
- H. M. Al-Saidi and S. S. Alharthi, Spectrochim. Acta, Part A, 2021, 253, 119552 CrossRef CAS PubMed.
- A. Torabi, M. Shirani, A. Semnani and A. Akbari, J. Iran. Chem. Soc., 2021, 18, 893–902 CrossRef CAS.
- M. De la Guardia and S. Garrigues, Challenges in Green Analytical Chemistry, Royal Society of Chemistry, Cambridge, UK, 2011 Search PubMed.
- W. H. Chan, A. W. M. Lee, J. Lu and X. Wu, Anal. Chim. Acta, 1998, 370, 259–266 CrossRef CAS.
- N. A. Yusof and M. Ahmad, Sens. Actuators, B, 2002, 86, 127–133 CrossRef CAS.
- H. H. El-Feky, S. El-Bahy and A. S. Amin, Anal. Biochem., 2022, 651, 114720 CrossRef PubMed.
- F. B. M. Suah, Anal. Chem. Res., 2017, 12, 40–46 CrossRef CAS.
- A. S. Amin, S. El-Bahy and H. H. El-Feky, Anal. Biochem., 2022, 643, 114579 CrossRef PubMed.
- H. H. El-Feky, A. M. Askar and A. S. Amin, RSC Adv., 2021, 11, 35300–35310 RSC.
- M. Gharehbaghi, F. Shemirani and M. D. Farahani, J. Hazard. Mater., 2009, 165, 1049–1055 CrossRef CAS PubMed.
- F. Bukhari and M. Suah, Anal. Chem. Res., 2017, 12, 40–46 CrossRef.
- A. A. Ensafi and A. Aboutalebi, Sens. Actuators, B, 2005, 105, 479–483 CrossRef CAS.
- I. M. Steinberg, A. Lobink and O. S. Wolfbeis, Sens. Actuators, B, 2003, 90, 230–235 CrossRef CAS.
- E. K. Paleologos, M. I. Prodromidis, D. L. Giokas, A. C. Pappas and M. I. Karayannis, Anal. Chim. Acta, 2002, 467, 205–215 CrossRef CAS.
- S. Rastegarzadeh and Z. Moradpour, Instrum. Sci. Technol., 2007, 35, 637–647 CrossRef CAS.
- A. S. Amin, I. S. Ahmed and M. E. Moustafa, Anal. Lett., 2001, 34, 749–759 CrossRef CAS.
- H. T. S. Britton, Hydrogen Ions, Chapman and Hall, London, 4th edn, 1952, p. 1168 Search PubMed.
- M. O. Luconi, R. A. Olsina, L. P. Fernandez and M. F. Silva, J. Hazard. Mater., 2006, 128, 240–246 CrossRef CAS PubMed.
- L. D. Coo and C. J. Belmonte, Talanta, 2002, 58, 1063–1069 CrossRef CAS.
- M. Shamsipur, T. Poursaberi, A. R. Karami, M. Hosseini and A. Momeni, et al., Anal. Chim. Acta, 2004, 501, 55–60 CrossRef CAS.
- M. Fouladgar and A. Ensafi, Sens. Actuators, B, 2010, 143, 590–594 CrossRef CAS.
- G. Absalan, M. Soleimani, M. Asadi and M. B. Ahmadi, Anal. Sci., 2004, 20, 1433–1436 CrossRef CAS PubMed.
- IUPAC, Spectrochim. Acta, Part B, 1978, 33, 241–245 CrossRef.
- B. K. Puri and S. Balani, Talanta, 1995, 42, 337–344 CrossRef CAS.
- M. P. San Andres, M. L. Marina and S. Vera, Talanta, 1994, 41, 179–183 CrossRef CAS PubMed.
- B. S. Chandravanshi and G. Asgedom, Chem. Anal., 1995, 40, 225–229 CAS.
- J. A. Murillo, J. M. Lemus, A. M. de la Pena and F. Salinas, Analyst, 1988, 113, 1439–1442 RSC.
- M. P. San Andres, M. L. Marina and S. Vera, Analyst, 1995, 120, 255–259 RSC.
- R. K. Sharma and S. K. Sindhwani, Talanta, 1988, 35, 661–663 CrossRef CAS PubMed.
- B. R. Reddy and P. V. R. Bhaskara Sarma, Talanta, 1994, 41, 1335–1339 CrossRef CAS PubMed.
- M. S. Carvalho, I. C. S. Fraga, K. C. L. Neto and E. Q. S. Filho, Talanta, 1996, 43, 1675–1680 CrossRef CAS.
- M. A. Taher and B. K. Puri, Analyst, 1995, 120, 1589–1592 RSC.
- R. K. Sharma, D. Sahadev and S. K. Sindhwani, Analyst, 1987, 112, 1771–1772 RSC.
- M. I. Toral, P. Richter and L. Silva, Talanta, 1993, 40, 1405–1409 CrossRef CAS PubMed.
- British Pharmacopoeia, HMSO Publication, London, 2017, vol. 4, https://www.pharmacopoeia.com/ Search PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Prentice-Hall, London, 5th edn, 2005 Search PubMed.
- E. A. Hutton, B. Ogorevc, S. B. Hocevar and M. R. Smyth, Anal. Chim. Acta, 2006, 557, 57–63 CrossRef CAS.
- M. Esteban and A. Castano, Environ. Int., 2009, 35, 438–449 CrossRef CAS PubMed.
- C. Timchalk, T. S. Poet, A. A. Kousba, J. A. Campbell and Y. J. Lin, J. Toxicol. Environ. Health, Part A, 2004, 67, 635–650 CrossRef CAS PubMed.
- J. P. Goullé, L. Mahieu, J. Castermant, N. Neveu, L. Bonneau, G. Lainé, D. Bouige and C. Lacroix, Forensic Sci. Int., 2005, 153, 39–44 CrossRef PubMed.
- V. N. Bulut, A. Gundogdu, C. Duran, H. B. Senturk, M. Soylak, L. Elci and M. Tufekci, J. Hazard. Mater., 2007, 146, 155–163 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |