
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase
Rami J. Obaida,
Nafeesa Naeem b,
Ehsan Ullah Mughal
*b,
Munirah M. Al-Rooqia,
Amina Sadiqc,
Rabab S. Jassasd,
Ziad Moussae and
Saleh A. Ahmed
*af
b,
Ehsan Ullah Mughal
*b,
Munirah M. Al-Rooqia,
Amina Sadiqc,
Rabab S. Jassasd,
Ziad Moussae and
Saleh A. Ahmed
*af
aDepartment of Chemistry, Faculty of Applied Sciences, Umm Al-Qura University, Makkah 21955, Saudi Arabia. E-mail: saahmed@uqu.edu.sa; saleh_63@hotmail.com
bDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
cDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
dDepartment of Chemistry, Jamoum University College, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
eDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, Abu Dhabi, United Arab Emirates
fDepartment of Chemistry, Faculty of Science, Assiut University, 71516 Assiut, Egypt
First published on 12th July 2022
Abstract
Heterocycles are the key structures in organic chemistry owing to their immense applications in the biological, chemical, and pharmaceutical fields. Heterocyclic compounds perform various noteworthy functions in nature, medication, innovation etc. Most frequently, pure nitrogen heterocycles or various positional combinations of nitrogen, oxygen, and sulfur atoms in five or six-membered rings can be found. Inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) enzymes is a popular strategy for the management of numerous mental diseases. In this context, cholinesterase inhibitors are utilized to relieve the symptoms of neurological illnesses like dementia and Alzheimer's disease (AD). The present review focuses on various heterocyclic scaffolds and their role in designing and developing new potential AChE and BChE inhibitors to treat AD. Moreover, a detailed structure–activity relationship (SAR) has been established for the future discovery of novel drugs for the treatment of AD. Most of the heterocyclic motifs have been used in the design of new potent cholinesterase inhibitors. In this regard, this review is an endeavor to summarize the biological and chemical studies over the past decade (2010–2022) describing the pursuit of new N, O and S containing heterocycles which can offer a rich supply of promising AChE and BChE inhibitory activities.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal is a Tenured Associate Professor of Organic Chemistry at the Department of Chemistry in University of Gujrat, Gujrat, Pakistan. He obtained his PhD from Bielefeld University, Germany in 2013 under the supervision of Prof. Dr Dietmar Kuck. For postdoctoral studies, he joined the group of Prof. Dr Xinliang Feng, Max-Planck-Institute for Polymer Research, Mainz, Germany. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metals-based terpyridine complexes and their uses in the fabrication of DSSCs and as efficient photo-catalysts, and organic functional materials for the applications in organic electronics. |
 Saleh A. Ahmed | Saleh A. Ahmed received his PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr in Saarland University, Saarbrücken, Germany. He has more than 20 years of experience as postdoctoral fellow, senior researcher and visiting professor in France, Japan, Germany, Italy and USA. Since 2010 he is a senior professor of organic chemistry (photochemistry) at Assiut University, Egypt. He had successfully published more than 200 publications. His current research interests include synthesis and photophysical properties of novel organic compounds, developments of synthetic methodologies for the synthesis of novel organic compounds with unique theoretical and biological applications. |
1 Introduction
Heterocyclic chemistry constitutes one of the most significant subclasses of organic chemistry. Heterocycles are cyclic organic compounds that contain at least one hetero-atom such as nitrogen, oxygen, sulfur etc.1,2 Among the heterocyclic compounds, five or six-membered heterocycles with one, or two, or three hetero atoms in their nucleus have attained special interest owing to their stability and ubiquitous occurrence in natural as well as synthetic compounds.3–5 Synthetic heterocyclic chemistry is used in a variety of domains including medicine, pharmacology, biocidal formulation, polymer science, electronics, agriculture, biology, optics, anticorrosive agents, agrochemicals, photo-stabilizers and material sciences.6–8 In particular, they considered one of the significant classes of organic compounds, which are used in many biological fields on account of their multiple uses in treating various illnesses.9,10 Many biological compounds, such as vitamins, hemoglobin, hormones, DNA, RNA, and others contain these heterocyclic rings as a key structural constituent. These structures can also be found in several FDA-approved medications that are used to treat a variety of disorders.11 Furthermore, heterocyclic compounds have a wide range of biological applications as antifungal, anticonvulsant, antibacterial, antioxidant, antidiabetic, anti-inflammatory, enzyme inhibitors, herbicidal action, antiallergic, anticancer tumor, anti-HIV, and insecticidal agents.12–22N-containing heterocycles are such organic compounds which contain one or more than one N-atom present in five- or six-membered ring systems (Fig. 1).23,24 Likewise, the O-containing heterocycles are also important scaffolds in organic chemistry mainly because of their diverse biological functions. The various subclasses of O-containing heterocycles are chromones, coumarins, furan, oxazole, and benzofuran etc. A substantial number of O-containing heterocycles exhibit a broad range of pharmacological activities such as anti-microbial, anti-HIV, antimalaria, anticancer, anti-tubercular, and diabetic activities.25–28 Moreover, S-containing heterocyclic compounds are often associated with foul odors but are widely used in various biological processes. Organosulfur compounds are fundamental entities in primary (cystine and methionine amino acids) and secondary metabolites (biotin and thiamine), and are also used in medicines, dyes, and agrochemicals etc.29,30 Many S-containing bioactive molecules, such as glutathione, hydrogen sulfide, and taurine play a crucial role in maintaining cellular redox equilibrium in living organisms. Additionally, S-containing β-lactam ring system is present in commercially available antibiotics such as penicillin and cephalosporin.31–35
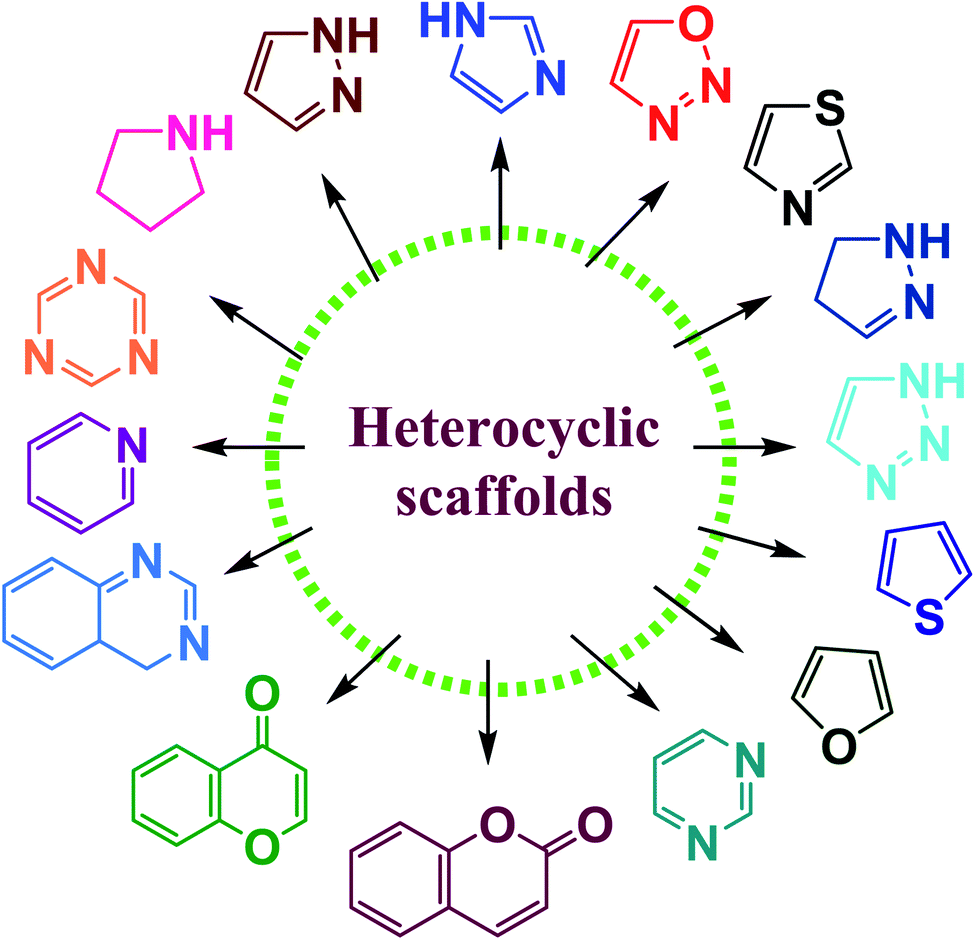 | ||
| Fig. 1 Various heterocyclic scaffolds. | ||
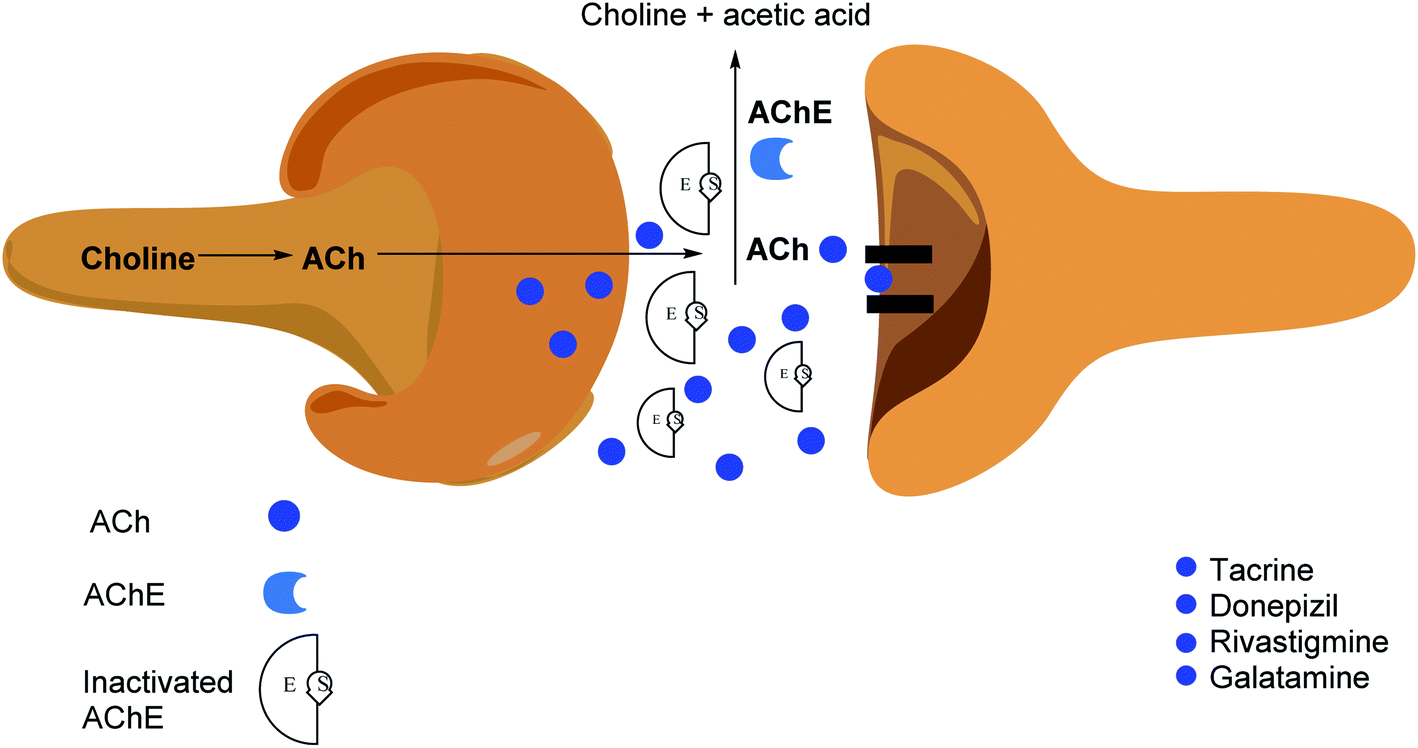
The applications of heterocyclic motifs in medicinal chemistry and chemical sciences are very vast as illustrated by the marketed medicines containing N, O, and S heteroatoms. The exceptional function of these heteroatoms in a variety of interactions with essential biological targets broadens the possibility of drug design and development. In this context, four cholinesterase (ChE) inhibitors (tacrine, donepezil, rivastigmine, and galantamine) have been approved as safe and non-toxic drugs for the symptomatic treatment of Alzheimer's disease (AD). Although, there are six types of these inhibitors which are commercially available in the market (Fig. 2).36–42 However, the drugs based-on edrophonium and pyridostigmine structural motifs have been found to show toxicity. Due to their toxic nature, such inhibitors have been commercially banned now.
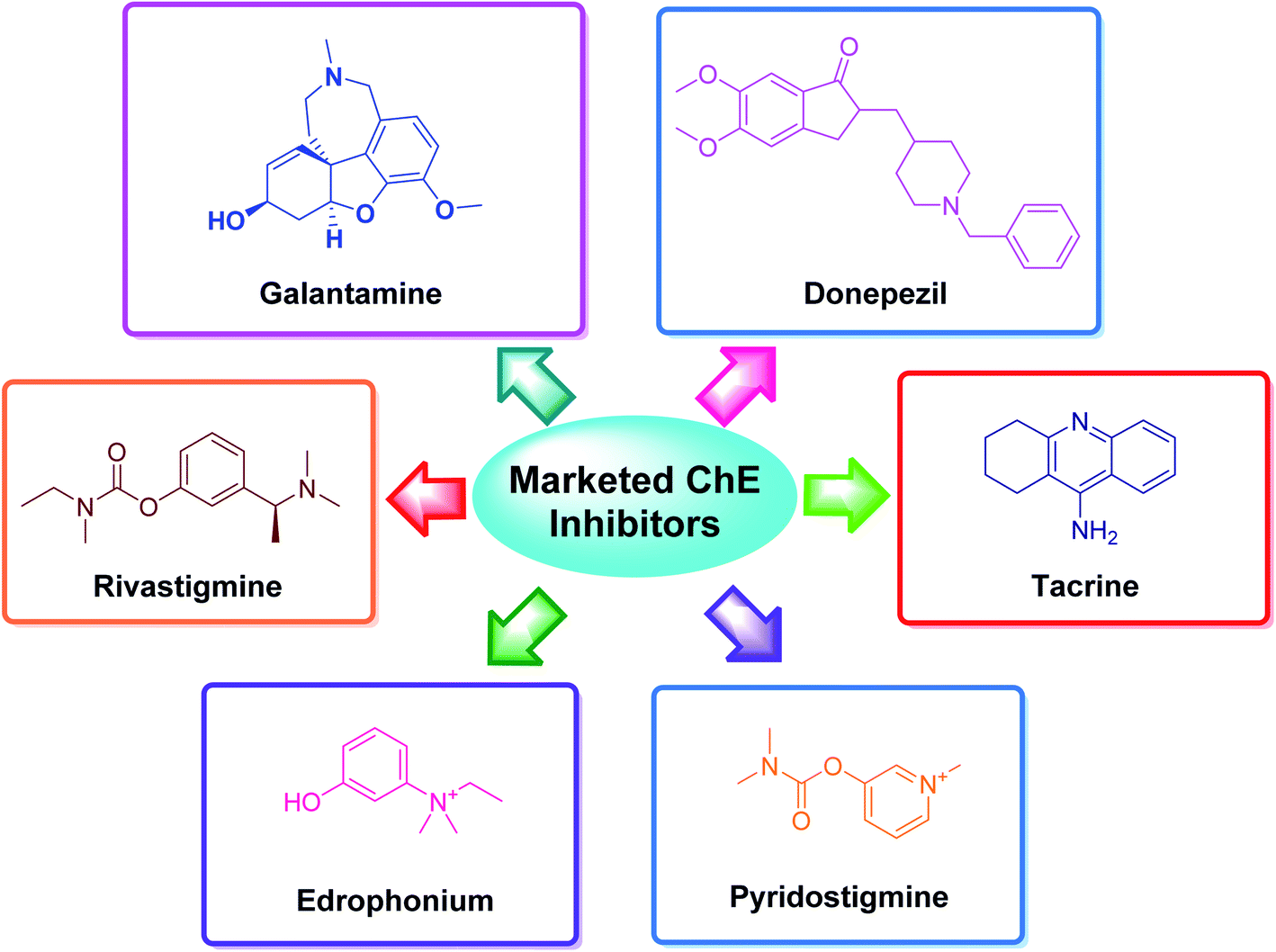 | ||
| Fig. 2 Selected examples of commercially available cholinesterase inhibitors. | ||
Since heterocyclic compounds occupy a crucial rank in organic chemistry43,44 and comprise a significant share of the chemical and biological sciences, they are used as potent motifs for many bio-evaluations.45–48 Such compounds play a vital role in the discovery of novel pharmacologically bioactive molecules.49 Indeed, with respect to the pharmaceutical industry, heterocyclic nuclei are remarkably ubiquitous with over 60% of the top most selling pills having at least one heterocyclic scaffold as part of the whole structure of the molecule.50 Furthermore, molecules containing heterocyclic scaffolds frequently demonstrate enhanced solubilities and can ease salt formation, both of which are recognized to be necessary for bioavailability and oral absorption.51 In this regard, various heterocycles show evidence of various biological and pharmacological activities partly due to certain parallels with many natural and synthetic molecules with known bioactivity.52
Therefore, there has been continuous research related to synthesis of more potent and highly efficacious cholinesterase inhibitors by modifying the main template moieties of available inhibitors for AD management. The present review discusses a variety of most significant heterocyclic structures, containing N, O and S atoms, exhibiting high potential as cholinesterase inhibitors in a concise way. Structure–activity relationship has also been established. To the best of our knowledge, such structures have not been considered before against acetylcholinesterase and butyrylcholinesterase enzymes.
2 Cholinesterase enzymes
Cholinesterases are essential enzymes present in vertebrates and insects that hydrolyze the acetylcholine (ACh) in the central and peripheral nervous system.53,54 In the body, ChE act as neurotransmitters responsible for the conduction of nerve impulses to the cholinergic synapses.Acetylcholinesterase (AChE; EC 3.1.1.7) and butyrylcholinesterase (BChE); (EC 3.1.1.8) are two forms of cholinesterases. BChE is an enzyme closely related to AChE and serves as a cholinergic neurotransmission co-regulator that hydrolyzes ACh. During the progression of Alzheimer's disease, investigations have revealed an increase in BChE activity (40–90%) in the most affected parts of the brain, such as the temporal cortex and hippocampus. During the early phases of senile plaque development, enhanced BChE activity is also significant in Aβ-aggregation. As a result, inhibition of AChE and BChE has been identified as a significant target for the effective management of AD, as evidenced by a rise in ACh availability in brain areas and a decrease in Aβ deposition.55 However, BChE is primarily found in peripheral tissues, including plasma, with only a trace amount present in the brain. Moreover, the possible benefit of selective inhibition of AChE over BChE may include a lower risk of peripheral cholinesterase enzyme inhibition and related side effects.56
Cholinergic theory states, Alzheimer's disease (AD) symptoms are largely produced by structural alterations in cholinergic synapses, the damage of subtypes of acetylcholine (ACh) receptors, the death of acetylcholine-generating neurons, and, as a result, cholinergic neurotransmission degradation. These problems cause AChE, an ACh-hydrolyzing enzyme to accumulate.57 Apart from a small number of familial instances caused by genetic abnormalities, no viable therapy options exist for most patients, and the illness's primary causes remain unknown. The following are the main categories of pharmacotherapeutic tactics for the treatment of AD: (i) therapies that prevent the commencement of the disease by isolating the main progenitors; (ii) disease-modifying treatments that stop or reverse disease development; and (iii) symptomatic treatments that target the disease's cognitive signs and protect patients from further cognitive decline.58,59 Because cholinergic neuron damage is prevalent in disease states, the present pharmacotherapeutic method founded on cholinesterase inhibitors offers a viable therapeutic aim for partial stabilization of cognitive function in AD patients. However, these compounds only have a short-term effect, usually 1–3 years, and they have no effect on disease progression.60–62
2.1 Alzheimer's disease and properties of various kinds of cholinesterase
Alzheimer's disease (AD) is a progressive chronic illness that causes gradual neurodegeneration. It was first characterized by Alois Alzheimer in 1907. Alzheimer's disease produces gradual cognitive dysfunction, including difficulty in making decisions, language problems, mood swings, learning, orientation, and other behavioral issues. Aging is the most important risk factor for Alzheimer's disease.63,64 Physical activity, on the other hand, can help to lower dementia rates. The enzyme cholinesterase (ChE) is a promising therapeutic target for Alzheimer's disease (AD). The loss of neurotransmission and the deterioration of cholinergic neurons in the brain are the main causes of cognitive decline in Alzheimer's patients.65,66According to the cholinergic hypothesis, the main cause of Alzheimer's disease is decline in acetylcholine synthesis. Brain atrophy is the most visible clinical feature in Alzheimer's disease as acetylcholine (ACh), a neurotransmitter involved in the transmission of electrical impulses from one nerve cell to another, is rapidly hydrolyzed by the acetylcholinesterase (AChE) enzyme.67,68 According to the amyloid hypothesis, AChE has non-cholinergic effects such as promoting the formation of β-amyloid (Aβ), a proteolytic fragment produced from amyloid precursor protein (APP), and deposition in the brain of afflicted persons in the form of senile neurofibrillary tangles. The accumulation of Aβ is thought to have a key role in the onset and progression of Alzheimer's disease.69–72
Cholinesterase (ChE) is a choline-based esterase that hydrolyzes choline-based esters like acetylcholine (ACh), a neurotransmitter. The hydrolysis of cholinergic neurotransmitters is catalyzed by two enzymes known as AChE and BChE. AChE activity is prominent in the healthy brain, whereas BChE, which works as a coregulator of cholinergic neurotransmission, plays a supporting role.73,74 AChE activity remains constant or decreases in individuals with AD, but BChE activity rises, resulting in an imbalance between BChE and AChE.75 Consequently, both enzymes are involved in the control of ACh levels and act as a useful therapeutic target for treating cholinergic deficits76 (Fig. 3). As a result, inhibiting both AChE and BChE at the same time may be beneficial in the latter phases of the illness.77 Accordingly, AChE and BChE inhibitors have surfaced as effective symptomatic therapies for AD. So far, four acetylcholinesterase inhibitors (AChEIs) have been authorized for commercial use: galantamine, rivastigmine, tacrine, and donepezil. Nevertheless, tacrine was detached from clinical use due to unadorned adverse effects accompanying with hepatotoxicity.78
 | ||
| Fig. 3 Brain cholinergic signaling. | ||
2.2 Properties of acetylcholinesterase
AChE is an esterase-like acetylcholine hydrolase enzyme (Fig. 4). Through cholinergic pathways, it plays an important role in brain function.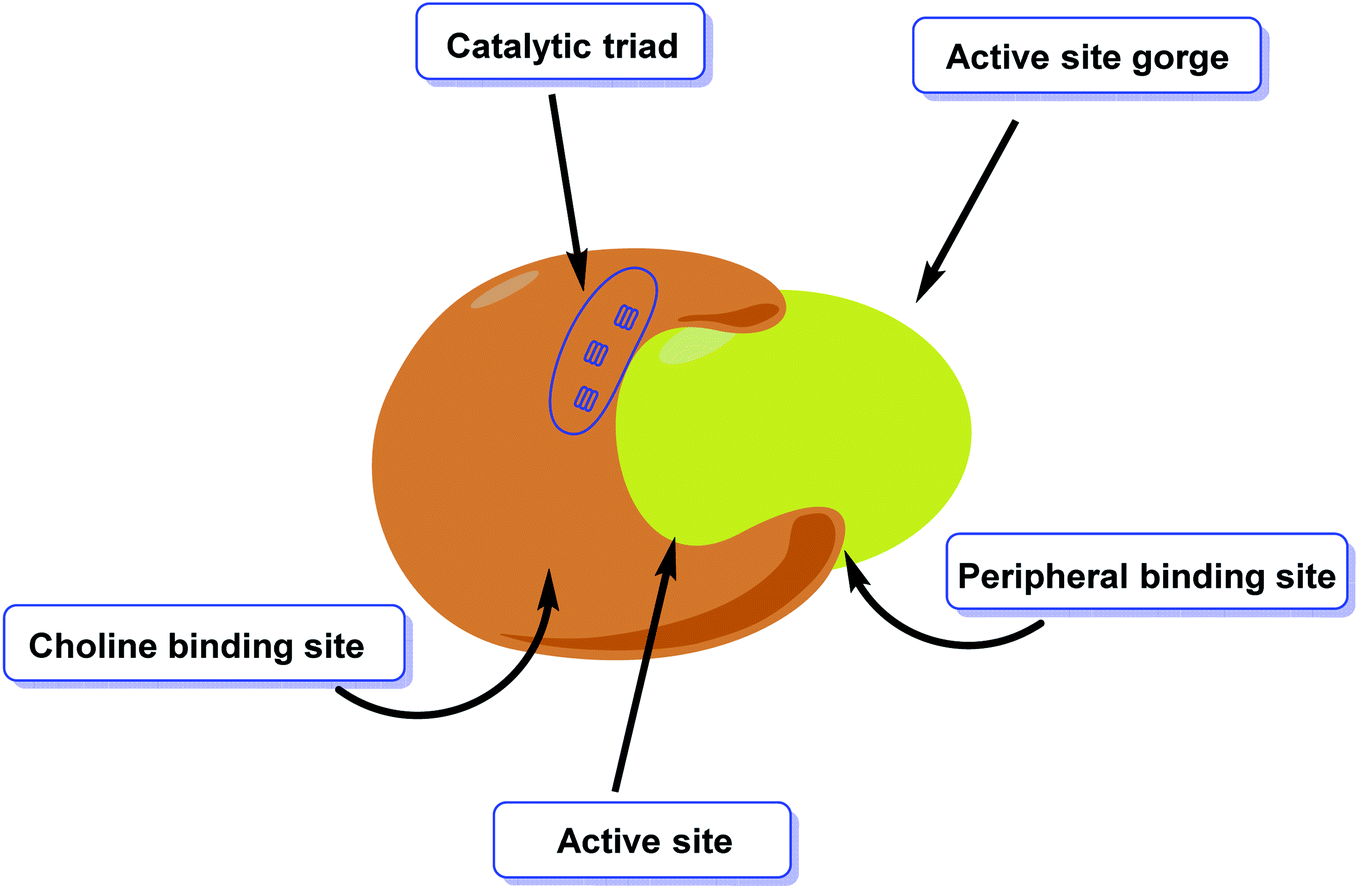 | ||
| Fig. 4 Diagrammatic representation of the active site of acetylcholinesterase. | ||
In 1991, the 3D structure of AChE was discovered in a Pacific electric ray (Torpedo californica (TcAChE)). The enzyme is mainly found in the central and peripheral nervous systems' synaptic gaps, and on red cell membranes.79–81 ACh is a cholinergic system neurotransmitter that regulates a variety of processes, including cognition.82 Botox, an exotoxin produced by the bacterium Clostridium botulinum, inhibits the discharge of ACh from cholinergic nerves blocking local neural conduction and muscle contraction. Botox is used in cosmetics to diminish facial creases, and for other therapeutic purposes.83,84
Inhibiting AChE by specific inhibitors is the therapeutic target to control Myasthenia gravis, glaucoma, Lewy body dementia, and AD.85 Enzyme inhibitors are crucial in a variety of disease management situations.86 AChE inhibitors are utilized in clinical practice to treat these problems, as they improve cholinergic function by increasing the amount of ACh in cholinergic synapses.87 AChEIs were initially utilized in the treatment of myasthenia gravis, a neuromuscular condition that generates skeletal muscle weakening, in 1932. These inhibitors were first approved (1938) for the treatment of individuals with myasthenia gravis.88 Antibodies targeting ACh receptors were developed later in 1960.89,90 AChEIs have been utilized to protect retinal ganglion cells from ocular hypertension in glaucoma. The intraocular pressure was reduced, the arteries were protected, and ocular blood flow was enhanced.91,92 AChEIs can also treat the symptoms of AD, known as the most frequent kind of dementia defined by the accumulation of amyloid plaques,93 which is triggered by the loss of ACh functions in the brain, which occurs most commonly in the elderly.94 Many compounds have been approved to prevent AChE breakdown in the brain, which can boost ACh activity and lessen AD symptoms,95 despite the fact that no treatments exist that to stop or reverse Alzheimer's progression (Fig. 5).
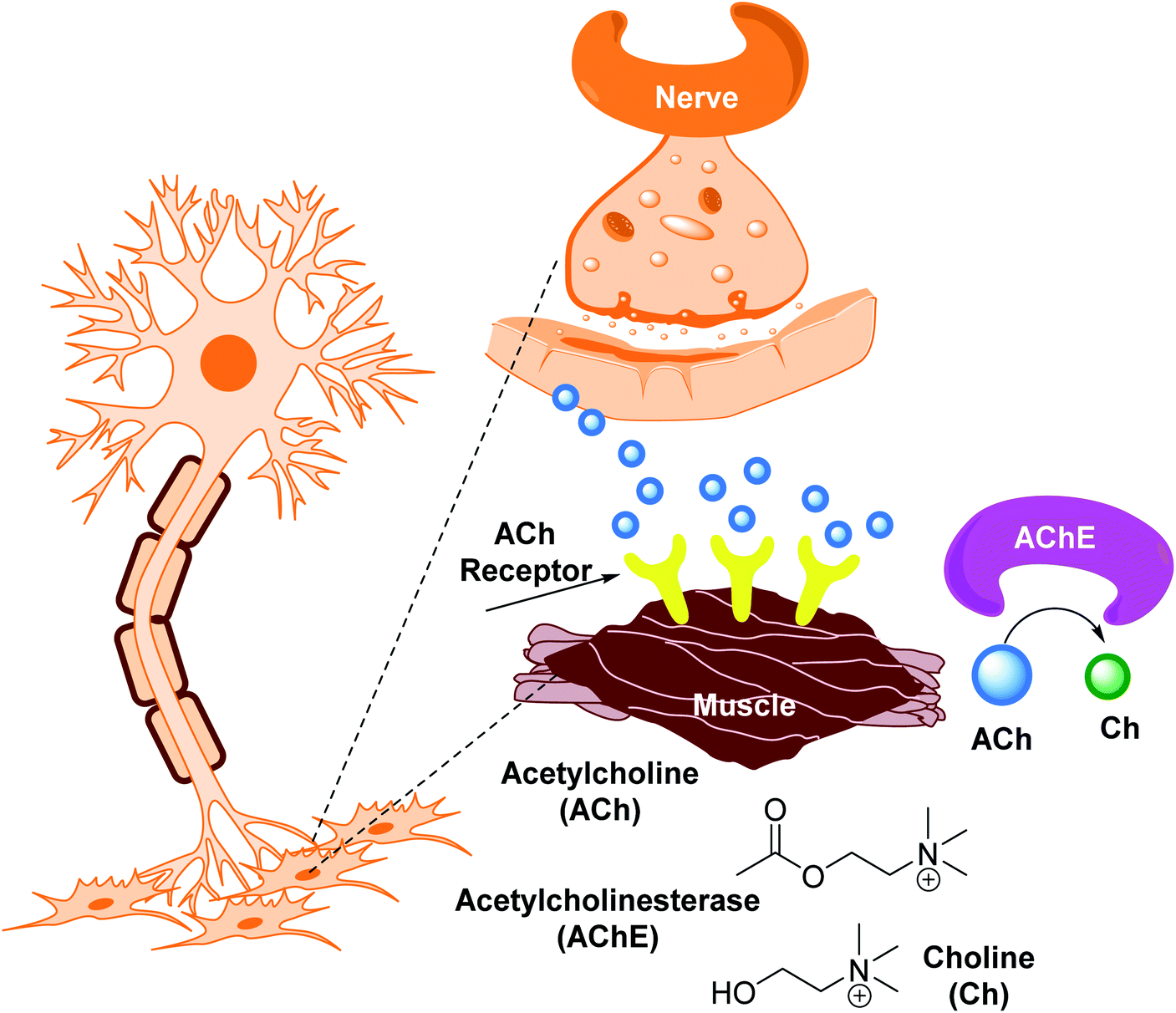 | ||
| Fig. 5 Synthesis of acetylcholine. | ||
2.3 Properties of butyrylcholinesterase
BChE is found in a variety of organs, including the liver, where it is produced and released into the bloodstream.96,97 BChE has a restricted neuronal distribution in the central nervous system as a pseudocholinesterase (CNS). In the human cerebral cortex, the number of BChE-rich neurons is around 2 orders of magnitude lower than the number of AChE-rich neurons. Glial origins are the most common.98,99 Although the role of BChE in normal conditions is unknown, it has been linked to lipoprotein, drug, and detoxification metabolism.100,101 It has a role in the breakdown of succinylcholine, an amyorelaxant used in surgical procedures. It also activates the antiasthmatic prodrug bambuterol and hydrolyzes medicines like heroin and physostigmine.102 As a result, the level of BChE has been demonstrated to play an important role in diabetes, obesity, hepatic steatosis, and other diseases.103 BChE knockout mice show no physiological abnormalities.104 Similarly, BChE-deficient persons can live long and healthy lives.105 BChE's compensating nature, on the other hand, is a prominent aspect. BChE compensates for the absence of AChE in the AChE-knockout mouse model, allowing normal cholinergic pathways to be maintained in AChE nullizygous animals.106 Due to highly damaged cholinergic neurons, the amount of AChE reduces by 90% in advanced AD. Meanwhile, BChE levels and function rise to 105–165% of normal, making it the primary ACh metabolic enzyme.107 Because of the critical role of BChE, ACh does not significantly increase in AChE knocked-out mice.108,109 As a result, licensed selective AChE inhibitors such as galantamine and donepezil have a very limited effect on severe AD, which is linked to a significant drop in AChE levels in individuals with serious AD. In recent years, a growing number of researchers have focused their research efforts on the design of BChE inhibitors for the treatment of advanced AD.110–1122.4 Mechanism of action of cholinesterase enzymes
As mentioned previously, there are two types of cholinesterase (ChE) enzymes: acetyl and butyrylcholinesterase (AChE and BChE). The ChE inhibitors are the most valuable agents for increasing ACh levels in neuronal cells by preventing the hydrolysis of ACh into choline and acetic acid. Clinical data suggest that BChE plays an significant role in the control of ACh and the maintenance of normal cholinergic activities, making it an additional intriguing target in the battle against AD.113–115 As a result, addressing both ChEs (AChE and BChE) simultaneously might provide a remedial benefit in advanced and late-stage AD. Furthermore, both ChEs are known to play an important role in Aβ aggregation.116 The Aβ is an insoluble protein fragment produced by beta secretase-1's catalytic proteolysis of amyloid precursor protein (BACE-1). The production of oligomers, fibrillary rods, or -sheets, which have been implicated in increasing neurotic damage and cognitive failure, is triggered by the buildup of Aβ aggregates.117 Furthermore, the buildup of Aβ aggregates in mitochondria may result in the formation of free radicals, resulting in oxidative stress.118 As a result, multitargeted therapeutics that inhibit ChEs (dual AChE and BChE), BACE-1, and Aβ aggregation while also having antioxidant potential could be useful in slowing the progression of AD instead of just delivering symptomatic relief (Fig. 6).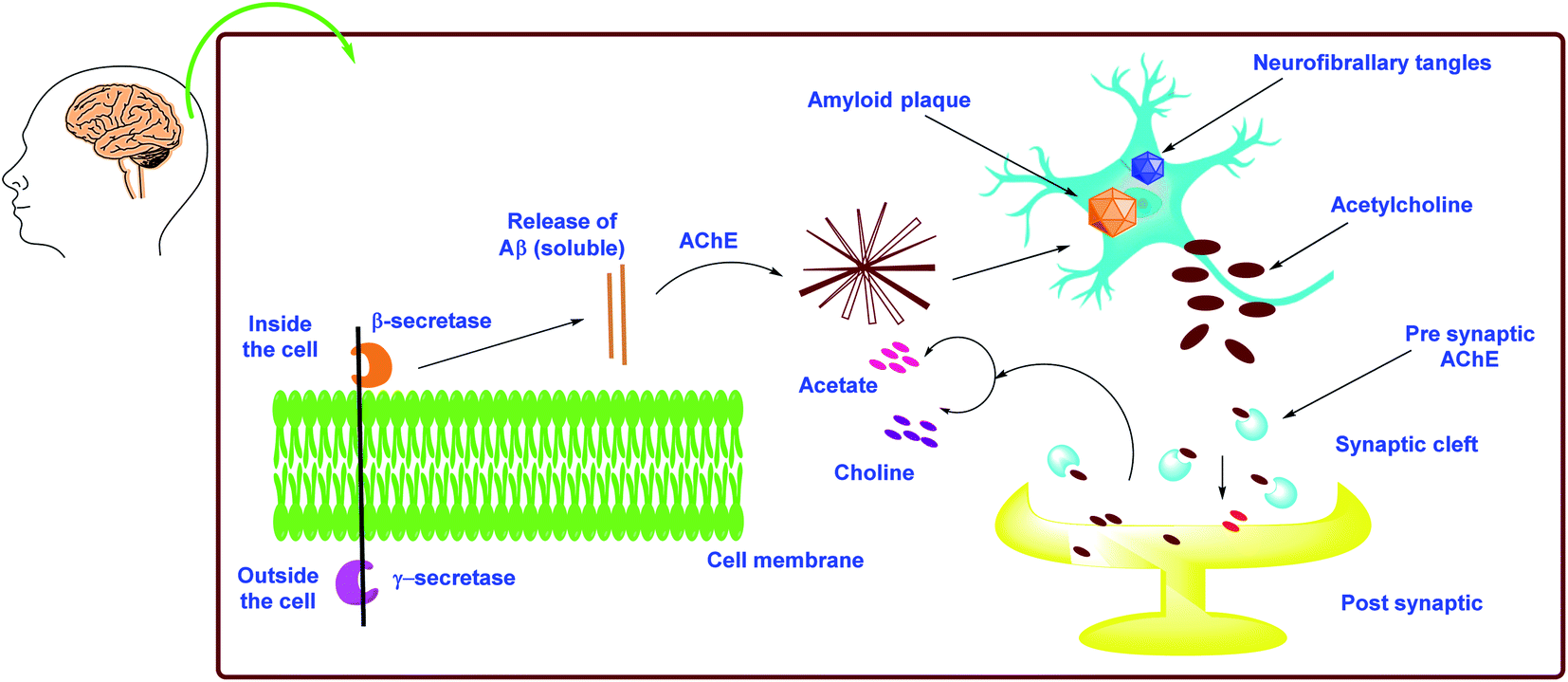 | ||
| Fig. 6 Schematic representation of AD pathogenesis in light of the cholinergic and amyloid hypothesis. | ||
3 Cholinesterase inhibition
Cognition is a blend of memory, attention, acquaintance, perception, skills, decision making, reminiscence, planning and judgment.119,120 The loss of memory coupled with cognitive impairment are noted in a variety of conditions such as aging, head injury and neurodegenerative disorders like depression, schizophrenia, AD and Parkinson's disease121–123 (Fig. 7). | ||
| Fig. 7 Factors involved in AD progression. | ||
Acetylcholine, glutamate, serotonin, and dopamine are neurotransmitters that regulate cognitive functioning. ACh, is an essential neurotransmitter in the control of learning and memory processes.124–127 Low acetylcholine concentrations in the cortex, hippocampus, and basal forebrain have been linked to cognitive impairment and short-term memory loss.128 By forming AChE-A complexes, AChE also causes the aggregation and deposition of A fibrils, resulting in cognitive dysfunction. As a result, increasing ACh by AChE inhibition and preventing Aβ aggregation are the best promising treatments to slowing dementia development.129 Oxidative stress is another harmful component where excessive reactive oxygen species (ROS) production causes lipid peroxidation and protein oxidation, which causes oxidative damage and impairs cognitive function.130–132
The United States Food and Drug Administration (US FDA) has licensed three AChE inhibitors (rivastigmine, galantamine, and donepezil) to treat the symptoms of AD, however these drugs have no effect on disease progression.133 These medications have several adverse effects, including urine incontinence and muscular cramping, which restricts their usage in the latter periods of the disorder.134,135 As a result, it is critical to develop novel drugs that will obstruct ACh metabolism by blocking AChE and inhibiting Aβ aggregation and should demonstrate antioxidant activity to slow disease progression.136,137
3.1 Cholinesterase inhibition mechanism
Aggregation of β-amyloid proteins and decreased cholinergic neurotransmission, which degrade the structural proteins of neurons, are the two major targets of treatment methods for cognitive decline.138 In the pathogenesis of cognitive decline, oxidative stress has also been implicated. ROS cause oxidative damage to the lipid and protein composition of neurons, resulting in changes in neuronal structure and function. The most severe and persistent biochemical alteration in cognitive impairment is cholinergic insufficiency (Fig. 8). Diminished levels of AChE, acetylcholine, and choline acetyltransferase have been reported in necropsy brain samples, indicating this. Choline esters are hydrolyzed by AChE, a hydrolase139 exhibiting high catalytic activity where each molecule is capable of degrading around 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 acetylcholine (ACh) molecules/second, reaching the substrate's diffusion limit. The anionic and esteratic subsites of AChE's active site are separated by a membrane. The crystal structure of AChE has revealed the shape and mechanism of action of the enzyme.140–150
000 acetylcholine (ACh) molecules/second, reaching the substrate's diffusion limit. The anionic and esteratic subsites of AChE's active site are separated by a membrane. The crystal structure of AChE has revealed the shape and mechanism of action of the enzyme.140–150
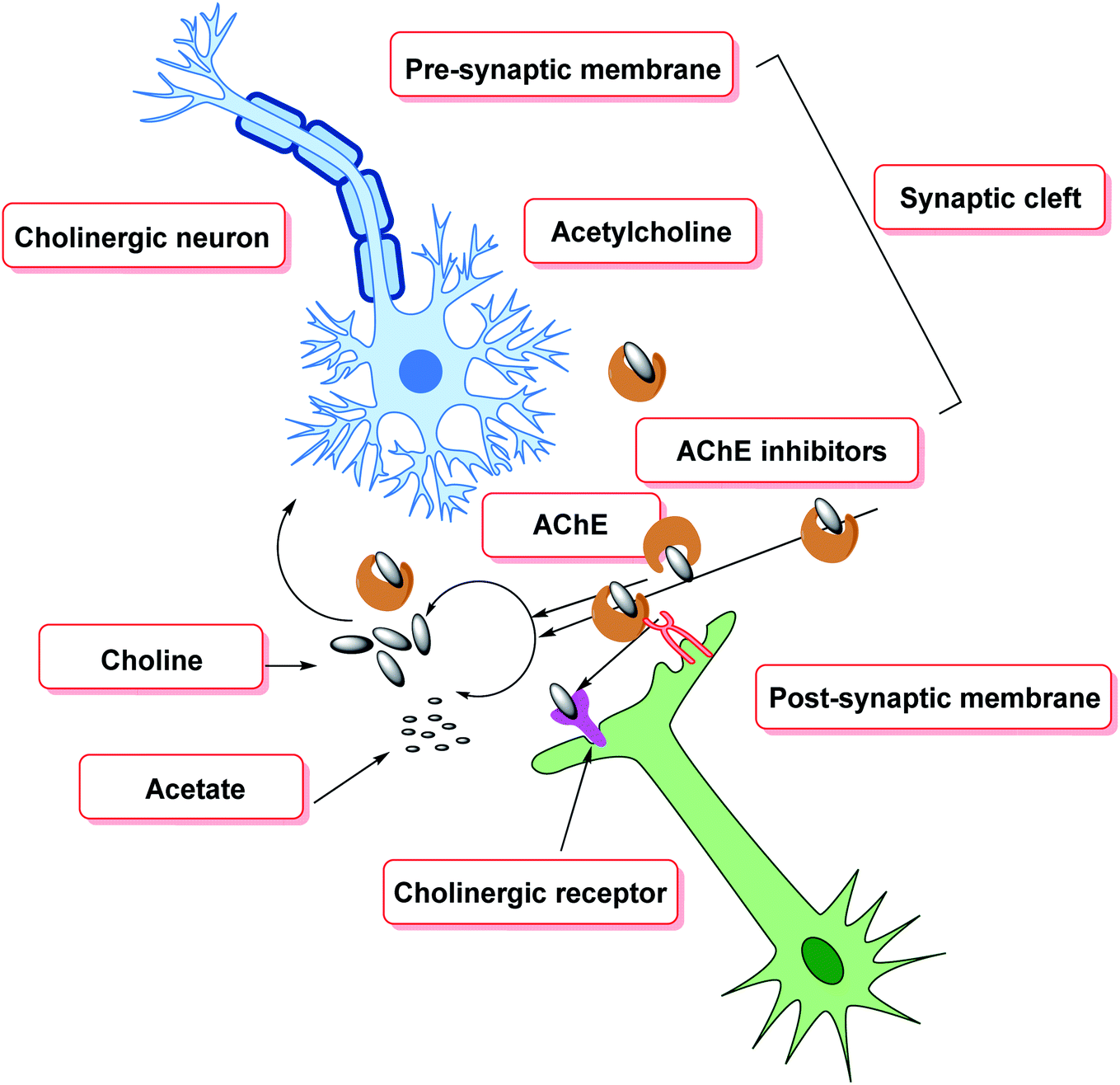 | ||
| Fig. 8 Therapeutic strategies towards cognitive decline. | ||
AChE is one of the most important enzymes in the serine hydrolase family, as it is engaged in the cleavage of ACh, exhausting ACh levels linked to memory and learning.151 As a result, the cholinergic hypothesis for cognition problems implies that degeneration in ACh-containing neurons play a significant role in the cognitive decline linked to old age. Multiple cognition enhancers have been developed throughout the years, but the discovery and development of several possible AChE inhibitors has cleared the path for a more effective therapy and treatment methodology to cognitive decline.152 As a result, cholinesterase inhibitors (ChEIs) are employed to treat cholinergic insufficiency. AChEIs have been designed using a variety of pharmacophoric scaffolds. The FDA has already authorized AChE inhibitors including physostigmine, rivastigmine, tacrine, and donepezil for the treatment of neurodegenerative illnesses like AD.153,154
4 Synthetic sources of ChE inhibitors
Donepezil and galantamine are agents that inhibit AChE function in a reversible and selective manner.155 Rivastigmine, on the other hand, connects covalently to the receptors' active site and is thus referred to as a pseudo-irreversible drug due to the sluggish release of the substrates. Rivastigmine is additionally a dual inhibitor of AChE and BChE.156,157 Rivastigmine is the only carbamate medicine that is prescribed to cure the symptoms of AD in patients. Other important functions of the carbamate group in pharmaceuticals include chemical stability, the ability to enhance permeability across cellular membranes, and participation in serine hydrolase inhibitors to cure asthma and pesticides for pest control.158 The amino acid sequences of human AChE and BChE are about 89% similar.159Surface specificity and gorge sensitivity, as well as the volume of the gorge exposed to entrance inhibitors, are the main differences between AChE and BChE. The entrance of the BChE gorge is noticeably broader than AChE, and the amino acid residues at the peripheral site of AChE and BChE are partly different, according to X-ray spectroscopic study (Fig. 9). Three aromatic amino acids are missing from the peripheral site of BChE, resulting in altered interactions between ligand and substrate.160,161 The role of BChE in normal conditions of healthy brain activity is overlooked, even though BChE level rises in tandem with AD progression in the latter phases of the illness. Remarkably, the level of AChE begins to decrease in these situations162–164 (Fig. 10).
 | ||
| Fig. 9 Synthesis, storage, discharge and termination of acetylcholine's action. | ||
 | ||
| Fig. 10 Mechanism of action of anti-AD drug. | ||
5 Synthetic cholinesterase inhibitors
5.1 Nitrogen-, oxygen- and sulfur-based heterocycles
Taslimi et al. (2018) reported a series of substituted pyrazol-4-yl-diazene derivatives 1 and their in vitro cholinesterase (ChE) studies and suggested that all the analogs were potent AChE (Ki = 44.66–78.34 nM) and BChE (Ki = 50.36–88.36 nM) inhibitors even better than tacrine (standard) for the treatment of AD (Table 1). Later, inhibition of such metabolic enzymes has emerged as a promising factor for pharmacologic intervention in a range of disorders like epilepsy, obesity, and neurodegenerative diseases.165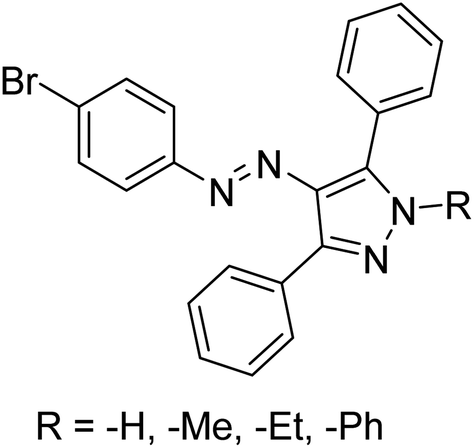
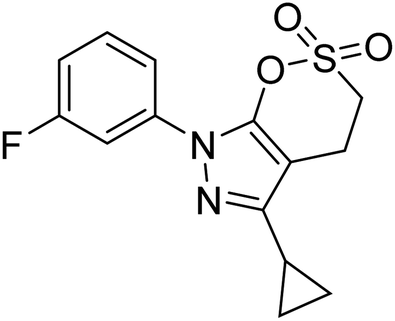
Qin et al. (2019) prepared a series of δ-sulfonolactone-fused pyrazole motif using sulfur(VI) fluoride exchange chemistry employing pyrazolones and aryl sulfonyl fluorides. The in vitro enzyme screening demonstrated their ChE inhibitory action. Among the synthesized compounds, compounds 2, 3, and 4 were recognized as selective BChE inhibitors with IC50 = 0.20, 0.46 and 0.42 µM, respectively. Kinetic studies showed that BChE inhibition of compounds 3 and 4 were reversible, mixed, and non-competitive (Ki = 145 nM and 60 nM respectively). This type of inhibition exhibited remarkable neuroprotective activity and developed as promising therapeutics for AD treatment166 (Table 1).
Şen et al. (2019) prepared a series of substituted pyrazole-based pyridazine derivatives and checked their inhibitory abilities against AChE inhibitors. These pharmacophores have gained special attention in many different synthetic drug designs due to their various bioactivities and the scaffolds of various bioactive natural compounds. All the synthesized analogs 5 and 6a–f exhibited excellent AChE inhibition (1–9 = 506 to 1022 nM). The discovery of novel inhibitors of AChE, one of the ChE enzymes, is particularly important for AD167 (Table 1).
Singh et al. (2019) prepared several 3,5-diaryl-1H-pyrazole derivatives and evaluated them for their potencies against AChE and BChE inhibitors. All analogs demonstrated mild to good activity compared to the reference standard donepezil. Compound 7 (p-Cl) was found to be potent inhibitor of AChE and BChE with 2-folds increase in IC50 (IC50 = 1.937 µM for AChE; 1.166 µM for BChE). These results represent a valuable milestone for the design of new agents against AD168 (Table 1).
Shaikh et al. (2020) reported novel scaffolds of N-substituted pyrazole derived α-amino phosphonates and evaluated their anti-ChE activity, where two of these analogs 8 (IC50 = 0.055 µM for AChE) and 9 (IC50 = 0.017 µM for AChE) showed strong efficacy against AChE. The remaining compounds possessed moderate to good BChE inhibition and performed better than the commercially available drugs galantamine and rivastigmine. This research produced promising lead compounds for further development as anti‐Alzheimer agents169 (Table 1).
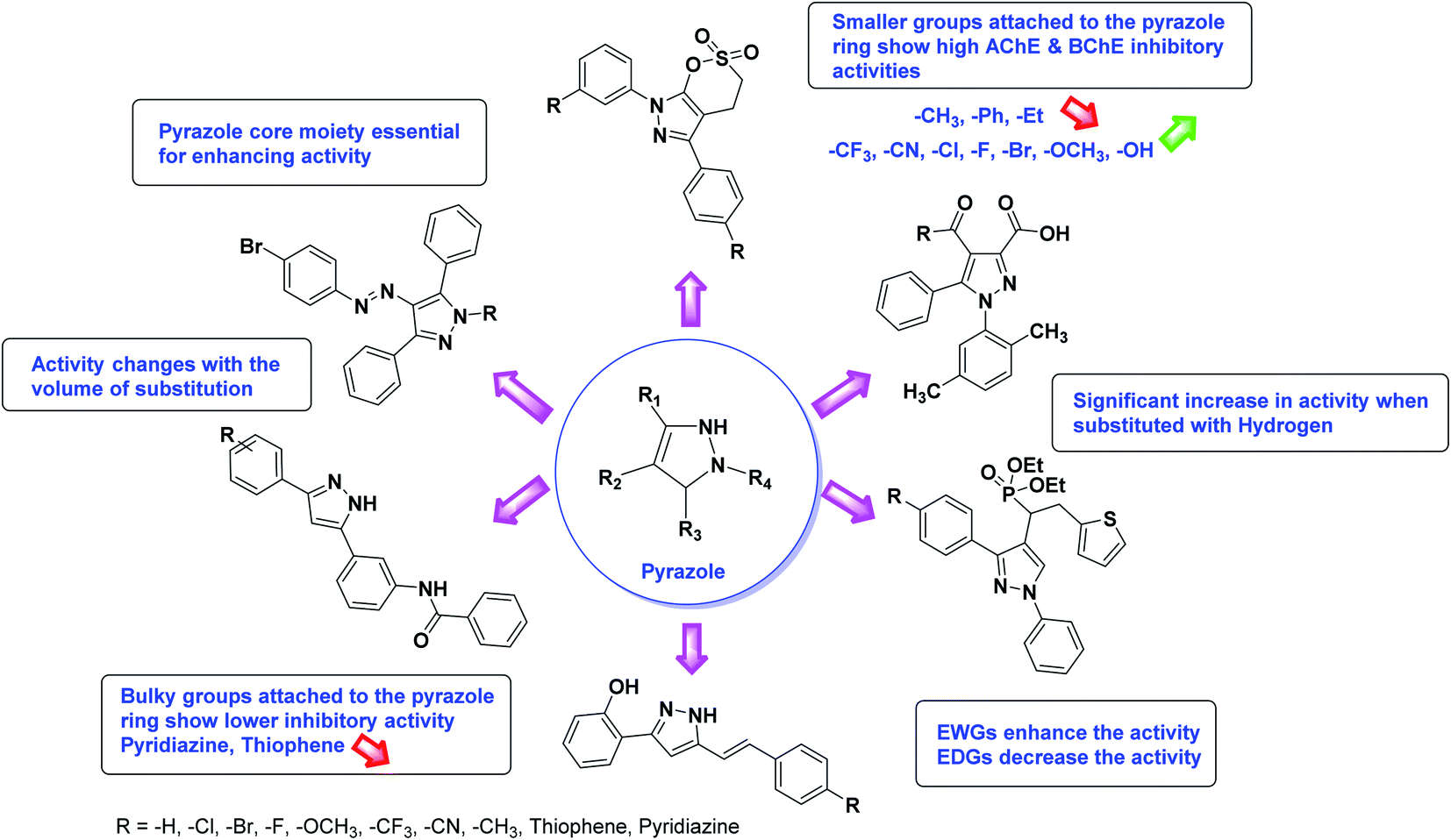
Limited SAR studies were conducted based on the central core and substitution pattern on the pyrazole scaffold (Fig. 11). Accordingly, it may be deduced that the variations observed in cholinesterase activity of the above-mentioned analogs is a consequence of variations in substitution pattern present on the main structural motif of the molecule. All these structural features are performing a considerable role in the inhibitory activity, though, observed variation in the activity of these analogs, which may be substantial in some cases, is ascribed to variability in the nature and positions of substituents on aryl rings. The smaller groups attached to the pyrazole ring promote higher AChE and BChE inhibitory abilities compared to bulky ones. Electron withdrawing atoms or groups (–F, –Cl, –Br, etc.) increase the activity whereas electron-donating groups (EDG) (–CH3, –OCH3 etc.) decrease the activity. Thus, ChE activity seems to be impacted by electronic and steric factors inherent to the inhibitor molecule.
 | ||
| Fig. 11 SAR analysis of different pyrazole derivatives as AChE and BChE inhibitors. | ||
In all the figures (Fig. 11 to 25) given below, the red arrow indicates the decrease in activity and green arrow indicates an increase in activity.
Ghalib et al. (2012) synthesized two indenoimidazoles by reacting ninhydrin with diphenylthiourea and diphenylurea for the treatment of AD. In vitro assays demonstrated that most of the compounds successfully inhibited ChEs in the µM range. Analogs 10 (IC50 = 177.69 µM for AChE and 90.20 µM for BChE) and 11 (IC50 = 274.69 µM for AChE and 101.20 µM for BChE) demonstrated good ChE enzyme. Moreover, 11 was found to be 3 times more selective BChE inhibitor, highlighting its potential for possible use in preventing further neurodegeneration as well for symptomatic treatment of Alzheimer patients. The AChE inhibitors are the mainstay drugs for the management of AD170 (Table 2).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 10 | 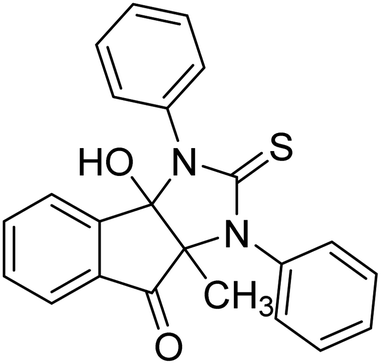 |
177.69 | 90.20 | 170 |
| 11 | 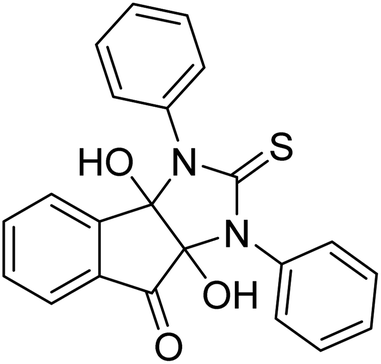 |
274.69 | 101.20 | 170 |
| 12 | 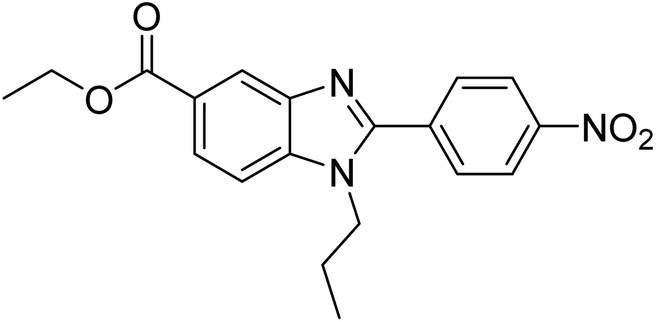 |
5.12 | 8.63 | 171 |
| 13 | 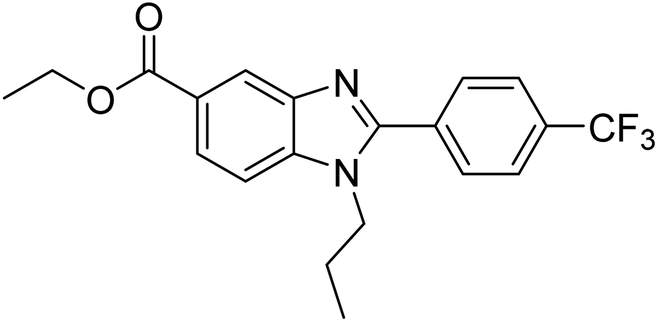 |
9.74 | 6.59 | 171 |
| 14 | 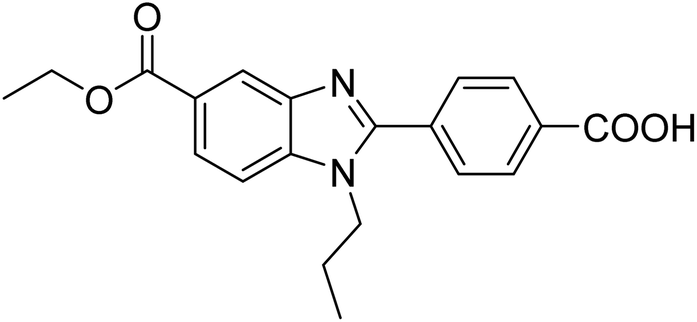 |
16.38 | 11.44 | 171 |
| 15 | 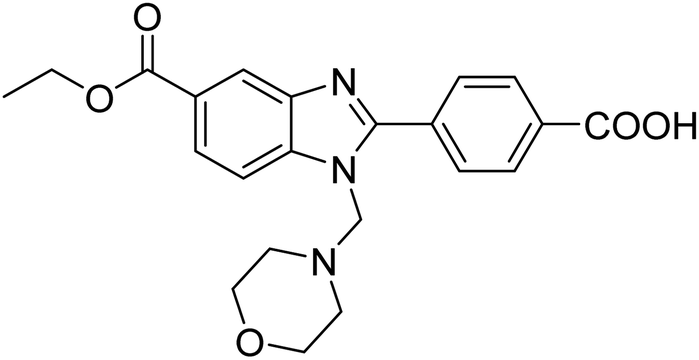 |
19.57 | 18.08 | 171 |
| 16 | 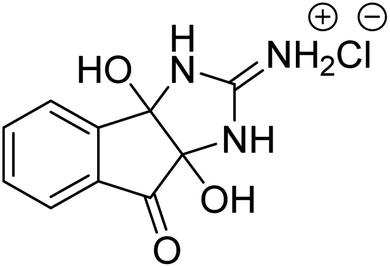 |
0.33 | — | 172 |
| 17 | 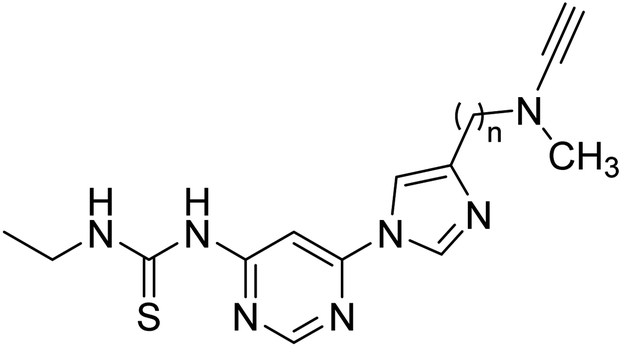 |
0.324 | — | 173 |
| 18 | 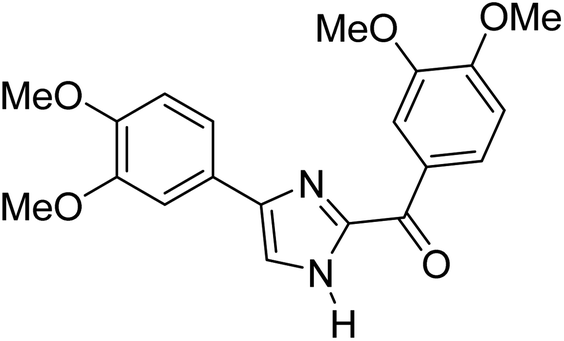 |
17.3 nM | 41.67 nM | 174 |
| 19 | 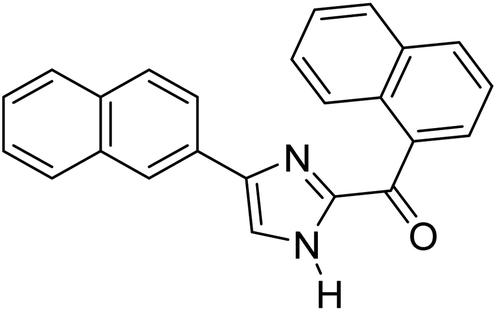 |
17.3 nM | 27.02 nM | 174 |
| 20 | 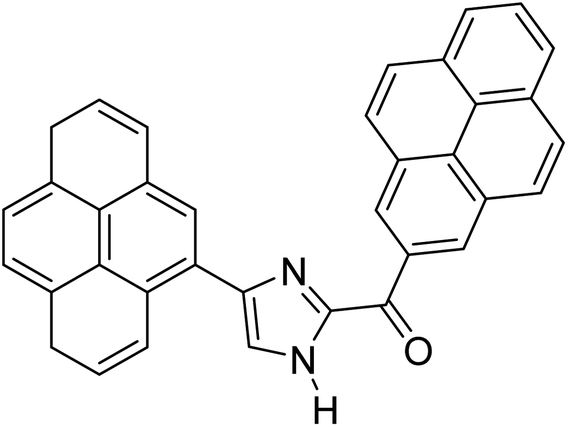 |
17.32 nM | 64.28 nM | 174 |
| 21 | 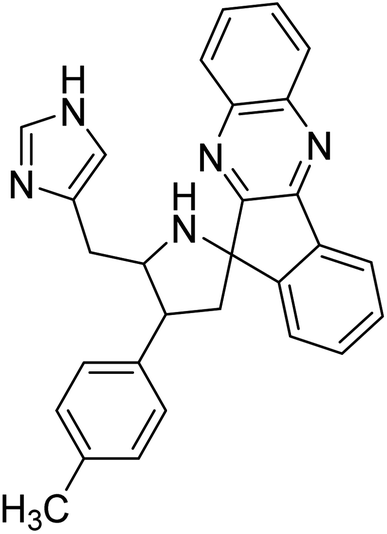 |
2.02 | 12.40 | 175 |
| 22 | 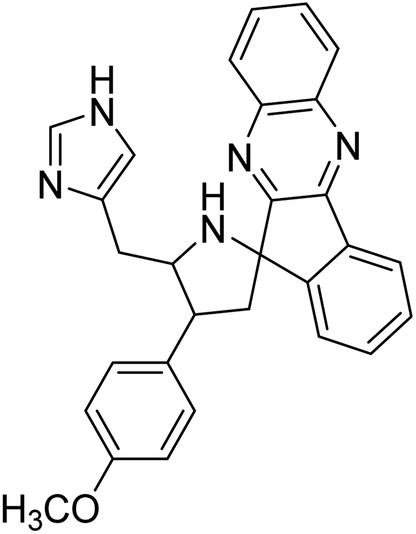 |
2.05 | 11.45 | 175 |
| 23 | 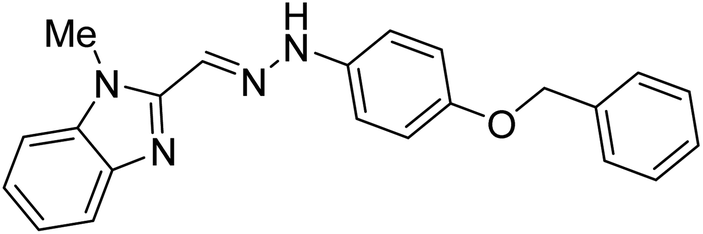 |
11.8 | — | 176 |
| 24 | 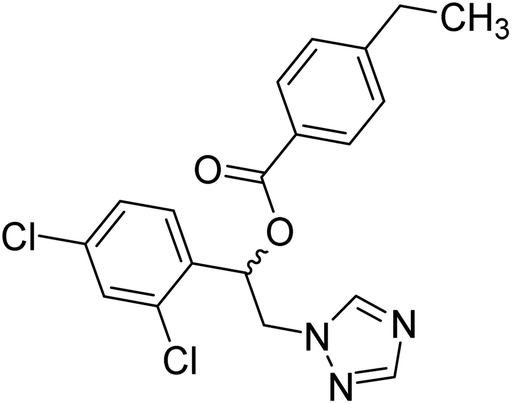 |
8.77 | — | 177 |
Yoon et al. (2013) synthesized a novel series of compounds containing benzimidazole core structure and evaluated their inhibitory potential against AChE and BChE inhibitors. Among the synthesized analogs, four benzimidazoles exhibited excellent AChE inhibition with IC50 < 10 µM. Compound 12 showed the highest inhibitory activity (–NO2, IC50 = 5.12 µM for AChE and 8.63 µM for BChE). This work demonstrated that manipulating substituents on the 2nd position on the benzimidazole core as well as the 4th position on the aryl ring moiety could potentially result in novel analogs with potent ChE inhibition activity. The compounds containing EWGs at the 4th position in the phenyl ring are important for better activities as shown by 13 (–CF3; IC50 = 9.74 µM for AChE and 6.59 µM for BChE), 14 (–COOH; IC50 = 16.38 µM for AChE and 11.44 µM for BChE) and 15 (–COOH; IC50 = 19.57 µM for AChE and 18.08 µM for BChE) compared to donepezil and rivastigmine. Compound 12–15 gave AChE inhibition activity with IC50 value <20 µM. The best inhibitor was 12 with IC50 of 8.63 µM (ref. 171) (Table 2).
Alam et al. (2018) described the preparation and AChE inhibition of imidazole iminium chloride derivatives. Among these, compound 16 was the best inhibitor of AChE with an IC50 value of 0.33 µM compared to the standard drug tacrine (IC50 = 0.20 µM)172 (Table 2).
Inspired by multi-target-directed ligands (MTDLs), Xu et al. (2018) described the preparation of a new series of propargylamine-modified imidazole substituted pyrimidinyl thiourea derivatives. All compounds successfully inhibited AChE but displayed poor inhibitory activity toward BChE. Compound 17 was the best and most selective inhibitor having an IC50 = 0.324 µM. SAR studies demonstrated that the size of the propargylamine N-substituent strongly influenced the inhibitory profile. Moreover, the outcomes revealed that analog 17 may represent a multifunctional agent for the therapy of AD173 (Table 2).
Menges et al. (2019) synthesized a series of mono and di-substituted imidazole derivatives and evaluated them in vitro for AChE and BChE inhibitory abilities. The synthesized derivatives exhibited excellent inhibition of AChE (IC50 = 17.3–120.9 nM) as well as BChE (IC50 = 27.02–151.2 nM) which was nearly equal to donepezil and 20–40 folds higher than the standard drug tacrine. Among them, compound 19 (substituted with α-naphthyl groups) displayed the most potent inhibition of both the esterases with IC50 values of 17.3 and 27.02 nM, respectively. The dimethoxy substituted imidazole derivative 18 and pyrene-substituted derivative 20 also displayed potent AChE and BChE inhibitory abilities with IC50 values of 17.3 and 17.32 nM (for AChE) and 41.67 and 64.28 nM (for BChE), respectively. SAR studies revealed that the nature of the substitution and their relative positions on the aromatic ring has a significant effect on the biological activity profile174 (Table 2).
Arumugam et al. (2020) synthesized a small library of spiropyrrolidine tethered imidazole heterocyclic hybrids. These were evaluated for their in vitro ChEs inhibitory abilities, where analogs possessing p-CH3 21 and p-OCH3 22 substituents displayed potent activities with IC50 values of 2.02 and 2.05 µM against AChE and 12.40 and 11.45 µM against BChE enzyme, respectively, compared to galantamine (IC50 = 2.09 µM for AChE and 19.34 µM for BChE). Therefore, novel heterocycles capable of suppressing the ChEs enzyme activity can compete with current ChEIs for promising AD treatments175 (Table 2).
Boulebd et al. (2020) synthesized 10 hydrazones bearing a benzimidazole nucleus and assessed them for their anti-ChE activities. Among them, compound 27 (IC50 = 11.8 µM) was the best AChE inhibitor with an IC50 value comparable to that of the standard galantamine (IC50 = 8.9 µM). Furthermore, docking studies results revealed that these analogs inhibited the AChE enzyme mainly through H-bonds, π–π stacking, and hydrophobic interaction. These researchers succeeded in incorporating these two independently biologically active moieties (imidazole and hydrazone) into one molecule to generate compounds with new and/or enhanced biological activities176 (Table 2).
Sari et al. (2021) reported a variety of azole antifungals like miconazole to possess ChE inhibitory effects. In this study, they have tested a set of azole antifungal analogs selected through virtual screening of an in-house library for their AChE and BChE inhibitory effects. Compound 24 showed potent and selective AChE inhibition, 70 times more potent (IC50 = 8.77 µM) than the standard. The study also yielded dual AChE/BChE inhibitors in addition to several potent AChE inhibitors. All the active compounds were imidazole derivatives and the modeling study showed that imidazole in the protonated state contributed greatly to the binding interactions with some key residues of AChE and BChE active site177 (Table 2).
According to SAR studies, as represented in Fig. 12, the imidazole core moiety is essential for cholinesterase activity. Imidazole-based cholinesterase inhibitors proved excellent drugs against neurological diseases i.e., Alzheimer's and Parkinson's disease. All the imidazole structural features perform a considerable role in the inhibitory activity, though, a slight variation in the activity of the reported analogs is attributed to variability in the nature and positions of substituents on aromatic rings. Smaller groups attached to the imidazole ring foster higher AChE and BChE inhibitory abilities compared to bulky ones. The AChE and BChE activities change with the volume of substitution. Electron withdrawing groups (–CF3, –Cl, –NO2, –COOH etc.) enhance the activity and electron-donating groups (–CH3, –OCH3 etc.) exhibit inhibitory effect. Thus, new imidazole derivatives are a gateway to many novel and cheap anticholinesterase drugs.
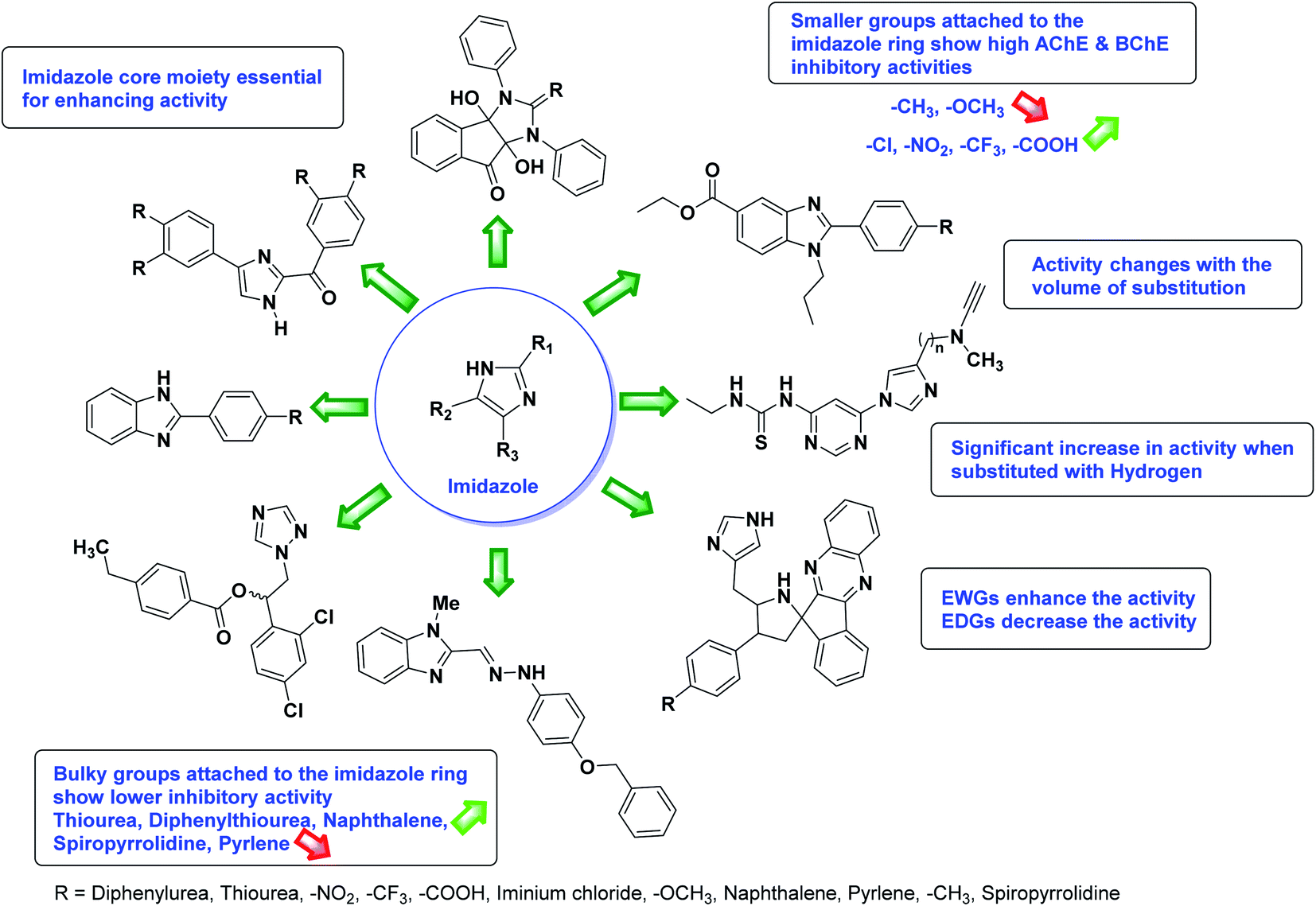 | ||
| Fig. 12 SAR analysis of different imidazole derivatives as AChE and BChE inhibitors. | ||
Abbasi et al. (2013) synthesized S-substituted analogs of 5-(2-nitrostyryl)-1,3,4-oxadiazole-2-thiol 25–30 and evaluated their ChE inhibitory activity. The analysis demonstrated that the synthesized analogs exhibit moderate to good activity against BChE and excellent inhibitory potential against AChE as evident from the IC50 values obtained for 25–30 (IC50 = 135, 254, 301, 289, >400, 101 µM for AChE and 132, 138, 74, 114, 80, 152 µM for BChE), respectively, relative to the reference standard eserine (IC50 = 0.04 mM for AChE and 0.85 mM for BChE). Hence, based on the outcome of this work, it was that halogenated analogs of 1,3,4-oxadiazoles 29 and 30 appear as good drug contenders for the treatment of AD.178 (Table 3).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 25 | 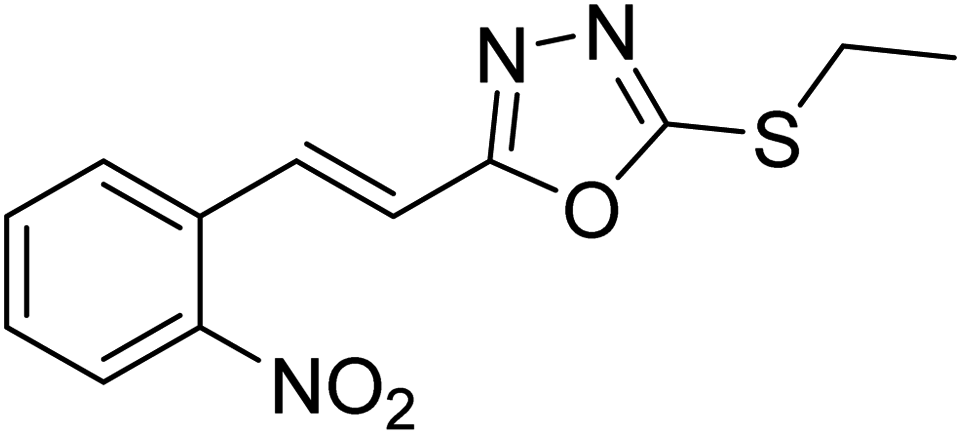 |
135 | 132 | 178 |
| 26 | 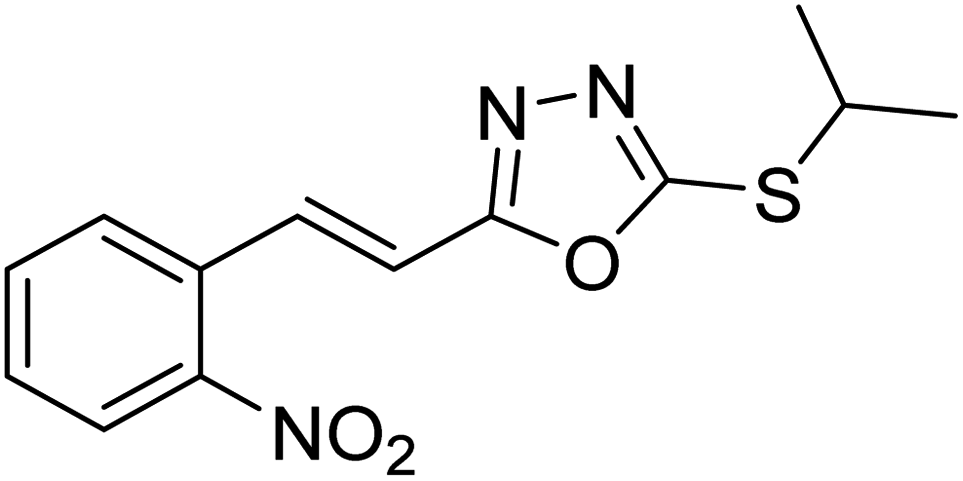 |
254 | 138 | 178 |
| 27 | 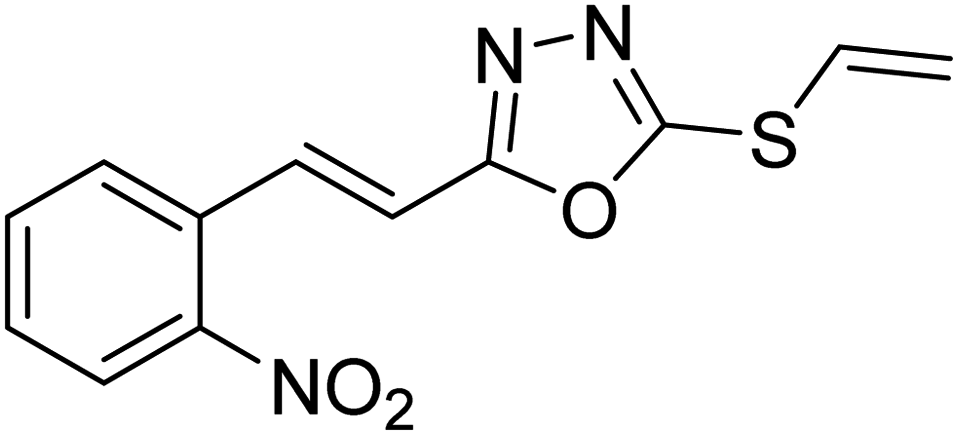 |
301 | 74 | 178 |
| 28 | 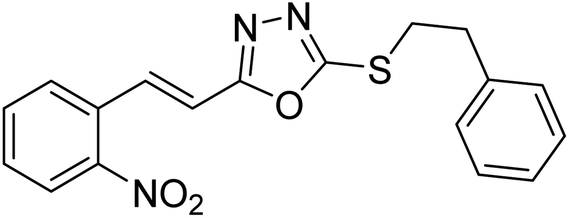 |
298 | 114 | 178 |
| 29 | 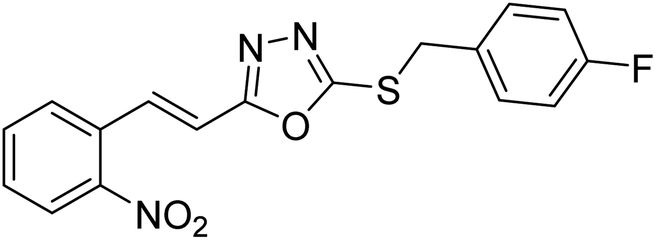 |
>400 | 80 | 178 |
| 30 | 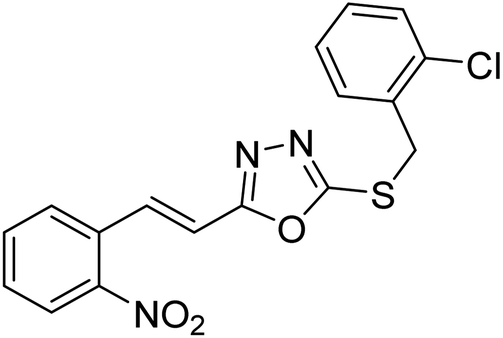 |
101 | 152 | 178 |
| 31 | 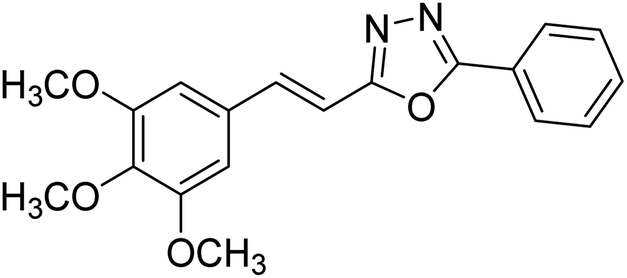 |
24.89 | — | 179 |
| 32 | 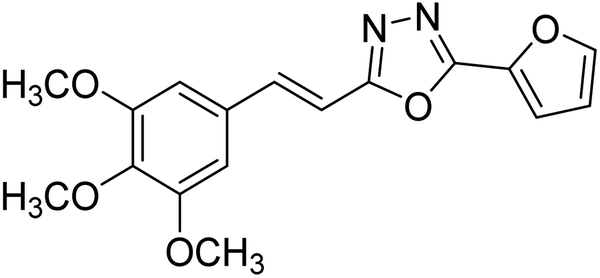 |
13.72 | — | 179 |
| 33 | 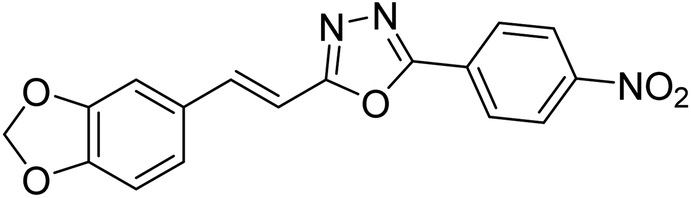 |
37.65 | — | 179 |
| 34 | 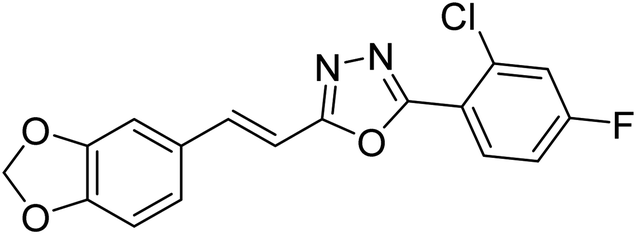 |
19.63 | — | 179 |
| 35 | 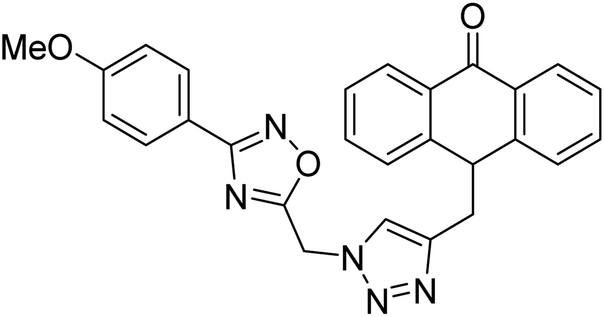 |
11.55 | — | 180 |
| 36 | 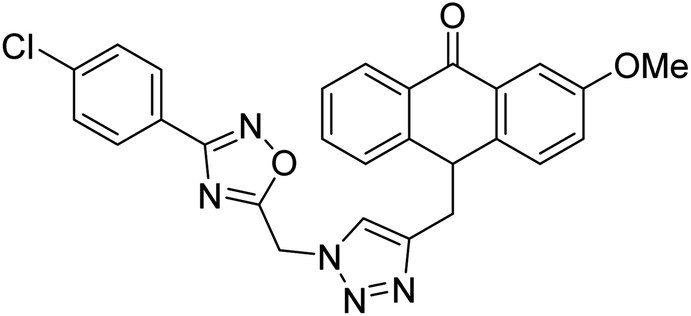 |
11.55–77.79 | — | 180 |
| 37 | 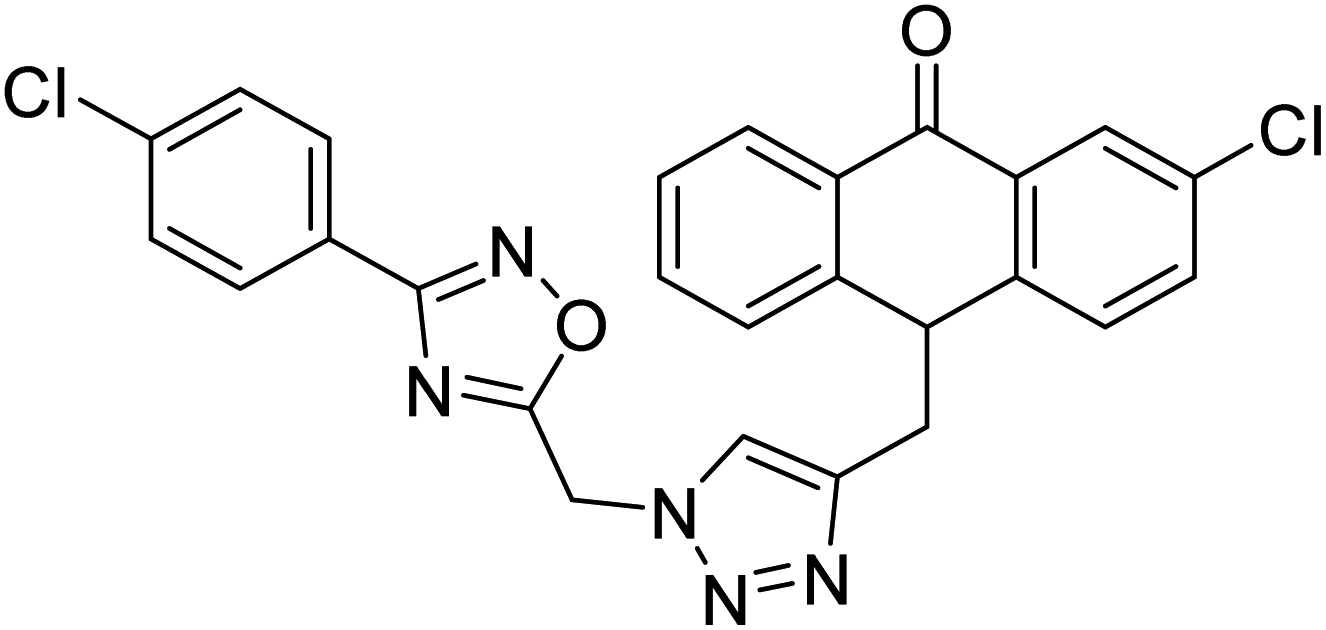 |
11.55–77.79 | — | 180 |
| 38 | 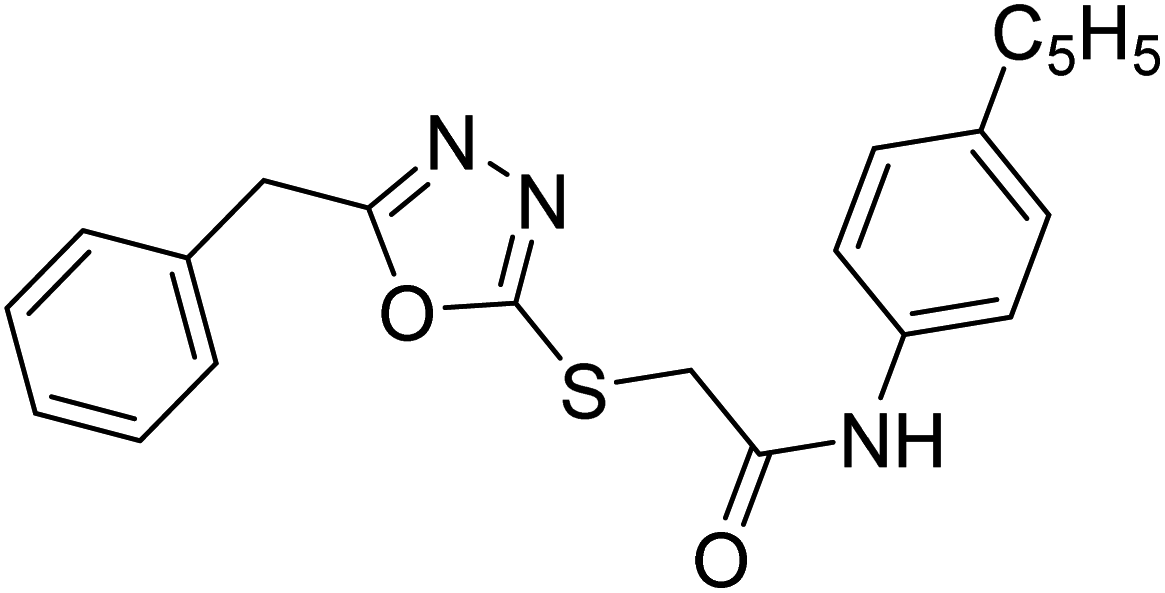 |
74.7 | 64.3 | 181 |
| 39 | 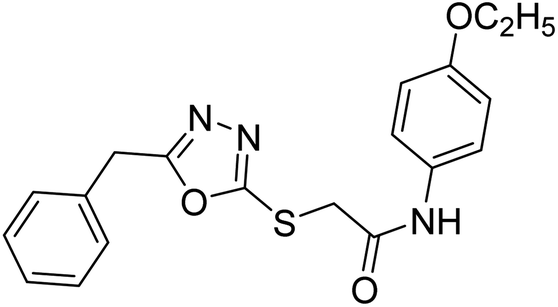 |
129.6 | 69.6 | 181 |
| 40 | 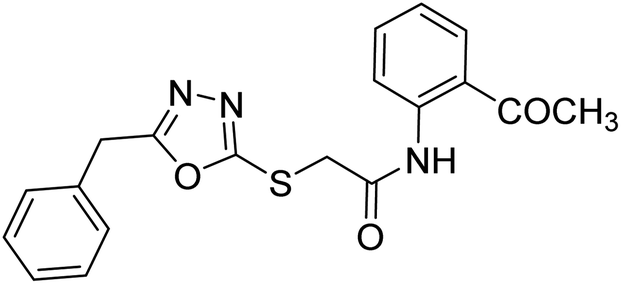 |
107.9 | 66.1 | 181 |
| 41 | 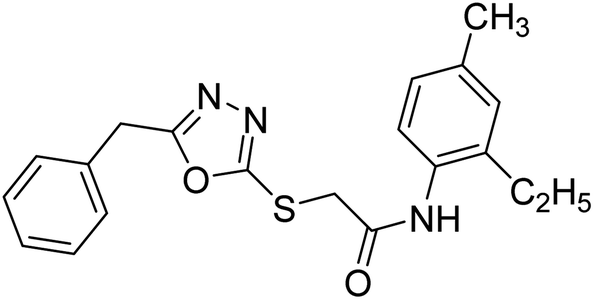 |
70.8 | 82.2 | 181 |
| 42 | 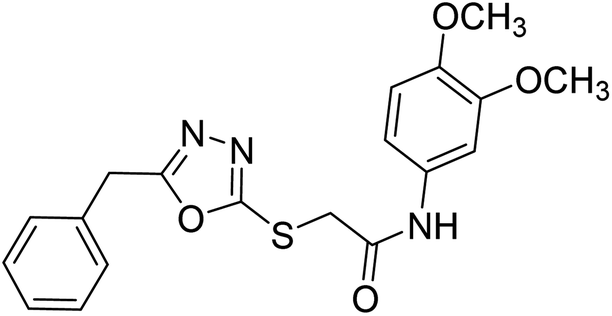 |
17.5 | 72.7 | 181 |
| 43 | 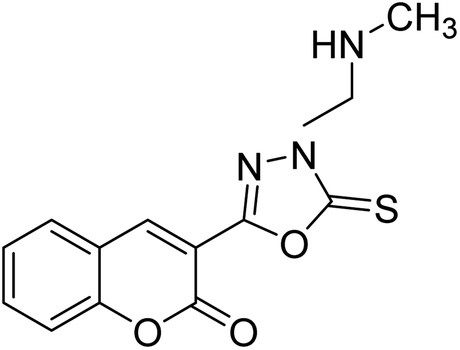 |
6.07 | 2.98 | 182 |
| 44 | 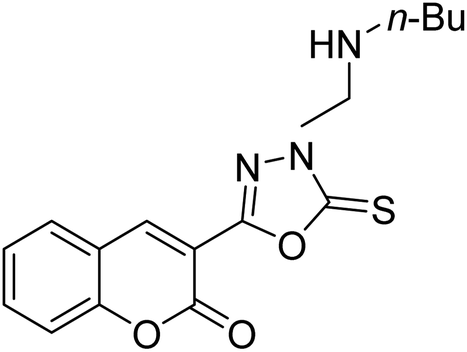 |
7.12 | 1.45 | 182 |
| 45 | 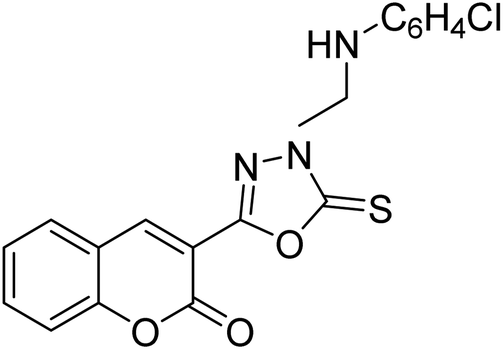 |
9.18 | 0.15 | 182 |
| 46 | 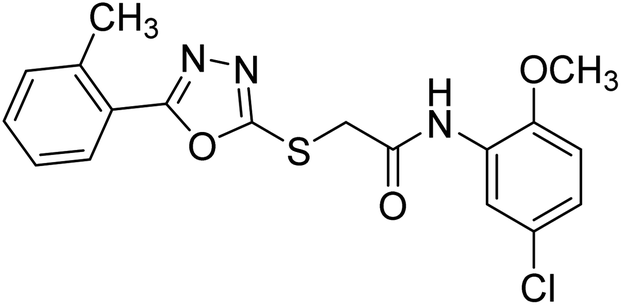 |
34.61 | — | 183 |
| 47 | 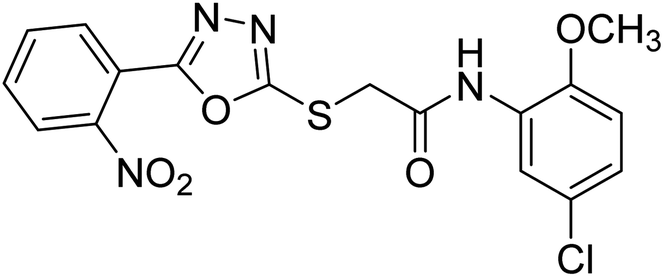 |
40.21 | — | 183 |
| 48 | 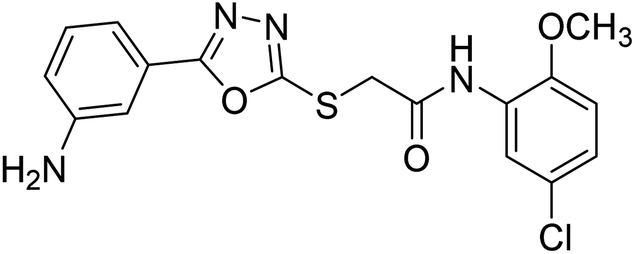 |
45.11 | — | 183 |
| 49 | 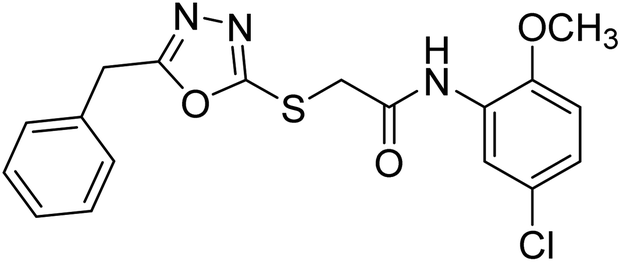 |
33.31 | — | 183 |
| 50 | 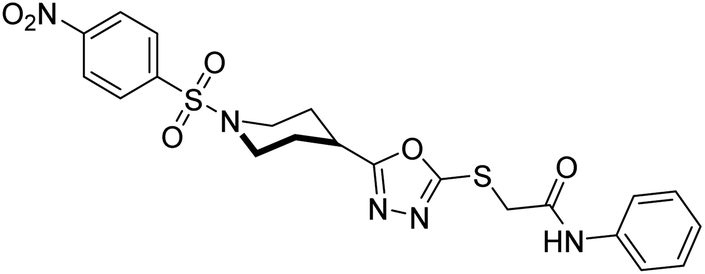 |
62.54 | — | 184 |
| 51 | 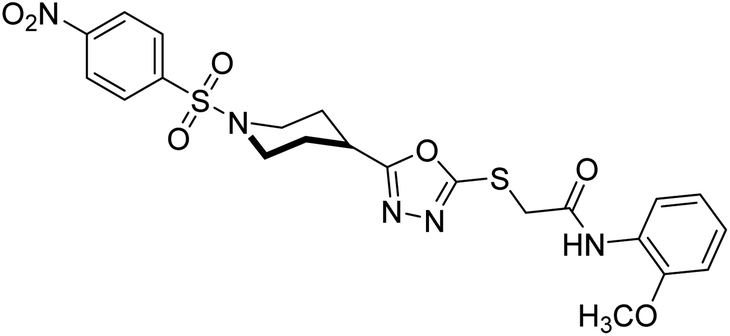 |
47.69 | — | 184 |
| 52 | 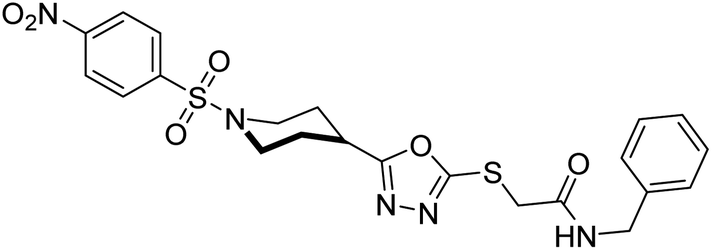 |
28.54 | — | 184 |
| 53 | 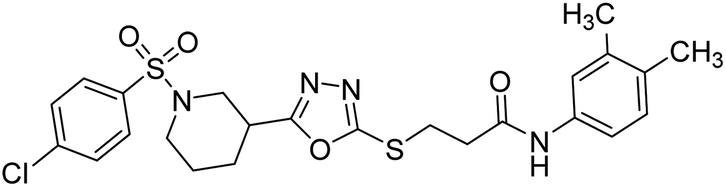 |
7.21 | — | 185 |
| 54 | 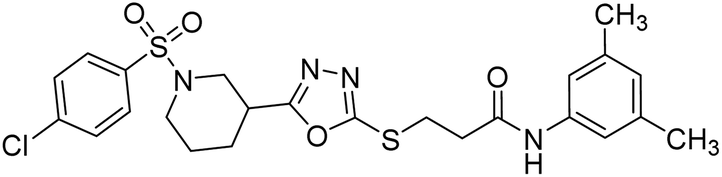 |
5.76 | — | 185 |
| 55 | 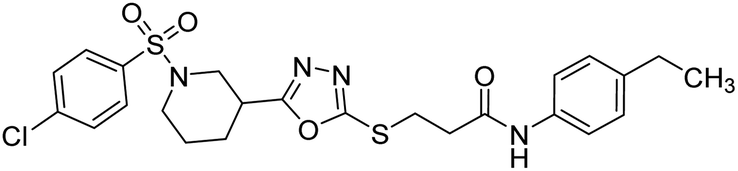 |
3.64 | — | 185 |
| 56 | 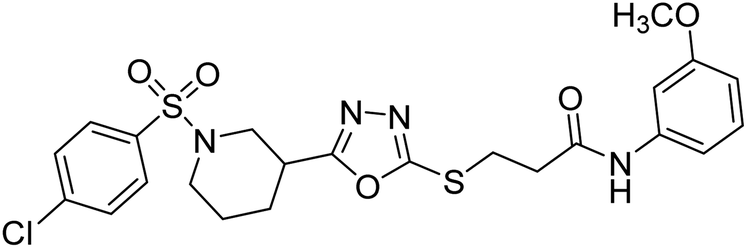 |
7.62 | — | 185 |
| 57 | 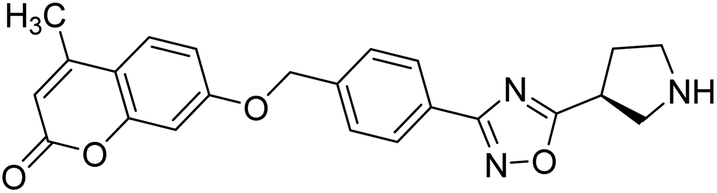 |
9.49 | 8.17 | 186 |
| 58 | 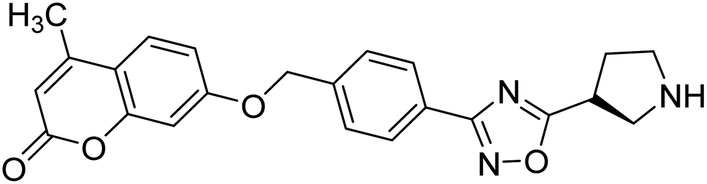 |
7.58 | 9.56 | 186 |
| 59 | 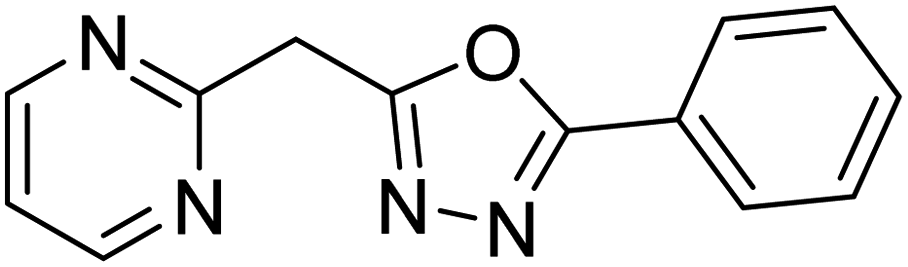 |
5.69 | — | 187 |
| 60 | 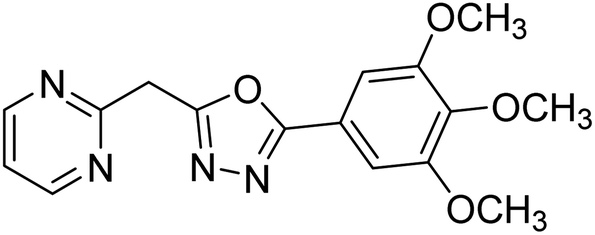 |
5.91 | — | 187 |
| 61 | 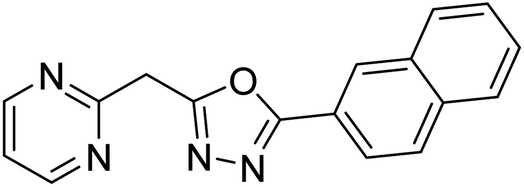 |
6.52 | — | 187 |
| 62 | 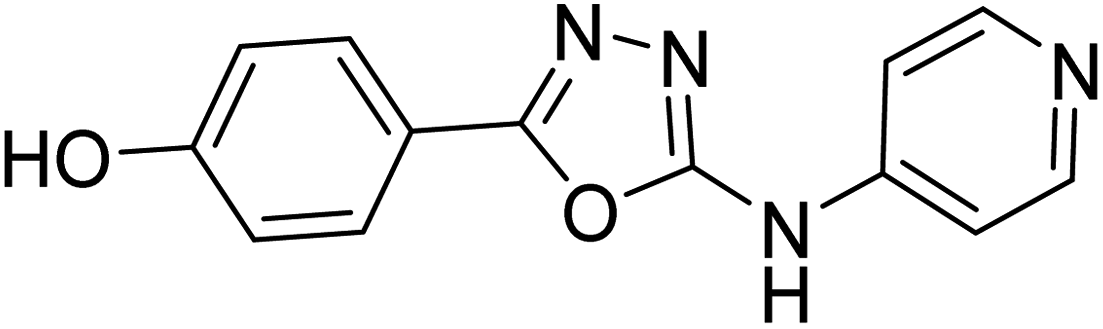 |
1.098 | — | 188 |
| 63 | 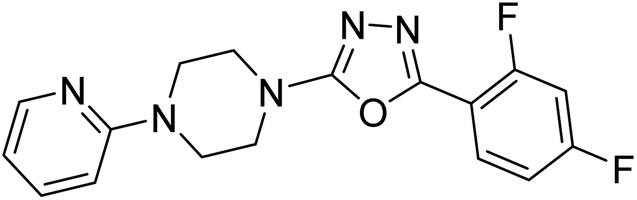 |
0.054 | 0.787 | 189 |
| 64 | 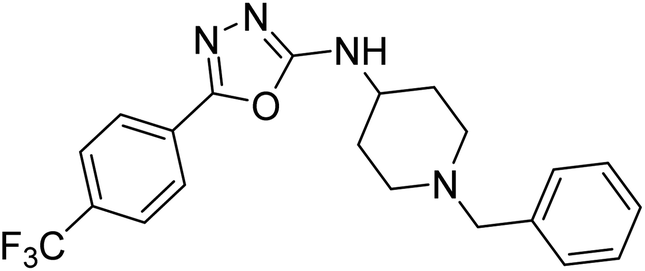 |
0.055 | 0.186 | 190 |
| 65 | 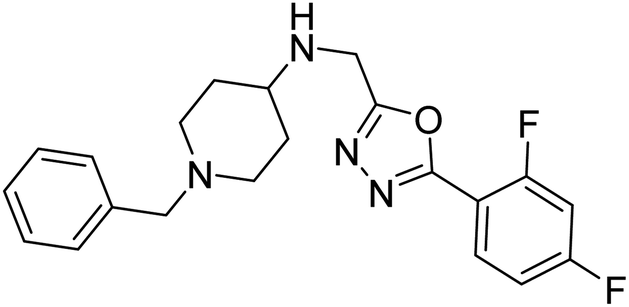 |
0.086 | 0.143 | 190 |
| 66 | 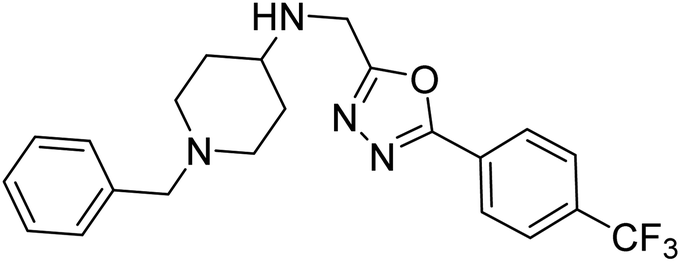 |
0.144 | 0.220 | 190 |
| 67 | 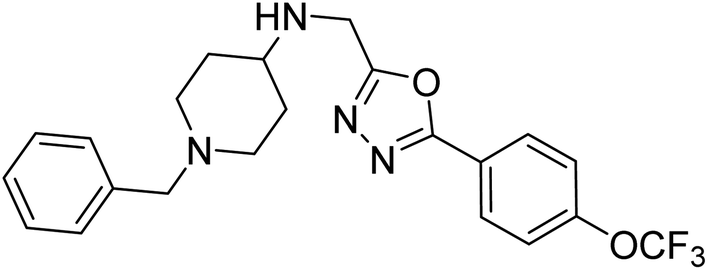 |
0.119 | 0.751 | 190 |
| 68 |  |
7.19 | 4.61 | 191 |
| 69 | 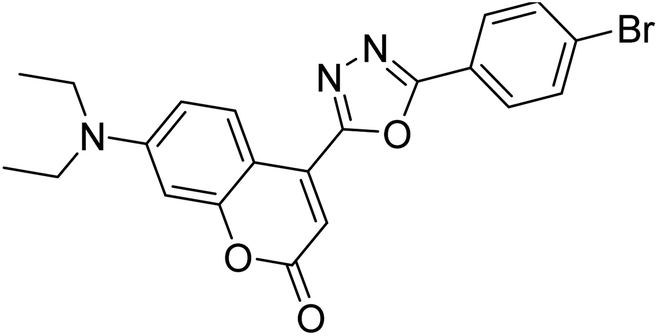 |
9.45 | 5.28 | 191 |
| 70 | 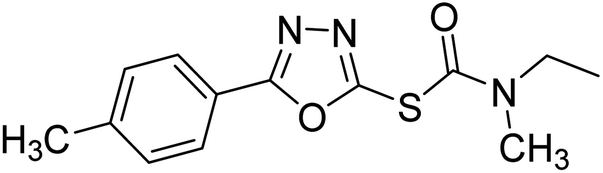 |
0.51–69.44 | — | 192 |
| 71 | 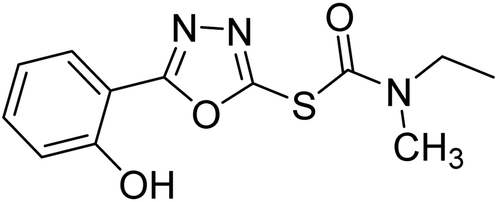 |
0.51–69.44 | — | 192 |
| 72 | 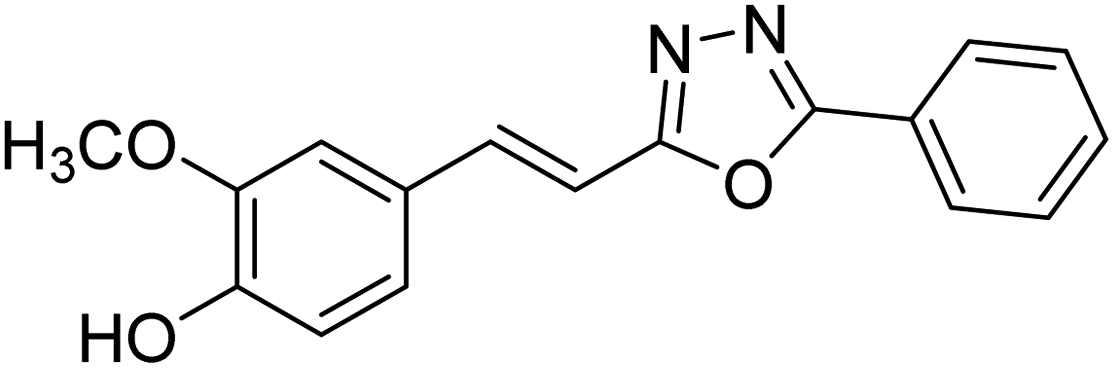 |
0.16 | 3.12 | 193 |
| 73 | 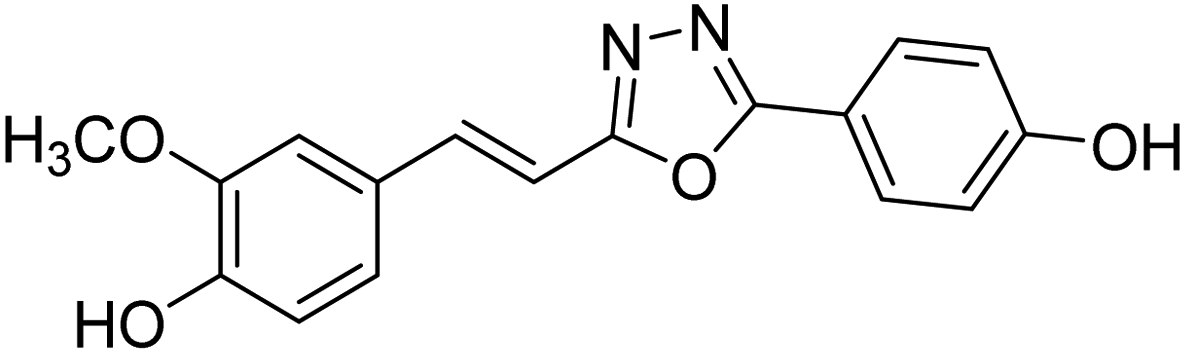 |
1.10 | 1.94 | 193 |
| 74 | 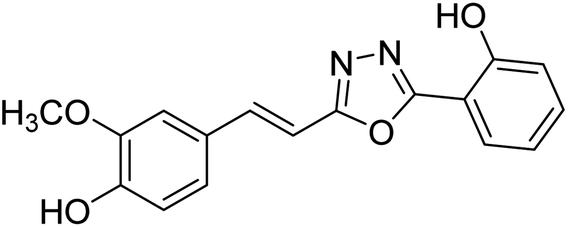 |
1.59 | 1.86 | 193 |
| 75 | 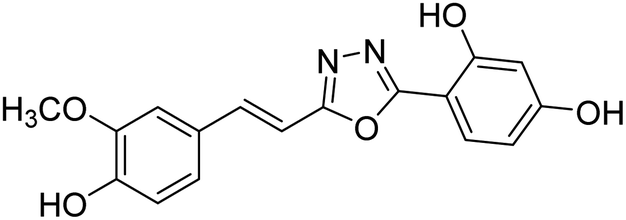 |
1.82 | 2.76 | 193 |
| 76 | 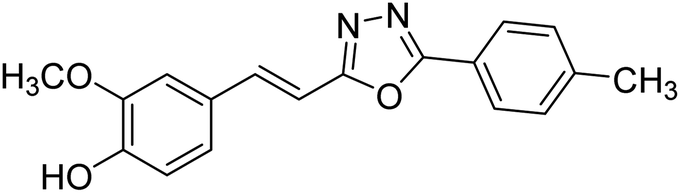 |
2.17 | 5.23 | 193 |
| 77 | 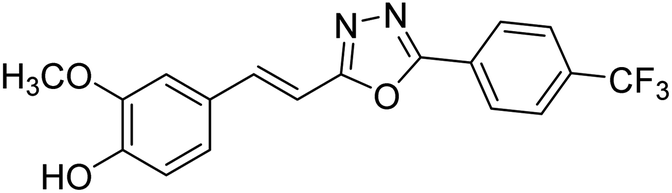 |
0.068 | 0.218 | 193 |
| 78 | 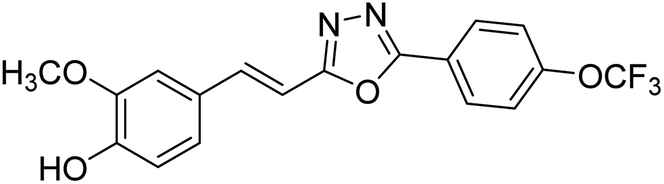 |
0.092 | 0.163 | 193 |
| 79 | 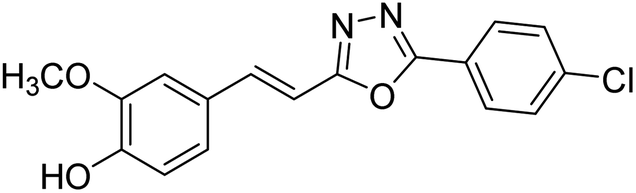 |
0.33 | 0.73 | 193 |
| 80 | 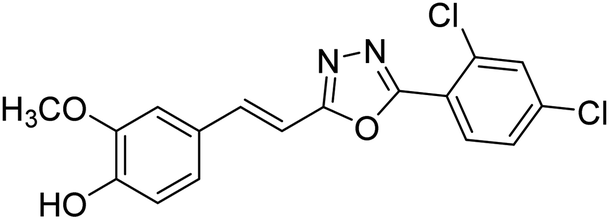 |
0.22 | 0.91 | 193 |
| 81 | 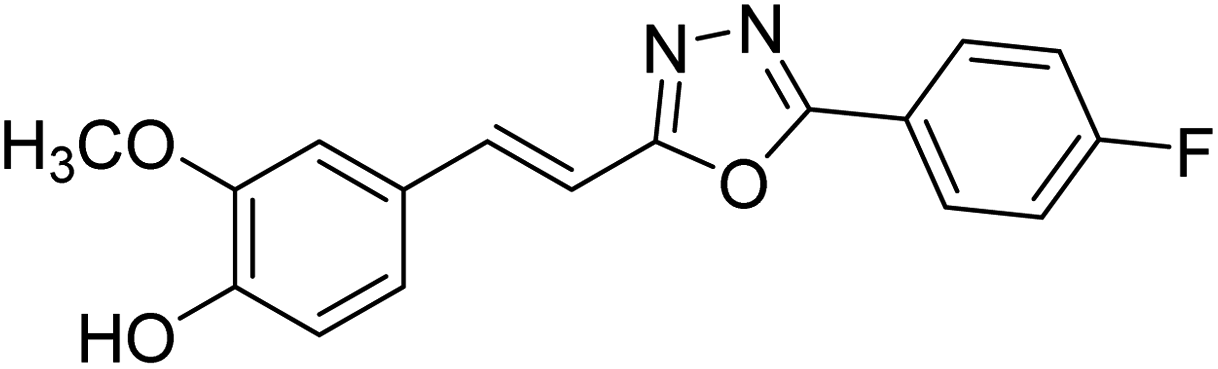 |
0.28 | 0.29 | 193 |
| 82 | 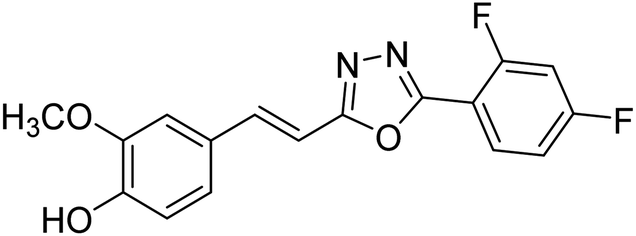 |
0.19 | 0.42 | 193 |
| 83 |  |
— | 0.463 | 194 |
| 84 |  |
— | 0.359 | 194 |
Kamal et al. (2014) prepared a library of 2,5-disubstituted 1,3,4-oxadiazole analogs and evaluated their AChE inhibitory activity in vitro. All compounds showed good to moderate inhibitory activity toward the AChE enzyme. Amongst the surveyed oxadiazole analogs, compounds 31, 32, 33 and 34 (IC50 = 24.89, 13.72, 37.65, and 19.63 µM, respectively) stood out as the most promising inhibitors of AChE. Based on molecular modeling results, it was observed that the compounds 31–34 bind to the AChE enzyme in a similar fashion to donepezil. This investigation provided an insight for the future direction in the development of conjugates as potential AChE inhibitors179 (Table 3).
Acridone-1,2,4-oxadiazole-1,2,3-triazole hybrids were prepared and assessed by Akbarzadeh et al. (2015) for their AChE and BChE inhibitory potential. Among the series, compound 35 was the most potent AChE activity (IC50 = 11.55 µM), being as potent as rivastigmine. Among all newly synthesized acridone-1,2,4-oxadiazole-1,2,3-triazoles, compounds 36 and 37 (IC50 = 11.55–77.79 µM) showed anti-AChE activity and 35 (IC50 = 11.55 µM) was found as potent as rivastigmine (IC50 = 11.07 µM). According to their findings, compound 35 possessing substitution-free acridone and 4-methoxyphenyl-1,2,4-oxadiazole groups displayed the best activity (IC50 = 11.55 µM)180 (Table 3).
Siddiqui et al. (2017) described the preparation of 5-benzyl-1,3,4-oxadiazole-2-thiol derivatives and screened all the synthesized analogs against AChE and BChE. Among these, 38–42 demonstrated moderate to good anti-ChE activity (IC50 = 74.7, 129.6, 107.9, 70.84, 17.50 µM for AChE; 41; 82.2, 42; 72.7 µM for BChE, respectively) compared to eserine (IC50 = 0.04 µM for AChE, 0.85 µM for BChE) and was credited to the presence of the 3,4-dimethoxyphenylacetamide moiety181 (Table 3).
Al-Harrasi et al. (2018) synthesized novel coumarin-oxadiazole hybrids and evaluated them against AChE and BChE in order to explore their potential for the prevention of AD. In the case of the coumarinyl oxadiazole series, 43 was lead candidate against AChE with an IC50 value of 6.07 µM, whereas compound 45 was found significantly active against BChE with an IC50 value of 0.15 µM. To realize the binding interaction of these compounds with AChE and BChE, molecular docking studies were performed. The docking studies of coumarinyl oxadiazole derivatives suggested that the compounds with high anti-BChE activity 43–45 provided MOE scores of −9.9, −7.4, and −8.2 kcal mol−1, respectively, with the active site of BChE building π–π stacking with Trp82 and water bridged interaction. In the future, these compounds and their functionalized derivatives may be helpful in the development of potent drugs for AD182 (Table 3).
Rehman et al. (2018) synthesized 5-substituted-1,3,4-oxadiazole-2yl-N-(2-methoxy-5-chlorophenyl)-2-sulfanyl acetamide derivatives and screened these derivatives against AChE and BChE. These findings revealed that the desired compounds were potent AChE inhibitors relative to eserine (IC50 = 0.04 µM for AChE, 0.85 µM for BChE). Compounds 46, 47 and 48 showed reasonably good inhibiting activity against AChE having an IC50 value of 34.61, 40.21 and 45.11 µM, respectively. Screening against the BChE enzyme showed that only one compound 49 exhibited excellent inhibitory potential having IC50 33.31 µM. The current study emphasizes the research and development of new therapeutic approaches for AD183 (Table 3).
Rehman et al. (2018) reported the green synthesis of N-(substituted)-2-(5-(1-(4-nitrophenylsulfonyl)piperidin-4-yl)-1,3,4-oxadiazol-2-ylthio) acetamide hybrids and their pharmacological applications to overcome enzymatic disorders. All the synthesized compounds were screened for AChE inhibition potential. Compounds 50, 51, and 52 were found to be very active AChE inhibitors having IC50 values 62.54, 47.69, and 28.54 and % inhibition values of 85.36, 88.72, and 89.75, respectively. Eserine (IC50 = 0.04 µM) was the reference drug for AChE inhibition184 (Table 3).
Rehman et al. (2018) synthesized N-substituted derivatives of 3-[(5-{1-[(4-chlorophenyl)sulfonyl]-3-piperidinyl}-1,3,4-oxadiazol-2-yl)sulfanyl]propenamide and assessed them as new drug contenders for AD. The synthesized products were tested for enzyme inhibition activity against the AChE enzyme. All the derivatives showed moderate to excellent inhibition activity against the ChE enzyme. Compounds bearing dimethyl phenyl groups, 53 and 54, showed enhanced inhibitory ability against AChE with IC50 values of 7.21 µM and 5.76 µM, respectively, yet were less efficient than the reference drug eserine (IC50 value = 0.04 µM). The improved activity may be credited to the presence of 3,4-dimethyl phenyl and 3,5-dimethylphenyl groups due to the collective electron-donating positive inductive effect of two methyl groups. Compounds bearing mono-substituted phenyl groups like 55 and 56 exhibited excellent AChE inhibitory activity with IC50 values of 3.64 µM and 7.62 µM, respectively, compared to the reference drug185 (Table 3).
Sun et al. (2018) synthesized a series of new chiral coumarin/1,2,4-oxadiazole hybrids and evaluated them for ChE inhibitory activity. Among them, enantiomers 57 and 58 showed potent BChE inhibitory activity with IC50 values of 8.17 and 9.56 µM, respectively, compared to tacrine (IC50 = 0.16 µM for AChE, 0.24 µM for BChE) and also exhibited good selectivity for BChE over AChE by 9.49- and 7.58-fold, respectively. In the current study, coumarin/1,2,4-oxadiazole hybrids 57 and 58 could be highlighted as a new chiral molecular template for developing multifunctional anti-AD drugs186 (Table 3).
Shrivastava et al. (2019) synthesized new hybrids bearing a 2-aminopyrimidine (2-AP) moiety linked to substituted 1,3,4-oxadiazoles and evaluated them biologically. Among the synthesized derivatives, compound 59, with a phenyl ring at the 5th position of the 1,3,4-oxadiazole core, exhibited considerable AChE inhibitory activity (IC50 = 5.69 µM). Compound 60, bearing an EWG 3,4,5-trimethoxyphenyl group, showed significant AChE inhibitory potential (IC50 = 5.91 µM). Among all the evaluated derivatives, compound 61, bearing a naphthyl ring, displayed the most significant AChE inhibitory activity (IC50 = 6.52 µM). The enhanced lipophilicity of compound 61 due to its naphthyl group may be the cause of its effective interactions with the active site residues of AChE. Thus, this study indicated that multitargeted N-(pyrimidin-2-yl)-1,3,4-oxadiazole derivatives are potential scaffolds for the treatment of dementia with compound 61 representing a promising lead for further research187 (Table 3).
Shrivastava et al. (2019) synthesized novel hybrid bearing 4-aminopyridine tethered with substituted 1,3,4-oxadiazole nucleus and evaluated them for their potential AChE inhibitory property and antioxidant potential. Among all the compounds, 62 with 4-hydroxyl substituent promoted optimum AChE inhibition with the non-competitive type of enzyme inhibition (IC50 = 1.098 µM; Ki = 0.960 µM). These findings highlighted the potential of compound 62 as significant lead for the development of orally active therapeutics in the treatment of AD188 (Table 3).
Shrivastava et al. (2019) designed and synthesized molecular hybrids of 2-pyridylpiperazine and 5-phenyl-1,3,4-oxadiazoles. Compound 63 comprising 2,4-difluoro substitution at the terminal phenyl ring emerged as the most promising AChE inhibitor lead (IC50 = 0.054 µM), BChE (IC50 = 0.787 µM). The enzyme kinetics study of 63 against AChE indicated a mixed type of inhibition (Ki = 0.030 µM). Compound 63 may be deemed as a notable lead with multifunctional actions against AD189 (Table 3).
Shrivastava et al. (2019) synthesized multitargeted hybrids of substituted 5-phenyl-1,3,4-oxadiazoles and N-benzylpiperidine and assessed them against AD. The analyzed compounds showed moderate to excellent enzyme inhibition against hAChE and hBChE. Among them, 64 (IC50 = 0.055 µM for hAChE; 0.186 µM for hBChE), 65 (IC50 = 0.086 µM for hAChE; 0.143 µM for hBChE), 66 (IC50 = 0.144 µM for hAChE; 0.220 µM for hBChE) and 67 (IC50 = 0.119 µM for hAChE; 0.751 µM for hBChE) displayed balanced and noteworthy inhibition of hAChE and hBChE in nanomolar concentration range compared to donepezil (IC50 = 0.046 µM for hAChE; 1.94 µM for hBChE) and rivastigmine (IC50 = 2.58 µM for hAChE; 1.07 µM for hBChE). The results of in vitro assays corroborated their results, indicating that an increase in the chain length and suitable placement of the 1,3,4-oxadiazole between the N-benzylpiperidine core and terminal phenyl group would significantly improve the inhibitory potential against target enzymes. In conclusion, all these results emphasized 64 as a potential candidate for the treatment of AD190 (Table 3).
Chen et al. (2020) synthesized 7-diethylaminocoumarin-based-1,3,4-oxadiazole analogs via I2-induced oxidative cyclization. The in vitro outcome of these compound's activities inhibiting AChE showed that 68 and 69 had moderate inhibitory abilities with 69.19% and 65.06%, respectively. The preliminary SAR showed that the introduction of halogen atom on the p-position of the aryl ring of oxadiazole derivatives could lead to a promising AChE inhibitor. Molecular docking study suggested that 69 possessed an optimal docking pose with interactions inside AChE191 (Table 3).
Safavi et al. (2020) synthesized a new series of 5-aryl-1,3,4-oxadiazole-2-carbamothioate compounds using structure-based drug discovery approaches. The potential of the synthesized compounds was evaluated against AChE and BChE to determine their IC50 values. The results of biological experiments demonstrated that most synthetic compounds exhibit moderate to excellent selective activity against BChE (0.51–69.44 µM). Docking studies showed the range of binding affinity for the best poses of individual conformers for any compound was between −7.81 70 and −6.75 71 kcal mol−1. Recent essay data survey indicates that BChE plays a significant interest role in AD, especially at the advance stage of the disease, therefore these selective BChE inhibitors can be favorable drug candidates in the future192 (Table 3).
Shrivastava et al. (2020) synthesized a series of molecular hybrids with ferulic acid and 1,3,4-oxadiazole framework for the treatment of AD and screened them for multifunctional inhibitory potential against AChE and BChE. Compound 77 was the most potent inhibitor of AChE (IC50 = 0.068 µM). It also showed equipotent inhibition of BChE with IC50 value of 0.218 µM. Compound 78 possessed the most significant inhibition of BChE with IC50 value 0.163 µM. Among all the tested compounds, analogs with 4-CF3 and 4-OCF3 substitution exhibited excellent AChE inhibitory profile 77, IC50 = 0.068 µM; 78, IC50 = 0.092 µM. Several findings suggested that inhibition of BChE is also a vital therapeutic strategy in the treatment of AD. The dual inhibition of AChE and BChE could be beneficial in halting the disease progression rather than providing symptomatic relief only. Therefore, the BChE inhibitory potential of all the target compounds was also evaluated. Several of the prepared compounds 72 (IC50 = 3.12 µM for BChE), 73 (IC50 = 1.94 µM for BChE), 74 (IC50 = 1.86 µM for BChE), 75 (IC50 = 2.76 µM for BChE) and 76 (IC50 = 5.23 µM for BChE) demonstrated micromolar inhibitory ability against BChE. The remaining compounds 79 (IC50 = 0.33 µM for AChE; 0.73 µM for BChE), 80 (IC50 = 0.22 µM for AChE; 0.91 µM for BChE), 81 (IC50 = 0.28 µM for AChE; 0.29 µM for BChE), and 82 (IC50 = 0.19 µM for AChE; 0.42 µM for BChE) prompted outstanding dual inhibitory ability against both cholinesterases compared to donepezil (IC50 = 0.046 µM for AChE; 1.94 µM for BChE) and rivastigmine (IC50 = 2.58 µM for AChE; 1.07 µM for BChE)193 (Table 3).
Shrivastava et al. (2021) synthesized a hybrid of substituted 5-phenyl-1,3,4-oxadiazole and N-benzylpyrrolidine and the derivatives were at first screened for ChE inhibition ability. The results indicated that the highest BChE inhibition was attained with analogs (IC50, 83: 0.463 µM 84: 0.359 µM) substituted by 4-CH3, and 2,4-difluoro on the phenyl ring, respectively. Compound 84 also exhibited outstanding oral absorption attributes in a primary pharmacokinetic study194 (Table 3).
SAR studies of different oxadiazoles demonstrate that various derivatives are active against AChE and BChE enzymes (Fig. 13). All the structural features contribute to the inhibitory activity in different capacities where any slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups attached to the oxadiazole ring seem to promote higher AChE and BChE inhibitory abilities compared to bulky groups. The ChE activity changes with the size of substitution. Electron withdrawing groups (–F, –Cl, –Br, –CN, –OH, etc.) increase the activity and electron-donating groups (–CH3, –OCH3, Et, n-Bu etc.) decrease it. All the presented analogues so far have shown good to excellent ChE inhibitory abilities with a low risk of harmful side effects. Moreover, these species are economical and easy to synthesize in the laboratory, making them attractive for commercial development and marketing as drugs against cholinesterase.
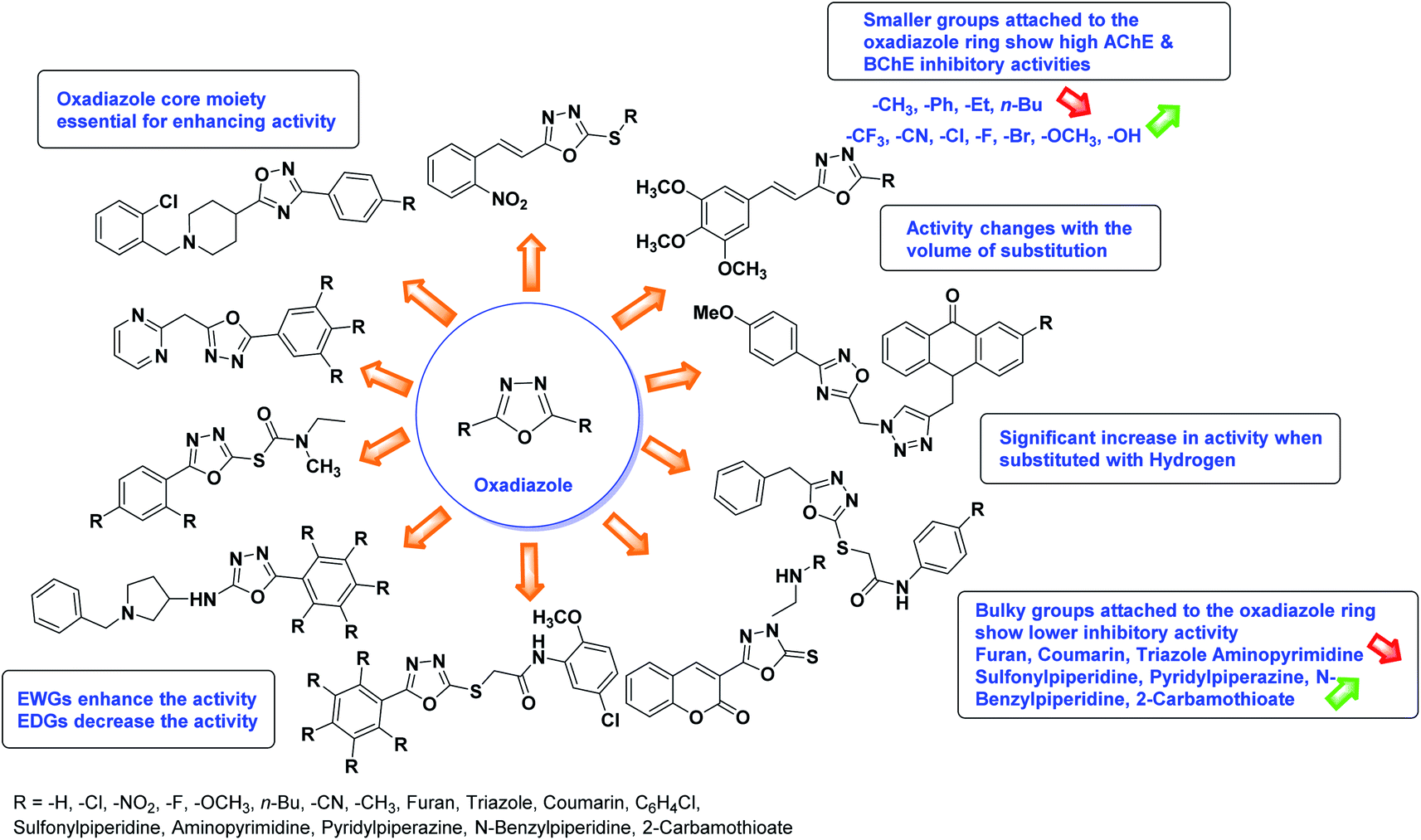 | ||
| Fig. 13 SAR analysis of different oxadiazole derivatives as AChE and BChE inhibitors. | ||
Ucar et al. (2005) prepared some 1‐N‐substituted thiocarbamoyl‐3‐phenyl‐5‐thienyl‐2 pyrazoline analogs and among the synthesized analogs, compound 85 selectively inhibited hAChE (IC50 = 0.09 µM) and is much more potent than rivastigmine (IC50 = 12.23 µM). Compound 85 carrying the p-OCH3 group on the phenyl ring inhibited the hAChE non-competitively and reversibly. The obtained results suggested that ChE inhibitors have promising features in the therapy of AD's195 (Table 4).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 85 | 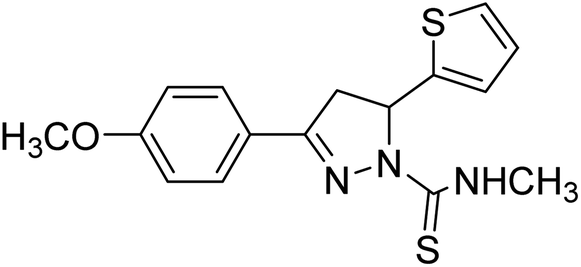 |
0.09 | — | 195 |
| 86 |  |
19.45 | — | 196 |
| 87 |  |
— | 6.31 | 196 |
| 88 |  |
0.72 µg mL−1 | 7.46 µg mL−1 | 197 |
| 89 |  |
7.2 µg mL−1 | >80 µg mL−1 | 197 |
| 90 |  |
3.2 µg mL−1 | 26.9 µg mL−1 | 197 |
| 91 |  |
48 µg mL−1 | — | 197 |
| 92 | 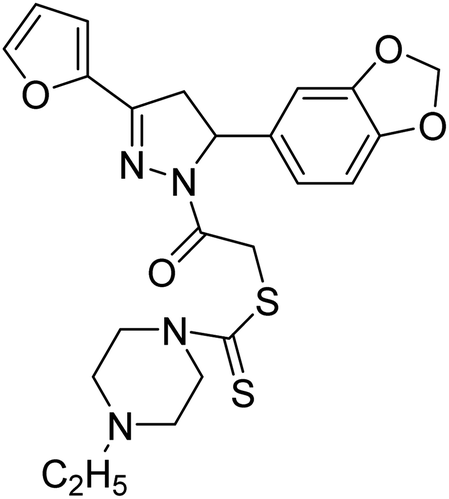 |
50.68 µg mL−1 | — | 197 |
| 93 | 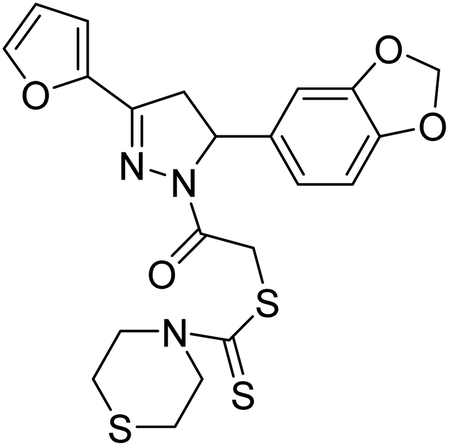 |
62 µg mL−1 | — | 197 |
| 94 | 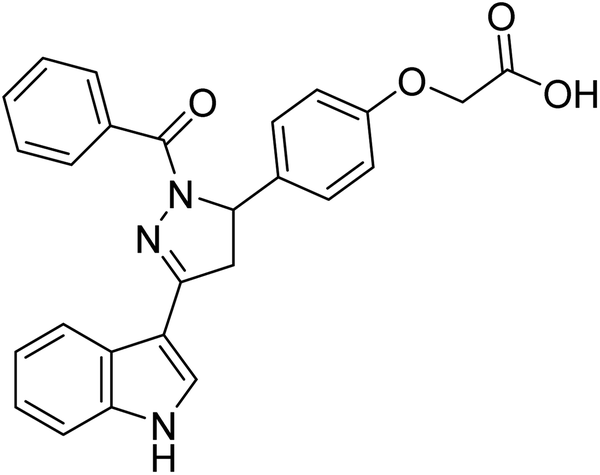 |
0.68 | — | 198 |
| 95 | 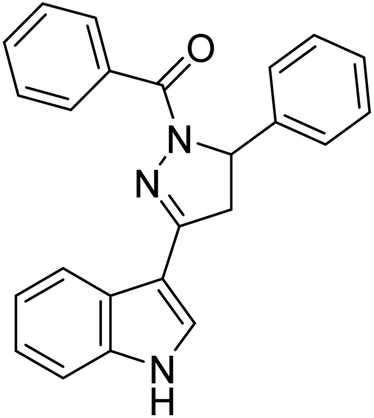 |
0.74 | — | 198 |
| 96 | 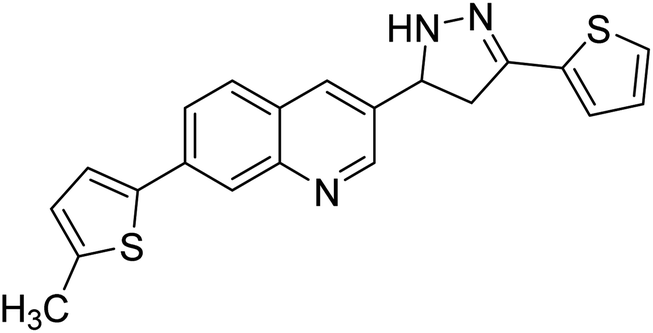 |
0.13 | — | 199 |
| 97 | 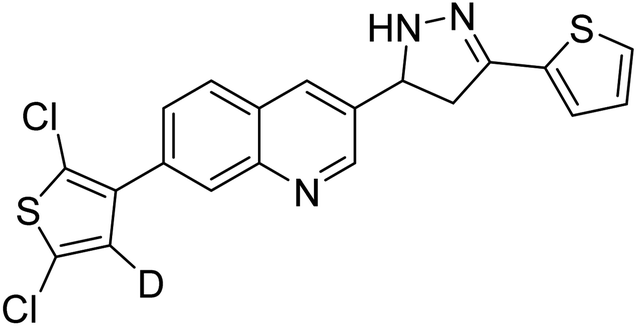 |
0.15 | — | 199 |
| 98 | 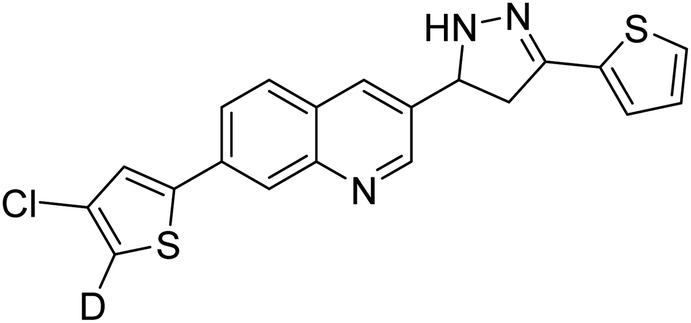 |
0.20 | — | 199 |
| 99 |  |
38.5 | — | 200 |
| 100 | 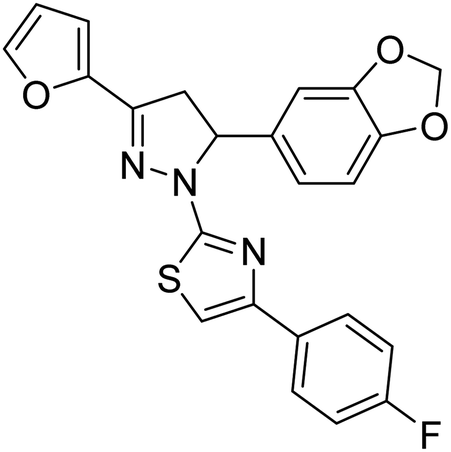 |
— | 43.02 | 200 |
| 101 |  |
48.15 | — | 201 |
| 102 |  |
52.65 | — | 201 |
| 103 |  |
123 nM | — | 202 |
| 104 | 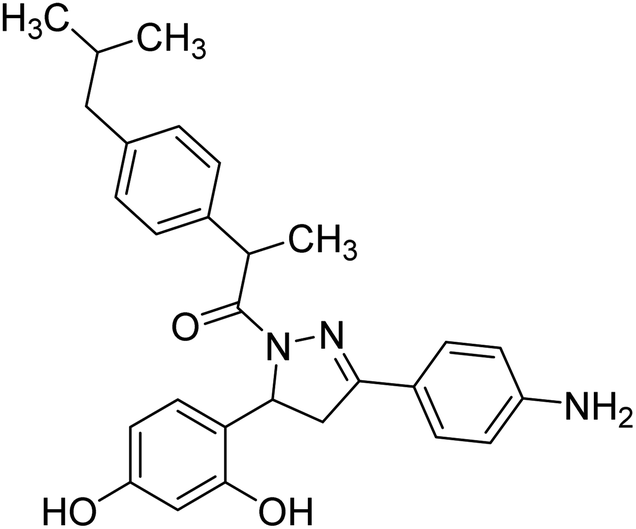 |
201 nM | — | 202 |
| 105 | 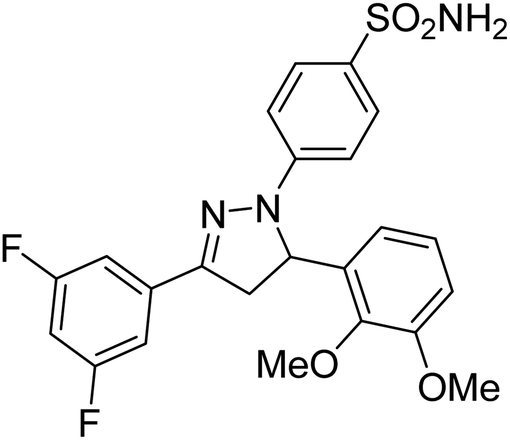 |
9.77 nM | — | 203 |
| 106 | 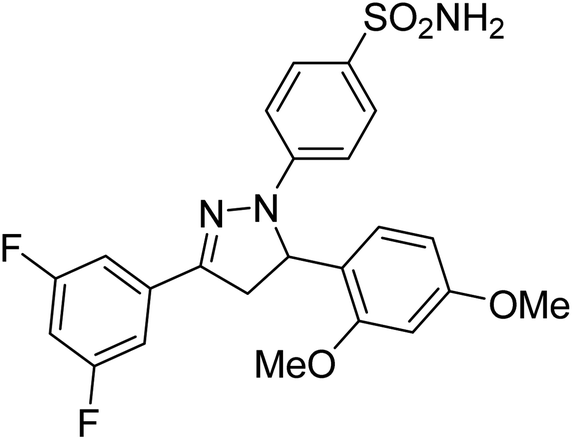 |
3.43 nM | — | 203 |
| 107 | 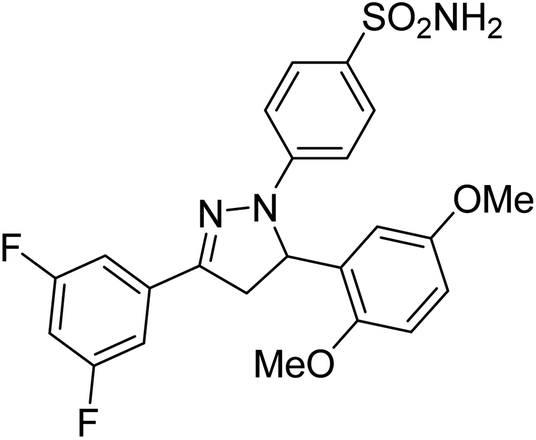 |
6.86 nM | — | 203 |
| 108 |  |
8.32 nM | — | 203 |
| 109 |  |
14.37 nM | — | 204 |
| 110 |  |
26.64 nM | — | 204 |
| 111 |  |
16.18 nM | — | 204 |
| 112 | 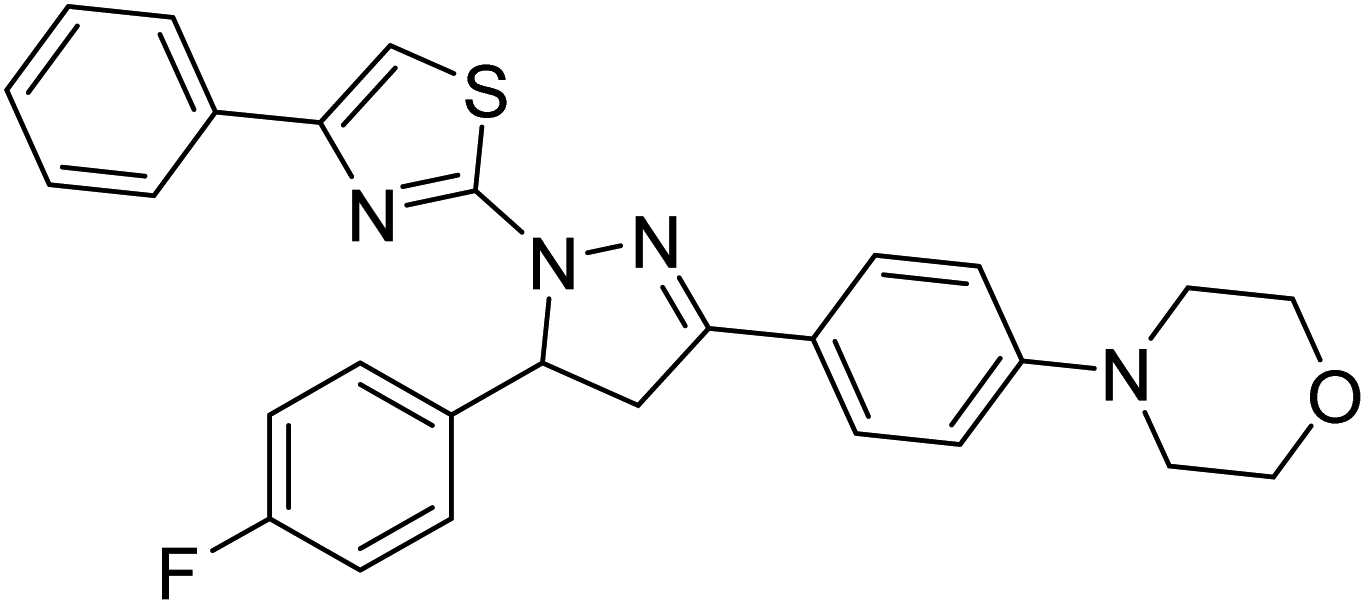 |
17.96 nM | — | 204 |
| 113 | 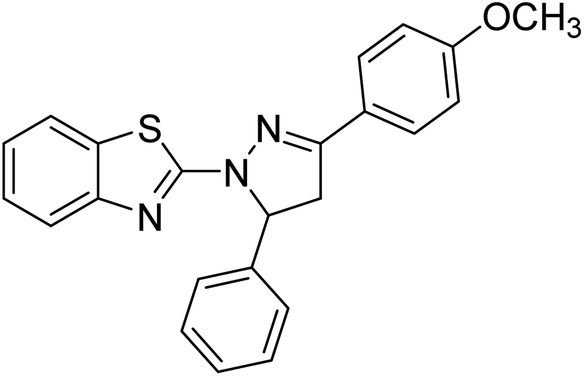 |
1.3 | — | 205 |
Jayaprakash et al. (2010) synthesized some 3,5‐diaryl‐2‐pyrazoline-1-carbothioamides and assessed their AChE inhibitory profile. Compound 86 showed outstanding AChE inhibitory activity (IC50 = 19.45 µM) and 87 displayed better BChE inhibitory ability (IC50 = 6.31 µM).
All analogs tested were potent inhibitors of ChE196 (Table 4).
Altintop et al. (2013) synthesized new pyrazoline derivatives and each analog was evaluated for its ability to inhibit AChE and BChE using a modification of Ellman's spectrophotometric method. The most potent AChE inhibitor was found to be compound 88 followed by compounds 89 and 90. Effective compounds against AChE are characterized by the presence of the 2-dimethylaminoethyl moiety, which resembles the trimethylammonium group and the ethylene bridge of acetylcholine. Among all compounds, compound 88 bearing 2-dimethylaminoethyl and 3,4-methylenedioxyphenyl moieties were also found to be highly effective inhibitor of BChE. Compound 88 can be regarded as the most promising anticholinesterase agent due to its inhibitory effect on AChE with an IC50 value of 0.72 µg mL−1 when compared with eserine (IC50 = 0.0013 µg mL−1). Compounds 89 and 90 exhibited AChE inhibitory activity with IC50 values of 7.2 µg mL−1 and 2.32 µg mL−1, respectively. Compounds 91, 92 and 93 exhibited AChE inhibitory activity with IC50 values of 48, 50.68 and 62 µg mL−1, respectively. Compound 88 also exhibited the highest inhibitory effect on BChE with an IC50 value of 7.46 µg mL−1 when compared with eserine (IC50 = 0.012 µg mL−1). Compound 90 exhibited BChE inhibitory activity with an IC50 value of 26.93 µg mL−1. Although compound 89 carries the 2-dimethylaminoethyl group, it was a weak inhibitor of BChE (IC50 > 80 µg mL−1)197 (Table 4).
Chigurupati et al. (2016) synthesized novel indolopyrazoline derivatives and assessed them as prospective anti-Alzheimer compounds through AChE inhibition study (in vitro). Specifically, 94 shows AChE inhibition (IC50 = 0.68 µM), while 95 ranked second best compound with AChE inhibition (IC50 = 0.74 µM). This study described the first use of indolopyrazoline compounds as potential anti-Alzheimer drugs198 (Table 4).
Iqbal et al. (2017) synthesized novel pyrazoline-based analogs and appraised their ChE inhibitory activity. Out of the synthesized compounds, compounds 96, 97 and 98 were the best inhibitors against AChE with an IC50 of 0.13, 0.15 and 0.20 µM, respectively. Compound 96 exhibited 173-fold higher inhibitory ability compared to neostigmine (IC50 = 22.2 µM). All the 2-pyrazoline analogs showed comparatively less (<50%) inhibitory capacity against BChE199 (Table 4).
Altintop et al. (2018) synthesized new thiazolyl-pyrazoline derivatives. The compounds were investigated for their inhibitory effects on AChE and BChE using a modification of Ellman's spectrophotometric method. As a part of this study, the compliance of the compounds to Lipinski's RO5 was evaluated. Naphthalene-substituted compound 99 was the most potent AChE inhibitor (IC50 = 38.5 µg mL−1), whereas fluoro-substituted compound 100 was the most effective BChE inhibitor (IC50 = 43.02 µg mL−1) in this series relative to the standard galantamine (IC50 = 97.17 µg mL−1 for AChE; 80.98 µg mL−1 for BChE)200 (Table 4).
Turkan et al. (2019) synthesized novel pyrazoline analogs and assessed their AChE inhibitory activity. These pyrazoline analogs were efficient inhibitors of the AChE, with Ki values ranging between 48.2–84.1 µM for AChE. In this study, all the evaluated pyrazoline derivatives showed potent inhibition against the AChE enzyme, but compounds 101 and 102 showed outstanding inhibition profiles against AChE with Ki values of 48.15 and 52.65 µM, respectively. Tacrine molecule was employed as a control compound for AChE inhibition201 (Table 4).
Mumtaz et al. (2019) synthesized a series of 1-(3-(4-aminophenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)-2-(4-isobutylphenyl)propan-1-one derivatives and evaluated their biological potential as potent ChE inhibitors. The top potent and most selective inhibitor for the AChE was analog 103 which had an inhibitory concentration of 123 nM. Compound 104 was discovered as a selective inhibitor of BChE with an IC50 value of 201 nM. The results showed that the attachment of different substituents at the para, ortho, and meta positions at the main moiety has a significant impact and contribution towards the inhibitory profile of ChE202 (Table 4).
Gul et al. (2020) synthesized a novel series of 4-(3-(difluorophenyl)-5-(dimethoxyphenyl)-4,5-dihydropyrazol-1-yl)benzenesulfonamides since sulfonamide and pyrazoline pharmacophores have garnered attention in drug design due to their wide range of bioactivities including AChE, hCA-I and hCA-II inhibitory potencies. In vitro enzyme assays showed that the novel compounds had a significant inhibitory profile against hCA I, hCA II and AChE enzymes at the nanomolar levels. When AChE inhibitory activity of the 3,5-difluorophenyl derivatives as assessed, IC50 values were calculated in the range of 8.66–15.07 nM. Compounds 105–108 having IC50 values 9.77, 3.43, 6.86 and 8.32 nM, respectively can be considered as promising AChE inhibitors for the development of novel bioactive molecules203 (Table 4).
Sever et al. (2020) prepared thiazolyl-pyrazolines analogs and evaluated their AChE inhibitory potential. In vitro studies demonstrated that all the compounds notably inhibited AChE even more than the reference drug tacrine. Compound 109 (IC50 = 14.37 nM for AChE) with the cyanophenyl substitution inhibited AChE with the lowest Ki value, whereas compound 110 (IC50 = 26.64 nM for AChE) with methyl substitution was determined as the most selective hCA I inhibitor. Compound 111 (IC50 = 16.18 nM for AChE) with the chloro substitution exhibited the most potent and selective inhibition towards AChE. In such a manner, compound 112 (IC50 = 17.96 µM) without any substitution was found as the best and most selective AChE enzyme inhibitor in this series. All compounds in the series stand out as excellent multi-targeted inhibitors for further investigations in AD treatment204 (Table 4).
Gül et al. (2020) assessed a novel series of pyrazoline compounds as potent AChE inhibitors. Compound 113 (IC50 = 1.3 µM; Ki = 0.13 µM) possessed the highest AChE inhibitory effect in the series, proving 2-fold more potent than the standard tacrine (IC50 = 0.84 µM; Ki = 0.26 µM)205 (Table 4).
The SAR analysis of pyrazoline derivatives summarized Fig. 14 has indicated that various derivatives are active against cholinesterase enzymes. All the structural features perform critical function in the inhibition where fine tuning of the activity of these analogs is possible with varying the nature and positions of substituents on aryl rings. The smaller groups attached to the pyrazoline ring foster higher AChE and BChE inhibitory abilities compared to bulky ones. The ChE activity changes with the size of substitution suggesting that steric and electronic factors are important in fine tuning the structure. In this context, electron withdrawing groups (–F, –Cl, –Br, etc.) have been found to increase the activity and electron-donating groups (–CH3, –(CH3)2, etc.) reduce it. All the presented analogues thus far have shown moderate to good ChE inhibitory abilities with a minimal risk harmful side effect. Moreover, these analogs are cost-effective and easy to prepare in the laboratory, making them attractive for commercial development and marketing as drugs against cholinesterase.
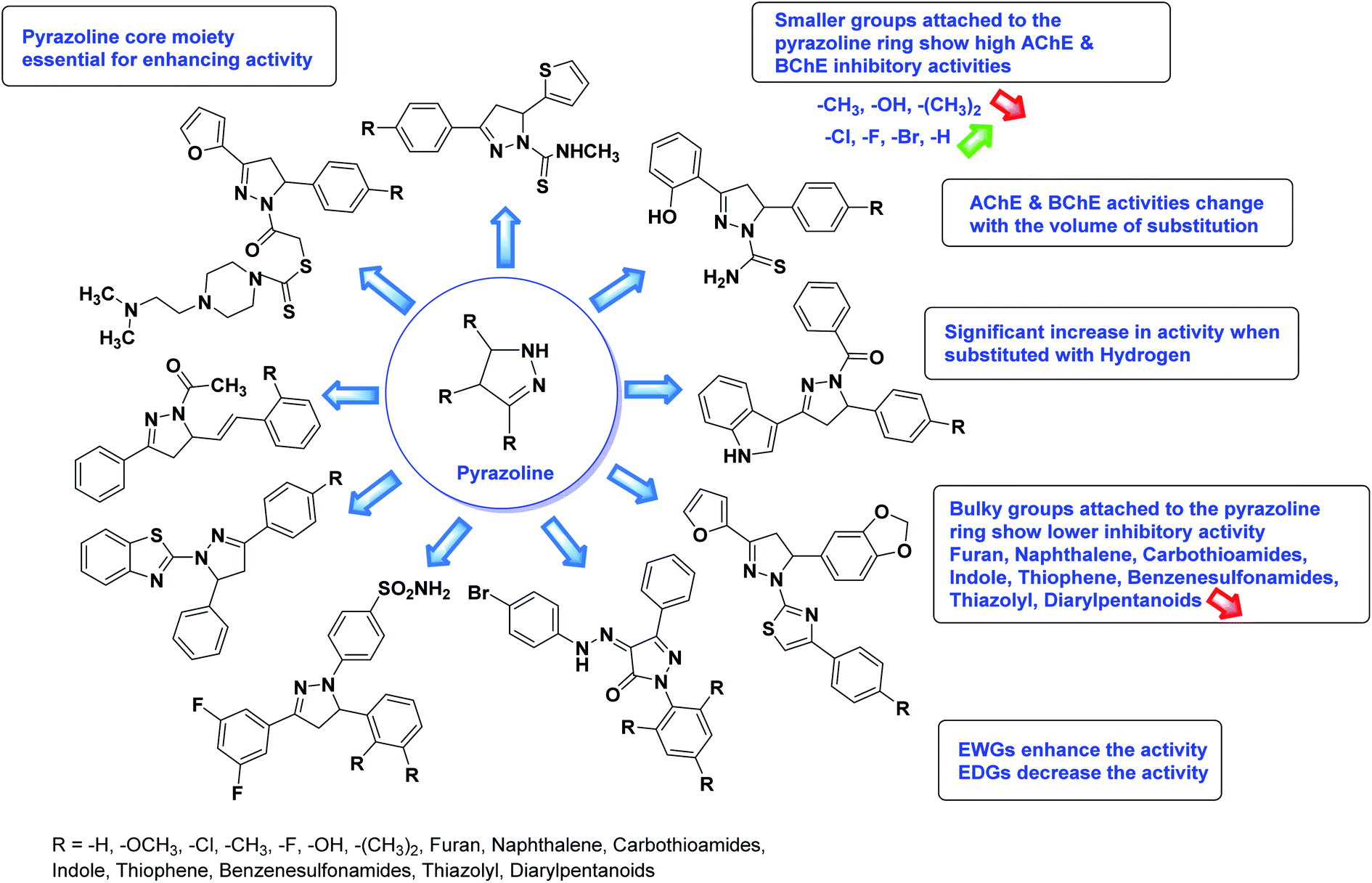 | ||
| Fig. 14 SAR analysis of different pyrazoline derivatives as AChE and BChE inhibitors. | ||
Alinezhad et al. (2015) synthesized various 1,2,3-triazole linked acridone analogs and evaluated for their proficiency to inhibit AChE using rivastigmine as a drug. Most compounds (IC50 ≥ 100 µM mL−1) were inactive, although compound 115 was very potent (IC50 = 7.31 µM mL−1) in comparison to rivastigmine (IC50 = 11.07 µM mL−1). The SAR study showed that the substituents' electronic properties on acridone and 1,2,3-triazole rings impacts the anti-AChE activity since no activity was observed for compound 114 (IC50 ≥ 100 µM mL−1). Notably, the existence of 4-substituted chlorine on the benzyl group 115 plays a substantial role in the anti-AChE activity. Compound 115 was the best contender against AChE, highlighting the usefulness of methoxy and chlorine groups to generate effective interactions with the active site of the enzyme and is in agreement with factors outlined earlier that play a crucial role in inhibitory activity206 (Table 5).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 114 | 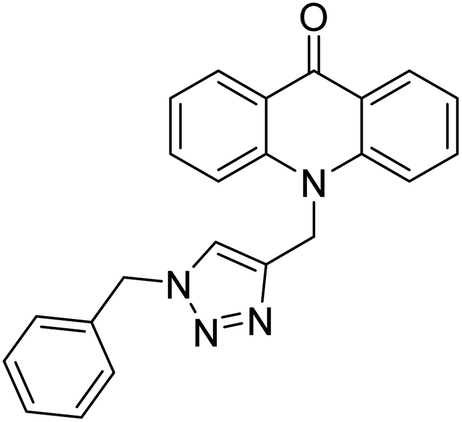 |
>100 µM mL−1 | — | 206 |
| 115 |  |
7.31 µM mL−1 | — | 206 |
| 116 |  |
4.52 | — | 207 |
| 117 |  |
5.31 | — | 207 |
| 118 |  |
114 | — | 208 |
| 119 | 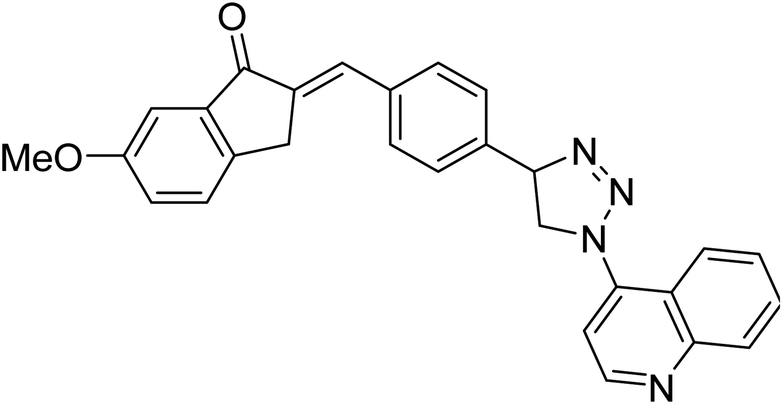 |
104 | — | 208 |
| 120 | 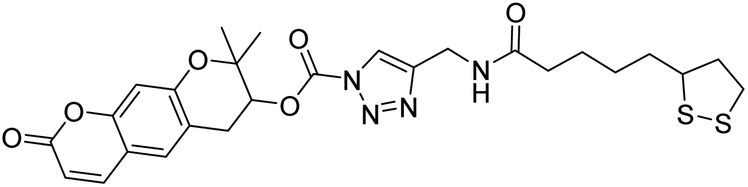 |
>350 | 5.98 | 209 |
| 121 | 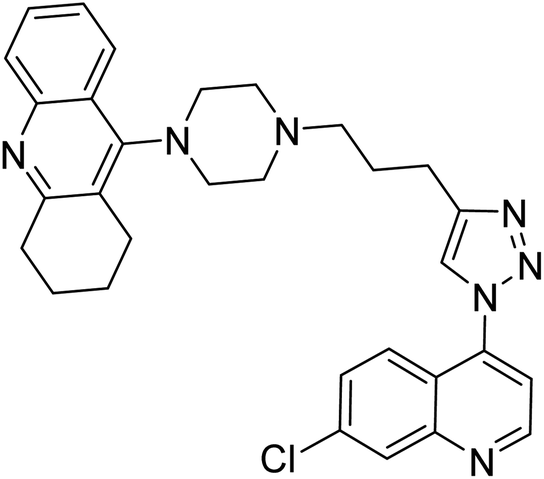 |
4.89 | 3.61 | 210 |
| 122 | 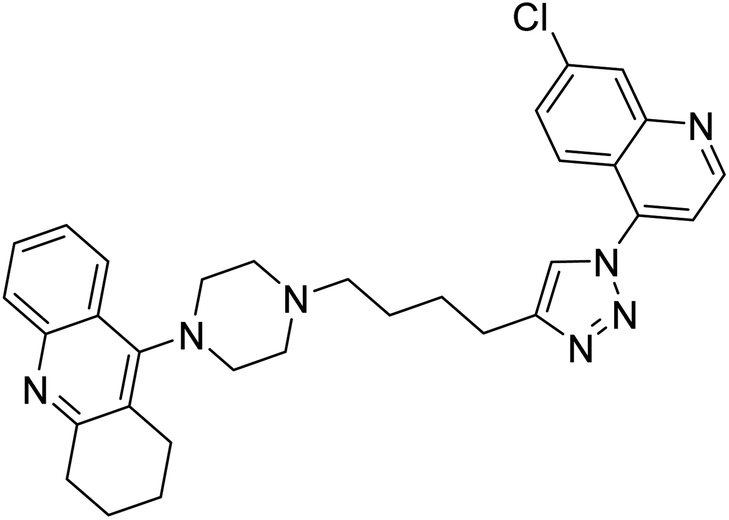 |
10 | 6.06 | 210 |
| 123 | 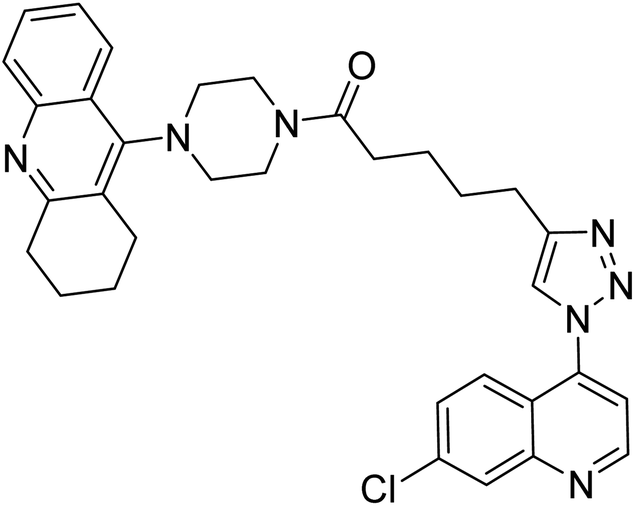 |
11.07 | 61.13 | 210 |
| 124 | 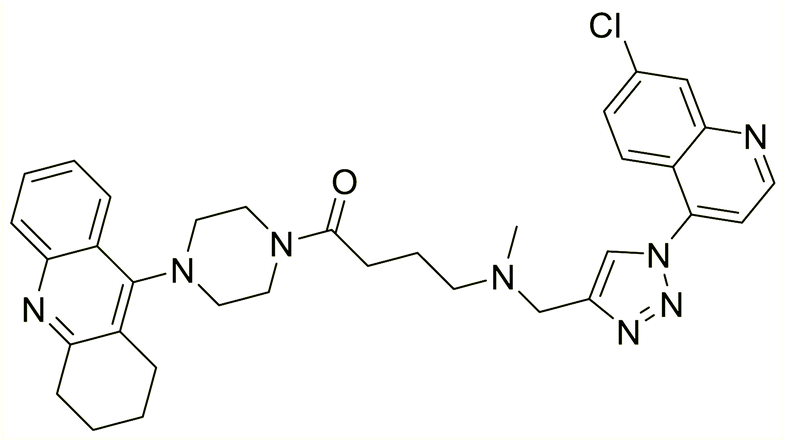 |
19.59 | 66.68 | 210 |
| 125 | 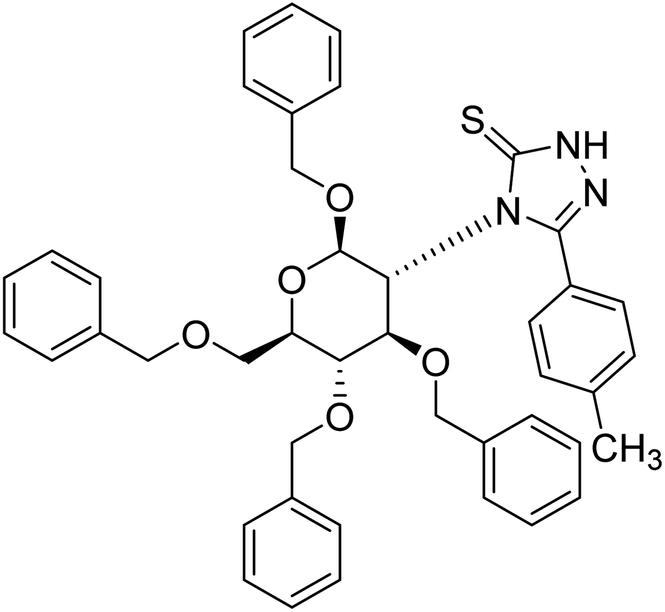 |
1.46 µg mL−1 | — | 211 |
| 126 | 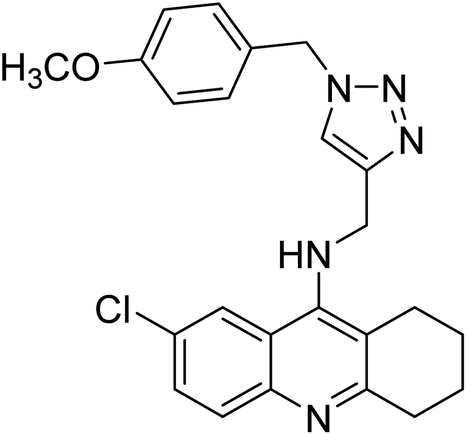 |
0.521 | — | 212 |
| 127 | 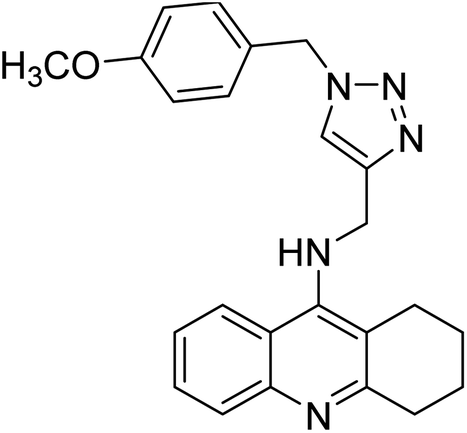 |
0.055 | — | 212 |
| 128 | 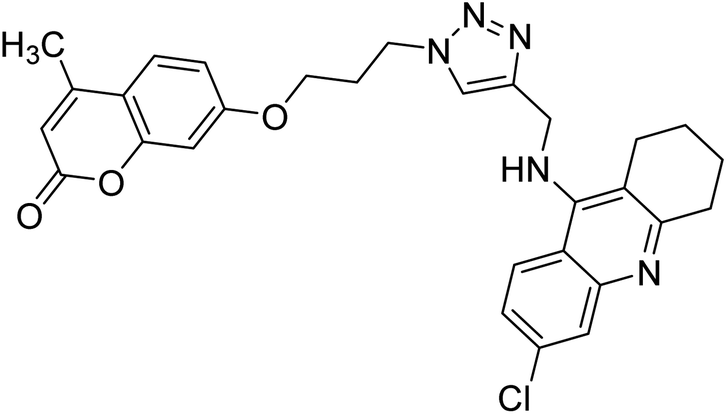 |
27 | — | 213 |
| 129 | 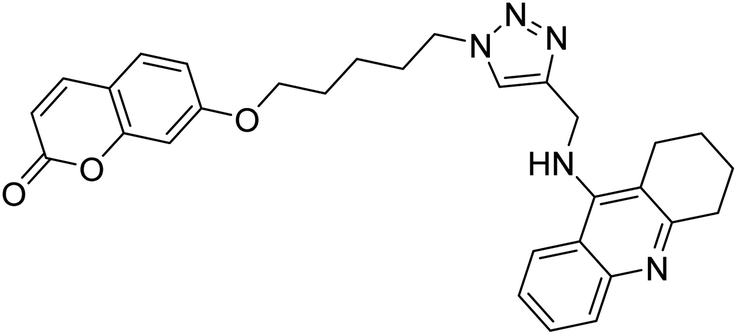 |
6 | — | 213 |
| 130 | 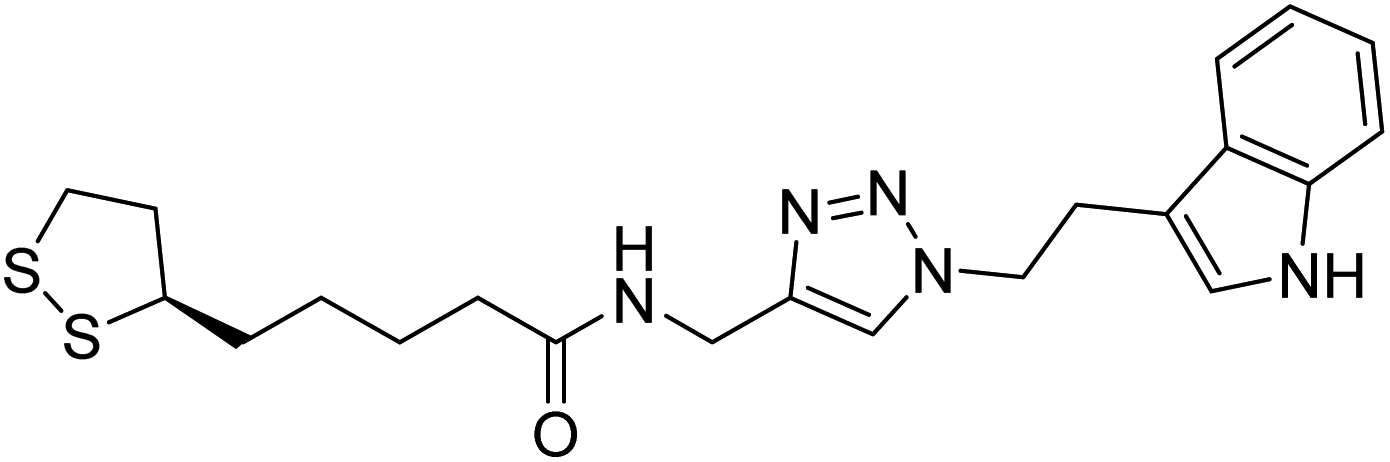 |
— | 0.42 | 214 |
| 131 | 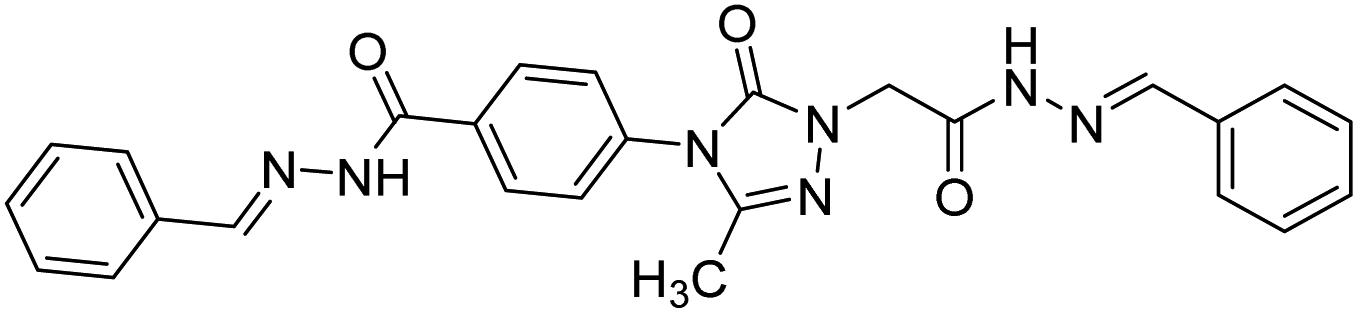 |
0.0876 | — | 215 |
| 132 | 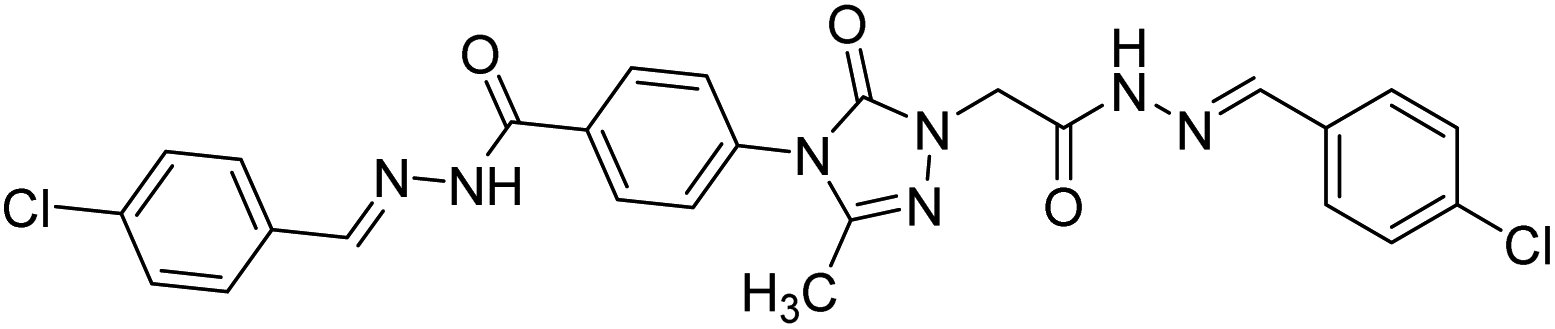 |
0.0574 | — | 215 |
| 133 | 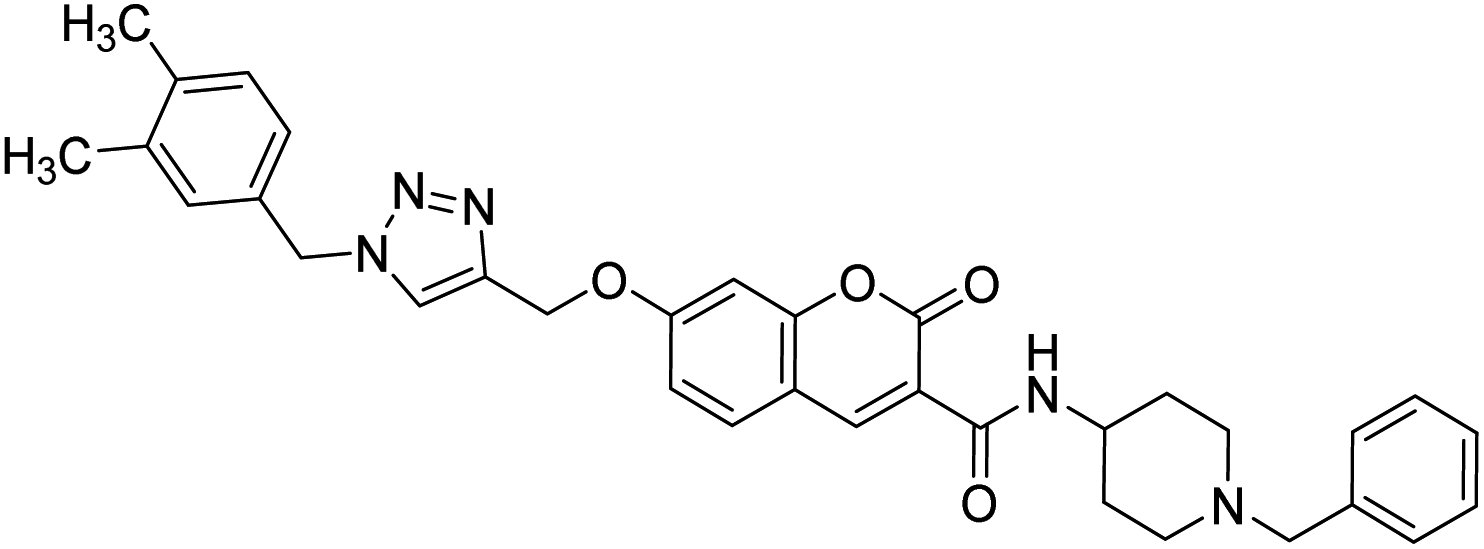 |
— | 1.80 | 216 |
| 134 | 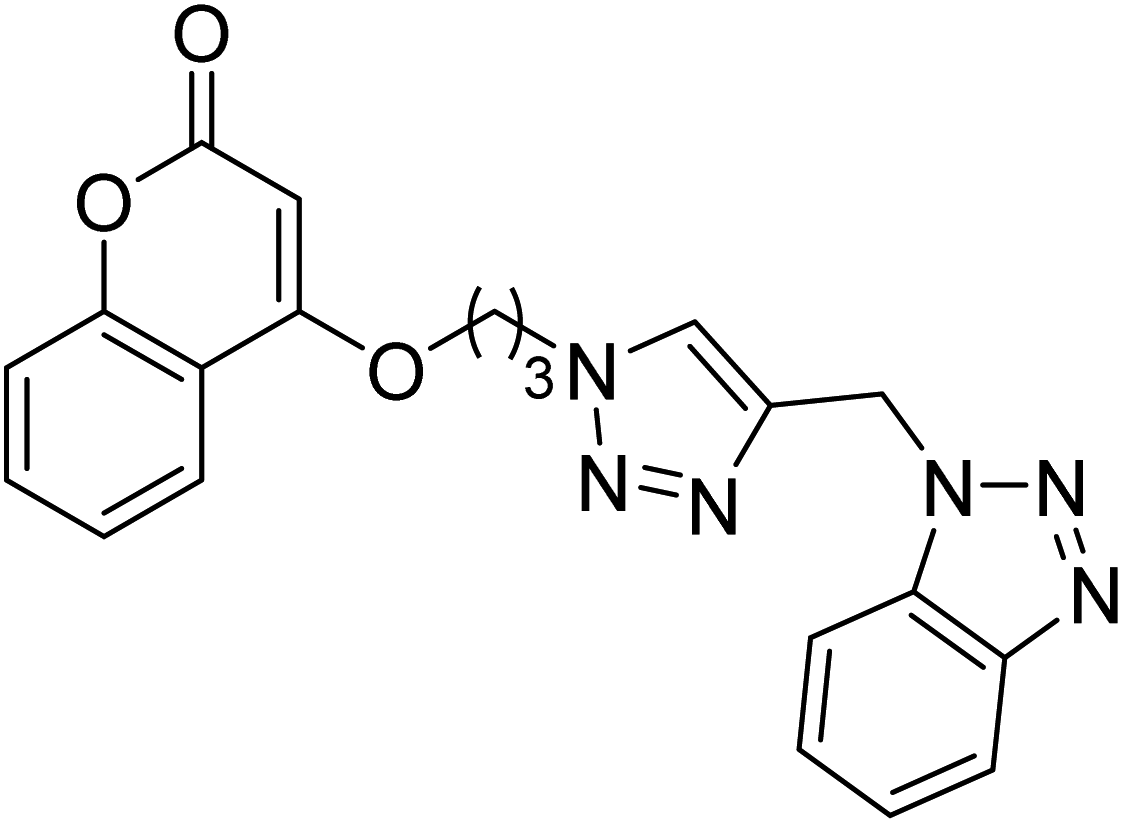 |
0.059 | — | 217 |
| 135 | 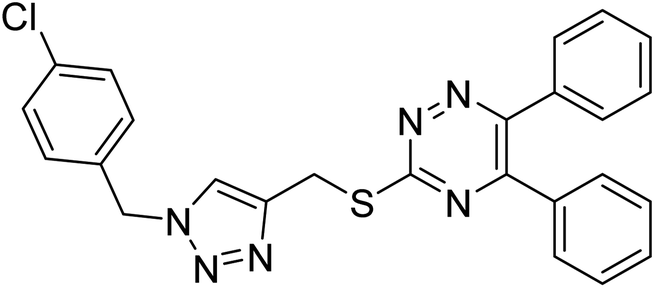 |
6.4 | — | 218 |
| 136 | 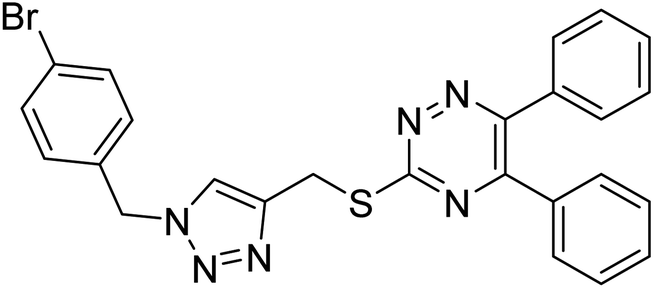 |
7.9 | — | 218 |
| 137 | 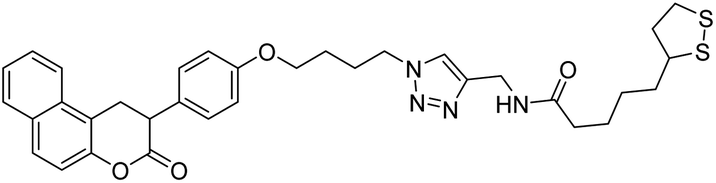 |
7.3 | 68.6 | 219 |
| 138 | 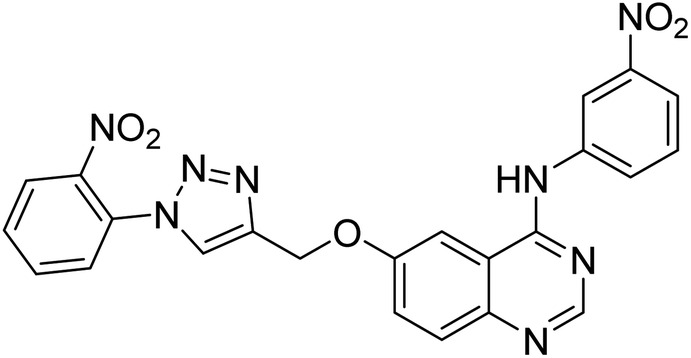 |
2.06 | — | 220 |
| 139 | 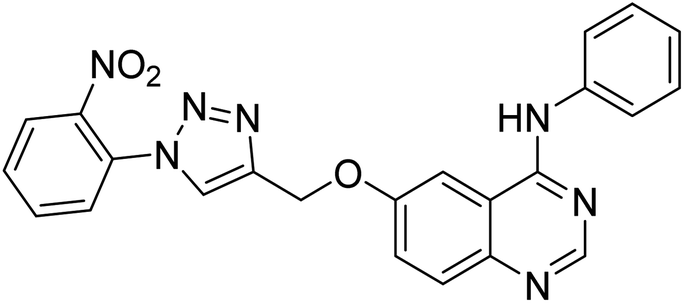 |
0.23 | — | 220 |
| 140 | 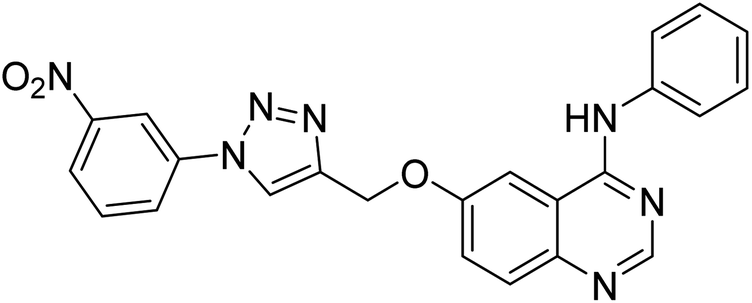 |
1.10 | — | 220 |
| 141 | 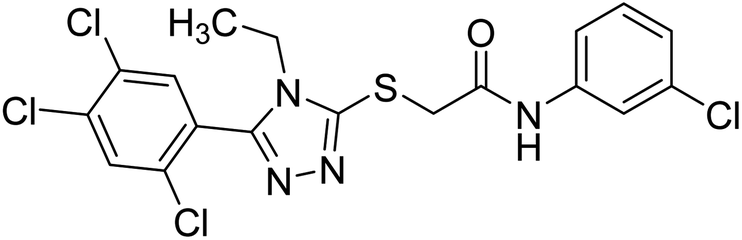 |
5.41 | 7.52 | 221 |
| 142 | 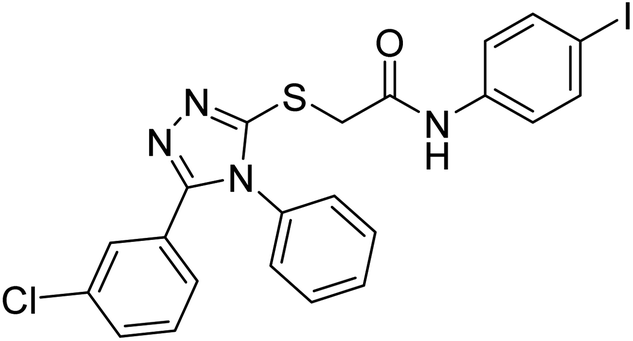 |
13.75 | — | 221 |
| 143 | 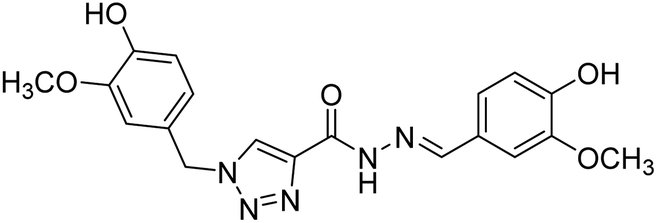 |
26.30 | — | 222 |
| 144 | 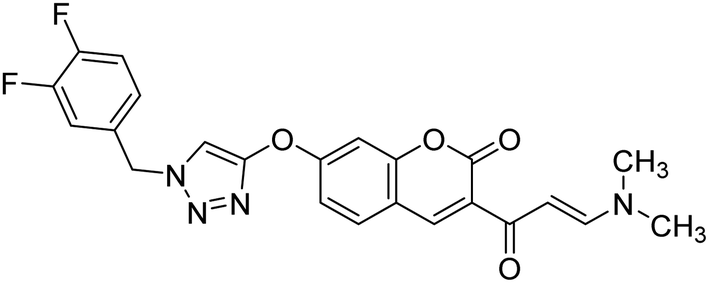 |
21.71 | >100 | 223 |
| 145 |  |
0.259 | — | 224 |
| 146 |  |
0.372 | — | 224 |
| 147 |  |
0.327 | — | 224 |
| 148 |  |
— | 4.78 | 225 |
Munawar et al. (2015) synthesized a variety of escitalopram triazoles and assessed them for their AChE and BChE inhibitory abilities. Most of these revealed moderate activities, and four of these analogs showed potent BChE inhibitory ability (IC50 = 4.52–9.52 µM) in comparison eserine (IC50 = 0.85 µM). The SAR showed that the escitalopram function was essential for the activity; 2-F 116 > 4-F 117 with optimum inhibition by ligands 116 and 117 that scored the lowest IC50 = 4.52 µM and 5.31 µM, respectively. The effect of substituent nature on the inhibition has been noted in the order of 2-F > 4-F > 4-OH-3-OCH3 > 2-I > 3-Cl > 3-F. Consequently, the escitalopram triazoles have shown decent inhibitory activity against BChE and can be utilized as an essential entry point for further analysis of the possible use of these compounds in the process of drug discovery against neurodegenerative diseases207 (Table 5).
Carvalho et al. (2016) reported the synthesis of 1,2,3-triazole-quinoline derivatives for use as selective dual binding site AChE inhibitors. All hybrids showed 0–55.7% growth inhibitory of hAChE at 100 µM level, but not as active as tacrine (92.8% growth inhibitory of hAChE at 100 µM) and donepezil (83.9% growth inhibitory of hAChE at 100 µM). Among these, products 118 (48.1% at 100 µM) and 119 (55.7% at 100 µM) demonstrated good hAChE inhibitory ability with IC50 values ranging between 114 and 104 µM, respectively. In contrast, compounds 118 (0.2% at 100 µM) and 119 (0% at 100 µM) proved inactive for hBChE enzyme208 (Table 5).
Park et al. (2016) synthesized 1,2,3-triazole linked decursinol hybrids 120 and tested them for their inhibitory ability against ChE and BChE for AD. Compound 120 (IC50 = 5.89 µM against BChE) showed more efficient inhibitory ability against BChE than galantamine (IC50 = 9.4 µM). In addition, compound 120 exhibited no inhibitory capacity against AChE (IC50 value > 350 µM). Triazole-linked decursinol derivative 120 can be deemed as a new class inhibitor for BChE and can be employed to be a new drug contender to treat AD209 (Table 5).
Liu et al. (2017) prepared tacrine-1,2,3-triazoles via a Cu(I)-catalyzed alkyne–azide 1,3-dipolar cycloaddition reaction. The compounds were assessed for their inhibition ability against AChE and BChE as prospective drug targets for AD. Among these, compound 121 displayed most potent and optimum inhibition against AChE and BChE with IC50 values of 4.89 µM and 3.61 µM, respectively. Besides, the inhibitory efficacy of 122, 123, and 124 produced IC50 values of 10, 11.07 and 19.59 µM against AChE, respectively. As for anti-BChE activity, IC50 value of 122 was 6.06 µM, followed by 123 (IC50 = 61.13 µM) and 124 (IC50 = 66.68 µM) were obtained. However, all compounds proved weaker inhibitors compared to tacrine. Further SARs and molecular modeling studies may offer invaluable insights to design and optimize better tacrine-triazole analogs with potential therapeutic uses for AD210 (Table 5).
Liu et al. (2017) synthesized new C2-glycosyl triazoles and assessed them as ChE inhibitors. The AChE inhibitory abilities of the derivatives were tested using Ellman's method. Those that displayed over 85% inhibition were consequently evaluated for their IC50. Compound 1 exhibited the best AChE inhibition ability with IC50 of 1.46 µg mL−1 (ref. 211) (Table 5).
Sharifzadeh et al. (2017) designed tacrine-1,2,3-triazole hybrids as dual ChE inhibitors. The majority of the synthesized compounds demonstrated good in vitro inhibitory abilities against both AChE and BChE. Amongst them, compound 126 proved the best potent anti-AChE derivative (IC50 = 0.521 µM) and compound 127 demonstrated the best anti-BChE activity (IC50 = 0.055 µM). Molecular modeling and kinetic investigations indicated that 148 and 149 bind concurrently to the peripheral anionic site (PAS) and catalytic sites (CS) of the AChE and BChE212 (Table 5).
Akbarzadeh et al. (2019) synthesized tacrine-coumarin hybrids linked to 1,2,3-triazole and verified them as potent dual binding ChEIs for the treatment of AD. Amongst them, 128 was the best potent anti-AChE species (IC50 = 27 µM) and 129 exhibited the optimum anti-BChE activity (IC50 = 6 µM) exceeding that of tacrine (IC50 = 0.048 µM for AChE; 0.01 µM for BChE) and donepezil (IC50 = 0.039 µM for AChE; 8.416 µM for BChE) as the reference drugs213 (Table 5).
Park et al. (2019) synthesized tryptamine-triazole hybrid compounds via the click reaction. Their ChE inhibitory ability was assessed. Amongst the synthesized analogs, compound 130 displayed the top potent inhibitory ability (IC50 = 0.42 µM) for horse BChE and 1.96 µM for human BChE. From the molecular modeling investigation, derivative 130 was bound to the catalytic anionic site, anionic subsite, peripheral anionic subsite, acyl-binding pocket, and oxyanion hole of BChE by forming a hairpin or U-shaped structure. The Lineweaver–Burk plot of 130 against BChE suggested a mixed type of inhibition which matches well with the molecular modeling study214 (Table 5).
Ozil et al. (2019) reported 1,2,4-triazole containing Schiff's bases and screened them for AChE and BChE activities. All compounds (IC50 = 0.0465–0.0966 µM for AChE, and IC50 = 0.0486–0.1253 µM for BChE) showed noteworthy potency against the two enzymes compared to neostigmine (IC50 = 0.136 µM for AChE and 0.084 µM for BChE). The SAR study showed that the aryl position substituent of the phenyl has a superior influence on AChE and BChE abilities than other groups, and EWGs as well as EDGs at the aryl position decrease the activity. The fact that only the phenyl group containing derivative 131 displayed the strongest inhibitory capacity may indicate that its overall geometry fosters strongest interactions with the enzyme's active site. A chlorine-containing analog 132 (0.0574 µM) substituted in the p-position appears to have an inhibitory value of 1.23 times less than 131. The results obtained in this study show that compounds can potentially be used to produce strong inhibitors that target AChE and BChE enzymes. These newly synthesized compounds can also be used as drug precursors or building blocks in the preparation of more effective drug molecules215 (Table 5).
Saeedi et al. (2019) synthesized 1,2,3-triazole-chromenone carboxamides and assessed their cholinesterase inhibitory ability. Amongst them, 133 showed the best BChE inhibitory activity (IC50 = 1.80 µM), though, it was not active against BChE. Noteworthy, 133 was appraised for its BACE1 inhibitory ability and the calculated IC50 = 21.13 µM confirmed its inhibitory activity216 (Table 5).
Singh et al. (2020) synthesized triazole tethered coumarin-benzotriazole hybrids based on donepezil framework multifunctional agents for the treatment of AD. Amongst the prepared compounds, 134 displayed the top potent AChE inhibition (IC50 = 0.059 µM) with mixed type inhibition scenario. Therefore, hybrid 134 may act as potential lead for further construction of selective AChE inhibitors as multifunctional anti-Alzheimer's agents217 (Table 5).
Edraki et al. (2020) synthesized a series of 5,6-diphenyl triazine-thio methyl triazole hybrid and assessed their ChE inhibitory activity, demonstrating that most of the derivatives displayed more selectivity against BChE than AChE. Compound 135 was determined as the top potent BChE inhibitor with an IC50 value of 6.4 µM, and 136 showed AChE inhibitory activity with 25.1% inhibition at 50 µM. Additionally, molecular docking investigations indicated that the thiazolidinediones function plays a key part in the inhibition mechanism by well-fitting into the enzyme binding pocket218 (Table 5).
Foroumadi et al. (2020) synthesized 1,2,3-triazole-containing 3-phenylcoumarin-lipoic acid conjugates which showed promising AChE and BChE activity, with IC50 at the µM level. Compound 137 displayed excellent AChE (IC50 = 7.3 µM) and BChE (IC50 = 68.6 µM) activity, indicating that it may act as promising multi-functional agent for additional development219 (Table 5).
Nguyen et al. (2020) synthesized a library of 12 quinazoline-triazole hybrids and tested them as AChE inhibitors to treat AD. The biological assay data confirmed the ability of several hybrid compounds to inhibit the AChE enzyme (IC50 range = 0.2–83.9 µM). To understand the high activity of these compounds, molecular docking simulations were carried out to get better insights into the mechanism of binding of these quinazoline-triazole hybrid compounds. As expected, compounds 138 (IC50 = 2.06 µM), 139 (IC50 = 0.23 µM) and 140 (IC50 = 1.10 µM) bind to both catalytic anionic site (CAS) and peripheral anionic site (PAS) in the active site of the AChE enzyme, suggesting that these compounds could act as dual binding site inhibitors. These compounds were not cytotoxic, and they also displayed appropriate physicochemical as well as pharmacokinetic profiles to be developed as new AD drug contenders220 (Table 5).
Riaz et al. (2020) synthesized two groups of N-aryl derivatives of 2-(4-ethyl-5-(3-chlorophenyl)-4H-1,2,4-triazol-3-ylthio) acetamide and 2-(4-phenyl-5-(3-chlorophenyl)-4H-1,2,4-triazol-3-ylthio)acetamide. All the compounds were assessed for their inhibitory ability against AChE and BChE, where these analogs exhibited moderate to good activities against the investigated enzymes. Compounds 141 and 142 showed strong inhibitory potential (IC50 = 5.41 and 13.57 µM, respectively) against AChE while 141 showed strong inhibitory activity (IC50 = 7.52 µM) against BChE. The remaining compounds displayed good to moderate inhibitory abilities against the enzymes in the range of IC50 14.29–43.94 µM for AChE and IC50 21.59–41.54 µM for BChE221 (Table 5).
Silva et al. (2020) synthesized a series of new triazole N-acylhydrazone hybrids and evaluated them for ChE inhibition. Compound 143 (IC50 = 26.30 µM) showed a potential profile of a multifunctional compound with the ability to inhibit AChE activity, though it was weaker than donepezil (IC50 = 0.026 µM). This compound also showed a good safety profile in the same neuronal model and in silico ADME parameters. Taken together, these results suggest that compound 143 could be considered as a lead compound for the development of further AD therapeutics222 (Table 5).
Saeedi et al. (2020) synthesized a set of novel 1,2,3-triazole-chromenone carboxamide derivatives and assessed them for their ChE inhibitory activity. Most of the prepared products were not active at a concentration of 100 µM, though analog 144 was the top potent AChE inhibitor (IC50 = 21.71 µM). However, it was inactive toward BChE (IC50 ≥ 100 µM). A kinetic study was undertaken to examine the mechanism of inhibition by 144 against BChE, revealing a mixed-type inhibition pattern based on graphical analysis of the reciprocal Lineweaver–Burk plot. Noteworthy, the butyrylcholinesterase inhibitor (BChEI) activity depended strongly on the electronic property of functional group substituents and their locations on the Bz group attached to the 1,2,3-triazole ring. For instance, changing the location of the methyl from the 2- to the 3-position destroyed the AChE inhibitory activity. The presence of EWGs (–Cl, –F and –Br) on the terminal phenyl ring was favorable in the 3-position yet detrimental in the o- and p-positions223 (Table 5).
Kumar et al. (2021) reported the preparation of 1H-1,2,3-triazole tethered tacrine-chalcone conjugates and measured their AChE inhibitory ability. In vitro AChE inhibition assay revealed three compounds, 145 (IC50 = 0.259 µM), 146 (IC50 = 0.372 µM) and 147 (IC50 = 0.327 µM), exceeding the activity of tacrine. The three active compounds 145–147 were further evaluated in vitro against AChE using the Ellman method with tacrine (IC50 = 0.375 µM) as a standard. Only compound 146 attained 50% inhibition at 10 µM concentration against the AChE enzyme. An IC50 value of 5.328 µM was obtained for compound 146, indicating that these hybrid analogs are selective inhibitors of the AChE enzyme224 (Table 5).
Mirfazli et al. (2021) prepared methylindolinone-1,2,3-triazole derivatives and assessed their in vitro ChE inhibitory activity. While most synthesized products exhibited weak AChE inhibitory activity, they showed moderate to good activity against BChE. The IC50 value for the anti-BChE activity of 148 was calculated as 4.78 µM which exceeded that of donepezil (5.19 µM). Based on the molecular docking assessment, compound 148 was found capable of binding at once to the peripheral and catalytic sites of BChE225 (Table 5).
According to SAR analysis, the aforementioned triazole derivatives are potent AChE and BChE inhibitors (Fig. 15). In general, the activity is highly dependent on the electronic property of substituents and their locations on the moieties linked to the 1,2,3-triazole ring. The smaller groups attached to the triazole ring show higher AChE and BChE inhibitory abilities as compared to the bulky groups present on the rings. The electron withdrawing groups (–F, –Cl, –Br, –CN, –OH, etc.) may enhance the activity, depending on their positions, and electron-donating groups (–CH3, –OCH3, Et, n-Bu etc.) may decrease the activity accordingly. All the presented analogues thus far have shown good to excellent ChE inhibitory abilities with a low risk of harmful side effects. Moreover, new modifications may be introduced in the main scaffold to design novel and more potent such types of compounds.
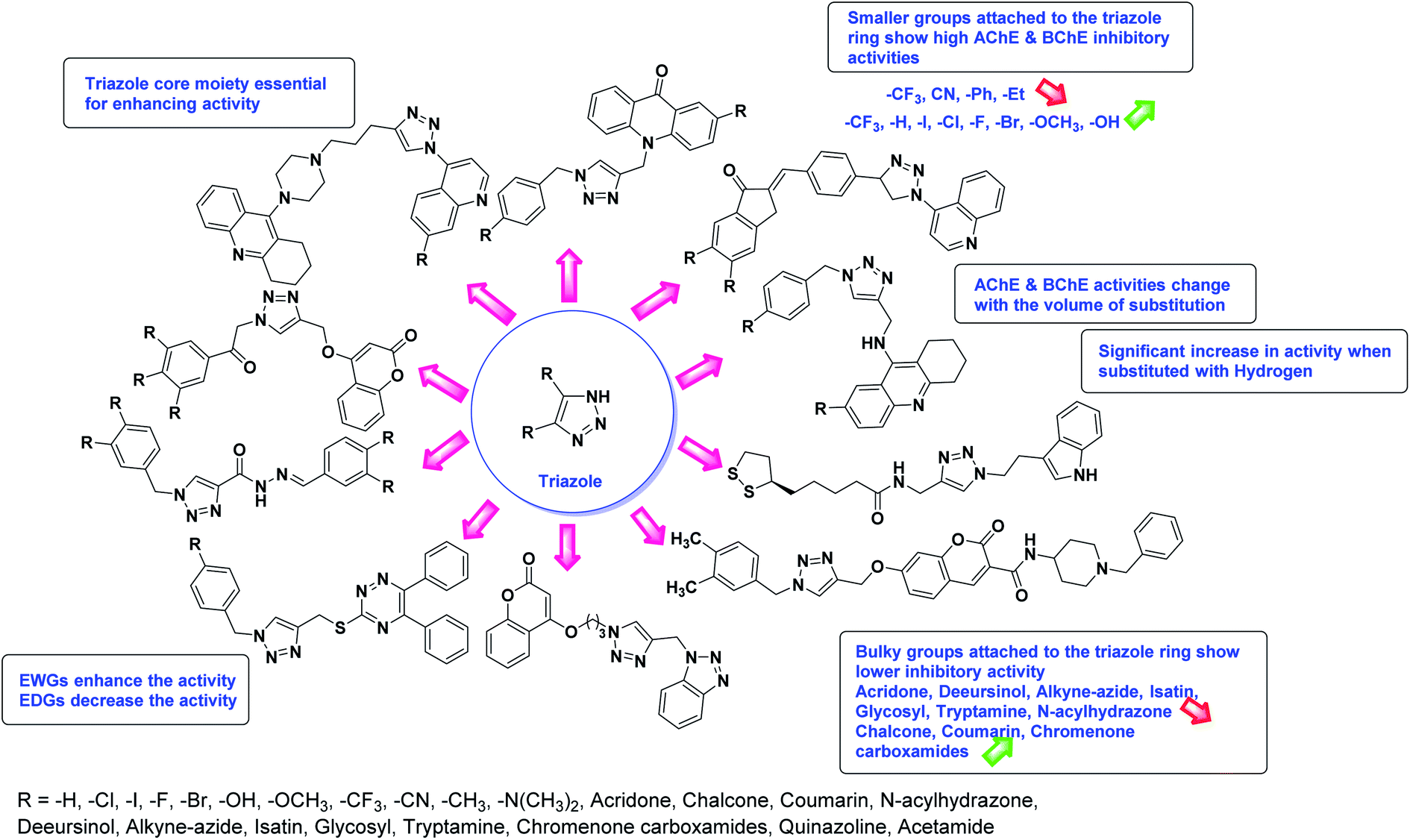 | ||
| Fig. 15 SAR analysis of different triazole derivatives as AChE and BChE inhibitors. | ||
Iqbal et al. (2012) reported a series of 2,5-disubstituted-1,3,4-thiadiazoles analogs and screened them for their AChE inhibition activity. Among the series, compound 149 showed excellent AChE inhibition (IC50 = 0.351 µM) due to the presence of the m-Cl group at the aryl ring226 (Table 6).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 149 | 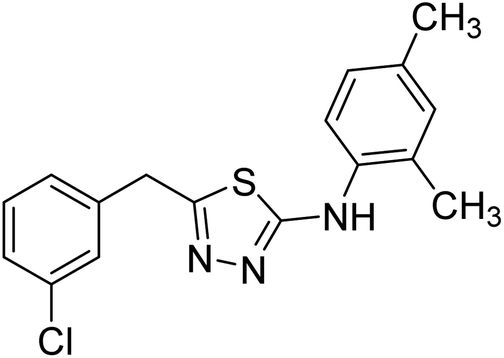 |
0.351 | — | 226 |
| 150 |  |
0.060 | — | 227 |
| 151 |  |
0.053 | — | 227 |
| 152 |  |
0.344 | — | 228 |
| 153 |  |
0.09 | — | 229 |
| 154 | 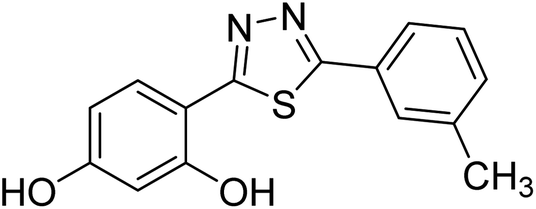 |
0.16 | — | 229 |
| 155 | 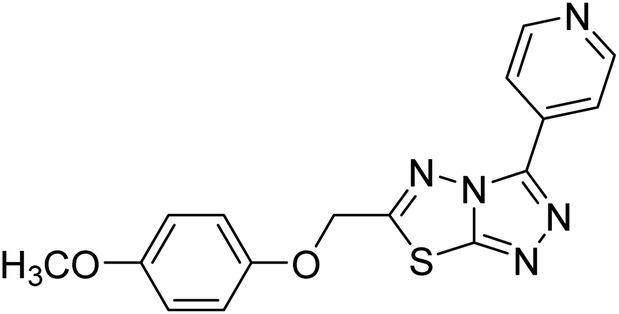 |
0.77 | — | 230 |
| 156 | 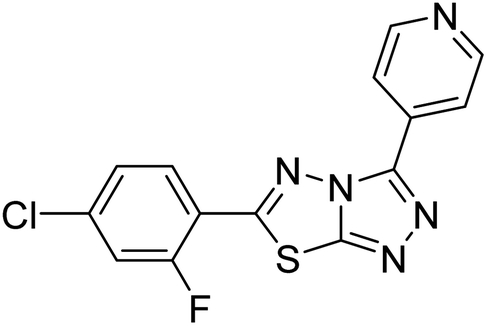 |
9.57 | — | 230 |
| 157 | 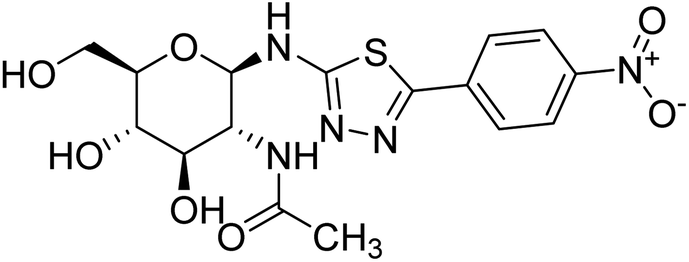 |
18.38 | — | 231 |
| 158 |  |
21.91 | — | 231 |
| 159 |  |
28.86 | — | 231 |
| 160 |  |
2.464 | — | 232 |
| 161 |  |
0.189 | — | 233 |
| 162 | 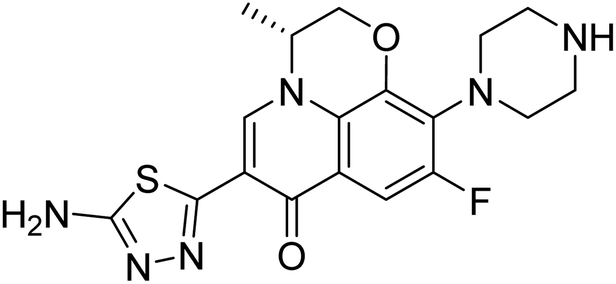 |
18.1 nM | — | 234 |
| 163 | 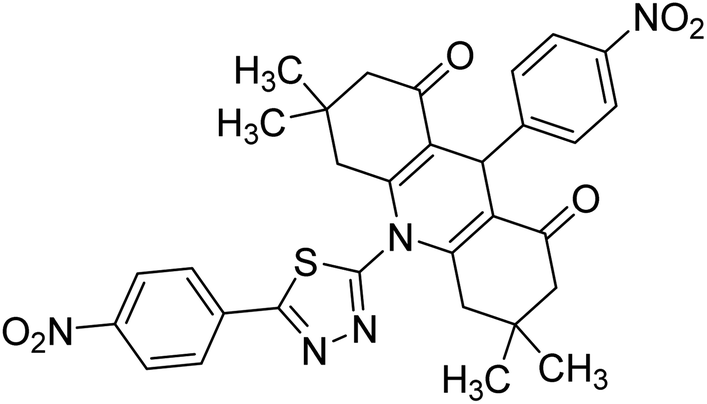 |
0.002 | — | 235 |
| 164 | 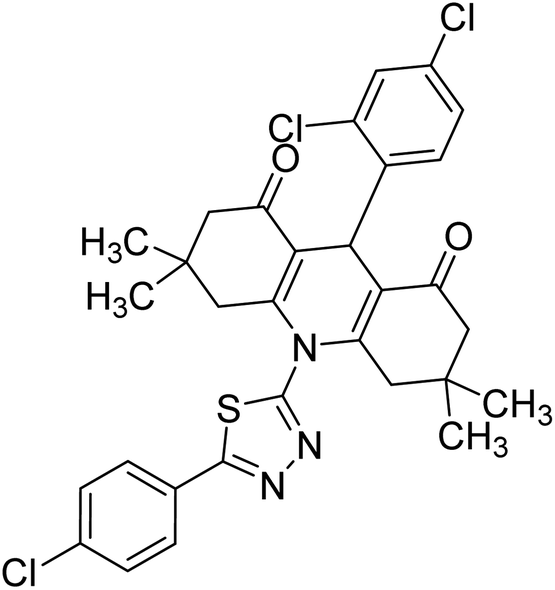 |
0.006 | — | 235 |
| 165 | 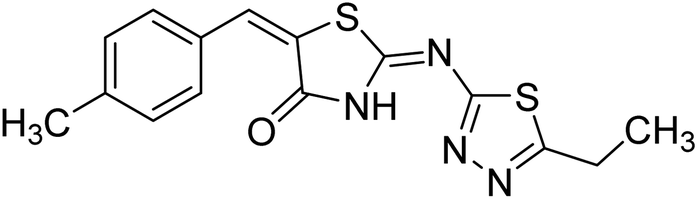 |
1.30 | — | 236 |
| 166 |  |
1.22 | — | 236 |
| 167 |  |
0.029 | 1.731 | 237 |
| 168 |  |
0.074 | 3.028 | 237 |
Matysiak et al. (2012) synthesized a new series of (1,3,4-thiadiazol-2-yl)benzene-1,3-diol based compounds and investigated their potential AChE properties using the modified of Ellman's spectrophotometric method. Most of the compounds acted as AChE and BChE inhibitors in vitro, with IC50 values ranging from >500 to 0.053 µM and from >500 to 0.105 µM, respectively. The most potent compound 151 (IC50 = 0.053 µM) proved to be selective toward AChE, exhibiting selectivity ratios versus BChE of ca. 950. The kinetic studies showed that it is a mixed type of AChE inhibitor. Another compound, 150 (IC50 = 0.060 µM), was active against both enzymes with IC50 values in the low µM range, quite comparable to donepezil (IC50 = 0.02 µM) and neostigmine (IC50 = 0.05 µM)227 (Table 6).
Seo et al. (2012) synthesized different 3,6-disubstituted 1,2,4-triazolo[3,4-b]1,3,4-thiadiazole analogs which were tested for their AChE inhibition activities. Neostigmine methyl sulfate with IC50 value of 69.1 µM and donepezil with IC50 value of 0.021 µM were used as reference drugs. Almost all compounds showed more activity than neostigmine methyl sulfate. The most active compound 152 with IC50 value of 0.344 µM showed comparable activity to that of donepezil. All other compounds showed IC50 values ranging from 1.78 to 78.21 µM (ref. 228) (Table 6).
Matysiak et al. (2013) synthesized a series of new 1,3,4-thiadiazole derivatives and evaluated them as AChE and BChE inhibitors. Some analogs showed promising inhibition of both enzymes in vitro in the µM range. Furthermore, the inhibitory potency of the compounds was stronger against AChE than BChE, where one analog was 1154-fold more active inhibiting AChE (IC50 = 0.17 µM) than BChE. The kinetic studies showed that one of the most active analogs 153 (IC50 = 0.09 µM, AChE) acted as a non-competitive AChE inhibitor and was characterized by a high selectivity index (300). The other derivative 154 (IC50 = 0.16 µM) exhibited a mixed type of AChE inhibition. Docking simulations enabled the detection of key binding interactions of the compounds with AChE and revealed that they occupied mainly the catalytic active site. The scoring function for the novel compounds was similar or higher than that of the reference drug229 (Table 6).
Iqbal et al. (2014) reported the preparation of 3,6-disubstituted-1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazoles. The newly synthesized analogs were assessed for AChE and BChE inhibition. Almost all the compounds showed outstanding activities against AChE even superior to the reference drug. Compound 155 showed IC50 = 0.77 µM against AChE and 156 showed IC50 = 9.57 µM against BChE, rendering these derivatives useful candidates for the therapy of AD230 (Table 6).
Liu et al. (2015) synthesized 4-substituted glycosyl-based thiadiazols and evaluated their AChE activity. The AChE inhibitory abilities of all the analogs were analyzed by Ellman's method. Amongst them, compound 157 displayed the best AChE-inhibition activity with IC50 of 18.38 µM and compounds 158, 159 with 4-nitrophenyl and 2-thienyl substituent exhibited a moderate inhibition of AChE with IC50 = 21.91 µM and 28.86 µM, respectively. Compared with tacrine (IC50 = 0.26 µM), known as the first medication used for the treatment of AD, the above species offered promising inhibition activity against AChE, potentially useful in the treatment of AD231 (Table 6).
Liu et al. (2017) prepared novel glycosyl containing 1,2,4-triazolo[3,4-b][1,3,4]thiadiazole compounds and evaluated their ChE inhibitory activity. The AChE inhibitory activity of the targeted compounds was screened by Ellman's method where the AChE extracts from Electrophorus electricus were used. The results indicated that all compounds had higher IC50 against AChE than the precursor D-glucosamine hydrochloride. The best compound 160 showed high activity with an IC50 of 2.464 µM against AChE232 (Table 6).
Shi et al. (2017) prepared 5-benzyl-1,3,4-thiadiazole derivatives and tested their AChE inhibitory activity. Bioassay results revealed that compound 161 was the best anti-AChE analog and potential multi-target lead candidate against AD233 (Table 6).
Saeed et al. (2019) synthesized a library of new 1,3,4-thiadiazole hybrids and measured their AChE enzyme inhibitory activity. The compounds demonstrated encouraging activities against AChE, particularly 162 (IC50 = 18.1 nM), which was the most promising analog in the library and considerably more active than the reference drug neostigmine methyl sulfate; (IC50 2186.5 nM)234 (Table 6).
Lotfi et al. (2020) synthesized new acridine derivatives containing substituted thiadiazol-2-amine moiety. Anticholinesterase (AChE) activity evaluation of the derivatives showed that all the derivatives are capable of inhibiting both enzymes and are highly selective towards AChE. Among them, the ability of 163 and 164 with respective IC50 values of 0.002 and 0.006 µM to inhibit AChE was higher than tacrine (IC50 = 0.016 µM)235 (Table 6).
Aggarwal et al. (2021) synthesized thiazolidin-4-one analogs with thiadiazole derivatives in appreciable yield. The in vitro AChE inhibitory activity of these compounds was assessed using Ellman's method spectrophotometer and donepezil as a standard drug. Compounds 165 and 166 were found to be potent AChE enzyme inhibitors, with IC50 values of 1.30 and 1.22 µM, respectively. Finally, these significant results could pave the way for the development of new AChE inhibitors236 (Table 6).
Matysiak et al. (2021) synthesized two series of novel 1,3,4-thiadiazole-resorcinol conjugates and evaluated them as ChE inhibitors. N-(Butyl- and N-chlorophenyl-5-amino-1,3,4-thiadiazol-2-yl)benzene-1,3-diols were identified as the most promising compounds of low nanomolar activity against AChE (IC50 = 29–76 nM) and moderate activity against BChE. The inhibition mechanism studies proved that the compounds are mixed-type inhibitors. IC50 values of the N-butyl thiadiazol and N-chloro thiadiazol derivatives are the most potent analogs against AChE and BChE, ranging from 0.029 to 0.085 µM and from 3.154 to 24.711 µM, respectively. The most potent compounds 167 has IC50 = 0.029 µM for AChE, 1.731 µM for BChE, 168 has IC50 = 0.074 µM for AChE, and 3.028 µM for BChE237 (Table 6).
SAR studies of different oxadiazoles demonstrate that the different derivatives are active against AChE and BChE enzymes (Fig. 16). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups attached to the thiadiazole ring show higher AChE and BChE inhibitory abilities as compared to the bulky groups present on the rings. The electron with-drawing groups (–F, –Cl, –NO2, –OH, –CF3 etc.) increase the activity and electron-donating groups (–CH3, –OCH3, etc.) decrease the activity. All the presented analogs thus far have shown good to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
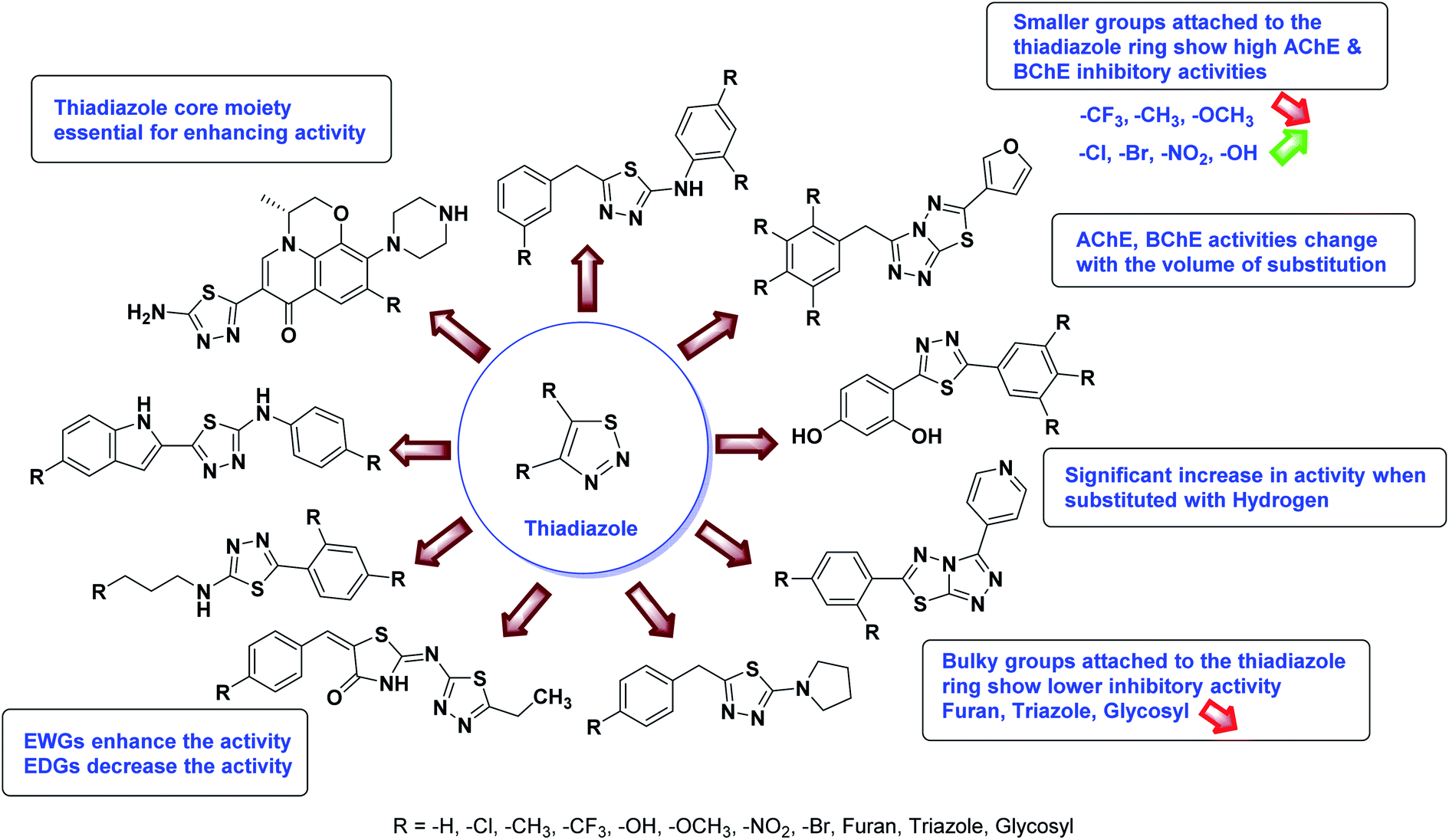 | ||
| Fig. 16 SAR analysis of different thiadiazole derivatives as AChE and BChE inhibitors. | ||
Lee et al. (2010) synthesized pyrrolidine analogs and examined these analogs as AChE inhibitors. Among the compounds, 169 ranked as the top potent inhibitors of the series. Compound 169 demonstrated effective inhibitory activity against the AChE enzyme with IC50 0.10 µmol L−1. Pyrrolidine analogs might be potential AChE agents for AD238 (Table 7).
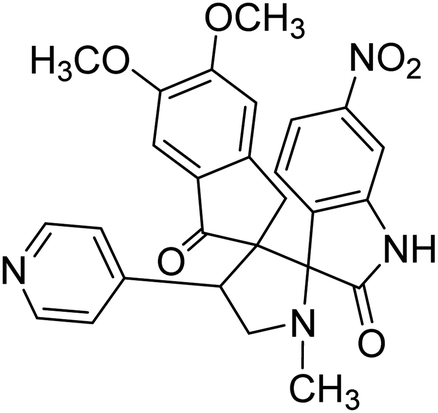
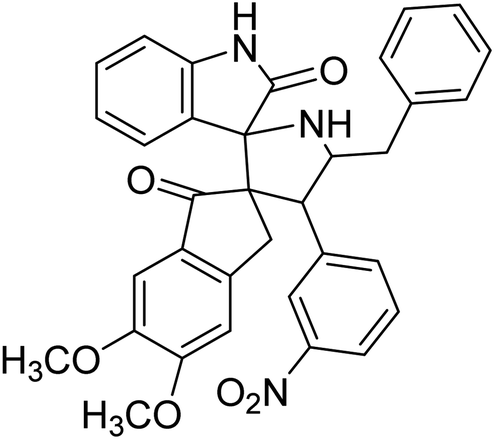
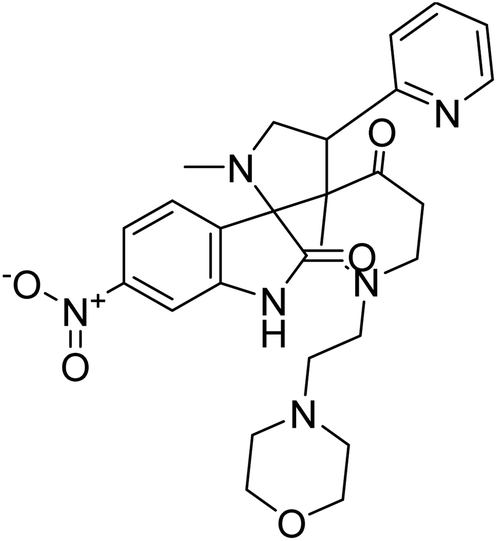
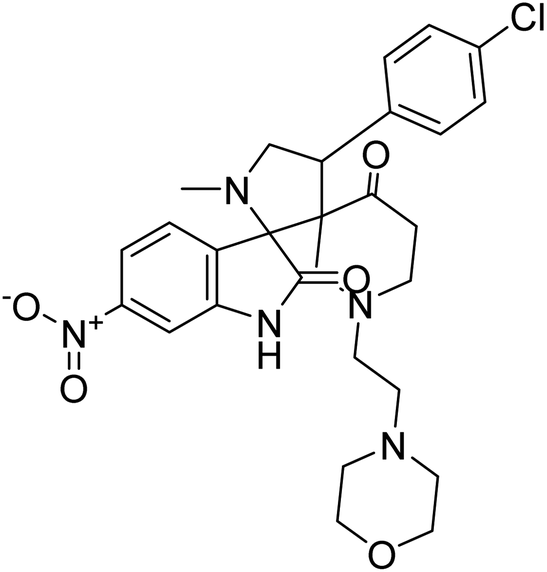
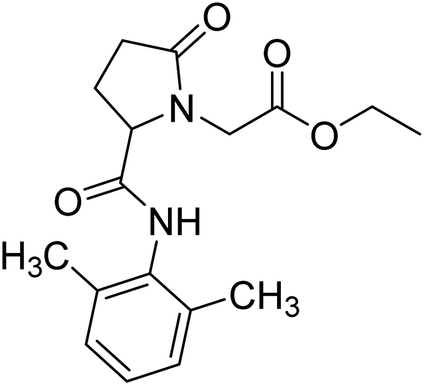
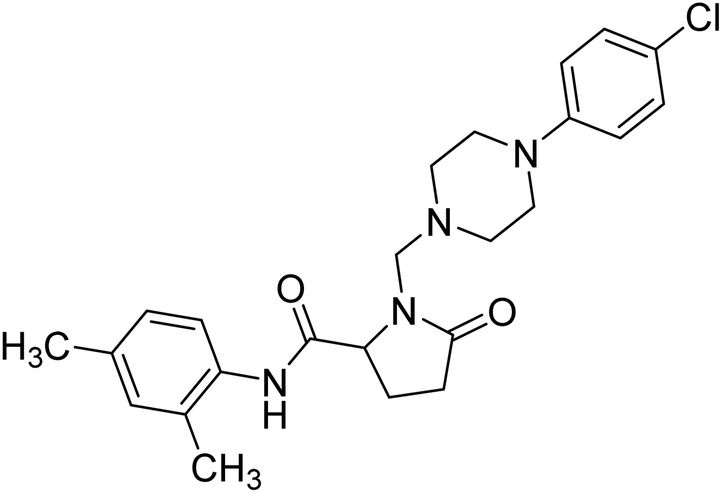
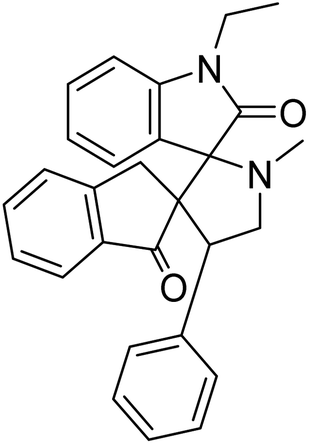
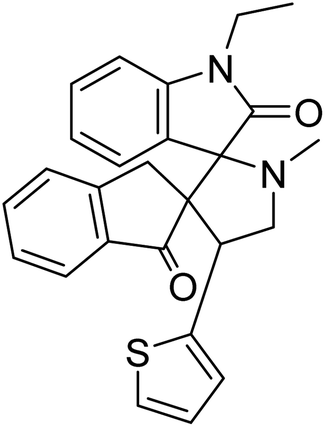
Kumar et al. (2015) synthesized a series of novel dimethoxyindanone embedded spiropyrrolidines in ionic liquid and were evaluated for their inhibitory abilities towards ChEs. Among the spiropyrrolidines, compound 170 exhibited the most potent activity with an IC50 value of 1.57 µM against AChE. Molecular docking simulation for the most active compound was employed to disclose its binding mechanism to the active site of the AChE receptor239 (Table 7).
Murugaiyah et al. (2017) synthesized a library of piperidone grafted spiropyrrolidines and assessed the compounds for their AChE and BChE inhibitory abilities. Within the series, species 171 and 172 was more potent against AChE than the standard drugs with IC50 values of 1.88 and 1.37 µM, respectively. Molecular docking simulations for 172 revealed its appealing binding templates to the active site channel of AChE enzymes. These analogs are astonishing AChE inhibitors and may serve as prospective AD drugs240 (Table 7).
Mohamed et al. (2018) prepared oxopyrrolidines and evaluated their effect on AD by measuring their inhibitory activity against AChE enzyme and amyloid β-42 protein. Most compounds showed decent inhibitory ability where compound 173 garnered the highest activity against AChE with IC50 value 1.84 ng g−1 tissue compared to the standard donepezil 3.34 ng g−1 tissue. Furthermore, compound 174 displayed the greatest activity against β-42 protein with IC50 value of 11.3 Pg g−1 tissue compared to 18.4 Pg g−1 tissue of donepezil241 (Table 7).
Srour et al. (2019) synthesized regioselectively dispiro[indene-2,3′-pyrrolidine-2′,3″-indoline]-1,2″(3H)-diones and explored them as inhibitors of AChE and BChE enzymes; although no substantial inhibitory activity for the tested analogs were detected on AChE, analogs 175, 176, 177, 178 and 179 proved best against BChE with IC50 = 13.7 µM, 21.8 µM, 22.1 µM, 22.9 µM and 24.9 µM respectively, compared to donepezil (IC50 = 0.72 µM). Compound 175 was determined to exhibit a mixed-type mode of inhibition242 (Table 7).
Girgis et al. (2020) synthesized a set of dispiro[indoline-3,2″-pyrrolidine-3′,3″-pyrrolidines] in a regioselective manner using multi-component azomethine cycloaddition reaction of 3-(arylmethylidene)pyrrolidine-2,5-diones, isatins and sarcosine. Compounds 180 (IC50 = 3.35 µM for AChE, 5.63 µM for BChE) and 181 (IC50 = 3.15 µM for AChE, 4.74 µM for BChE) exhibited cholinesterase inhibitory abilities with promising inhibition of both AChE and BChE and were most selective towards AChE than BChE, showing consistent selectivity index trend to that donepezil (IC50 = 0.59 µM for AChE, 0.77 µM for BChE)243 (Table 7).
SAR studies of pyrrolidine derivatives describe above indicate that the various analogous are potent against both AChE and BChE enzymes (Fig. 17). Limited SAR is establish based on the substitution pattern on the pyrrolidine motif and are accountable for influencing bioactivities. The smaller groups (–CH3, –Ph, –NO2, –Cl, –F, –Br, –OCH3, –OH, –H, etc.) attached to the pyrrolidine ring promote higher AChE and BChE inhibitory abilities compared to the bulky (imidazole) groups present on the rings. All the analogs presented thus far have shown good ChE inhibitory abilities with minimum toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
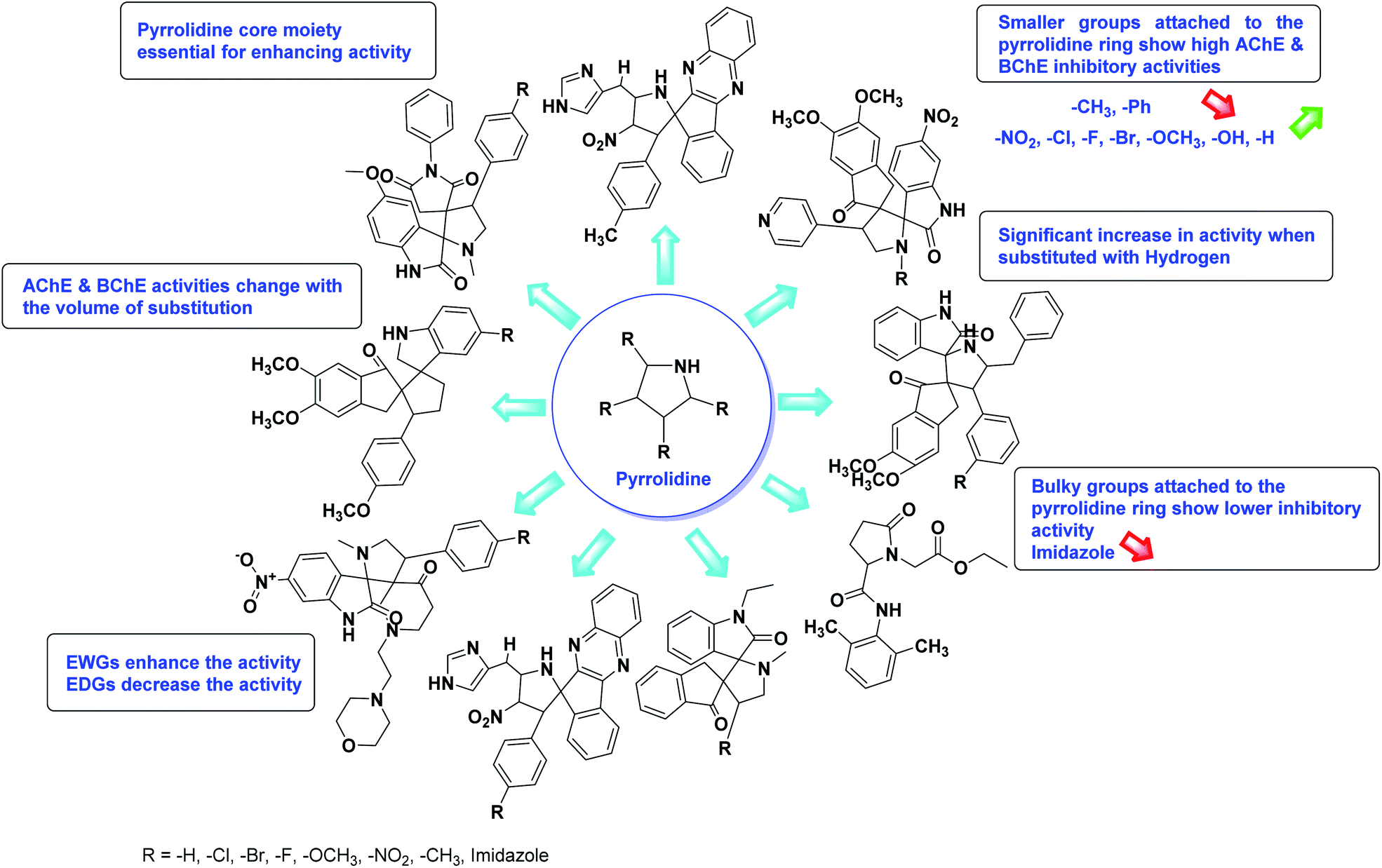 | ||
| Fig. 17 SAR analysis of different pyrrolidine derivatives as AChE and BChE inhibitors. | ||
Rangappa et al. (2009) developed many lead compounds including piperidine derivatives to enhance the efficacy and reduce the general side effects of these AChE inhibitors. Donepezil is a regularly recommended AChE inhibitor having a piperidine ring in its structural composition. The group synthesized cis-2,6-dimethyl piperidine sulfonamides 182–187 in the presence of triethylamine using a nucleophilic substitution procedure between cis-2,6-dimethyl piperidine and alkyl/aryl sulfonyl chlorides and docked the products on the AChE enzyme. These piperidine sulfonamides were used to inverse scopolamine-induced memory loss in rats through in vitro AChE enzyme inhibition trials and in vivo antiamnesic studies. The SAR of the synthesized piperidine derivatives 182–187 based on in vitro findings indicated that adding a methyl group to sulfonyl-cis-2,6-dimethyl piperidine 182 inhibits AChE moderately, whereas adding an electronegative chlorine atom to positions 2 and 5 inhibits AChE. Inhibition was also suppressed by the electronegative NO2 function at the m-position of the phenyl ring of 183 and the nitro group at the ortho position 184 of the phenyl ring (IC50 = 186, 192, 200 and 195, 185, 180 nM, respectively). The nitro group at the p-position of the phenyl ring 185 is detrimental to activity (IC50 = 120, 1150, and 1210 nM). Also, having a chlorine atom at the p-position of the phenyl group 186 reduces inhibitory activity (IC50 = 325, 318, and 312 nM) when compared to ortho, meta substitution of chlorine atoms as in 187. Alkyl substitution at the para position (methyl and tert-butyl) inhibits AChE (IC50 = 362, 368, 365, 463, 458, and 450 nM, respectively)244 (Table 8).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 182 |  |
186 nM | — | 244 |
| 183 |  |
192 nM | — | 244 |
| 184 | 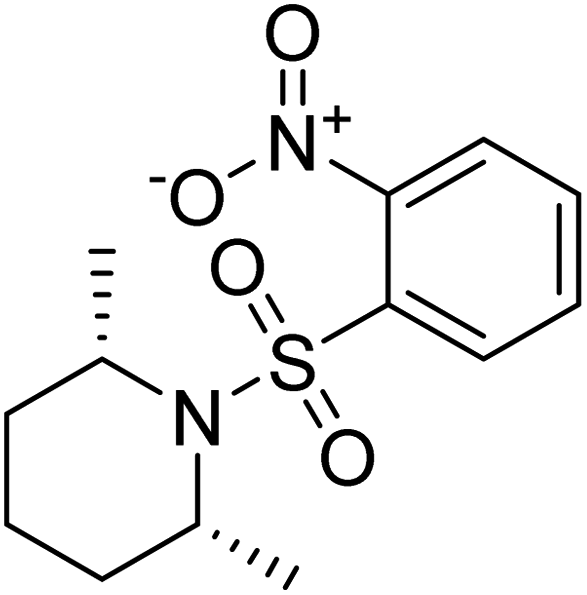 |
200 nM | — | 244 |
| 185 | 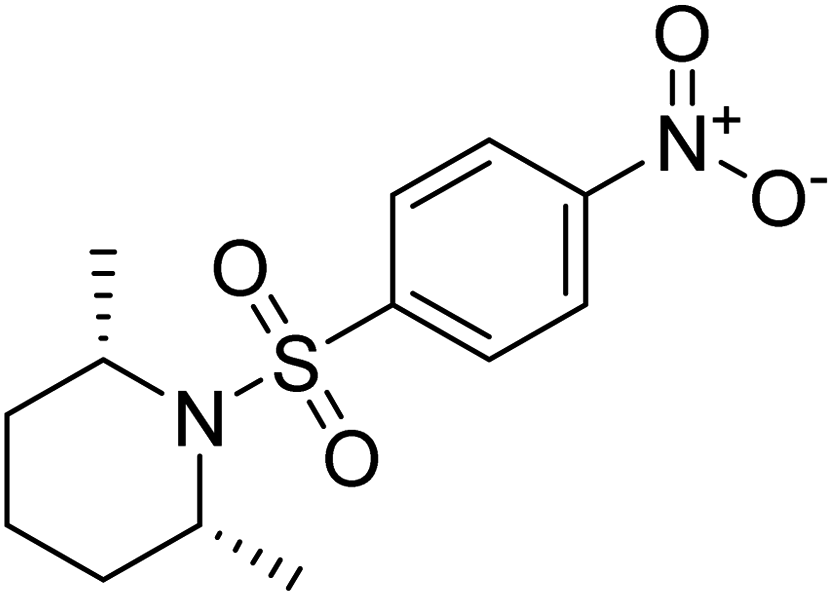 |
120 nM | — | 244 |
| 186 | 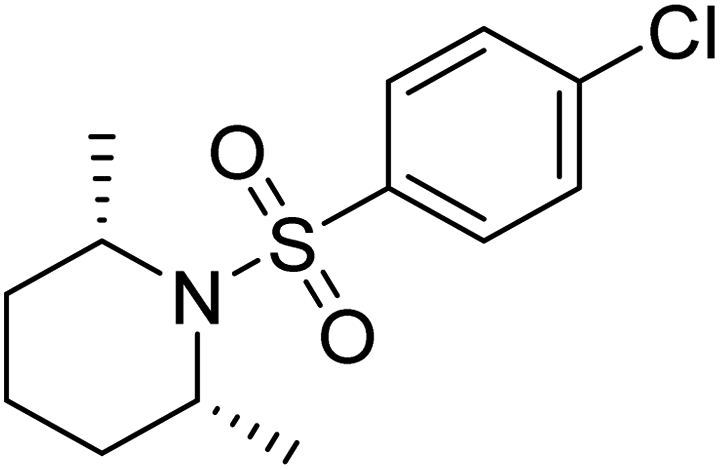 |
325 nM | — | 244 |
| 187 | 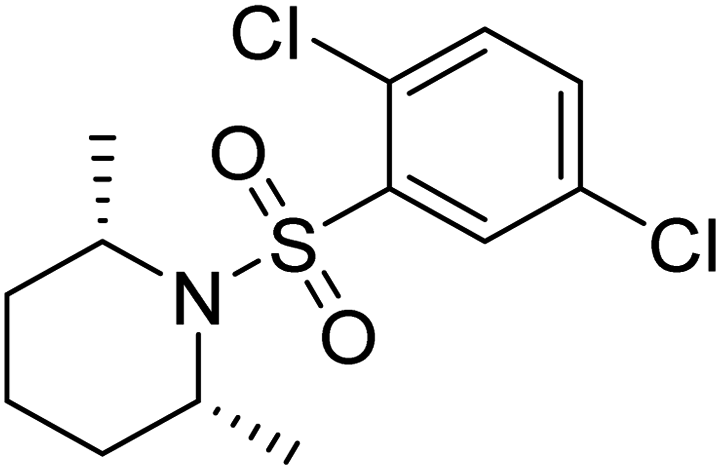 |
362 nM | — | 244 |
| 188 | 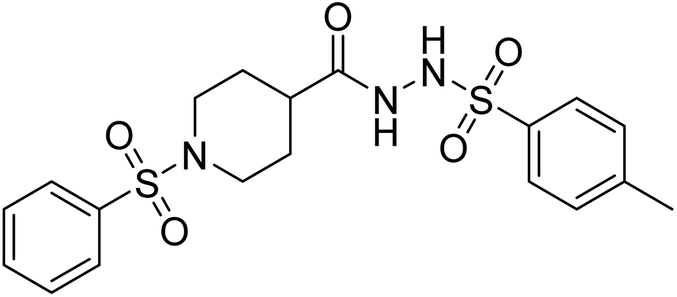 |
157 | — | 245 |
| 189 | 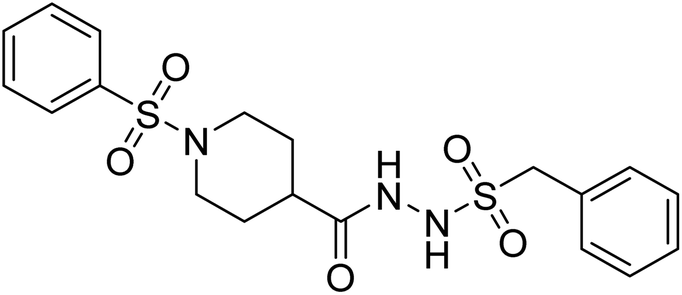 |
219 | — | 245 |
| 190 | 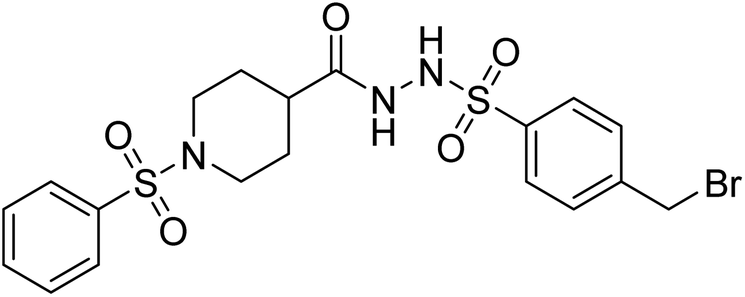 |
145 | — | 245 |
| 191 | 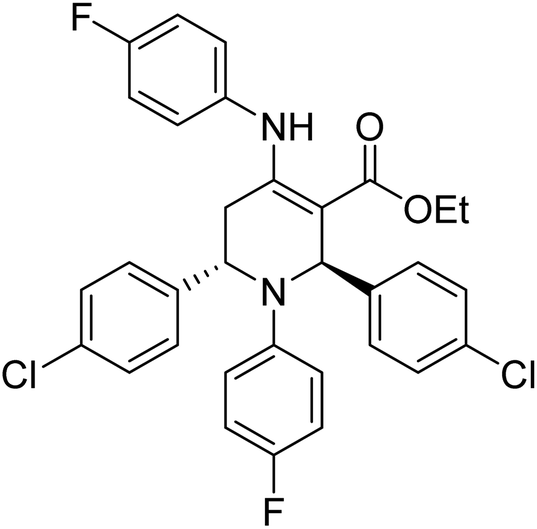 |
0.71 | — | 246 |
| 192 | 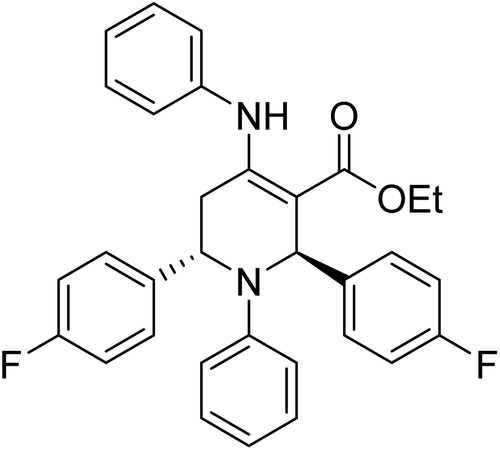 |
0.26 | — | 246 |
| 193 | 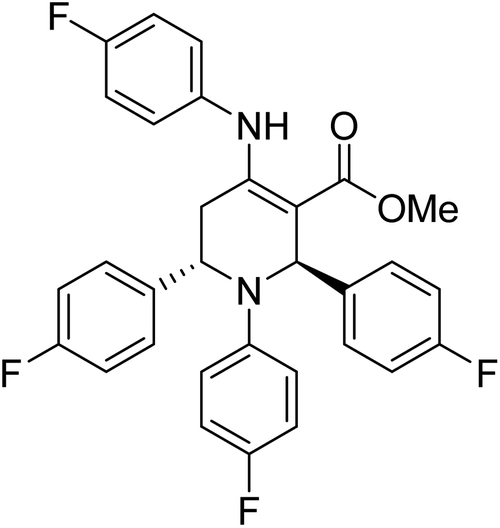 |
0.88 | — | 246 |
| 194 | 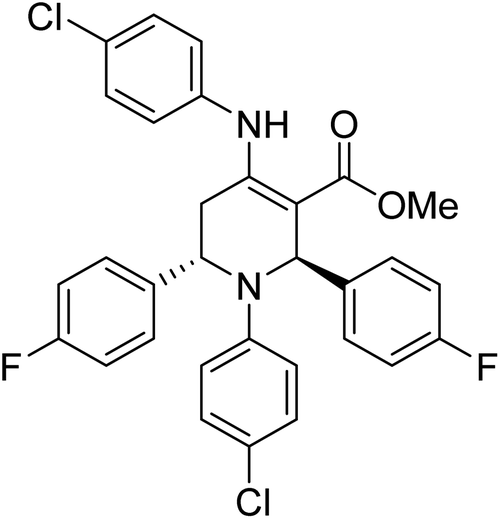 |
0.05 | — | 246 |
| 195 | 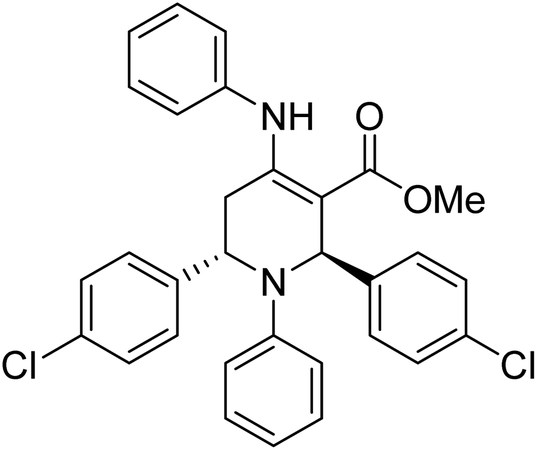 |
0.02 | — | 246 |
| 196 | 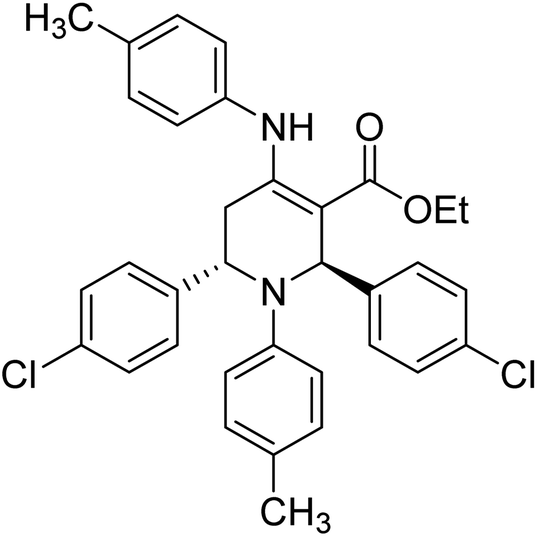 |
0.13 | — | 246 |
| 197 | 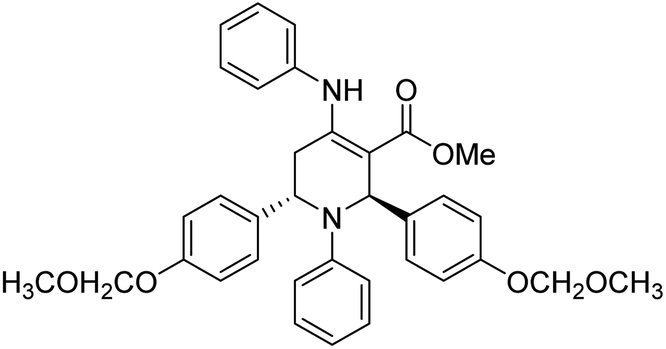 |
0.17 | — | 246 |
| 198 | 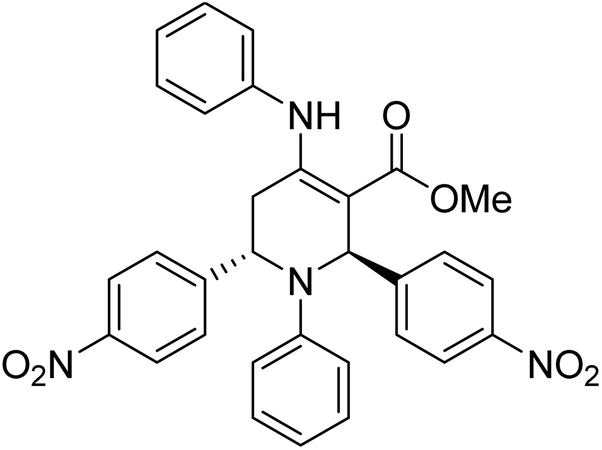 |
0.01 | — | 246 |
| 199 | 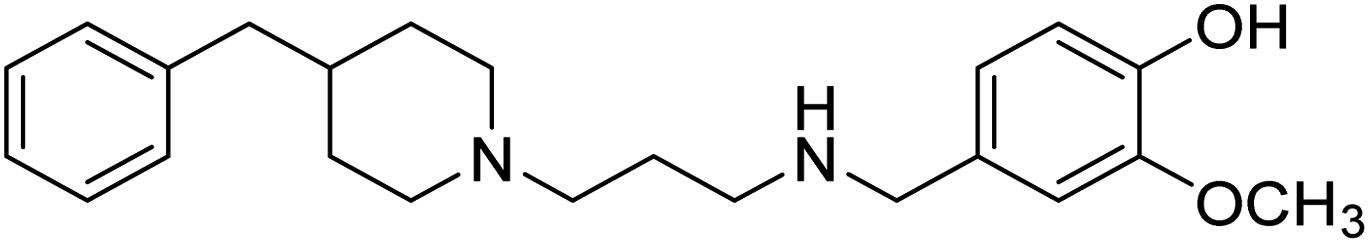 |
6.83 nM | — | 247 |
| 200 | 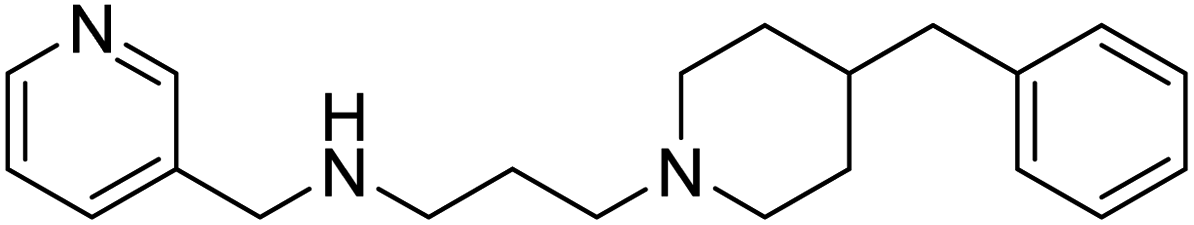 |
2.13 nM | — | 247 |
| 201 | 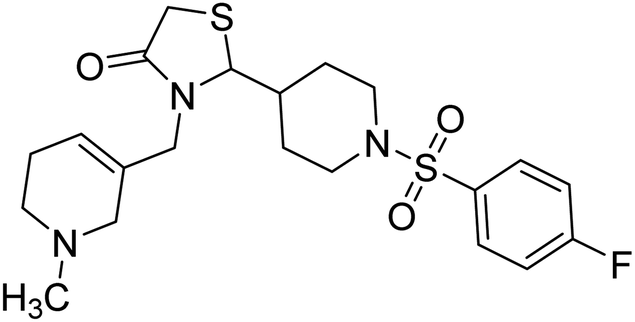 |
6.62 | 13.78 | 248 |
| 202 | 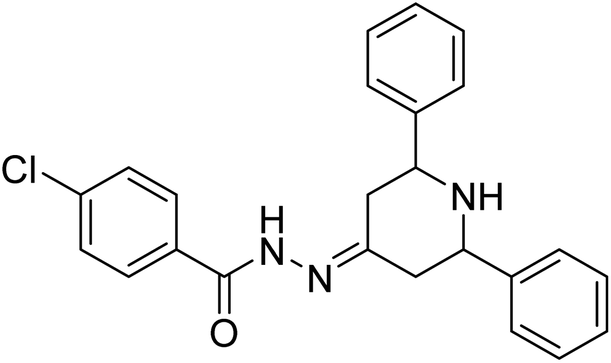 |
— | 35.30 | 249 |
Rehman et al. (2014) synthesized N′-[(alkyl/aryl)sulfonyl]-1-(phenylsulfonyl)piperidine-4-carbohydrazide analogs using ethyl piperidine-4-carboxylate to generate 2-(phenylsulfonyl)piperidine-4-carbohydrazide 188 N′-[(alkyl/aryl)sulfonyl]piperidine-4-carbohydrazide 189, and 4-(bromomethyl)-N′-(1-(phenylsulfonyl)piperidine-4-carbonyl)benzenesulfonohydrazide 190. The structures were confirmed using IR, 1H-NMR, and EI-MS spectra, and they were all examined for their capacity to inhibit the enzymes AChE and BChE. The interactions of these chemicals with the human proteins AChE and BChE were studied using molecular docking. The binding mechanisms of the inhibitors under investigation were identified and compared to anti-enzymatic IC50 values using an automated docking program (AutoDock). Both studies determined that the compounds are potentially effective AChE and BChE inhibitors245 (Table 8).
Brahmachari et al. (2015) used a diastereoselective one-pot multicomponent approach to synthesize a range of densely functionalized piperidine scaffolds under eco-friendly conditions. The piperidines were tested in vitro for inhibitory activity against AChE, and in silico studies were carried out for all analogs using molecular docking, pharmacophore mapping, and QSAR investigation to better appreciate the structural topographies mandatory for interaction with the AChE enzyme and the main active site residues implicated in intermolecular interactions. Nitration, halogenation, or 3,4-methylenedioxy-substitution at the benzene ring linked to the 2- and 6-carbons of the 1,2,5,6-tetrahydropyridine nucleus significantly improved the AChE inhibitory effect of compounds 191–198. The IC50 values of the reported analogs 191 (IC50 = 0.71 µM), 192 (IC50 = 0.26 µM), 193 (IC50 = 0.88 µM), 194 (IC50 = 0.05 µM), 195 (IC50 = 0.02 µM), 196 (IC50 = 0.13 µM), 197 (IC50 = 0.17 µM) and 198 (IC50 = 0.01 µM) showed that these derivatives are more potent than the standard galantamine (IC50 = 0.09 µM). According to docking studies, the inhibitors fit neatly in the active sites. Researchers will be able to better grasp how to alter scaffolds for improved therapeutic effectiveness against AD based on in silico tests246 (Table 8).
According to Tiwari et al. (2015), the creation of multi-target directed ligands (MTDLs) has emerged as a potential strategy for addressing the complex etiology of AD. Using this approach, a novel set of N′-(4-benzylpiperidin-/piperazin-/benzhydrylpiperazin-1-yl)alkylamine derivatives were designed, synthesized, and physiologically assessed as inhibitors of cholinesterase (ChEs), amyloid-beta (Aβ) self-aggregation, and radical scavenging activity. According to in vitro experiments, the majority of the compounds produced inhibited AChE and BChE with IC50 values in the low nanomolar range and were more powerful than the reference drug donepezil in this regard. With IC50 values of 6.83 nM and 2.13 nM, respectively, inhibitors 199 and 200 significantly suppressed AChE, while compound 200 was shown to be exceptionally selective for AChE (38-fold). Furthermore, both the kinetic analysis of AChE inhibition and the docking study revealed that 200 binds to both the catalytic active site and the peripheral anionic site of AChE. Additionally, at 25 µM, these compounds reduced self-induced Aβ1–42 aggregation with percentage inhibition ranging from 54 to 89%, with compound 200 offering the highest inhibition (88.81%). The ORAC of the compounds with methoxy and hydroxy groups ranged from 2.2 to 4.4 times that of Trolox. Furthermore, ADMET tests demonstrated that all compounds have sufficient drug-like properties. These findings suggest that 200 could be a promising lead chemical for the development of future Alzheimer's therapies or their AChE inhibiting activity247 (Table 8).
Kumar et al. (2015) disseminated a study aiming to discover, manufacture, and test novel AChE/BChE inhibitors. A series of 4-thiazolidinone and piperidine substituted arecoline compounds with inhibitory action against AChE and BChE was revealed, and the chemical structures of all compounds were validated using IR, 1H NMR, 13C NMR and mass spectroscopy. Only a few of the compounds generated demonstrated substantial activity for AChE over BChE at micromolar doses according to the cholinesterase inhibition studies. Compound 201 inhibits AChE the most, with IC50 values of 6.62 µM for AChE and 13.78 µM for BChE, which are similar to the normal neostigmine's IC50 values of 2.05 µM for AChE and 3.64 µM for BChE248 (Table 8).
Emre et al. (2016) presented hydrazones and piperidines as suitable substrates for drug development. The objective of their study was to prepare benzoyl hydrazones from 2,6-diphenylpiperidin-4-one and ethyl 4-oxopiperidine-1-carboxylate. The antioxidant, anticholinesterase, and anticancer activities of the synthesized compounds were investigated. Anticholinesterase activity was measured using the enzyme BChE. Compound 202 (IC50 = 35.30 µM) inhibited BChE more efficiently than galantamine (IC50 = 46.03 µM), pointing to 202 as a more suitable BChE inhibitor. The docking method was also used to determine the BChE inhibitory mechanism of analog 202. A molecular docking investigation revealed that analog 202 connected to the BChE enzyme more efficiently than AChE owing to its orientations and various sorts of interactions with the enzyme249 (Table 8).
SAR studies of piperidine explore that the various derivatives are potent against AChE and BChE enzymes (Fig. 18). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–F, –Cl, –NO2, –OH, Br, –I, –CH3) attached to the piperidine ring show higher AChE and BChE inhibitory abilities as compared to the bulky (sulfonamide) groups present on the rings. The electron withdrawing groups (–F, –Cl, –NO2, –OH, –CF3 etc.) increase the activity and electron-donating groups (–CH3, –OCH3, etc.) decrease the activity. All the presented analogs thus far have shown good to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these analogs are less toxic and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
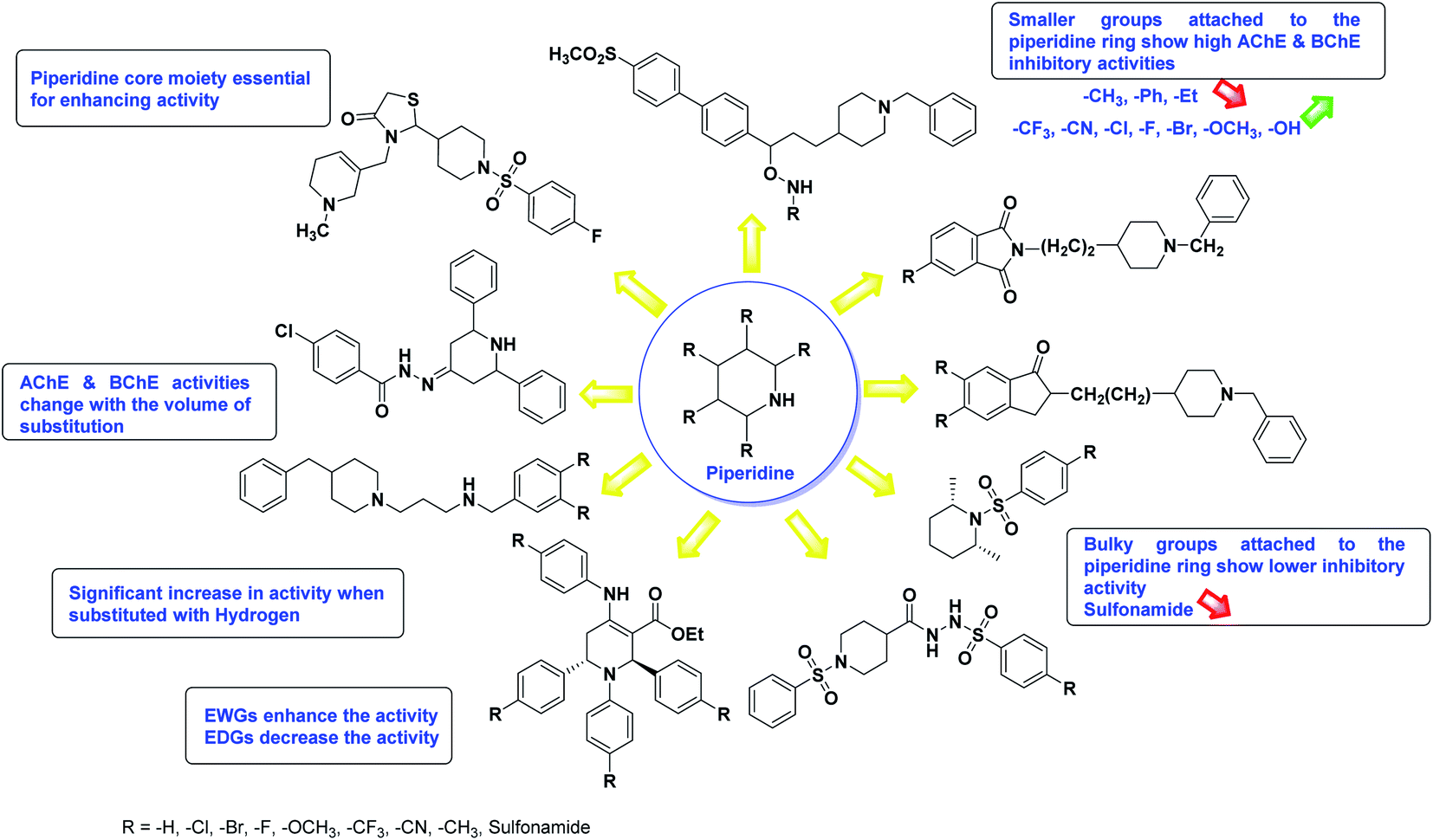 | ||
| Fig. 18 SAR analysis of different piperidine derivatives as AChE and BChE inhibitors. | ||
Kaplanckl et al. (2013) described a process for making novel 2-(4-substituted piperazine-1-yl)-N-[4-(2-methylthiazol-4-yl)phenyl]acetamide derivatives which were subsequently tested for anticholinesterase activity on AChE and BChE enzymes using Ellman's approach. The structures of all compounds were deduced based on IR, 1H-NMR, 13C-NMR, and MS spectral data, as well as elemental analysis measurements. The most powerful compounds against AChE at 0.1 µM concentration were 203 which showed 89.70% inhibition rates, respectively, in the described assay. Furthermore, the IC50 value of compound 203 was revealed to be 0.011 µM, whereas the IC50 value of the reference drug donepezil was 0.054 µM. The IC50 values of compounds, on the other hand, are in the range of 6.34–8.42 µM. The molecules which were comparable to donepezil, also have AChE inhibitory action. The chemical 203 has the same inhibitory efficacy as the reference drug because it contains a 2-pyridyl moiety in the fourth position of the piperazine ring. Compound 203, which included a 4-benzylpiperazine fragment, also demonstrated a five-fold lower IC50 (0.11 µM) than donepezil. There is no evident inhibitory effect of the compounds generated on BChE250 (Table 9).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 203 | 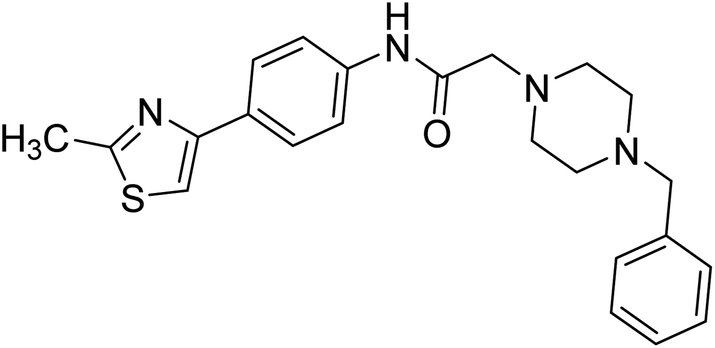 |
0.11 | — | 250 |
| 204 | 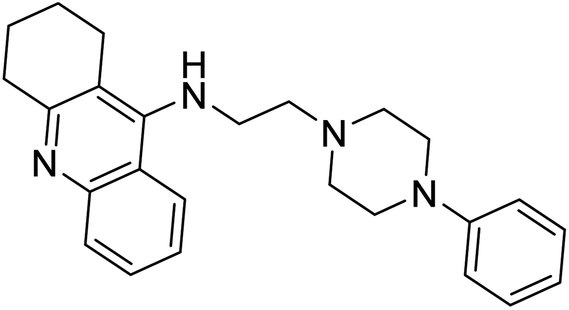 |
2 nM | — | 251 |
| 205 | 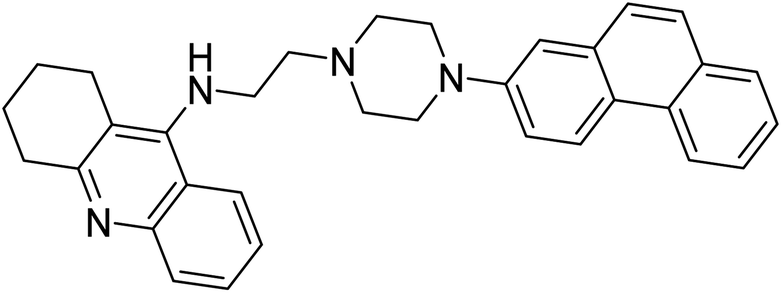 |
260 nM | — | 251 |
| 206 | 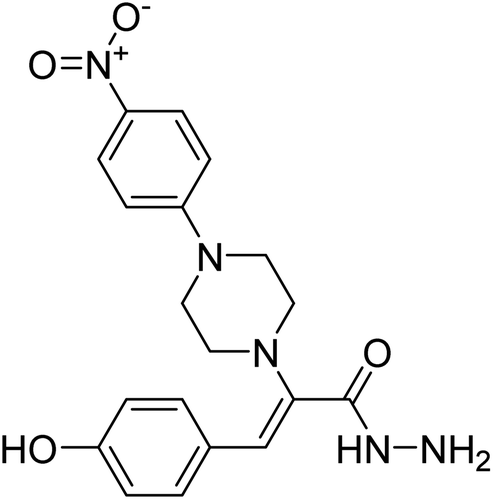 |
29.5 | — | 252 |
| 207 |  |
0.092 | — | 253 |
| 208 |  |
51.66 | — | 254 |
| 209 |  |
79.12 | — | 254 |
| 210 |  |
0.6 | — | 255 |
| 211 | 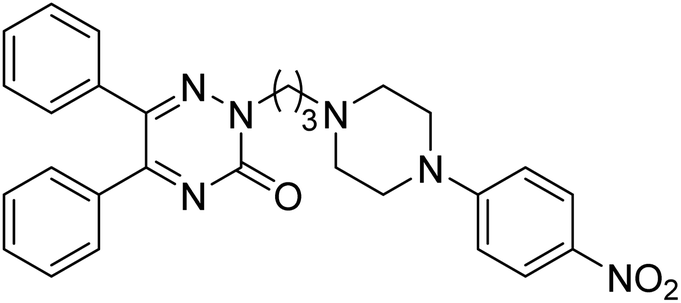 |
0.5 | — | 255 |
| 212 | 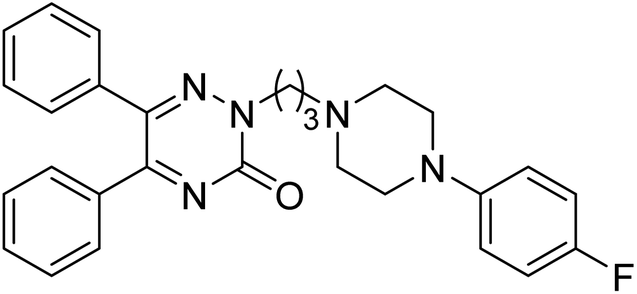 |
0.4 | — | 255 |
| 213 | 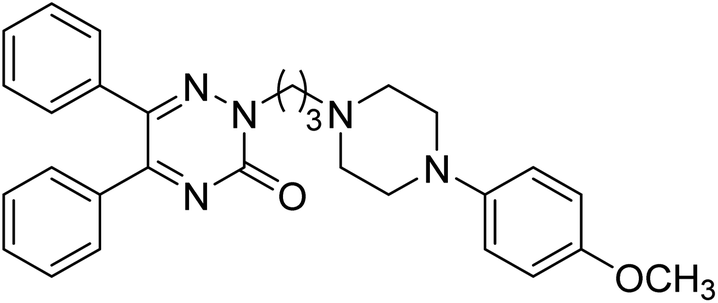 |
0.3 | — | 255 |
| 214 | 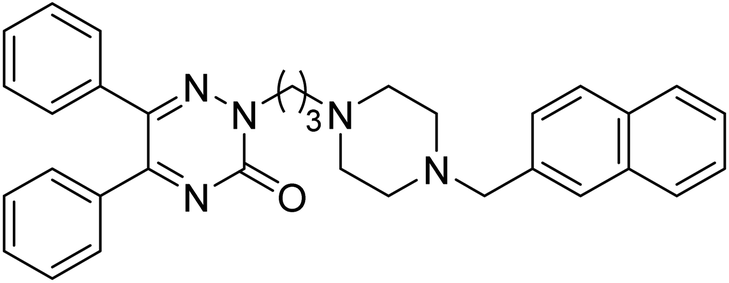 |
0.2 | — | 255 |
| 215 | 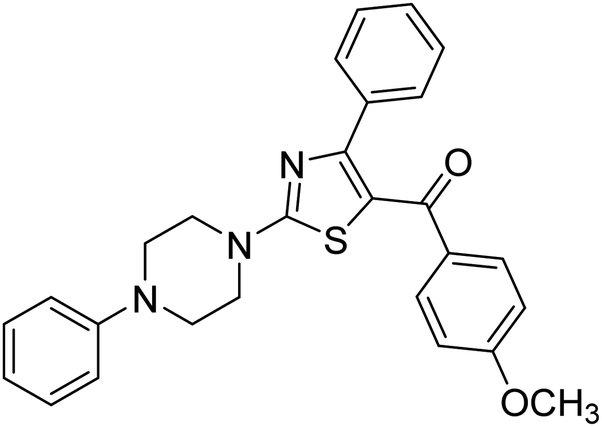 |
0.268 | — | 256 |
| 216 | 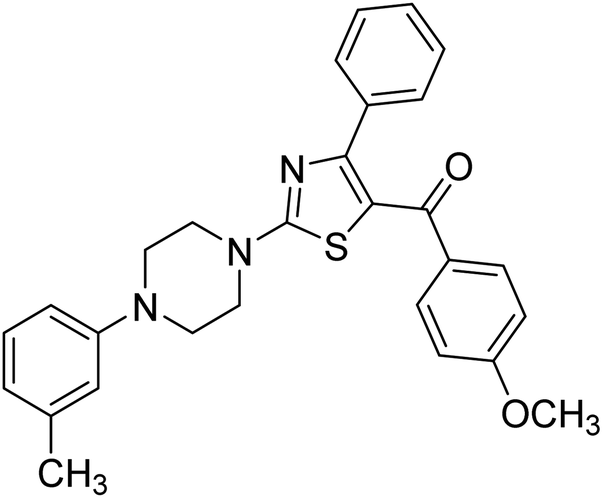 |
0.286 | — | 256 |
| 217 | 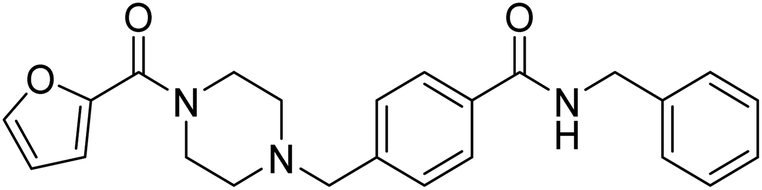 |
289.61 | — | 257 |
| 218 | 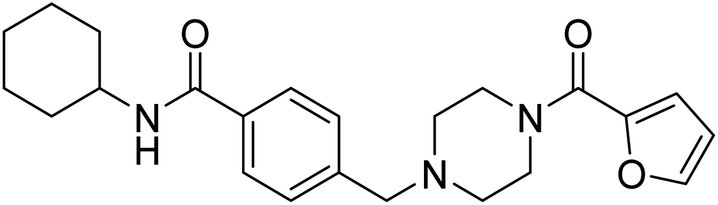 |
252.36 | — | 257 |
| 219 |  |
412.71 | — | 257 |
| 220 |  |
54.81 | — | 257 |
According to Hamulakova et al. (2014) the potential of synthetic piperidine derivatives as novel AChE and BChE inhibitors with nanomolar inhibitory activity was investigated using a new series of substituted tacrine/acridine and tacrine/tacrine dimers with aliphatic or alkylene-thiourea linkers. The most potent AChE inhibitor was found to be homodimeric tacrine derivative 204, which had an IC50 value of 2 nM, indicating a 250-fold higher activity rate than tacrine and 7500-fold higher activity rate than the study's standard, 7-MEOTA. Dual site binding is evident in the generated compounds with two tacrines or tacrine and acridine as terminal moieties, signifying a second binding site. According to the IC50 values obtained throughout the study, all compounds had an inhibitory effect on both AChE and BChE. Among the synthesized derivatives 204 and 205 displayed the highest levels of nanomolar AChE inhibition. The findings reveal that combining terminal acridine or tacrine units with alkylene-piperazine or alkylene-thiourea linkers is highly successful. Inhibitors of both mono and dual binding sites are present in ligands 204 and 205. A study of inhibitory efficacy within the series revealed the importance of linker length. The solitary ethylene unit in 204 provides only medium-nanomolar activity, but the two ethylene units in 205 ensure low-nanomolar inhibitory action (260 nM). This demonstrates that the length of molecule 204 is insufficient to simultaneously reach both enzyme binding sites. The tacrine derivatives 204 and 205, which have an ethylene unit attached to them, have a low, even sub-nanomolar effectiveness (for BChE: 0.4–20 nM), which is superior to tacrine itself. In this family of compounds, the presence of another aromatic rings on the opposite side of the molecules is a crucial factor that promotes binding (Bn or THA). Finally, a comparison of AChE/BChE selectivity within the series shows that compound 205 (despite its low activity) is the most selective for AChE, whereas compound 204 is the most selective for BChE251 (Table 9).
Ozkay et al. (2017) established a procedure for evaluating the anticholinesterase effects of different hydrazone derivatives. A sequence of eleven new N-(2,4-disubstitutedbenzylidene)-2-(4-nitrophenyl-piperazin-1-yl)acetohydrazide analogs were prepared by reacting 2-[4-(4-nitrophenyl)piperazin-1-yl]acetohydrazide with aromatic aldehydes. The inhibitory efficacy of compounds against AChE and BChE was tested and assessed using a modified version of Ellman's spectrophotometric approach. The most active derivative of the compounds studied was found to be compound 206. The drug galantamine was used as a reference drug. All the compounds had lower anticholinesterase potency than the reference drug. Only compound 206, which had a hydroxyl substituent at the para position, inhibited AChE, with an IC50 of 29.5 µM (ref. 252) (Table 9).
Ozkay et al. (2018) discussed a study in which they synthesized 2-(9-acridinylamino)-2-oxoethyl piperazine/piperidine/morpholinecarbodithioate analogs to investigate anticholinesterase activity. All the compounds exhibited unique and encouraging anti-BChE activity. The first class of compounds inhibit BChE with IC50 values ranging from 0.014 to 2.097 µM. Piperazine derivatives including 2-dimethylaminoethyl, 3-dimethylaminopropyl, 2-hydroxyethyl, 4-chlorophenyl, 4-(trifluoromethyl)benzyl and 4-methylbenzyl exhibited higher inhibitory activity against BChE than the other compounds in the group. Additionally, it inhibited BChE 15.4-fold better than the positive control, donepezil (IC50 = 1.419 µM), and the highest active chemical, 207 (IC50 = 0.092 µM)253 (Table 9).
Akbarzadeh et al. (2019) outlined a technique for developing, synthesizing, and testing a novel family of arylisoxazole-phenylpiperazines against AChE and BChE. [5-(2-Chlorophenyl)-1,2-oxazol-3-yl](4-phenylpiperazin-1-yl)methanone 208 was determined to be the most potent AChE inhibitor, with an IC50 of 21.85 µM. It should be noted that most of the compounds synthesized exhibited little anti-BChE activity, with the most active being [5-(2-fluorophenyl)-1,2-oxazol-3-yl](4-phenylpiperazin-1-yl)methanone 208 (IC50 = 51.66 µM). Additionally, kinetic studies of the inhibitory activity of compounds 208 and 209 on AChE and BChE indicated that they bind to both the catalytic site (CS) and the peripheral anionic site (PAS) of AChE and BChE. Additionally, docking analysis demonstrated that compound 208 formed favorable contacts with amino acid residues in the active and peripheral anionic sites. Finally, compound 208 had a negligible neuroprotective effect against Aβ-induced neurotoxicity in PC12 cells254 (Table 9).
Tripathi et al. (2019) reported the synthesis of several piperazine-tethered biphenyl-3-oxo-1,2,4-triazine analogs. In comparison to donepezil (AChE, IC50 = 0.1 µM), compound 210 demonstrated substantial non-competitive inhibitory activity against AChE (IC50 = 0.2 µM). In in vivo behavioural studies, compound 210 significantly improved cognitive dysfunctions in scopolamine-induced amnesia animal models. Ex vivo tests also demonstrated that compound 210 suppressed AChE and repaired the oxidative damage caused by scopolamine. The PAS and active catalytic site (CAS) residues of AChE displayed a reciprocal binding affinity and active site interactions, according to docking and dynamics simulations of 210. The compound 210 with a propyl (n = 3) linker have potential AChE inhibitory action. Compound 210, which contains a phenylpiperazine moiety, was found to have satisfactory AChE inhibitory activity (IC50 = 0.6 µM). Notably, EWG (nitro, 211; and fluoro, 212) and electron-releasing (methoxy, 213) substituents increased AChE inhibition at the p-position of the phenylpiperazine ring (211, IC50 = 0.5 µM; 212, IC50 = 0.4 µM; 213, IC50 = 0.3 µM). Compound 213, which contains a 4-methoxyphenyl group, inhibited AChE more effectively than compounds 211 and 212. The OCH3 group's non-polar properties, which interacted with AChE's hydrophobic pocket, may have improved AChE inhibitory potential. In a study of compounds with various electron-withdrawing groups, analog 211 with a fluoro atom demonstrated somewhat greater inhibitory capability against AChE than compound 212 with a nitro group. The increased inhibitory effectiveness might be explained by the fluorine's high electronegativity, which changes the molecule's lipophilicity. Of all the derivatives tested, compound 214, which contains 3 carbon atoms linked to the benzylpiperazine end group, exhibited the most potent inhibition of AChE (IC50 = 0.2 µM). Because of the engagement of the Bz group at the bottom of the enzyme gorge and improved interaction with AChE CAS residues, the AChE inhibitory activity of compound 214 matched that of donepezil (IC50 = 0.01 µM). The ability of all the produced species to inhibit BChE was then tested, but none of them showed any discernible effect. Because two aromatic amino acids have been replaced with smaller aliphatic amino acids, BChE has a wider gorge than AChE255 (Table 9).
Yurttaş et al. (2019) described a method for synthesizing [2-(4-(2,3,4-substituted phenyl)piperazin-1-yl)-4-phenylthiazol-5-yl][3,4-substituted phenyl]methanone derivatives 297 and investigated their anticholinesterase properties. The molecular interactions as well as the kinetic mode were examined. The Ellman approach was used to look at how AChE and BChE enzymes were inhibited. The activity of 44 compounds was assessed on AChE and BChE enzymes at doses of 103 and 104 µM. The inhibitory dosage for six compounds on AChE varied from 0.268 µM to 2.104 µM. Compound 215 with the 4-methoxy group (IC50 = 0.268 µM) and compound 216 with the 4-methoxy and 3-methyl substituents had the greatest AChE inhibitory action (IC50 = 0.286 µM). Hydrogen bonding with Arg296 and Ar interactions with Trp286 were also investigated256 (Table 9).
Abbasi et al. (2020) described a technique for synthesizing and testing multifunctional compounds 217–220 against BChE. The BChE enzyme has been shown to be inhibited by two of these compounds 217 and 219. Assessment of the hemolytic activity potential of 218 indicated that it possesses low toxicity level. An approach previously published for the BChE. Enzyme was used to measure the activity of the enzyme inhibitor. The data reveal that several of the chemicals have potential inhibitory capacity against this enzyme. Though the observed activity is related to the combined effect of all functionalities embedded in a molecule's entire framework, the effects of different entities in these four molecules, such as furoyl, piperazine, and benzamide functionalities, were examined to establish a brief SAR. N-Cyclohexyl-4-[4-(2-furoyl)-1-piperazinyl]methyl-benzamide 218 and N-cyclohexyl-3-[4-(2-furoyl)-1-piperazinyl] methylbenzamide were the most effective inhibitors of BChE enzyme 220. The other two compounds, 217 and 219, remained the least efficient due to their higher IC50 values. Eserine was utilized as the reference drug, attaining IC50 value of 0.85 µM. The difference between these compounds can be seen in the N-substituted groups such as aralkyl/aryl and the location of the methylene group on the benzamide moiety, as shown in the structures. In compounds 218 and 220, the cyclohexyl group is linked to the nitrogen of the benzamide functionality, but in compounds 217 and 219, a benzyl group is attached to the amide nitrogen. In molecules 217 and 218, the methylene group is perpendicular to the benzamide moiety, but in molecules 219 and 220, it is in the third position. When compared to the reference drug, eserine, which has an IC50 of 0.85 µM, the addition of methylene at the third position of the benzamide moiety reduces the IC50 from 252.36 µM in 218 to 54.81 µM in 220. Compounds 217 and 219 both had an evident impact due to the existence of a methylene group in p-position to the benzamide, as indicated by the IC50 of compound 217 (289.61 µM), which exhibited stronger enzyme inhibition than 219, which had an inhibitory potential of 412.71 µM. Eserine was utilized as a positive control for cholinesterase enzymes. The inhibitory potential could be improved even further by replacing a substituted cycloalkyl or straight chain alkyl group for the cycloalkyl group. Compounds 218 and 220, as a result, might be viable novel therapeutic candidates for inhibiting the BChE enzyme257 (Table 9).
SAR studies of piperazine analogs demonstrate that the reported derivatives are active against AChE and BChE enzymes (Fig. 19). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–CH3, –Ph, –Et, –CF3, –CN, –Cl, –F, –Br, –OCH3, –OH) attached to the piperazine ring show higher AChE and BChE inhibitory abilities as compared to the bulky groups (furan, urea, thiazole) present on the rings. All the presented analogs thus far have shown excellent ChE inhibitory abilities with minimum toxic side effects. Moreover, these analogs are easy to synthesize in the laboratory, making them appealing for viable development and marketing as drugs against cholinesterase.
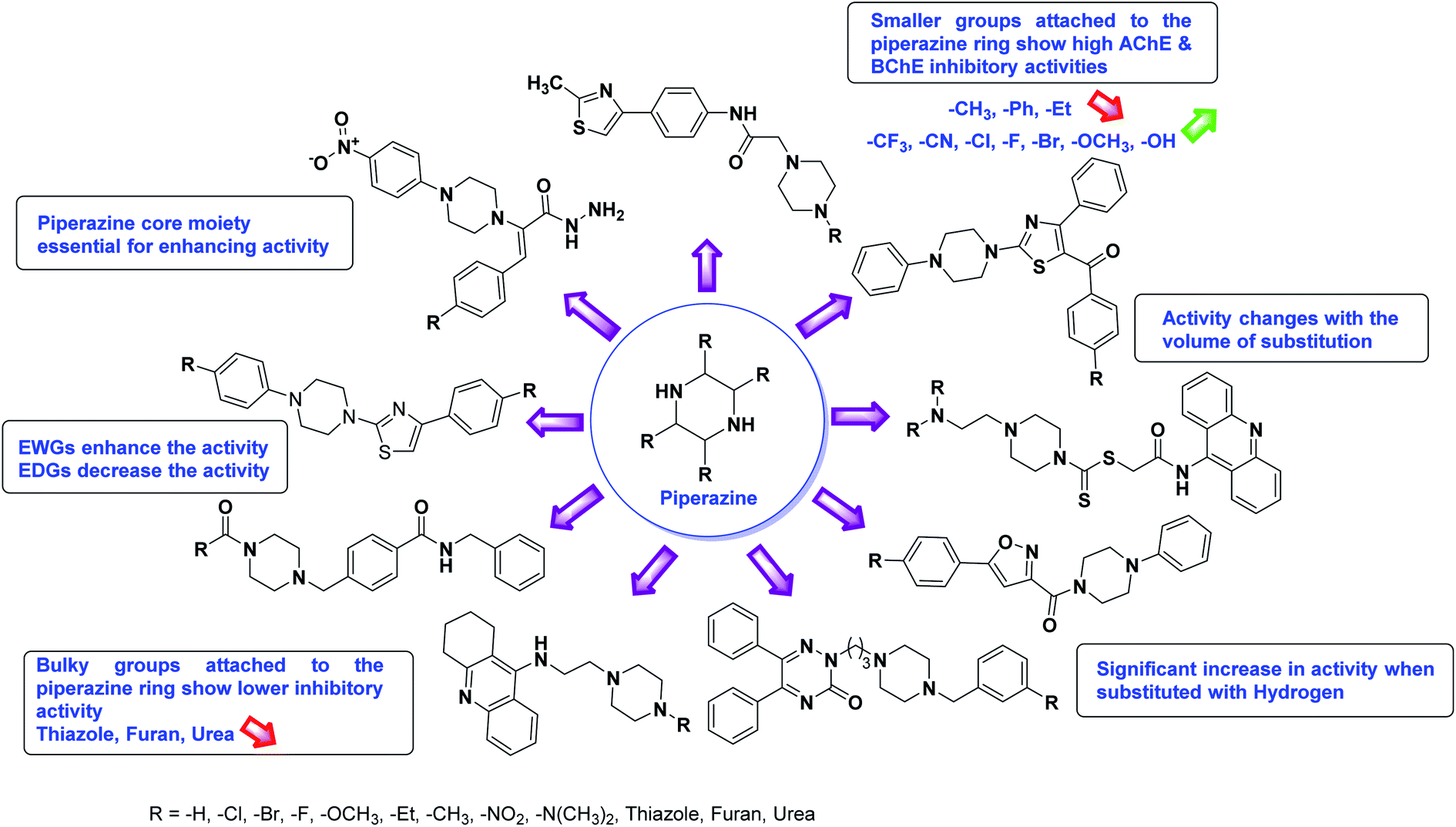 | ||
| Fig. 19 SAR analysis of different piperazine derivatives as AChE and BChE inhibitors. | ||
Szymanski et al. (2012) present a method for synthesizing and testing 4-fluorobenzoic acid and 2,3-dihydro-1H-cyclopenta[b]quinoline derivatives for AChE and BChE inhibition. The compounds were made by condensing amino derivatives of 2,3-dihydro-1H-cyclopenta[b]quinoline and 4-fluorobenzoic acid. The Ellman spectrophotometric method was used to conduct biological testing for cholinesterase inhibition. Compounds 222 (IC50 = 10.80 µM for AChE; 4.70 µM for BChE) and 224 (IC50 = 153 µM for AChE; 559 µM for BChE) proved less active than tacrine (IC50 = 0.016 µM for AChE; 0.0026 µM for BChE). The activity of compounds 223 (IC50 = 5.83 µM for AChE; 14.00 µM for BChE), 226 (IC50 = 5.20 µM for AChE; 699 µM for BChE), and 227 (IC50 = 5.48 µM for AChE; 744 µM for BChE), on the other hand, is comparable to that of tacrine. Compounds 221 (IC50 = 1.07 µM for AChE; 4.59 µM for BChE), 224 (IC50 = 0.49 µM for AChE; 5.91 µM for BChE), and 225 (IC50 = 2.77 µM for AChE; 717 µM for BChE) were shown to be more effective in deactivating AChE than tacrine. When compared to tacrine, all the generated compounds displayed better selectivity for AChE than BChE, except for compound 221. This compound was like tacrine in terms of AChE selectivity, although it was more selective for BChE258 (Table 10).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 221 |  |
1.07 | 4.59 | 258 |
| 222 | 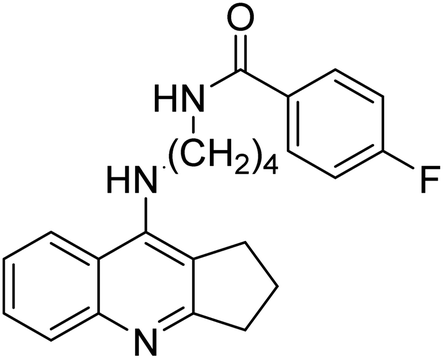 |
10.80 | 4.70 | 258 |
| 223 | 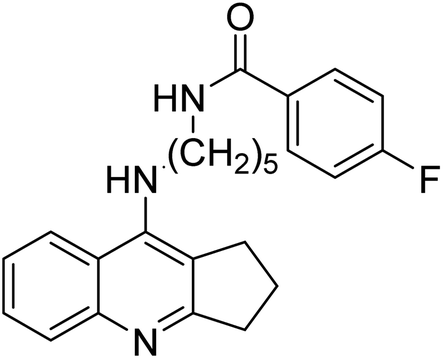 |
5.83 | 14.00 | 258 |
| 224 | 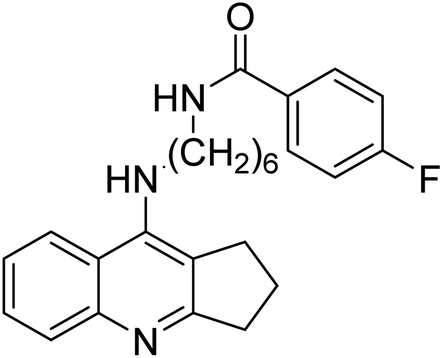 |
0.49 | 5.91 | 258 |
| 225 | 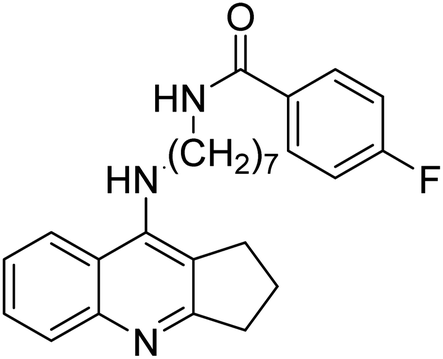 |
2.77 | 717 | 258 |
| 226 | 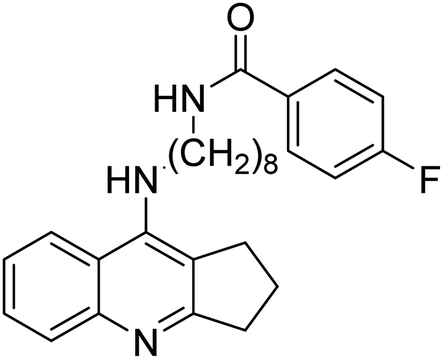 |
5.20 | 699 | 258 |
| 227 | 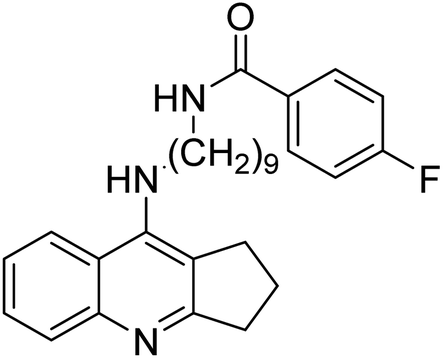 |
5.48 | 744 | 258 |
| 228 | 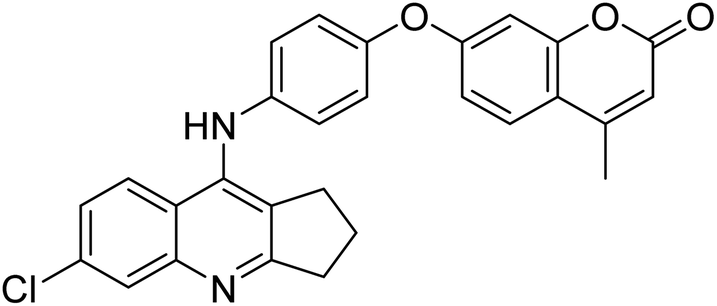 |
16.17 | — | 259 |
| 229 | 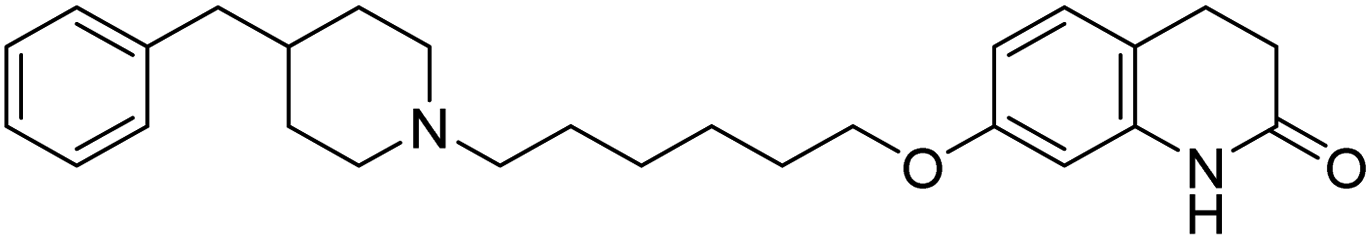 |
0.56 | 2.3 | 260 |
| 230 | 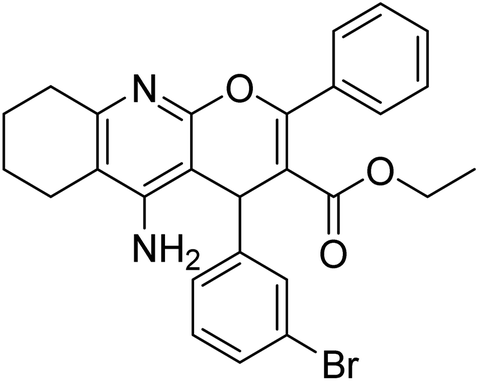 |
0.069 | 1.35 | 261 |
| 231 | 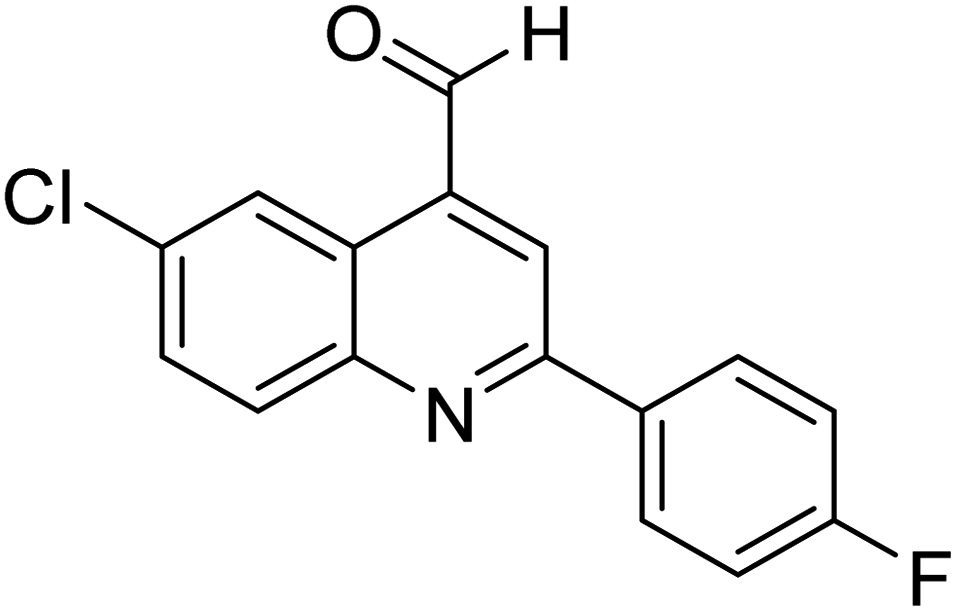 |
4.36 | — | 262 |
| 232 | 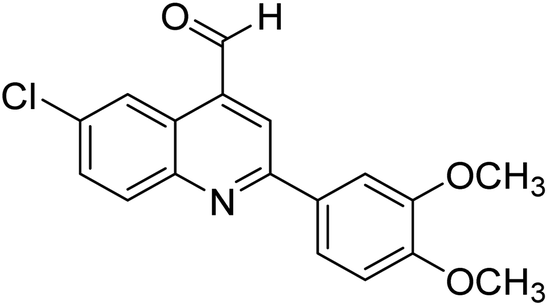 |
7.10 | — | 262 |
| 233 | 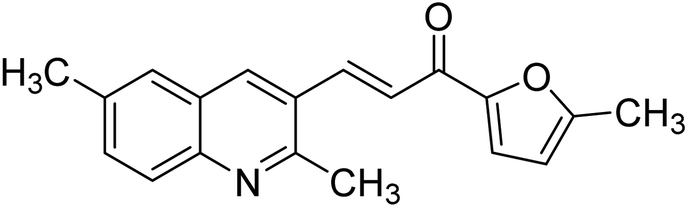 |
0.032 | 0.90 | 263 |
| 234 | 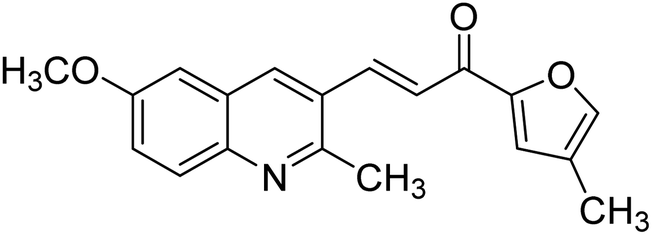 |
2.99 | 0.11 | 263 |
| 235 | 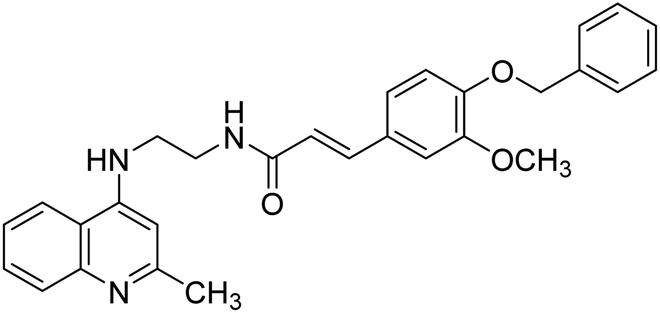 |
0.62 | 0.10 | 264 |
| 236 | 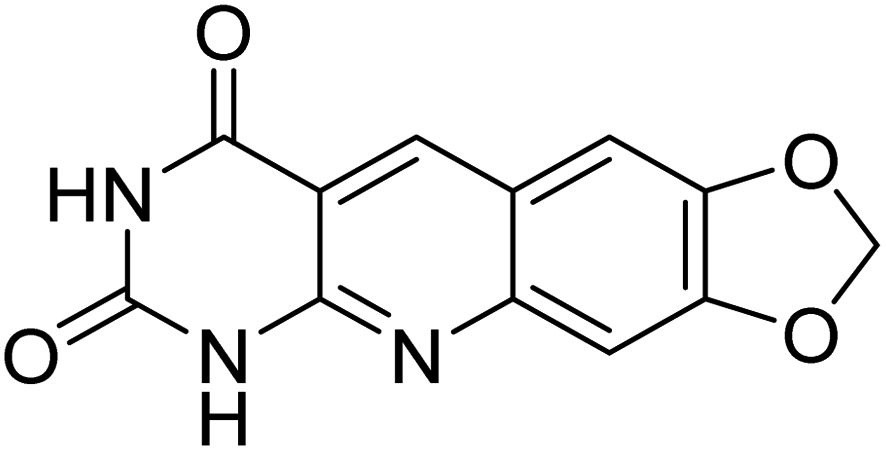 |
32.06 | — | 265 |
| 237 | 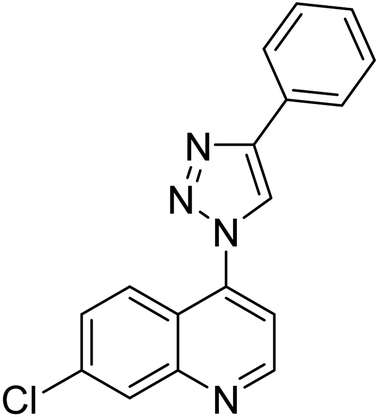 |
10.24 | — | 266 |
| 238 | 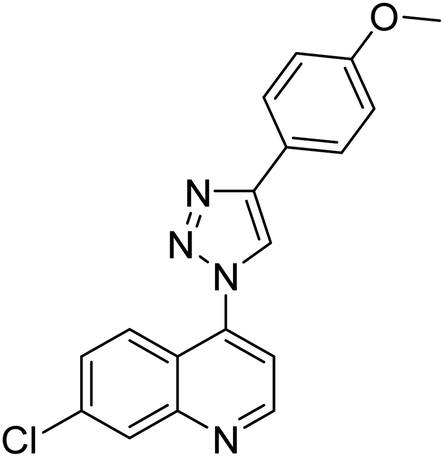 |
11.89 | — | 266 |
| 239 | 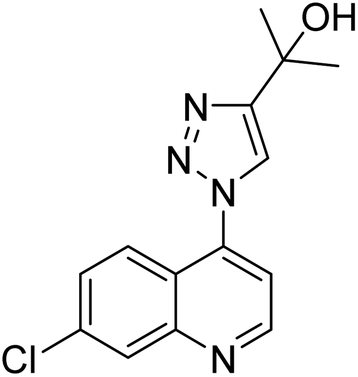 |
17.74 | — | 266 |
| 240 | 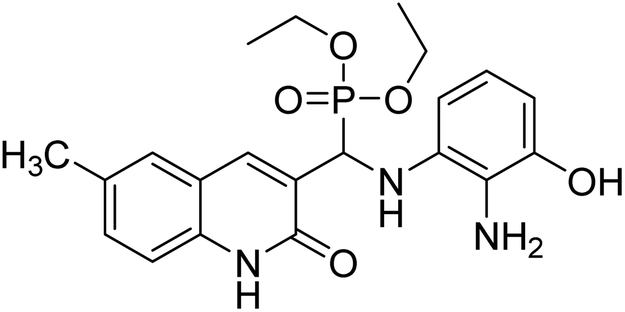 |
28.42 | — | 267 |
| 241 | 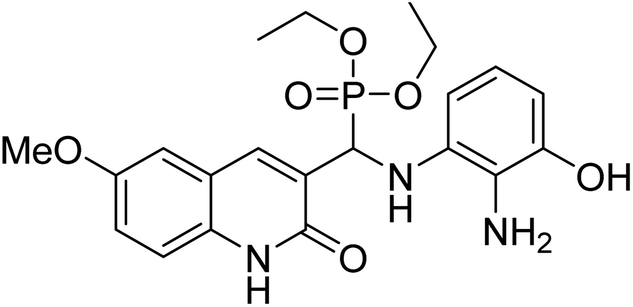 |
38.39 | — | 267 |
| 242 | 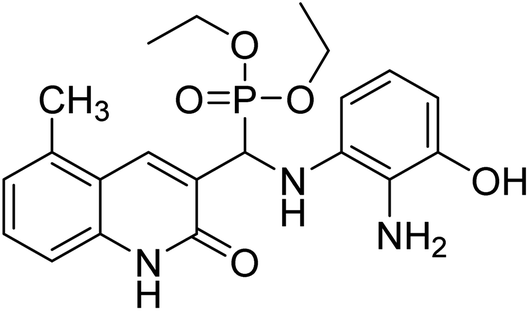 |
52.55 | — | 267 |
| 243 | 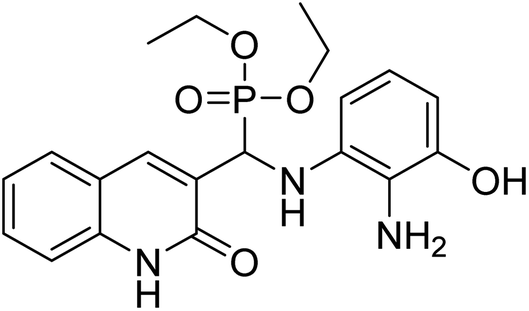 |
55.23 | — | 267 |
| 244 | 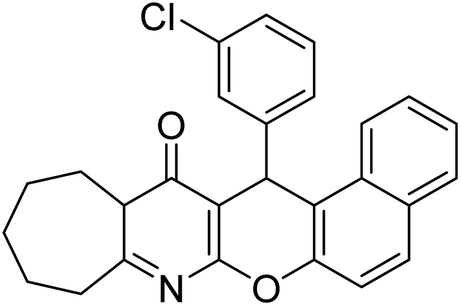 |
0.65 | 1.32 | 268 |
| 245 | 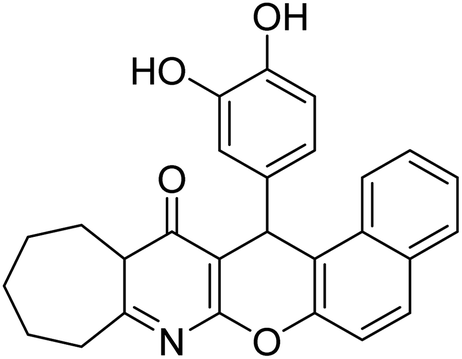 |
0.85 | 1.65 | 268 |
| 246 | 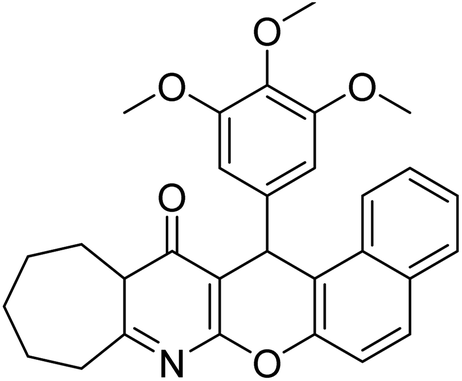 |
0.92 | 1.91 | 268 |
| 247 |  |
0.71 | 0.04 | 269 |
| 248 |  |
1.01 | 0.03 | 269 |
Najafi et al. (2016) reported a collection of acridine-chromenone and quinoline-chromenones and evaluated them for AD. 7-(4-(6-Chloro-2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)phenoxy)-4-methyl-2H-chromen-2-one 228 had the most anti-AChE inhibitory action (IC50 = 16.17 µM) compared to rivastigmine (IC50 = 11.07 µM). It is worth noting that kinetic studies and molecular modelling demonstrated that 228 interacted with both the catalytic active site and the peripheral anionic site of AChE simultaneously. The anti-AChE activity of 228 was the greatest (IC50 = 16.17 µM), while the rest of these compounds were only moderately effective in suppressing AChE. This compound has a 4-methylchromenone group as a peripheral site interaction unit and a quinoline structure as a catalytic site binding unit. Based on kinetic studies and molecular modelling, analog 228 has enough span to bind to both the CAS and the PAS of AChE as a dual binding inhibitor. In the PAS, the 4-methylchromenone group of 228 showed parallel π–π stacking with Trp279 at the gorge's mouth, but in the CAS, parallel π–π stacking with Trp84 was seen at the gorge's bottom. The neuroprotective effect was low as compared to quercetin259 (Table 10).
Liu et al. (2017) developed a novel class of multitarget drugs that bind to AChE, BChE and monoamino oxidase (MAO) A and B. Novel 3,4-dihydro-2(1H)-quinoline-O-alkylamine compounds were prepared using a conjunctive technique combining the JMC49 (MAO-B inhibitor) and donepezil. The study described the synthesis of a novel family of 3,4-dihydro-2-(1H)-quinoline-O-alkylamine derivatives that can be used to treat AD. According to both kinetic and molecular modelling studies, the most promising compound 229 exhibited powerful and balanced inhibitory effects against AChE, BChE, hMAO-A and hMAO-B. With IC50 values of 0.56 µM and 2.3 µM, compound 229 exhibited the highest inhibitory action against AChE and BChE and showed even better inhibitory effectiveness against hMAO-A (IC50 = 0.3 µM) and hMAO-B (IC50 = 1.4 µM). The kinetic study showed that 229 inhibited AChE in a mixed-type manner and that it could bind to both the CAS and the PAS, which was consistent with the molecular modelling studies. Furthermore, their findings showed that the compound could cross the blood–brain barrier (BBB) in vitro and adhered to Lipinski's rule of five. The findings of this study imply that 229 might be a potential multi-target lead chemical for further development into advanced AD therapy260 (Table 10).
Sarrafi et al. (2017) described a procedure for developing and manufacturing cholinesterase inhibitors, which consisted of polyfunctionalized tacrine-derived compounds, most notably 5-amino-2-phenyl-4H-pyrano[2,3-b] quinoline-3-carboxylates. In vitro inhibition studies against AChE and BChE established that most compounds were efficient AChE inhibitors, with the potential for BChE inhibition remaining. The most potent compound against AChE/BChE was compound 230, which bears a 4-(3-bromophenyl) moiety (IC50 = 0.069 µM and 1.35 µM, respectively). The anti-AChE activity of 230 was 5 times that of tacrine. Furthermore, the promising chemical 230 demonstrated lower cytotoxicity in HepG2 cells when compared to tacrine. According to the SAR study, a chloro/bromo atom at the o- or m-position of the 4-phenyl ring can enhance anticholinesterase activity. With respect to modification of substituents on the 4-phenyl ring, while electron releasing and EWG are not favorable for para-position, electron withdrawing groups such as chloro and bromo located at the ortho or meta positions may improve the cholinesterase inhibition potential261 (Table 10).
Iqbal et al. (2018) synthesized a series of quinoline carboxylic acids and investigated their inhibitory capabilities against monoamine oxidase and cholinesterase in vitro to identify novel and efficient Parkinson's disease inhibitors. The strongest inhibitors were subjected to molecular docking and in silico studies to uncover the likely binding mechanisms in the active site of monoamine oxidase enzymes. To measure the compounds' drug-likeness, molecular properties were also assessed. The examined compounds were shown to be particularly active against monoamine oxidase (A and B), with IC50 values of 0.51 and 0.51 µM for both isoforms of MAO, respectively. The tested compounds displayed a significant and completely specific inhibitory action on AChE, with IC50 values ranging from 4.36 to 89.24 µM (AChE). Quinoline 231 was shown to be the most powerful inhibitor of AChE among the compounds examined, with an IC50 value of 4.36 µM. Docking experiments confirmed strong binding site interactions with the inhibitors. Such quinolines hold promise as potential agents to treat neurodegenerative illnesses although they will require additional pharmacophore tweaking to boost their binding affinities. Given the importance of this target in PD pathogenesis, compound 232 might be a promising new chemical entity for the creation of multi-target directed ligands262 (Table 10).
According to Iqbal et al. (2018) cholinesterases (ChEs) play an important role in cholinergic transmission control. By restoring ACh levels in the brain, inhibition of ChEs is believed to embody a novel and potentially therapeutic target for neurodegenerative illnesses like AD. To increase the chemical diversity of cholinesterase inhibitors, a variety of quinoline chalcones derivatives were tested against AChE and BChE isoenzymes. These were discovered to exhibit strong anti-AChE and anti-BChE activity. Homology models were used to conduct molecular docking experiments on both AChE and BChE isoenzymes with the objective of establishing the likely binding mechanisms of the strongest inhibitor in the series. To analyze the pharmacological similarity of newly studied compounds, they employed in silico ADME assessment. The ADME results for compounds 233 (IC50 = 0.032 for AChE and 0.90 µM for BChE) and 234 (IC50 = 2.99 for AChE and 0.11 µM for BChE) were positive, indicating that these derivatives will have high oral bioavailability and projected to be more potent than donepezil (IC50 = 0.02 for AChE and 0.23 µM for BChE). Because of their favorable ADME profiles, the investigated analogs are expected to be a safer class of cholinesterase inhibitors263 (Table 10).
Chen et al. (2019) presented the development, production, and testing of a variety of new multifunctional quinoline-ferulic acid hybrids with cholinesterase inhibitory characteristics. Both AChE and BChE were inhibited by most of the compounds. The most effective inhibitor of AChE and BChE was determined to be 235 (AChE IC50 = 0.62 µM; BChE IC50 = 0.10 µM). According to molecular docking and dynamic modelling, the generated compounds connect to the target by simultaneously interacting with the catalytic active site (CAS) and the peripheral anionic site (PAS) of both AChE and BChE. The U-shaped conformation of 235 coupled to BChE was preferred over the linear conformation of 235 bound to AChE. According to cell-based experiments, compound 235 exhibit moderate neuroprotective effects against H2O2-induced oxidative damage in PC12 cells. Furthermore, 235 had lesser hepatotoxicity than tacrine, suggesting that it might be a safer alternative to treat AD264 (Table 10).
Brum et al. (2019) developed and evaluated quinoline-piperonal hybrids as prospective AD therapies. Theoretical examination of the compounds' pharmacokinetic and toxicological attributes revealed that they have excellent oral bioavailability and can pass the blood–brain barrier to reach their target. Three compounds were shown to exhibit inhibitory action against acetyl- and butyrylcholinesterase in the Ellman's test, with one analog capable of deactivating both enzymes. Further molecular docking studies of the 6 analogs developed aided in clarifying the primary interactions that might be accountable for the detected inhibitory effects. The existence of aromatic rings in the quinolines resemble the base structure of tacrine (IC50 = 0.0414 µM) allows enables π–π interactions with the amino acids present in the active site, while the guanyl hydrazone is able of interacting with the CAS of AChE via the guanidine function. In comparison to rivastigmine, compound 236 (IC50 = 32.06 µM) showed promise as AChE inhibitors265 (Table 10).
Kouznetsov et al. (2019) reported the preparation of triazolyl-quinoline hybrids and described their modest inhibitory action against commercial AChE from Electrophorus electricus (electric-eel AChE) (IC50 = 27.7 g mL−1) and low anti-ChE activity on S. frugiperda larval homogenate (IC50 = 68.4 g mL−1). Molecular docking simulations revealed that hybrid 237 binds to the enzyme's catalytic active site (CAS) and the rim of the cavity, operating as a mixed (competitive and noncompetitive) inhibitor like methomyl. Triazolyl-quinolines 238 and 239 act as non-competitive inhibitors of AChE by binding around the periphery of the enzyme cavity266 (Table 10).
Bazine et al. (2020) employed the Kabachnik–Fields reaction to develop and synthesize a series of novel amino phosphonate derivatives with quinoline moiety. The derivatives 240 (IC50 = 28.42 µM), 241 (IC50 = 38.39 µM), 242 (IC50 = 52.55 µM), and 243 (IC50 = 55.23 µM) demonstrated the highest inhibitory activity against AChE when compared to galantamine standard (IC50 = 21.81 µM). All tested compounds showed strong anti-BChE activity when compared to galantamine standard (IC50 = 120.93 µM)267 (Table 10).
Garlapati et al. (2020) prepared several novel tacrine analogues and evaluated them for cholinesterase inhibitory activity. Most synthesized compounds showed inhibitory action against AChE and BChE enzymes in vitro. With IC50 values of 0.65, 1.32 and 0.85, 1.65 and 0.92, 1.91 µM against AChE and BChE, compounds 244, 245 and 246, which have a larger saturated carboxylic ring tethered to the pyridine group and 3,4-dihydroxy, 3,4,5-trimethoxy substituents on the aryl ring attached at the stereogenic centre, have shown identical potency to tacrine. Tacrine's IC50 values against AChE and BChE were 0.47 and 0.65 µM, respectively, while donepezil had IC50 values of 0.71 and 0.31 µM respectively. All the analogs had hydrogen bond interactions with the binding site, according to docking experiments268 (Table 10).
Garlapati et al. (2021) prepared a new family of AChE and BChE inhibitors based on the structure of tacrine using a multicomponent Friedlander reaction between 2-aminobenzonitrile and cycloalkanones. The Ellman technique was used to test their inhibitory ability against AChE and BChE. When compared to the standard tacrine and rivastigmine, which had IC50 values of 0.23, 0.47 µM for AChE and 0.31, 0.65 µM for BChE, respectively, compounds 247 and 248, which contained piperazine containing acetamide and butyrylamide chains, had IC50 values of 0.71, 0.04 µM for AChE, 1.01, 0.03 µM for BChE, 0.52, 0.03 µM for AChE, and 0.73, 0.04 µM for BChE, respectively. As a result, these five membered-ring novel tacrine analogs have emerged as possible cholinesterase inhibitors that might one day be employed as Alzheimer's anti-drugs. Docking studies on all the compounds demonstrated tight hydrogen bond interactions inside the binding area. With IC50 values of 0.97 and 1.74 µM, the unsubstituted compound 247 (n = 3 and n′ = 1), which has a five-membered ‘C’ ring and a two-carbon linker, has demonstrated significant activity against AChE and BChE. These findings pointed to a larger nonpolar area in the binding pocket that might accommodate the larger cycloalkyl ring. The inhibitory action of the preceding species found active in vitro on AChE and BChE in the brain was investigated in vivo. To further understand the binding interactions inside the active site of cholinesterases, the compounds were also subjected to molecular docking investigations. Except for a few compounds, docking tests revealed that the developed molecules interacted with the nicotinic receptor via at least two hydrogen bond interactions269 (Table 10).
The SAR studies of the aforementioned quinolines demonstrate that the various analogs are active AChE and BChE inhibitors (Fig. 20). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–N(CH3)2, –CH3, –Ph, –Et, –CF3, –Cl, –F, –Br, –OCH3, –OH, etc.) attached to the quinoline ring show higher AChE and BChE inhibitory abilities compared to the bulky groups (tacrine, furan urea, etc.) present on the rings. All the presented analogs thus far have shown good to excellent ChE inhibitory abilities with a minimum toxic side effect. Furthermore, these are easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
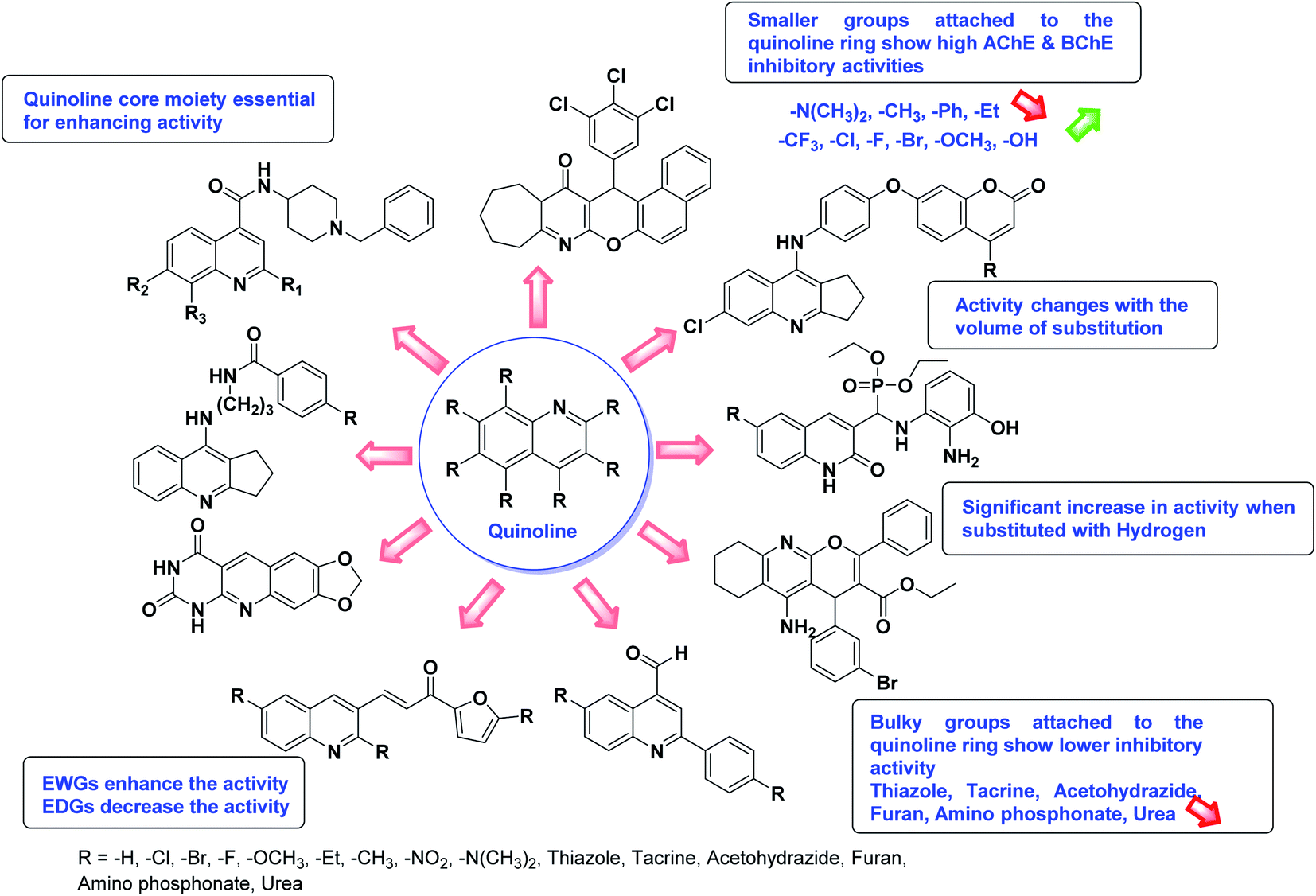 | ||
| Fig. 20 SAR analysis of different quinoline derivatives as AChE and BChE inhibitors. | ||
Rao et al. (2011) created a novel class of 2,4-disubstituted pyrimidines and investigated them as dual cholinesterase and amyloid-β (Aβ)-aggregation inhibitors. In vitro, the most potent AChE inhibitor was determined to be N-(naphth-1-ylmethyl)-2-(pyrrolidin-1-yl)pyrimidin-4-amine 249 (IC50 = 5.5 µM). The most effective and selective BChE inhibitor was discovered to be 2-(4-methylpiperidin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine 250, which was roughly 5.7 times more potent than the commercially available, authorized reference drug galantamine (BChE; IC50 = 12.6 µM). Furthermore, N-benzyl-2-(4-methylpiperazin-1-yl)pyrimidin-4-amine 251, a specific AChE inhibitor, effectively reduced hAChE-induced Aβ1–40 fibril aggregation (59% inhibition). Furthermore, molecular modelling studies revealed that a central pyrimidine ring can be employed as a template for generating dual inhibitors of cholinesterase and AChE-induced Aβ aggregation, allowing for the targeting of a variety of pathogenic pathways in AD. ChE inhibition against AChE was enhanced by the addition of a five-membered heterocycloalkyl C-2 group, such as pyrrolidine 251 (AChE IC50 = 8.7 µM; BChE IC50 = 26.4 µM). The ChE inhibitory potency was lowered when the C-2 five-membered pyrrolidine was replaced with a six-membered ring. On the other hand, the addition of a C-2 4-methylpiperidine 250 increased BChE inhibitory potency and selectivity. Compound 250 had a 5.7-fold higher BChE inhibition (IC50 = 2.2 µM) and selectivity (S.I. = 11.7) than the reference drug galantamine (BChE IC50 = 12.6 µM; S.I. = 0.27) and was much more potent than donepezil (BChE IC50 = 3.6 µM), relative to 250 (AChE IC50 = 25.8 µM)270 (Table 11).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 249 | 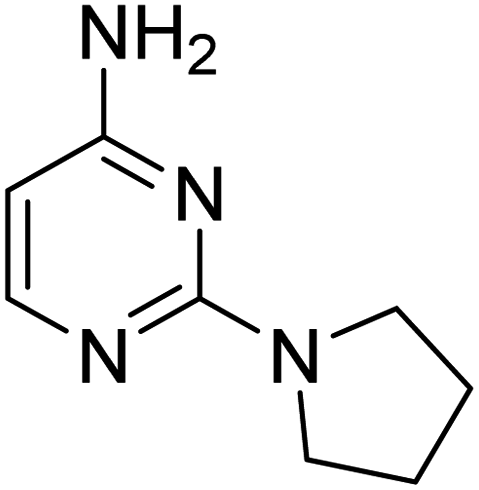 |
5.5 | 56.9 | 270 |
| 250 | 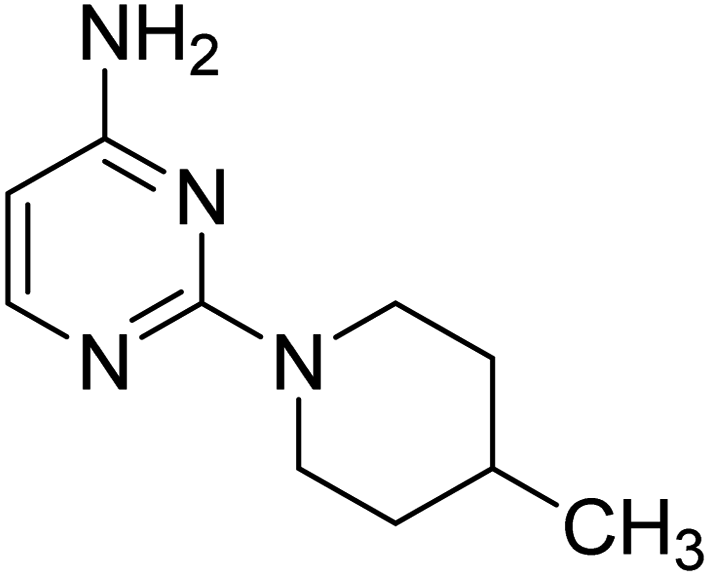 |
8.7 | 26.4 | 270 |
| 251 | 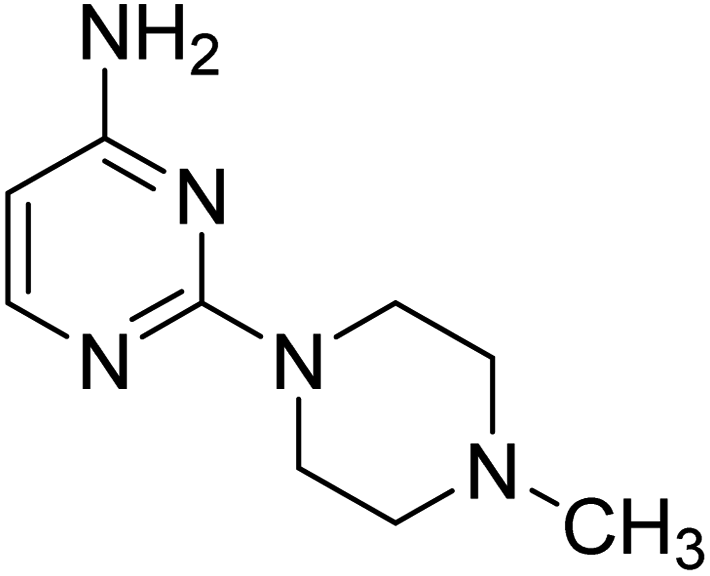 |
7.9 | 2.2 | 270 |
| 252 | 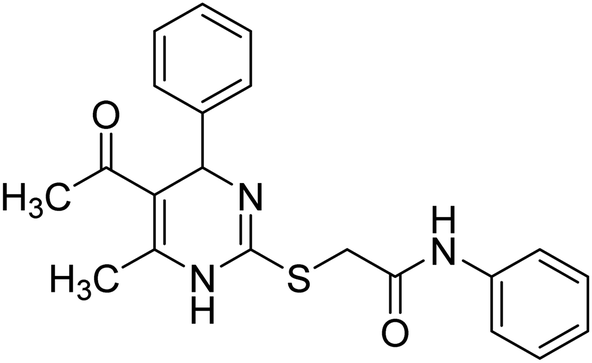 |
0.17 | 2.37 | 271 |
| 253 | 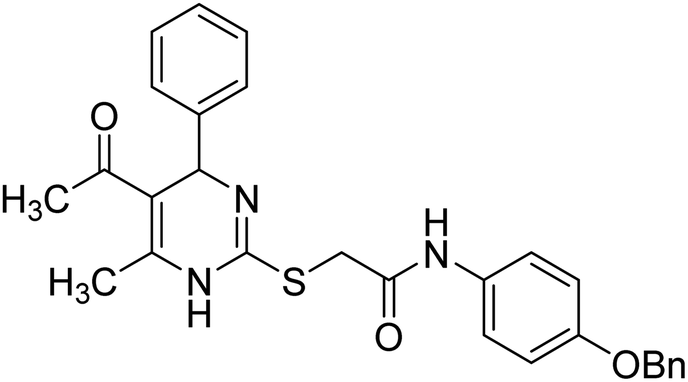 |
0.39 | 5.69 | 271 |
| 254 | 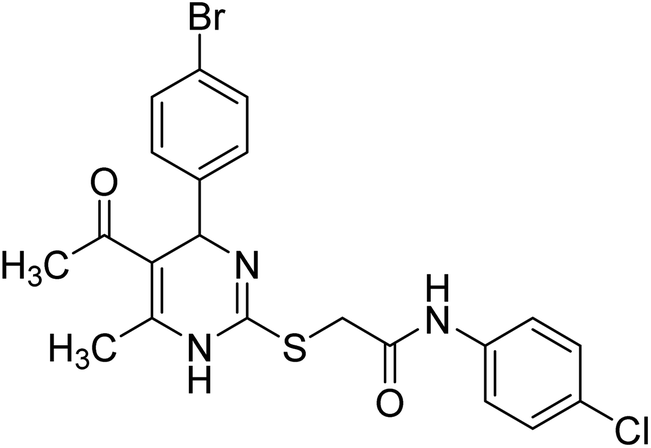 |
2.01 | — | 271 |
| 255 | 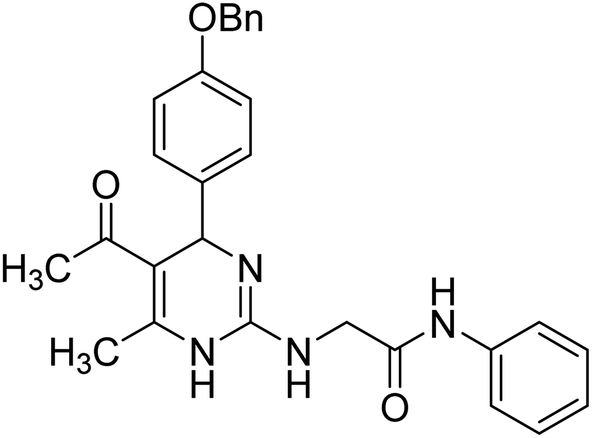 |
— | 30 | 271 |
| 256 | 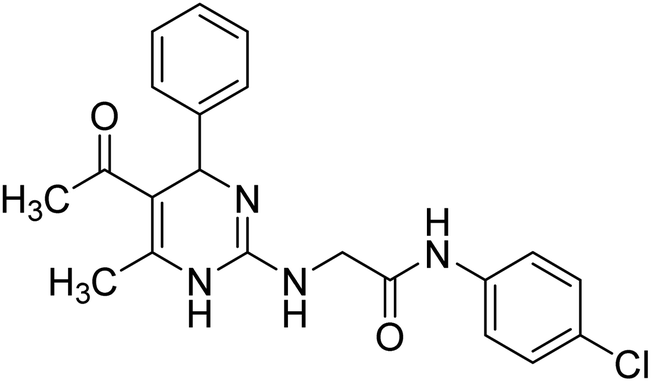 |
2.01 | — | 271 |
| 257 | 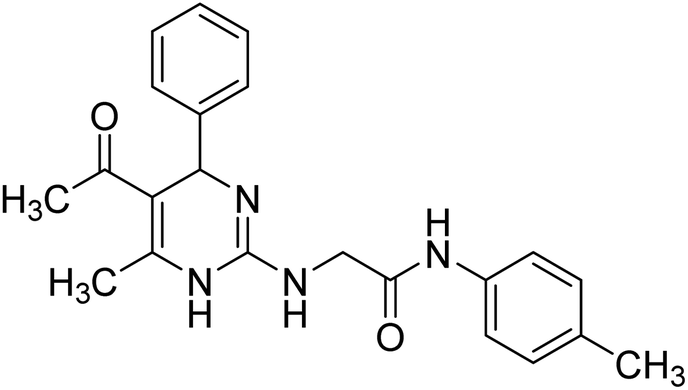 |
15.01 | — | 271 |
| 258 | 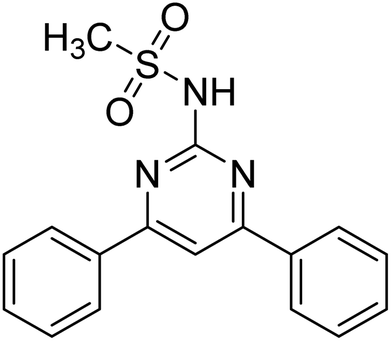 |
143.73 | 144.81 | 272 |
| 259 | 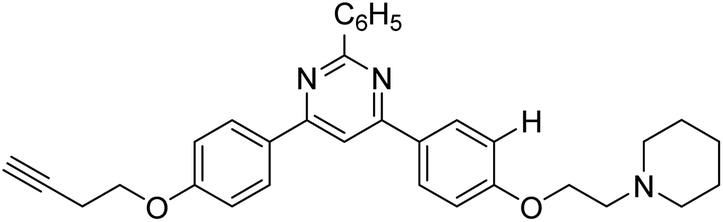 |
2.84 | — | 273 |
| 260 | 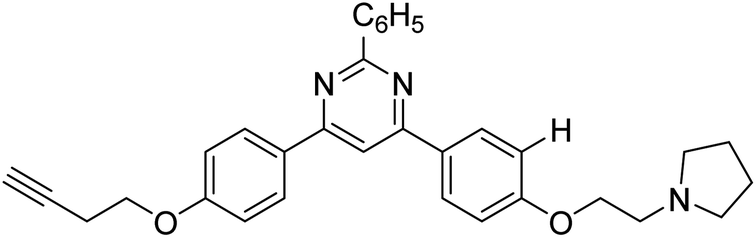 |
2.19 | — | 273 |
| 261 |  |
47.33 nM | 159.43 nM | 274 |
| 262 |  |
51.36 nM | 153.3 nM | 274 |
Based on the pharmacological relevance of the dihydropyrimidine (DHPM) scaffold, Rashid et al. (2016) synthesized substituted DHPMs coupled with an acetamide linker to substituted aromatic anilines and tested their effectiveness as AChE and BChE inhibitors. Among the 4-dihydropyrimidine-2-thione and 2-amino-1,4-dihyropyrimidines series, compounds 355 and 356 having the highest IC50 values of 0.17 and 0.39 µM, respectively, inhibited AChE effectively. BChE inhibition was seen at higher doses (2.37–56.32 µM). The compounds were tested for AChE (Electrophorus electricus) and BChE inhibition using Ellman's approach (equine serum). The most efficient ChE inhibitor was identified to be compound 252, which has a 14-fold selectivity for AChE. (IC50 values for AChE 0.17 µM and BChE 2.37 µM). With an IC50 of AChE 0.39 µM and BChE IC50 = 5.69 µM, S.I = 15, compound 253, which includes a benzyloxy phenyl ring, is likewise more selective against AChE. Compound 254 demonstrated significant AChE activity with IC50 values of 2.01 µM. 255, on the other hand, had a low inhibitory effect on BChE, with an IC50 of 30 µM (S.I. = 15). With an IC50 of 1.07 µM (S.I. = 17), the 4-benzoxypehnyl derivative 256, which belongs to the 2-amino-1,4-dihyropyrimidine family, is the strongest and selective AChE inhibitor. The 4-chloro substituted derivative 256 demonstrated strong AChE activity with an IC50 of 2.01 µM and a substitution pattern on the aniline ring. The IC50 value of the compounds in 2-amino-1,4-dihyropyrimidine series for BChE inhibition is seen over a greater concentration range (15.01–38.53 µM) than for AChE inhibition. Compound 257 has emerged as the most potent BChE inhibitor in this series, with an IC50 of 15.01 µM (ref. 271) (Table 11).
Khan et al. (2017) synthesized novel pyrimidine-based sulfonamides with fair to excellent yield (54–86%) in short period of time under microwave conditions. Structurally, these heterocycles have a core pyrimidine ring with a phenyl group and pyrimidine groups with sulfonamide functions. The capacity of these compounds to suppress the enzymes AChE and BChE, which are key in AD therapy, was investigated. When compared to the reference drug eserine, the IC50 values of the synthesized compounds varied from 3.73 µM to 57.36 µM for AChE and 4.81 µM to 111.61 µM for BChE. Among the compounds tested, compound 258 with a –CH3 group was shown to be the most effective against both enzymes (AChE, IC50 = 143.73 µM; BChE, IC50 = 144.81 µM). Molecular docking and QSAR studies were also conducted on the synthesized molecules. The goal of the molecular modelling work on sulfonamide 258 was to anticipate how it would bind to the active sites of the relevant enzyme. This study presents a straightforward approach for generating compounds with high yields and a fast response time that are helpful against AD272 (Table 11).
Kumar et al. (2020) investigated AD as a complicated neurological ailment in which single-targeted treatments failed to reduce or reverse disease progression. In recent years, multi-target medicines have been explored as a therapeutic strategy for the successful treatment of AD. A variety of (4-(pent-4-yn-1-yloxy)phenyl)-2-phenylpyrimidine analogs have been examined for their effects on AChE enzyme. The bulk of the synthesized compounds were discovered to be effective AChE inhibitor, with IC50 values in the low micromolar range. Analogs 259 and 260 were the most effective inhibitors of AChE enzyme. The former inhibited AChE with IC50 value of 2.84 µM, respectively. The AChE enzyme was likewise inhibited by 260, with IC50 value of 2.19 µM. Consequently, 259 and 260 may be employed as lead compounds in the design of more effective AD treatments273 (Table 11).
According to Kumar et al. (2021) AD is a complicated neurological condition characterised by impaired behavioural and cognitive functions. Multitarget-directed ligand (MTDL) approaches are a potential drug development paradigm that might lead to new AD therapy alternatives. A series of new MTDLs phenylsulfonyl-pyrimidine carboxylate derivatives were designed and synthesized for the treatment of AD. To investigate the likely binding affinity of the prepared pyrimidines, a molecular docking study was done, and the findings revealed a significant interaction with the active sites of AChE and BChE. The synthesized compounds show moderate to excellent in vitro enzyme inhibitory activity against AChE and BChE at nanomolar (nM) doses. The drug 262 inhibited AchE non-competitively with Ki 14 nM in an enzyme kinetics study. The hybrids generated demonstrated moderate to good in vitro inhibitory effectiveness against AchE and BchE at nanomolar doses. 261 and 262 substances inhibited AchE and BChE, respectively, with IC50 values of 47.33, 51.36 nM and IC50 159.43, 153.3 nM, respectively. Compounds 261 and 262 had the strongest AChE inhibitory action, which can be attributable to the presence of electron-donating dimethoxy and chloro groups, which enhance AChE binding affinity. These derivatives were able to attach to both the CAS and PAS of AChE at the same time274 (Table 11).
SAR studies of pyrimidines demonstrate that the various derivatives are potent against AChE and BChE enzymes (Fig. 21). All the structural features perform significant role in the inhibitory activity, though, the observed variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–CH3, –Et, –Ph, –OCH3, –CF3, –NH2, –CN, etc.) attached to the pyrimidine ring show higher AChE and BChE inhibitory abilities as compared to the bulky group (sulfonamide) present on the rings. All the presented analogs thus far have shown to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
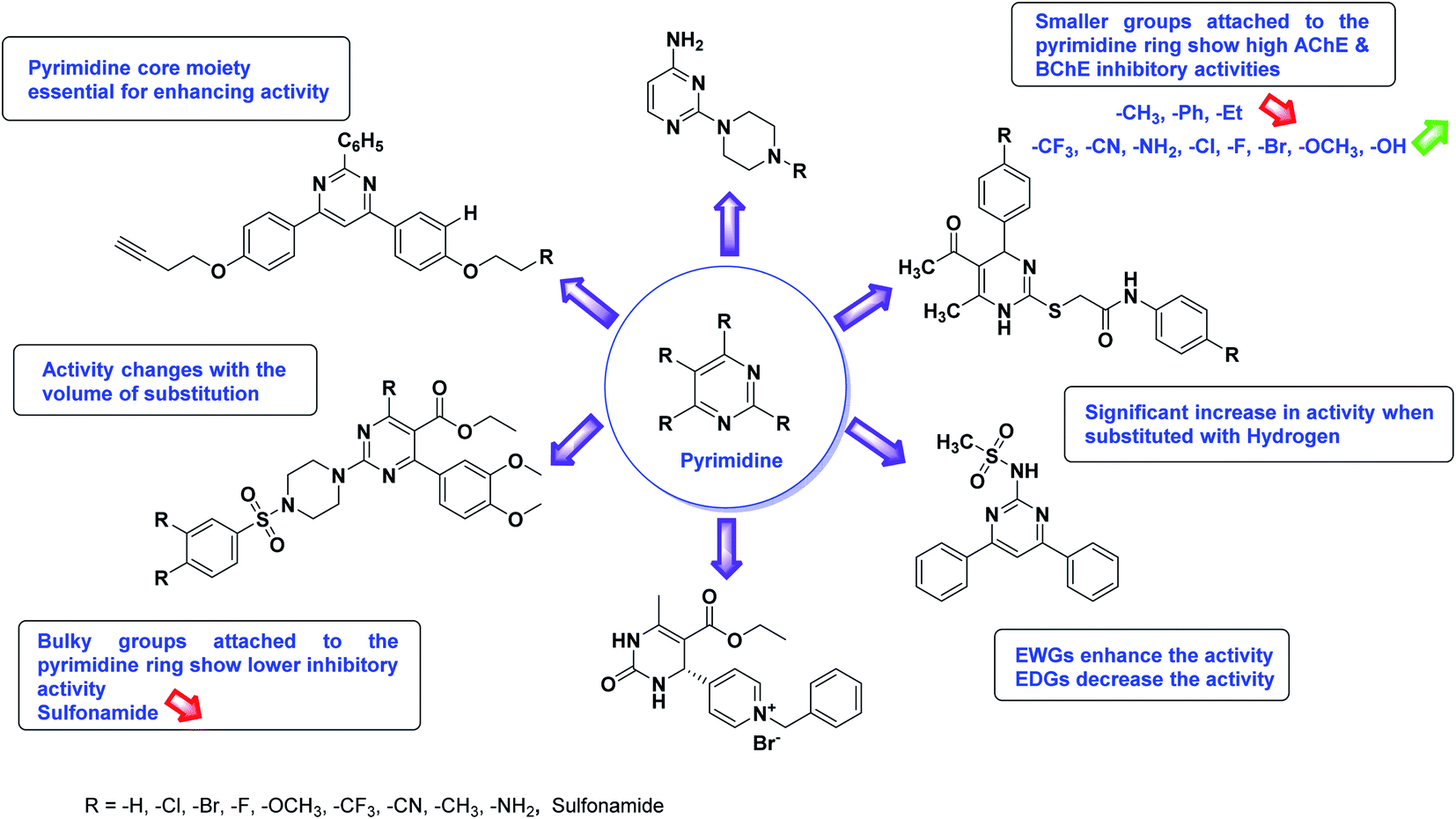 | ||
| Fig. 21 SAR analysis of different pyrimidine derivatives as AChE and BChE inhibitors. | ||
A series of new cyanopyridine–triazine hybrids were synthesized and assessed by Hoda et al. (2016) as multitarget agents. These molecules were created with the aid of computational tools and synthesized utilizing a feasible and efficient synthetic approach. The inhibitory potencies of the triazines against cholinesterases were investigated. Compounds 263 and 264 showed high inhibitory action against AChE, as well as good inhibition selectivity towards AChE over BChE, with IC50 values of 0.059 and 0.080 µM, respectively. According to molecular modelling, these compounds interacted with AChE's catalytic active site (CAS) and peripheral anionic site (PAS) simultaneously. The mixed type inhibition mode of compound 263 in kinetic studies further confirmed the dual binding nature. Furthermore, an in silico analysis of the absorption, distribution, metabolism, and excretion (ADME) profiles of the best compounds 263 and 264 revealed that they had drug-like properties. Overall, these cyanopyridine–triazine hybrids seem to be a decent candidate for further pharmacological investigation in the treatment of AD. Ellman's approach was utilized to evaluate the inhibitory activity of synthesized compounds against AChE (from Electrophorus electricus) and BChE (from horse serum) with donepezil (IC50 = 0.038 µM for AChE; 3.14 µM for BChE) and tacrine (IC50 = 0.13 µM for AChE; 0.054 µM for BChE) acting as positive controls. To improve the inhibitory effect of the generated compounds on ChEs, substituents with varied electronic properties were added to the core triazine scaffold. Compounds with 3-(trifluoromethyl) aniline connected to the triazine moiety inhibited AChE more efficiently than those with 2-fluoro and 3-chloro-4-fluoro substituents, i.e. 265 (IC50 = 1.412 µM) and 266 (IC50 = 1.701 µM), respectively. Within the subgroup of triazines containing 3-(trifluoromethyl) aniline, the inhibitory effect against AChE improved as the electron withdrawing properties of the 5-(substituted) aniline moiety increased. Compound 263, which included 3-chloro-4-fluoroanaline, showed the highest inhibitory potency (IC50 = 0.059 µM). Furthermore, substituting cyclopropylamine for the modified aniline group in compound 267 resulted in a 9-fold drop-in inhibitory activity (IC50 = 0.528 µM), compared to the most powerful compound 263. All the target compounds demonstrated a greater selectivity for AChE than for BChE, with the best derivative 263 having a 61-fold selectivity ratio. In addition, the inhibitory efficacy of 263 was shown to be comparable to conventional donepezil (IC50 = 0.038 µM) and superior to tacrine (IC50 = 0.038 µM). Additionally, docking experiments involving cholinesterases were carried out to better understand their method of action275 (Table 12).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 263 |  |
0.059 | — | 275 |
| 264 | 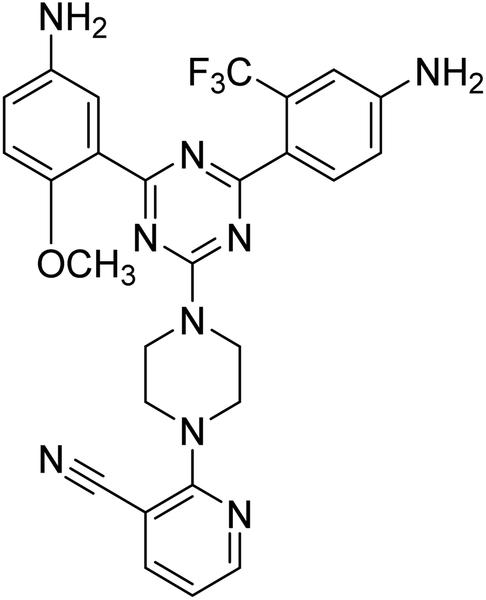 |
0.080 | — | 275 |
| 265 | 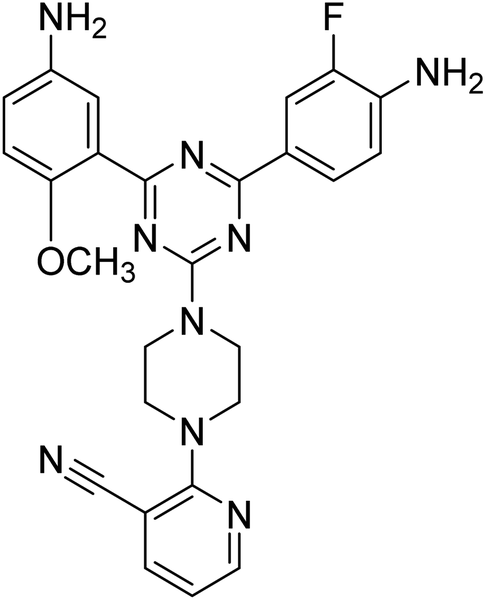 |
1.412 | — | 275 |
| 266 | 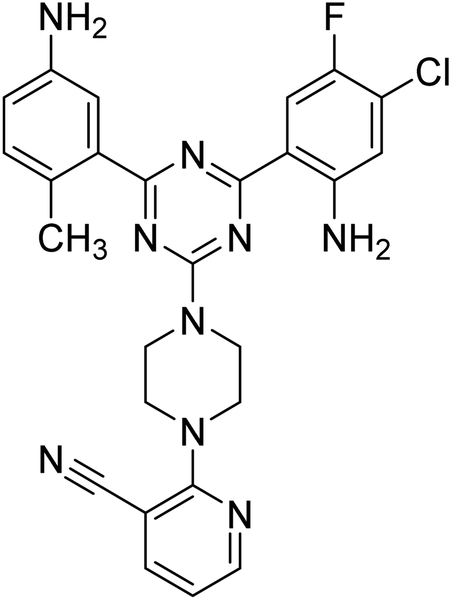 |
1.701 | — | 275 |
| 267 | 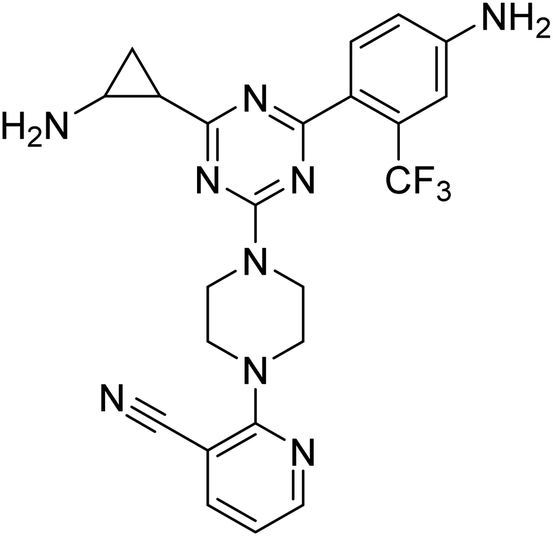 |
0.528 | — | 275 |
| 268 | 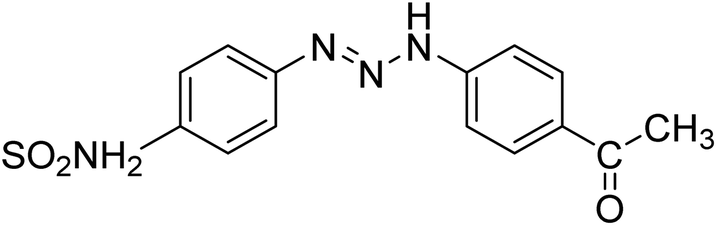 |
84.48 | — | 276 |
| 269 | 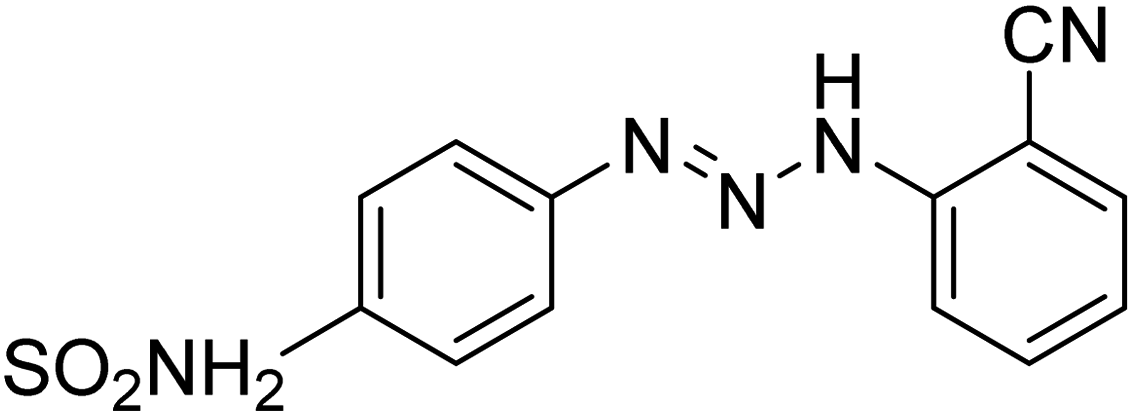 |
69.60 | — | 276 |
| 270 | 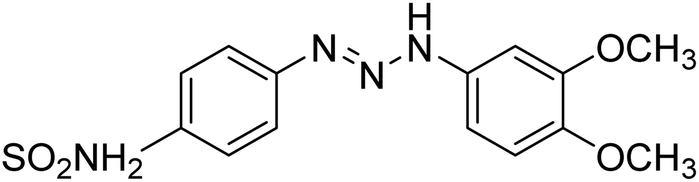 |
85.01 | — | 276 |
| 271 | 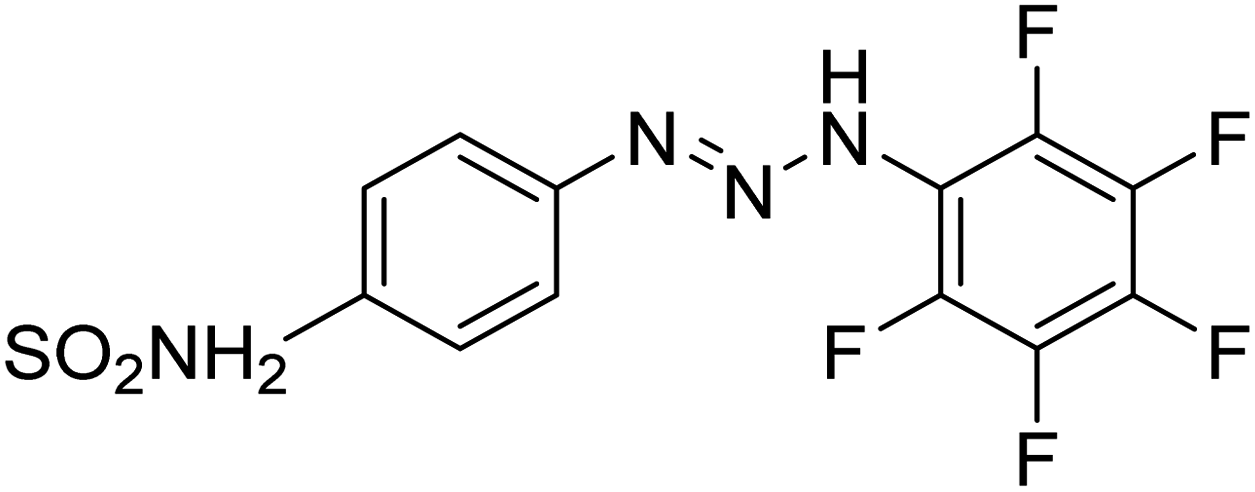 |
93.67 | — | 276 |
| 272 | 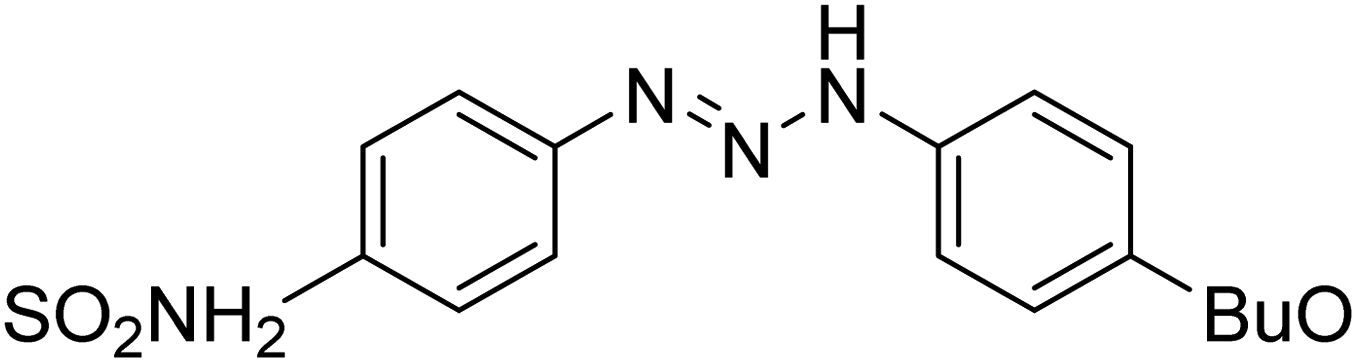 |
98.47 | — | 276 |
| 273 | 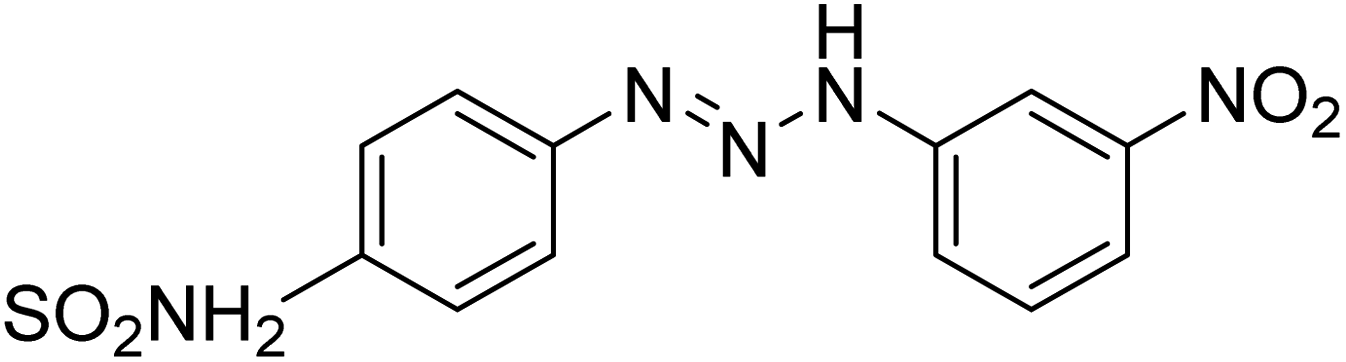 |
55.35 | — | 276 |
| 274 | 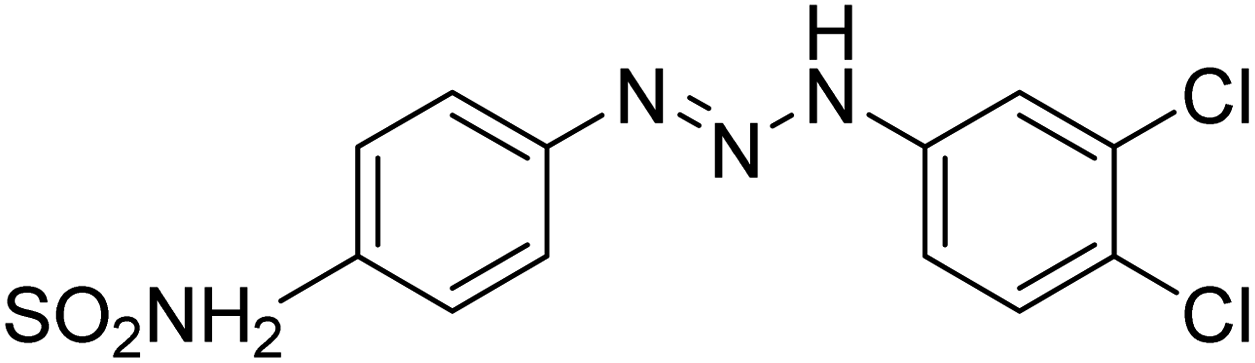 |
97.00 | — | 276 |
| 275 |  |
96.37 | — | 277 |
| 276 | 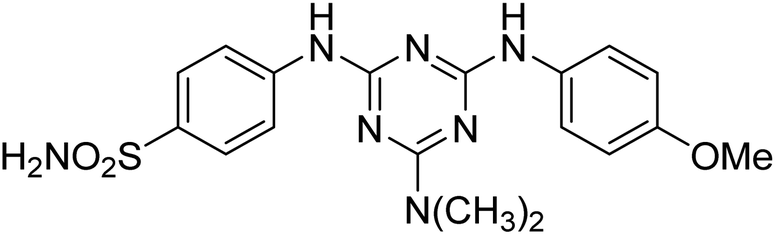 |
91.10 | — | 277 |
| 277 | 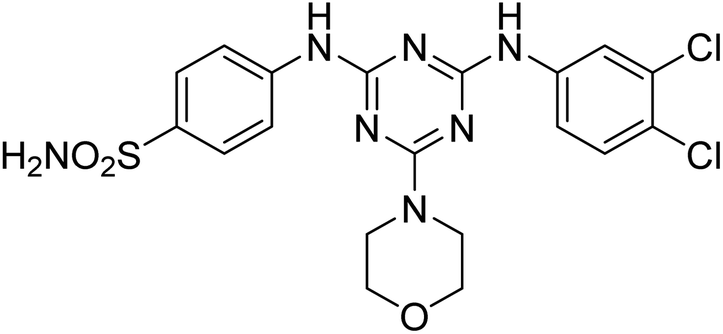 |
93.19 | — | 277 |
| 278 | 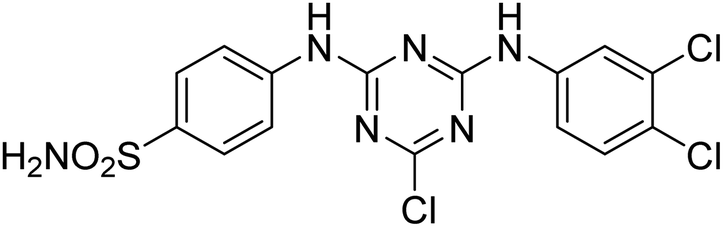 |
88.48 | — | 277 |
According to Boga et al. (2019), 1,3-diaryltriazenes are useful linkers for diverse pharmacological applications. In their investigation, the diazonium salt of sulfanylamide and substituted aryl amines were used to synthesize a series of 1,3-diaryltriazene sulfonamides. Most of the 1,3-diaryltriazene sulfonamides in the present series showed high activity against both cholinesterases, AChE and BChE. All chemicals, except for 268 and 269, showed stronger BChE inhibitory activity than AChE inhibition activity. With % inhibition values of 84.48, 85.01 and 93.67, the compounds 268, 270 and 271 inhibited AChE more efficiently than the standard drug galantamine. The compounds 269, 272, 273 and 274 were moderate inhibitors of this enzyme, with % inhibition values ranging from 55.35 to 69.60. With 98.47 and 97.00% inhibition, compounds 271 and 274 reduced BChE activity the highest at 200 µM. Because the AChE and BChE enzymes are associated to neurodegenerative diseases and their inhibition is important for various sorts of brain problems, these 1,3-diaryltriazene sulfonamides could serve as useful in in vivo research276 (Table 12).
Supuran et al. (2020) investigated novel triazine benzenesulfonamides containing aromatic amines, dimethylamine, morpholine, and piperidine as substituents on the 1,3,5-triazine moiety. They compounds were investigated as AChE, BChE, and tyrosinase inhibitors. Compounds 275, 276 and 277 demonstrated inhibitory activity against AChE with % inhibition values >90. Most of the synthesized compounds also successfully inhibited BChE, with inhibitory potencies >90%. The most potent AChE inhibitors were compounds 275, 276 and 277 with % inhibition values of 96.37, 91.10 and 93.19, respectively, exceeding the standard medicine galantamine. The bulk of the generated compounds, on the other hand, effectively inhibited the BChE enzyme. On the % inhibition scale, several of the remaining compounds 275, 276, 277 and 278 exhibited % inhibition values of 87.44, 88.76, 89.21 and 88.48, respectively, which were quite comparable in values to the standard drug. The three compounds have anticholinesterase activity, suggesting that they may be produced and used as effective cholinesterase inhibitors277 (Table 12).
SAR studies of triazines demonstrate that the different derivatives are active as AChE and BChE inhibitors (Fig. 22). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aromatic rings. The smaller groups (–Ph, –Me, –Et, –OMe, –CF3, –OH, Cl, –Br, –CN, etc.) attached to the triazine ring show higher AChE and BChE inhibitory abilities as compared to the bulky groups present on the rings. All the presented analogs thus far have shown excellent ChE inhibitory abilities with less toxic side effects. Moreover, these analogs are easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
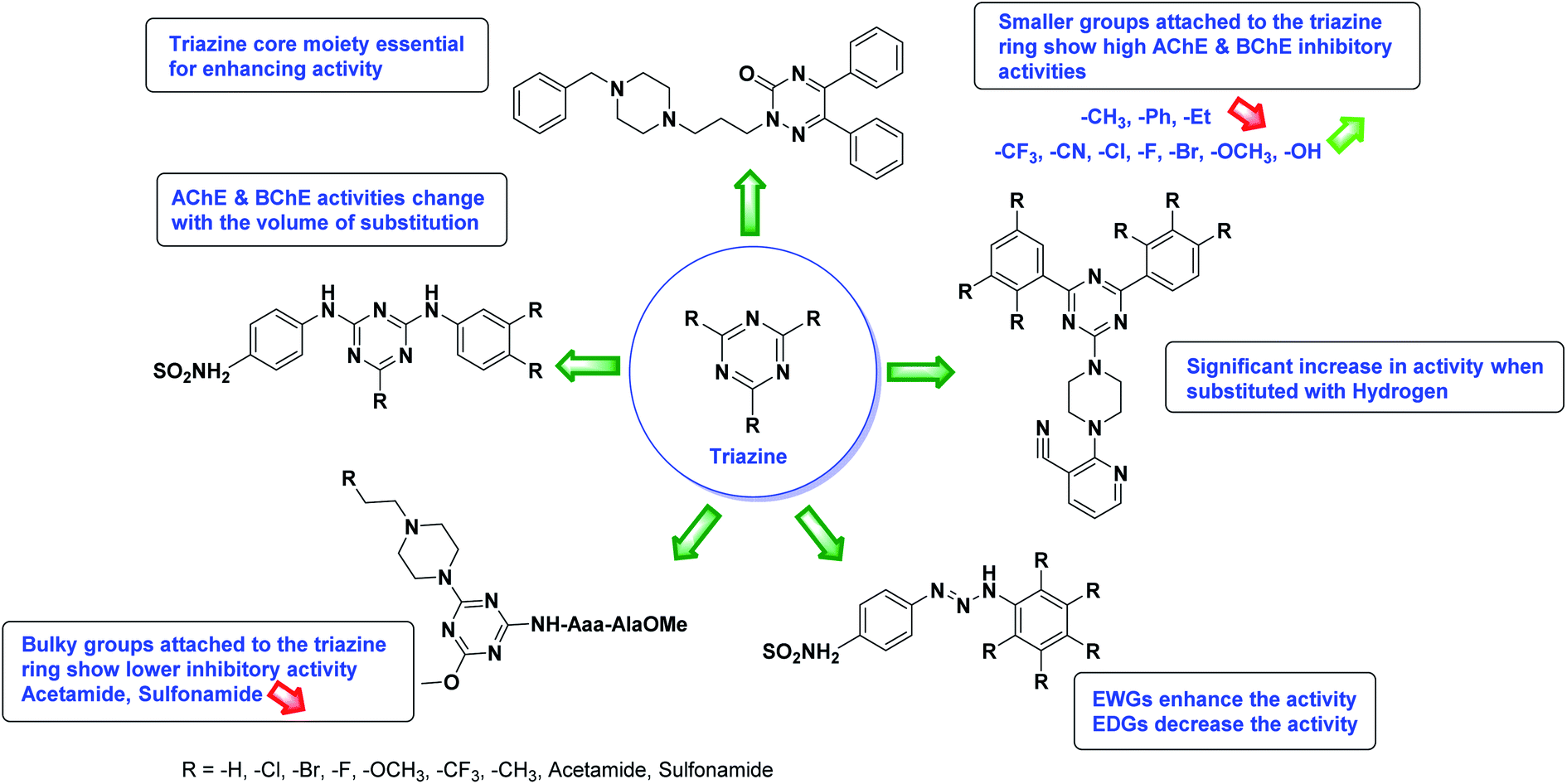 | ||
| Fig. 22 SAR analysis of different triazine derivatives as AChE and BChE inhibitors. | ||
Alptüzün (2016) used a series of pyridinium salts with alkylphenyl groups at position 1 and hydrazone structure at the 4th position of the pyridinium ring to inhibit both AChE and BChE enzymes. The inhibitory activity of cholinesterase was measured using Ellman's colorimetric method (ChE). All the compounds displayed considerable AChE and BChE inhibitory activity when compared to galantamine, the reference drug, and some of them had exceptional anti-AChE activity. The series of title compounds with a benzofuran aromatic ring had the highest inhibitory action on both AChE and BChE enzymes. With an IC50 value of 0.23 µM, 1-(3-phenylpropyl)pyridinium bromide 280 was the most effective molecule against AChE (hAChE), whereas 4-[2-(1-(benzofuran-2-yl)ethylidene)hydrazinyl]-1-phenethylpyridinium bromide 279, with an IC50 of 0.95 µM, was the most effective compound against BChE. Pyridinium ions 279 and 280 were also found to be more active than galantamine (AChE (hAChE) IC50 0.43 µM; BChE IC50 14.92 µM). Compound 280, which demonstrated the highest inhibitory action against AChE, was the subject of molecular docking investigations. Several hydrazone-containing pyridinium salts were tested for their ability to inhibit cholinesterase. All compounds demonstrated inhibitory activity ranging from excellent to moderate on both AChE and BChE enzymes. In the title compounds, the length of the side chain and its hydrophobic feature appear to be important for AChE activity. The scaffold of benzofuran-pyridiniumhydrazone can be utilized as a starting point for further study and a template for further structural adjustments278 (Table 13).
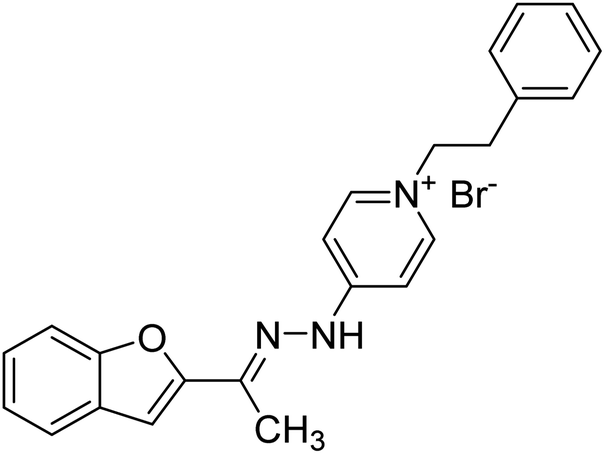
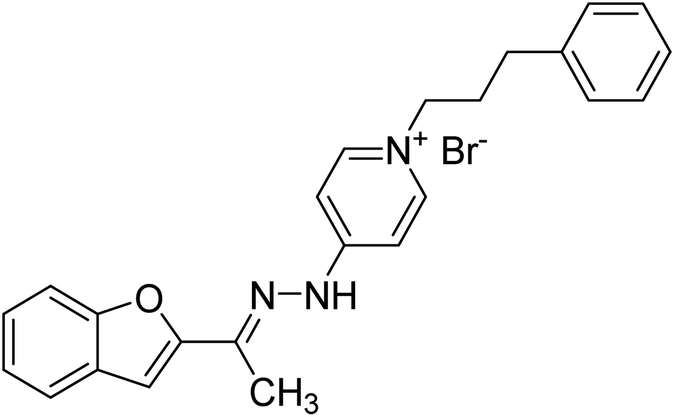
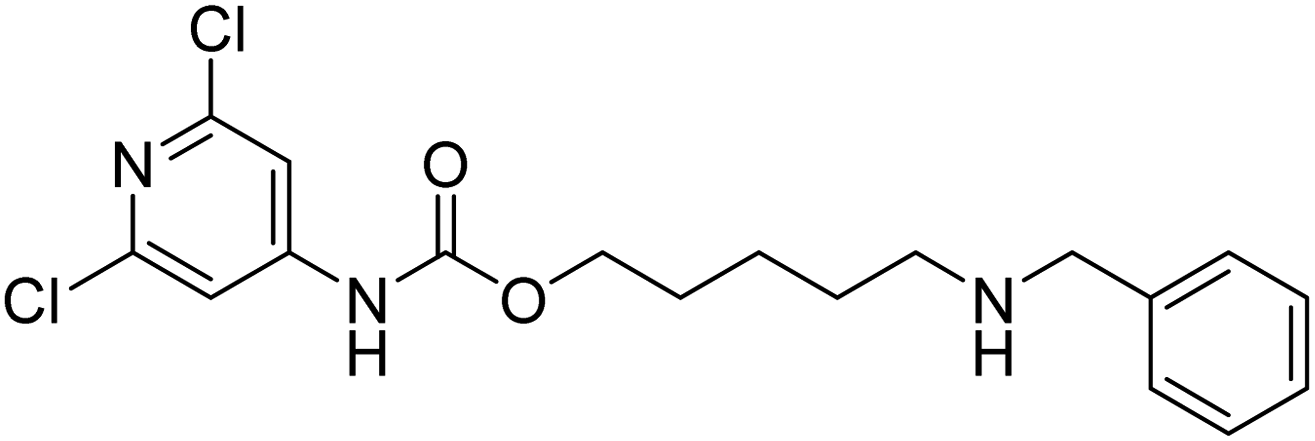
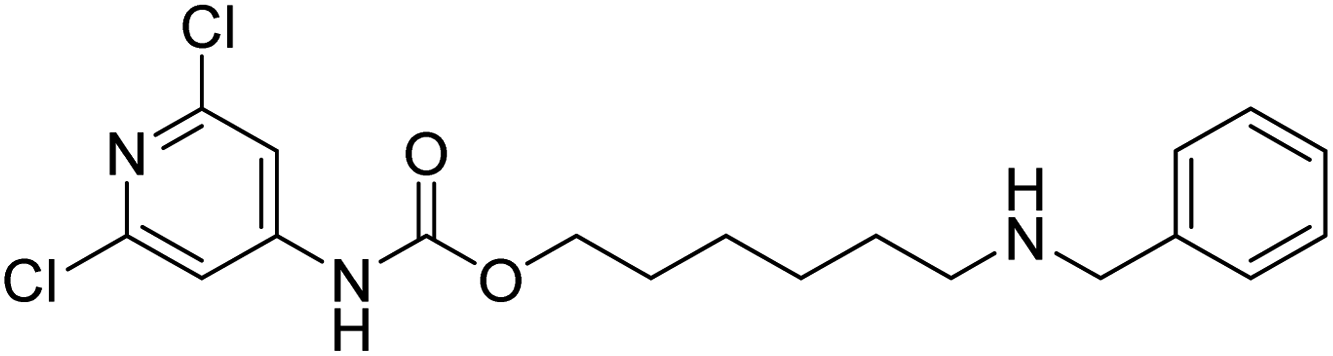
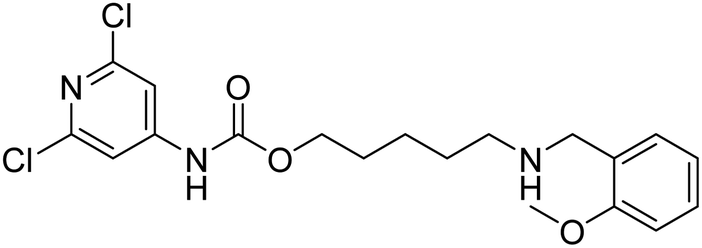
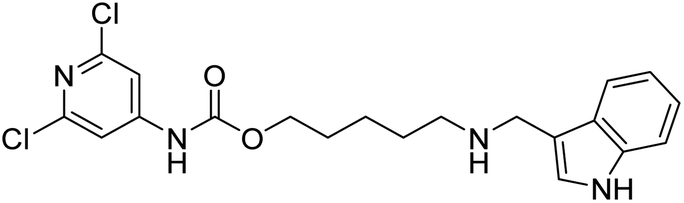
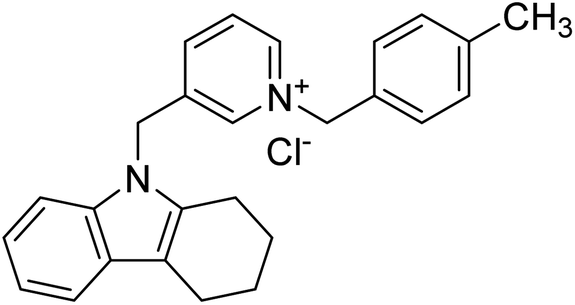
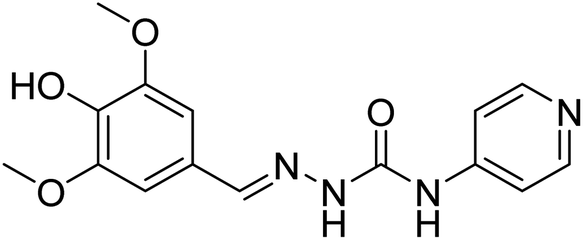
Bergamini et al. (2017) developed and synthesized a novel class of pyridine compounds with carbamic or amidic functions that can inhibit cholinesterase. The molecules feature two aromatic ends connected by a flexible alkyl chain of variable length. AChE and hAChE, as well as BChE and hBChE, were used to evaluate the produced compounds. Compound 281 was the best inhibitor of hAChE (IC50 0.153 nM), whereas compound 282 was the most effective inhibitor of hBChE (IC50 0.828 nM). According to molecular docking study, 281 may bind AChE through interacting with both CAS and PAS, confirming a mixed-type inhibition mechanism. The most active compounds against EeAChE were 281 and 282, which had Ki values of 38.6 nM and 52.8 nM, respectively. Further research on the human isoforms of AChE and BChE revealed that, despite some differences in enzyme source, both carbamate and amide derivatives were effective inhibitors of hChE, with 283 being the most potent inhibitor of hAChE (IC50 153 nM) and 284 being the best inhibitor of hBChE (IC50 828 nM). Molecular docking experiments on hAChE (co-crystallized with donepezil, PDB code 4EY7) provided more insight on the most powerful ChEI in the series' method of interaction. The study found that 281 may form a variety of connections with the amino acids of the CAS area via the benzyl group and protonated amine nitrogens, whereas the 2,6-dichloro-pyridine moiety can contact Trp286 inside the PAS. In the amyloid self-aggregation experiment, 281, the most potent hAChEI in this investigation, inhibited Aβ42 self-aggregation by 26.5% at 50 nM. This compound was also less toxic to T67 and HeLa cells, with IC50 values of 24.1 and 20.8 nM, respectively. Finally, it appears that 281 has a high inhibitory efficacy against hAChE, as well as a high selectivity against hBChE and the ability to prevent Aβ42 self-aggregation. These findings, together with the low toxicity and strong projected BBB permeability properties, make this chemical a promising candidate for the creation of novel multifactorial cholinesterase inhibitors that might help cure AD279 (Table 13).
According to Amini et al. (2018), BChE inhibitors have emerged as a viable target for AD therapy. A class of dual binding site BChE inhibitors was designed and synthesized using 2,3,4,9-tetrahydro-1H-carbazole linked benzyl pyridine moieties. In an in vitro investigation, all the prepared compounds were found to be selective and potent BChE inhibitors. The most potent BChE inhibitor with mixed-type inhibition was compound 285 (IC50 0.088 µM). According to docking studies, 285 is a BChE inhibitor with two binding sites. Furthermore, 285 pharmacokinetic characteristics were in accordance with Lipinski's rule. In addition, 285 is neuroprotective and inhibits the enzyme β-secretase (BACE1). This compound may also inhibit AChE-induced and self-induced Aβ peptide aggregation at doses of 100 µM and 10 µM. The findings suggest that selective BChE inhibitors hold therapeutic promise in the treatment of AD. Because intact BChE compensates for the loss of AChE activity as AD progresses, they may have therapeutic use in the treatment of the condition. According to molecular modelling studies, compound 285 had strong interactions with BChE's choline binding site, peripheral binding site, and catalytic site. It was long enough to contact the acyl pocket as well. BACE1 inhibitory and neuroprotective effects were also found in compound 285. The pharmacokinetic properties of 285 were also confirmed using Lipinski rules of 5. In summary, the study has discovered new highly selective BChE inhibitors with therapeutic promise for AD therapy280 (Table 13).
Shirsat et al. (2018) synthesized and characterized new 4-aminopyridine analogs using analytical methods such as UV, IR, NMR, elemental analysis, and assessed their inhibitory character on AChE activity using molecular docking studies and Ellman's spectrophotometric method for enzyme kinetics. The antiamnesic and cognition-enhancing properties of the synthesized analogues were then evaluated using a passive avoidance test. The AChE inhibitory activity of all produced analogs was measured using the Ellman spectrophotometric technique. The inhibitory concentration of produced analogs (IC50) for inhibiting AChE was calculated using Graph Pad Prism. The IC50 values for all the analogs ranged from moderate to excellent. Compounds 286 and 287 produced IC50 values of 6.32 µM and 5.58 µM, respectively, compared to rivastigmine (6.15 µM). The most active compounds 286 and 287 inhibited AChE in a non-competitive manner (Ki = 11.23 and 6.44, respectively). The non-competitive inhibition is attributed to a probable contact of the analog with the peripheral anionic site (PAS) of AChE and docking investigations have supported this. In vitro studies of the synthesized analogs revealed that compounds 286 and 287 had the highest activity when compared to the standard drug rivastigmine, whereas enzyme kinetic studies demonstrated a non-competitive inhibition of AChE, which was attributed to a possible interaction of the analogs with AChE's peripheral anionic site (PAS) and confirmed by molecular docking studies. The hydroxyl group of one of these compounds' phenyl rings was observed forming an H-bond with Tyr-70 in docking studies, and Tyr-70 appears to play a dual role in the active centre: (a) its hydroxyl appears to maintain the functional orientation of Phe-288 and Tyr-70 by hydrogen bonding, and (b) its aromatic moiety appears to maintain the functional orientation of the anionic subsite Trp-84. Compounds 286 and 287 were discovered as the most potent species in an AChE inhibition and passive avoidance test, which could lead to the discovery of new cognition enhancers soon281 (Table 13).
The SAR studies of the aforementioned pyridines demonstrate that the different derivatives are potent as AChE and BChE inhibitors (Fig. 23). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–CH3, –OCH3, –CF3, –F, –Cl, –NO2, –OH, etc.) attached to the pyridine ring show higher AChE and BChE inhibitory abilities as compared to the bulky group (benzofuran) present on the rings. All the presented analogs thus far have shown good to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
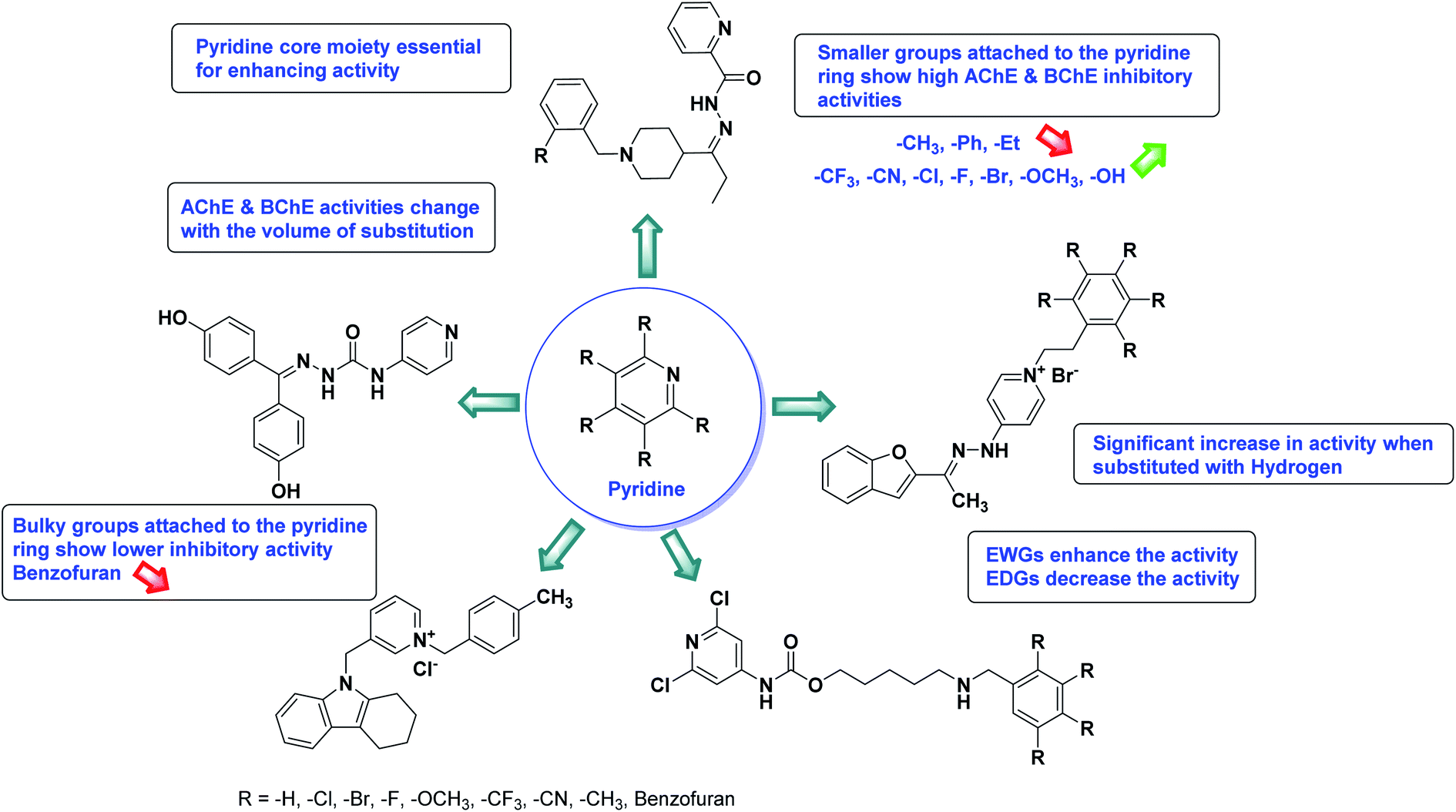 | ||
| Fig. 23 SAR analysis of different pyridine derivatives as AChE and BChE inhibitors. | ||
Sultana et al. (2017) described using the N1-substitution of the 2,3-dihydroquinazolin-4(1H)-one nucleus to make AChE inhibitors that were effective. A set of N-alkylated/benzylated quinazoline derivatives were made and evaluated to see if they could inhibit cholinesterases. N-Alkylation improved the activity of a series of compounds previously described (N-unsubstituted). All the substances inhibited both enzymes in the micromolar to submicromolar range. According to the SAR of the synthesized derivatives, N-benzylated compounds have higher activity than N-alkylated compounds. The N-benzylated compounds 288 and 289 were found to be extremely effective against AChE, with IC50 values in the micromolar range (0.8 µM and 0.6 µM, respectively). According to ADMET computational projections, all the substances exhibited good pharmacokinetic properties and no AMES toxicity or carcinogenicity. Furthermore, all the chemicals were expected to be absorbed and cross the blood–brain barrier in humans. Generally, the prepared compounds have paved the path for the development of new cholinesterase inhibitors282 (Table 14).
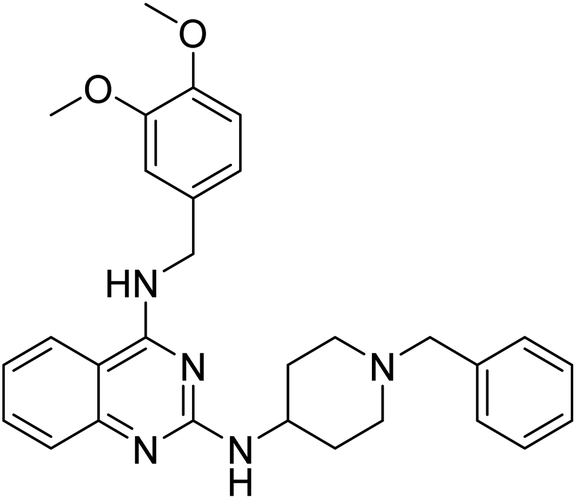
Rao et al. (2017) created and appraised a series of 2,4-disubstituted quinazoline analogs as a new type of AD multi-targeting therapy (AD). The results of the biological assays show that several quinazoline derivatives can inhibit both AChE and BChE enzymes (IC50 range 1.6–30.5 µM). Compound 290 was reported to be a dual cholinesterase inhibitor (AChE IC50 = 14.3 µM; BChE IC50 = 8.3 µM) that also inhibited Aβ aggregation (Aβ40 IC50 = 2.3 µM). A 2,4-disubstituted quinazoline ring can be utilized as a template for generating multi-targeting drugs to treat AD, according to these thorough SAR investigations. Compound 290 demonstrated a 4.7-fold increase in AChE potency and a 1.4-fold rise in BChE potency when compared to the pyrimidine derivative previously disclosed (AChE IC50 = 9.90 µM; BChE IC50 = 11.40 µM)283 (Table 14).
Sarfraz et al. (2017) developed and evaluated several cholinesterase inhibitors based on 2,3-dihydroquinazolin-4(1H)-one analogs. In vitro experiments demonstrated that all compounds were active against both AChE and BChE enzymes, where analogs with a longer alkyl chain at the C-2 position displayed stronger inhibitory activity than galantamine. Bromo derivatives were also more active than their non-substituted counterparts and nitro derivatives. Dihydroquinazolinone 291 proved a potent cholinesterase (AChE/BChE) inhibitor with a higher selectivity for BChE, with IC50 values of 4.8 µM for AChE and 11.1 µM for BChE. Synthetically, the reaction of 2-aminobenzamide with butyrylchloride produces quinazolinone, which is subsequently brominated to generate inhibitor 291 (ref. 284) (Table 14).
SAR studies of quinazoline demonstrate that the different derivatives are potent AChE and BChE inhibitors (Fig. 24). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–F, –Cl, –NO2, –OH, –CH3, –OCH3, –CF3 etc.) attached to the quinazoline ring show higher AChE and BChE inhibitory abilities as compared to the bulky group (triazole) present on the rings. All the presented analogs thus far have shown good to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
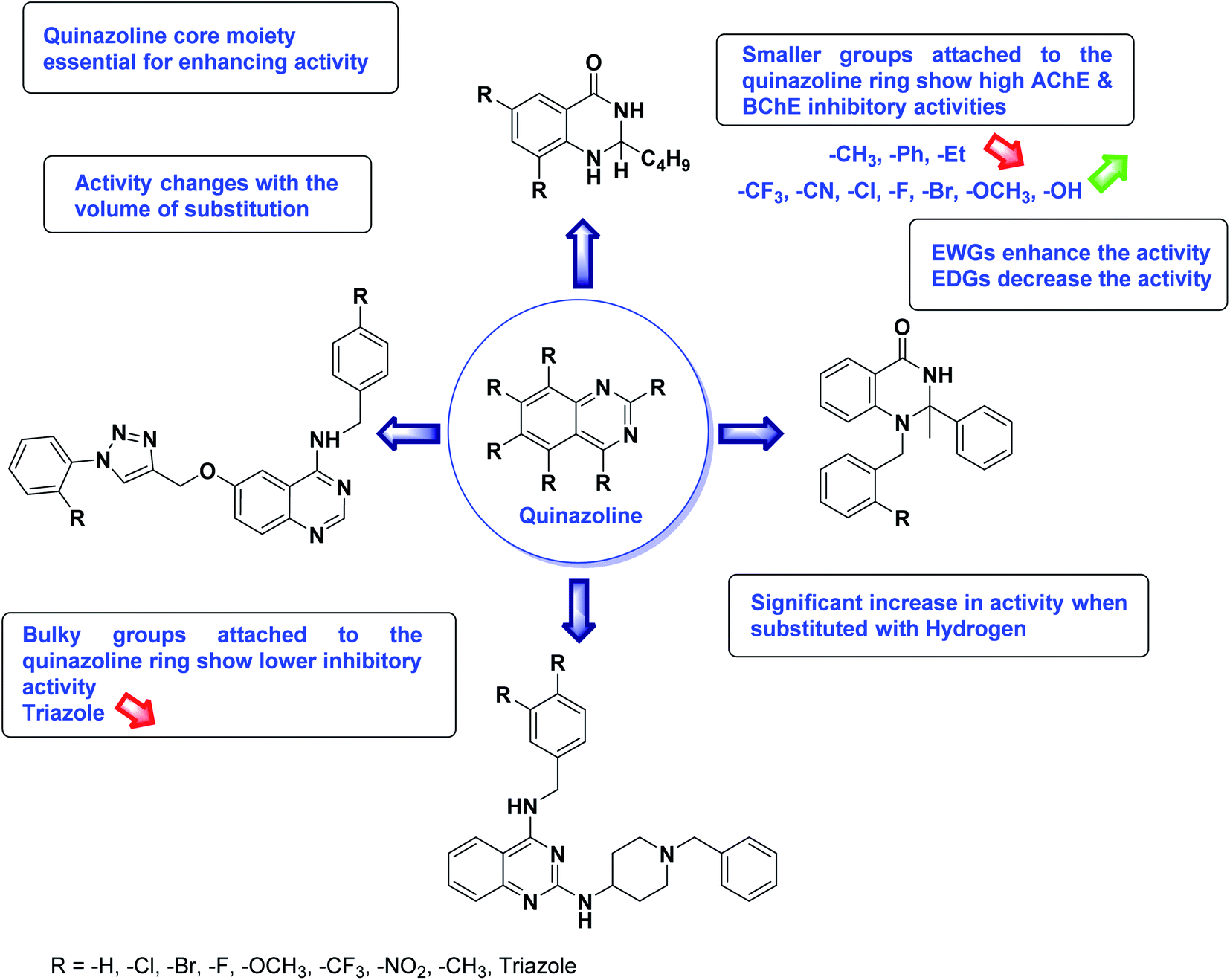 | ||
| Fig. 24 SAR analysis of different quinazolines derivatives as AChE and BChE inhibitors. | ||
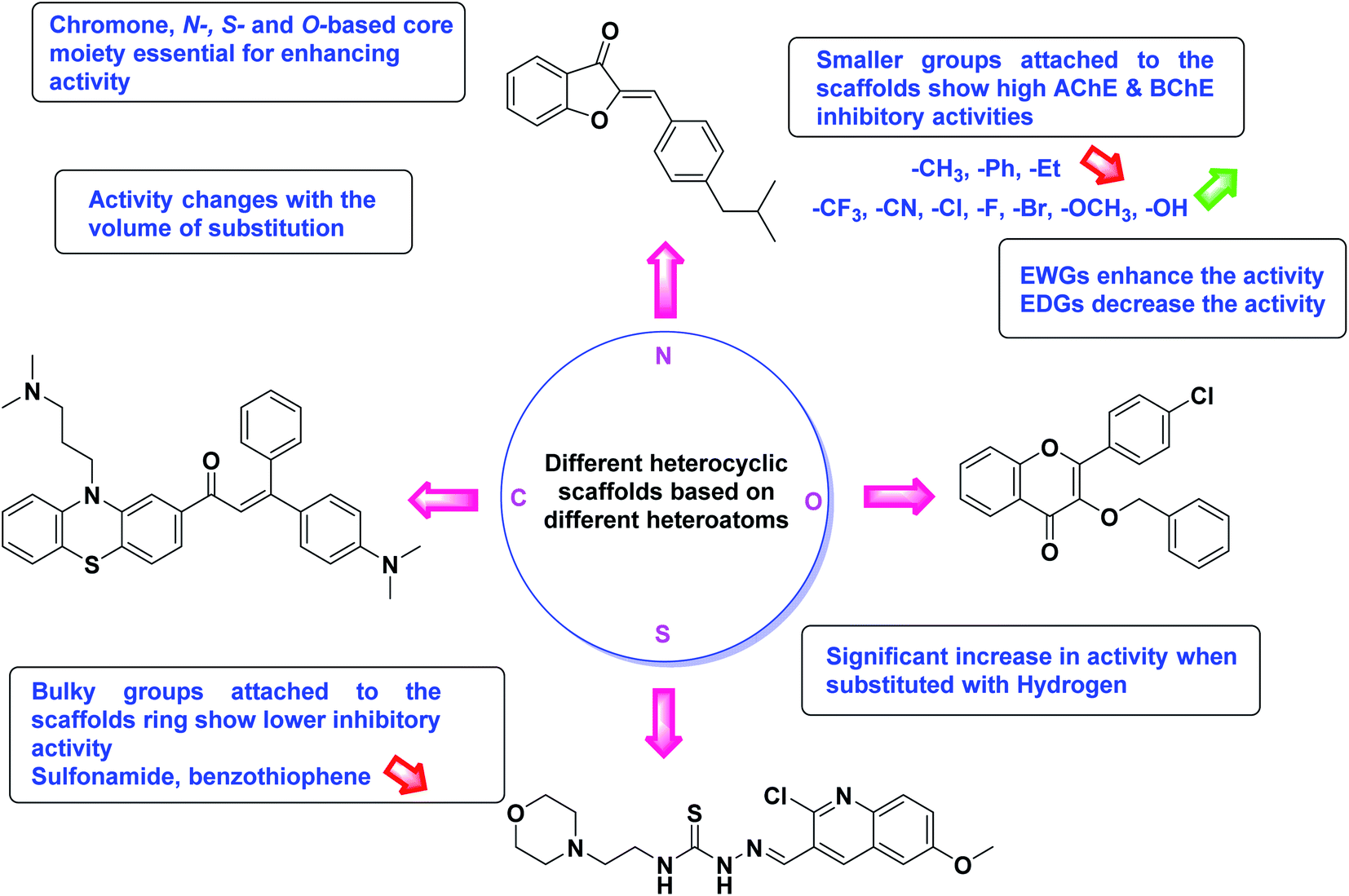 | ||
| Fig. 25 SAR analysis of different N, O and S based heterocycles as AChE and BChE inhibitors. | ||
Gulcin et al. (2017) designed and synthesized a series of new 1-(4-(3-(aryl)acryloyl)phenyl)-1H-pyrrole-2,5-diones and screened them for ChE inhibitory activities. All the target compounds exhibited AChE inhibiting activity with IC50 values in the range of 139.43–244.70 nM. Chalcone-imide derivatives effectively inhibited AChE enzyme with Ki values in the range of 70.470–229.42 nM. All the synthesized chalcone-imide derivatives showed similar inhibition profile against AChE. Compound 292, which showed the weakest AChE inhibition, had two times AChE inhibition effects than that of tacrine. The best inhibition for AChE enzyme was determined by compound 293 with Ki value of 70.470 nM (ref. 285) (Table 15).
| Compound no. | Chemical structure | IC50 values (µM) | References | |
|---|---|---|---|---|
| AChE | BChE | |||
| 292 | 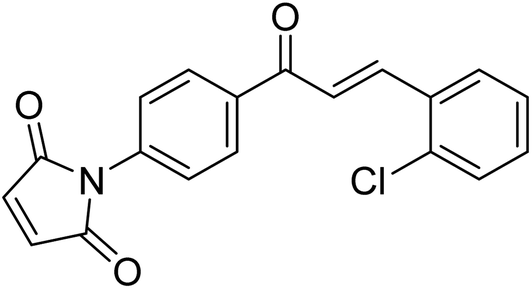 |
210.56 | — | 285 |
| 293 | 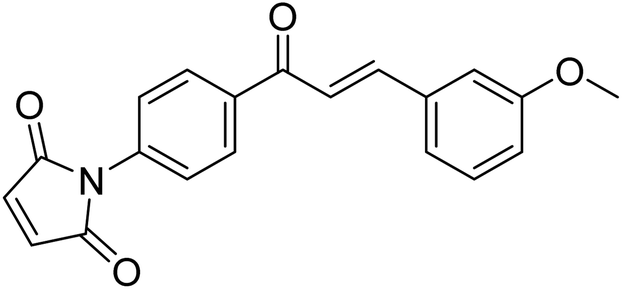 |
70.470 | — | 285 |
| 294 | 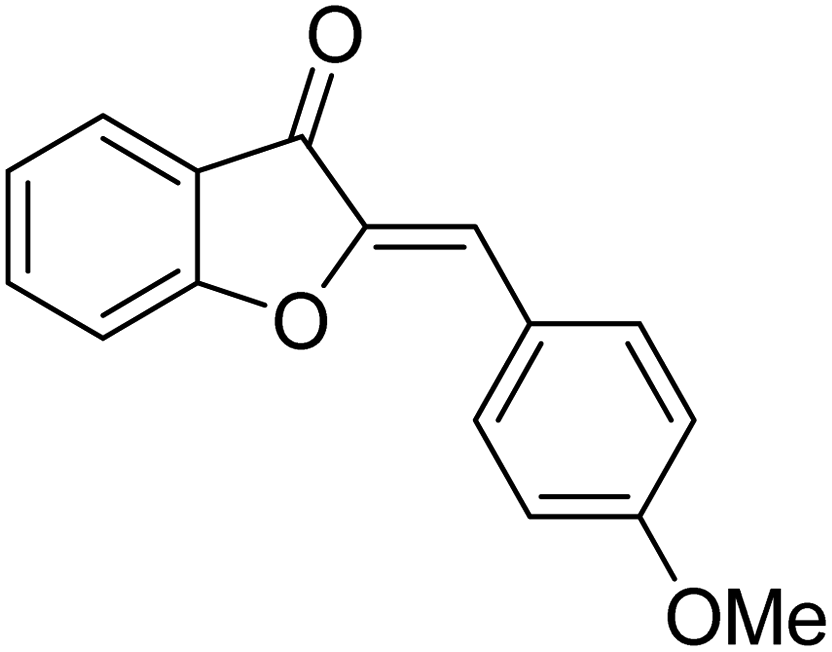 |
1.26 | — | 19 |
| 295 | 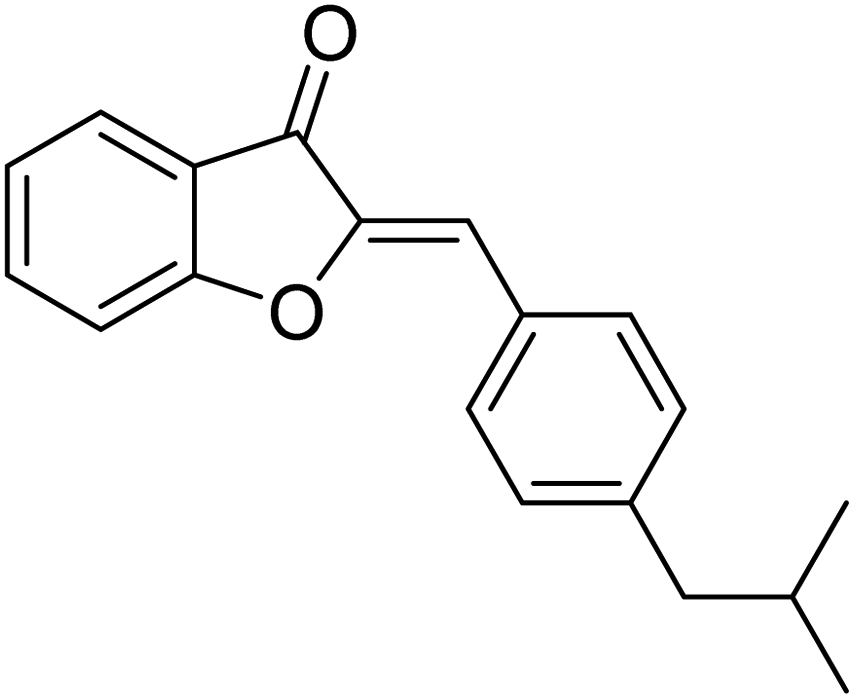 |
0.98 | 1.02 | 19 |
| 296 |  |
6.39 | — | 19 |
| 297 |  |
5.27 | — | 19 |
| 298 |  |
2.05 | 5.42 | 18 |
| 299 |  |
10.2 | 3.12 | 18 |
| 300 | 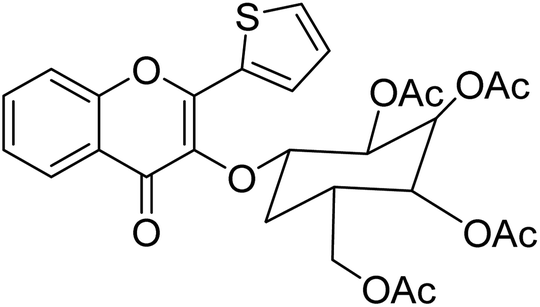 |
18.52 | 25.51 | 18 |
| 301 | 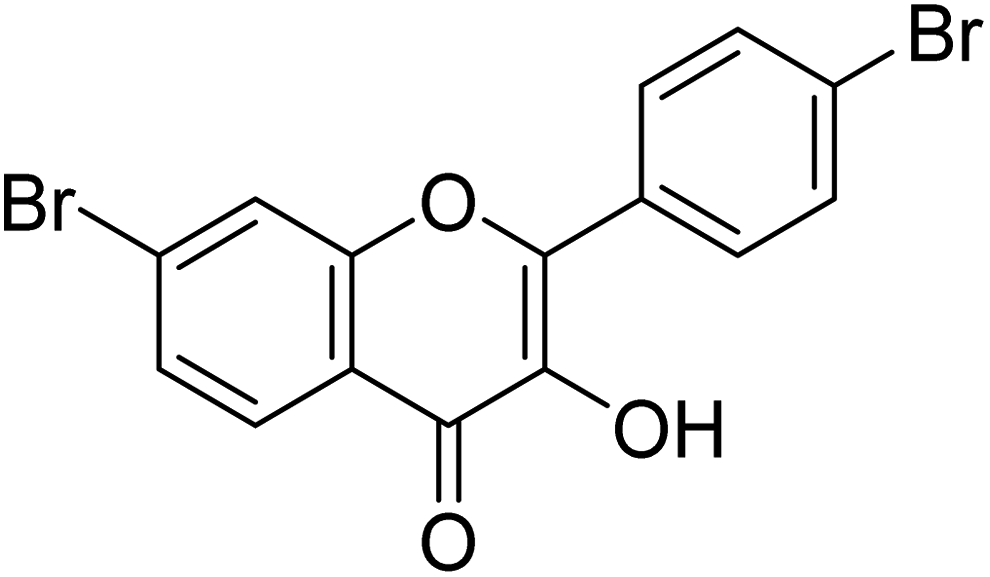 |
2.01 | 1.40 | 13 |
| 302 | 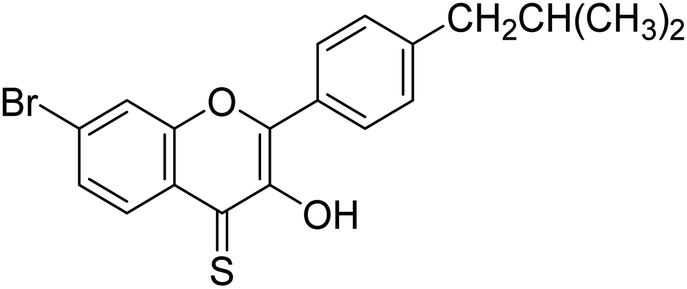 |
0.08 | 0.12 | 13 |
| 303 | 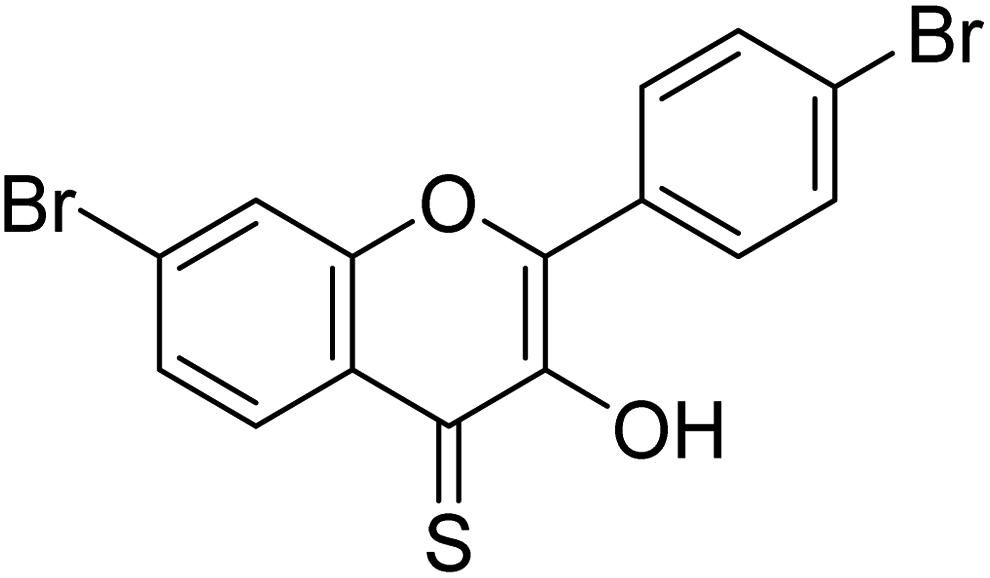 |
0.07 | 0.15 | 13 |
| 304 | 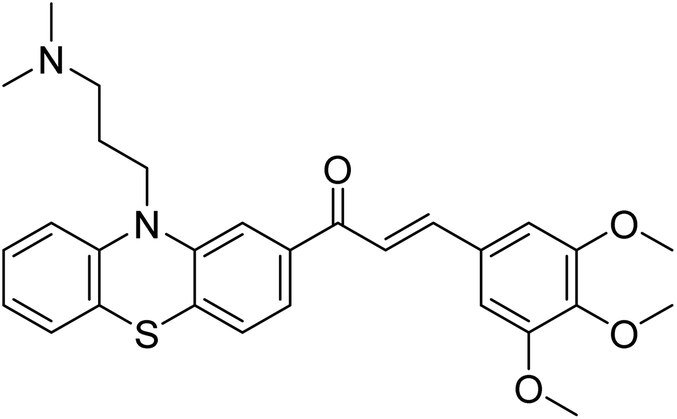 |
3.63 | — | 286 |
| 305 | 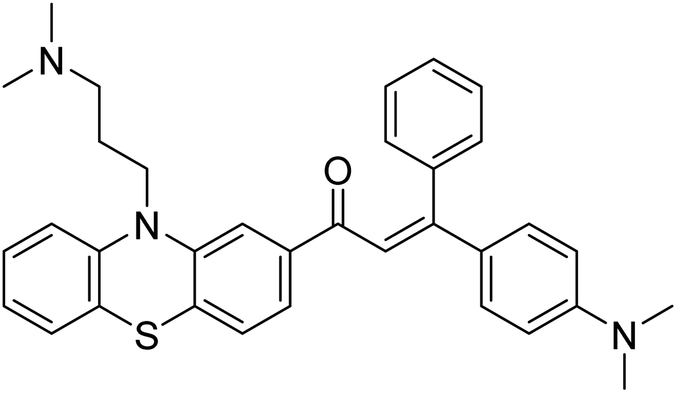 |
1.10 | — | 286 |
| 306 | 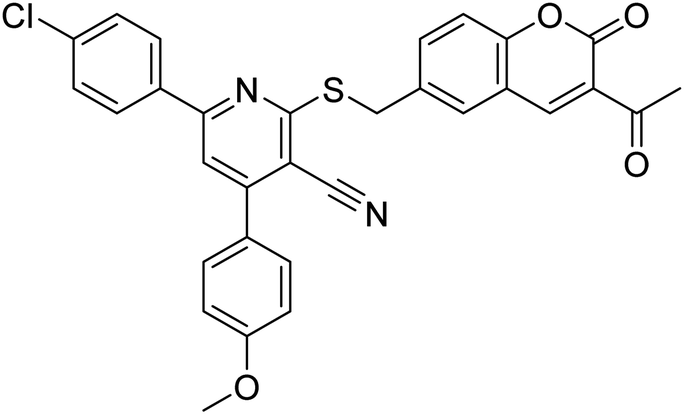 |
324 nM | — | 287 |
| 307 | 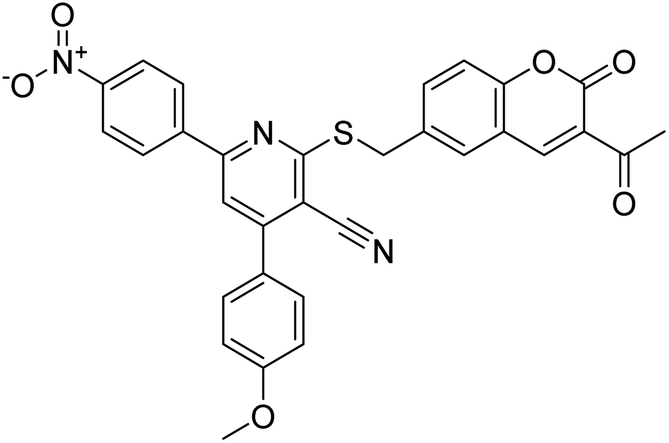 |
226 nM | — | 287 |
| 308 | 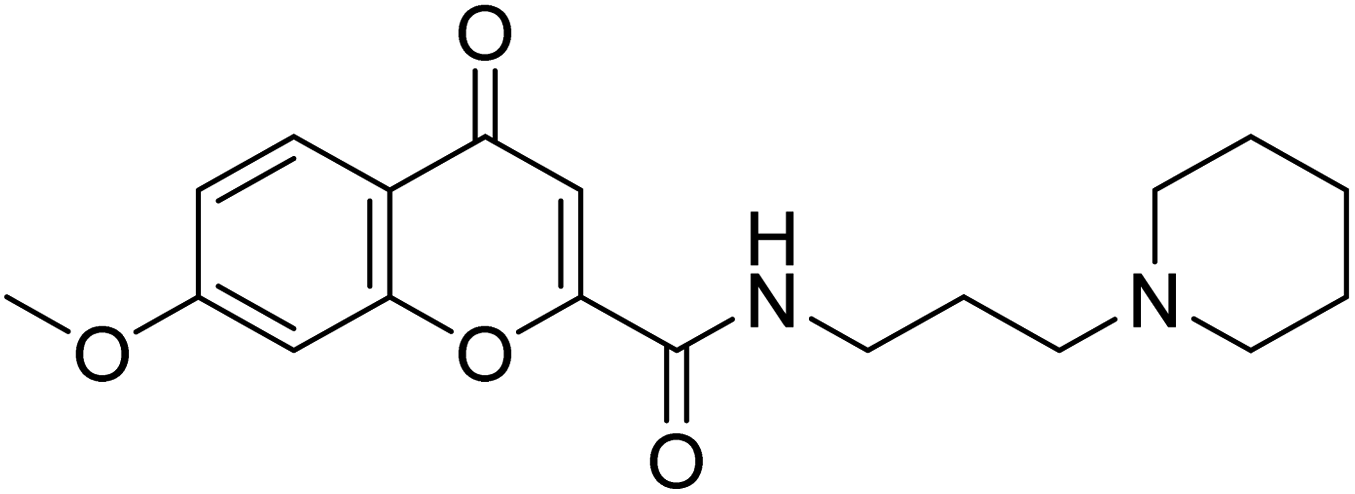 |
0.09 | 27.91 | 288 |
| 309 | 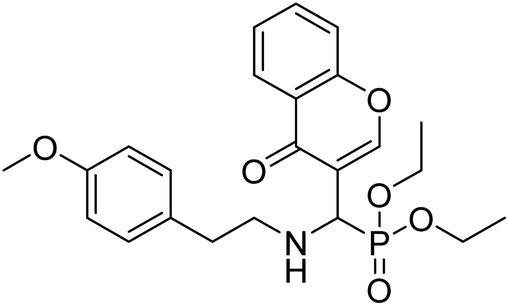 |
0.103 | — | 289 |
| 310 | 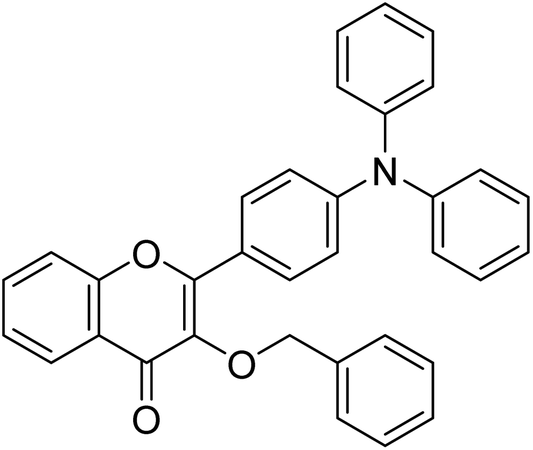 |
0.05 | 0.09 | 21 |
| 311 | 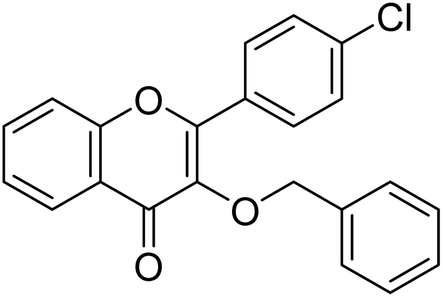 |
0.07 | — | 21 |
| 312 | 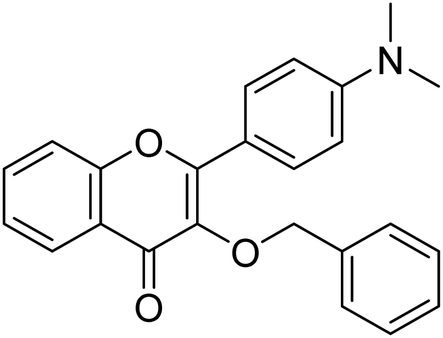 |
0.08 | — | 21 |
| 313 | 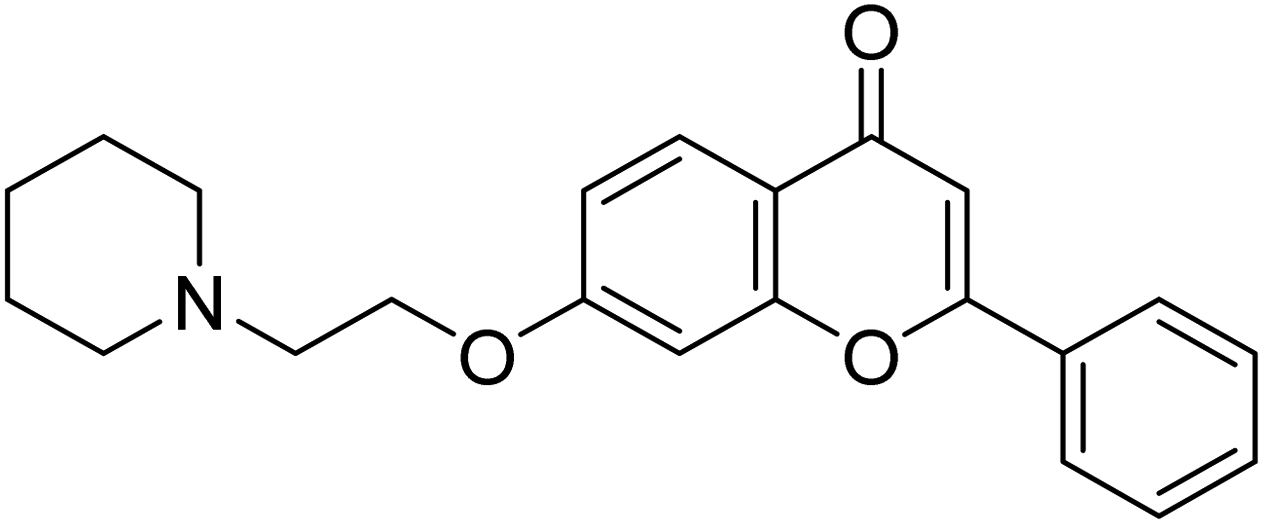 |
1.29 µmol L−1 | — | 290 |
| 314 | 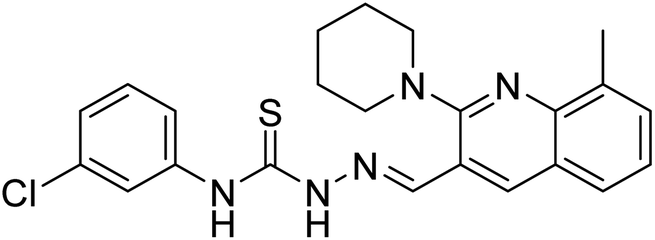 |
9.68 | 11.59 | 291 |
| 315 | 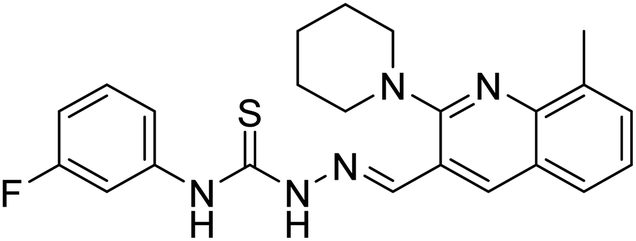 |
15.8 | — | 291 |
| 316 | 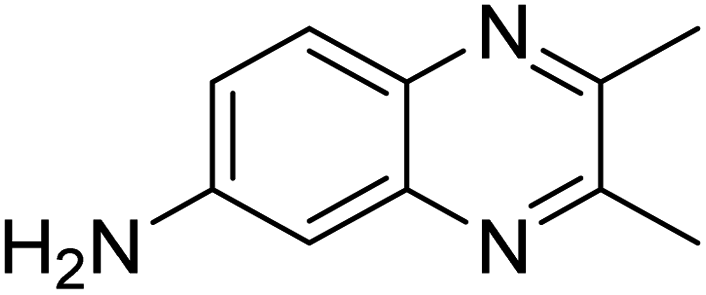 |
0.077 | — | 292 |
| 317 | 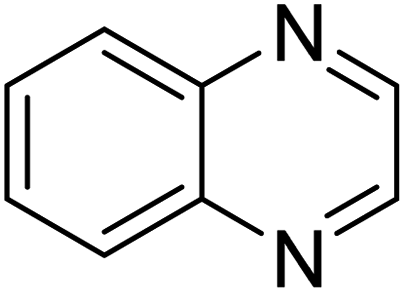 |
40.64 | — | 292 |
| 318 | 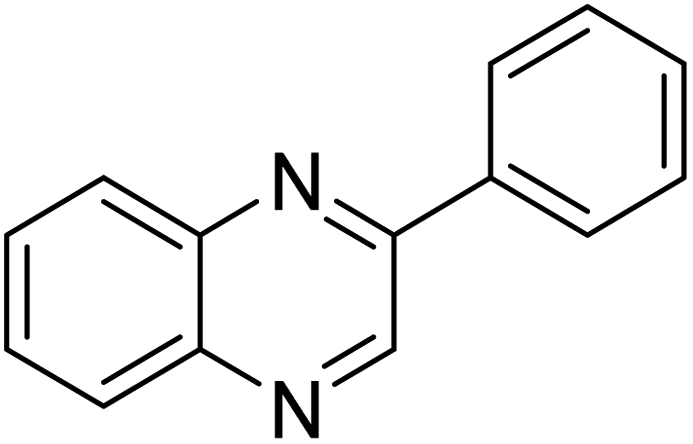 |
14.91 | — | 292 |
| 319 | 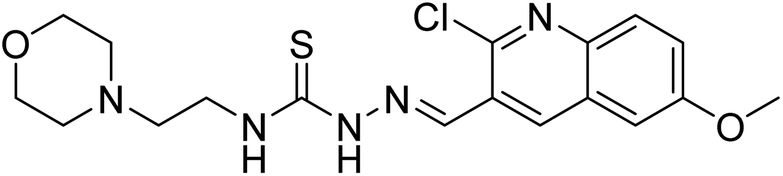 |
0.12 | — | 293 |
| 320 | 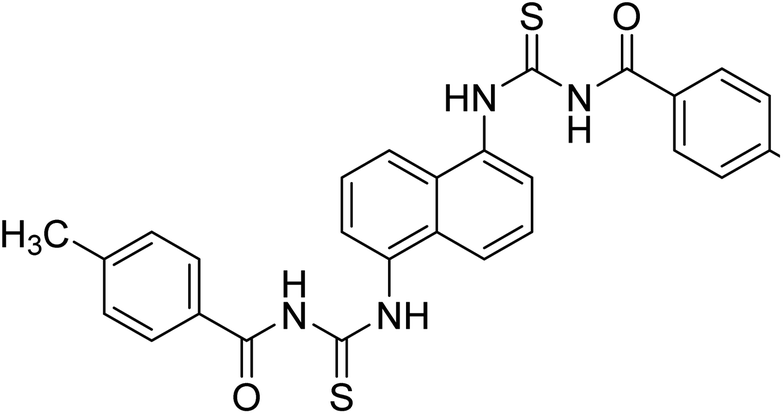 |
0.1761 | — | 294 |
| 321 |  |
3.63 | 56.01 | 295 |
| 322 |  |
52.50 | — | 296 |
| 323 |  |
0.42 | — | 297 |
Mughal et al. (2017) reported a series of 3-oxoaurones and 3-thioaurones. All the synthetic compounds were screened for their inhibitory potential against in vitro AChE and BChE enzymes. The results showed that compounds 294 (IC50 = 1.26 µM), 295 (IC50 = 0.98 µM) and 296 (IC50 = 6.39 µM) showed AChE inhibition activities whereas, compounds whereas 295 (IC50 = 1.02 µM) and 297 (IC50 = 5.27 µM) against BChE. Noteworthy, the compound 295 was found to be the potent dual inhibitor of AChE (IC50 = 0.98 µM) and BChE (IC50 = 1.02 µM) as compared to the standard donepezil (IC50 = 0.09 µM for AChE; 0.13 µM for BChE). Furthermore, compound 294 (IC50 = 1.26 µM) found to have considerable selective activity against AChE and may serve as lead compound for the development of powerful inhibitor for AChE. Overall, it is concluded from the study that 3-oxoaurones are appeared to be more potent as compared to 3-thioaurones. The replacement of oxygen with sulfur caused decreased in the activities such compounds 294 (IC50 = 1.26 µM for AChE) and 295 (IC50 = 0.98 µM for AChE; 1.02 µM for BChE) when transformed their thio derivatives (IC50 = 0.00 µM for AChE; 0.10 µM for BChE) and (IC50 = 0.00 µM for AChE; 0.00 µM for BChE) they completely lost the AChE activity. These compounds may serve as a lead-candidates in near future for the development of new drugs to treat AD19 (Table 15).
Mughal et al. (2018) designed and synthesized 3-O-flavonol glycosides, which were evaluated for inhibition potential against ChE enzymes. The results displayed that most of the derivatives were potent inhibitors of AChE and BChE with varying degree of IC50 values (IC50 = 0.205 to 102.91 µM for AChE; 0.13to 49.83 µM for BChE). Donepezil was used as standard (IC50 = 0.09 µM for AChE; 0.13 µM for BChE). Among the series, the compound 298 was found to be the most active dual inhibitor (IC50 = 02.05 µM for AChE and 05.42 µM for BChE) having isobutyl at p-position. The next most potent dual inhibitor is compound 299 (IC50 = 10.20 µM for AChE and 03.12 µM for BChE) with bromo group at p-position of the phenyl ring. Moreover, sulfur containing ring, for example compound 300, makes strong interaction with active pockets of AChE (IC50 = 18.52 µM) rather than BChE (IC50 = 25.51 µM). These compounds could be used for the development of new drugs for the cure of AD18 (Table 15).
Similarly, Mughal et al. (2019) reported a series of substituted flavonols and 4-thioflavonols potent as ChE inhibitors. Therein, the results unveiled that these derivatives were potent selective inhibitors of acetylcholinesterase (AChE), except the compound 301 (IC50 = 2.01 µM for AChE; 1.40 µM for BChE) which was selective inhibitor of butyrylcholinesterase (BChE), with varying degree of IC50 values. Remarkably, the compounds 302 (IC50 = 0.08 µM for AChE; 0.12 µM for BChE) and 303 (IC50 = 0.07 µM for AChE; 0.15 µM for BChE) have been found the most potent dual inhibitors of ChE amongst the series with IC50 values even less than the standard drug donepezil (IC50 = 0.09 µM for AChE; 0.13 µM for BChE)13 (Table 15).
Thai et al. (2020) synthesized a series of N-substituted-4-phenothiazine-chalcones and tested them for AChE inhibitory activity. All the synthesized target analogs showed IC50 values in the range of 186.21 to 1.10 µM for AChE. Among all the synthesized compounds, two substances 304 (IC50 = 3.63 µM for AChE) and 305 (IC50 = 1.10 µM for AChE) exhibited the most potent AChE inhibitory activity as compared to the standard galanthamine (IC50 = 1.26 µM for AChE). These synthesized compounds could be used as AChE inhibitors to cure the AD286 (Table 15).
Mekky et al. (2020) explored a new series of nicotinonitrile-coumarin hybrids as potent AChE inhibitors. The in vitro AChE inhibitory activity was examined for the new nicotinonitrile-coumarin hybrid molecules, when compared with donepezil as a standard drug with IC50 of 14 nM. Coumarin derivative, linked to 6-(4-nitrophenyl)-4-phenylnicotinonitrile, showed more effective inhibitory activity than the reference donepezil with IC50 of 13 nM. Compounds 306 and 307, with p-Cl and p-NO2 linked to 6-aryl moiety, respectively, exhibited the best AChE inhibitory activities with IC50 of 324 and 226 nM, respectively287 (Table 15).
Lomlim et al. (2020) designed and synthesized chromone-2-carboxamido-alkylamine derivatives and evaluated their ChE inhibitory activities. The compounds exhibited potent AChE inhibitory activities at micromolar range (IC50 = 0.09–9.16 µM) and demonstrated weak BChE inhibitory activities (IC50 = 12.09–44.56 µM). Compound 308 (IC50 = 0.09 µM) was the most potent AChE inhibitor in this series; it showed higher activity than the clinical used drug tacrine (IC50 = 0.13 µM for AChE) and weak BChE inhibitor (IC50 = 27.91 µM) as compared to the tacrine (IC50 = 0.01 µM for AChE). Chromone-2-carboxamidoalkylamines can be promising lead compounds for development of anti-Alzheimer's agents288 (Table 15).
Jadhav et al. (2020) synthesized a series of N-substituted α-aminophosphonates-bearing chromone moiety evaluated for ChE activities. Inhibitory activity against AChE ranged between 0.103 and 5.781 µM, whereas for BChE, activities ranged between 8.619 and 18.789 µM. The results showed that among the different synthesized analogs, strongest AChE inhibition was found for the compound containing aliphatic amine analogs, while in case of BChE, aromatic amines showed better activity as compared to aliphatic amines. Compound 309 was found to be the most potent inhibitor of AChE with an IC50 value of 0.103 µM and inhibited AChE through mixed-type inhibition. Compound 309 was 2-folds more potent than tacrine (IC50 = 0.289 µM), 35-folds potent than galantamine (IC50 = 3.643 µM) and 50-folds potent than rivastigmine (IC50 = 5.207 µM)289 (Table 15).
Mughal et al. (2020) explored 3-benzyloxyflavones as the potent ChE inhibitors. The findings showed that all the synthesized target compounds were dual inhibitors of AChE and BChE enzymes with varying IC50 values. In comparison, they are more active against AChE than BChE. Fascinatingly, amongst the series, the compound 310 was identified as the most active inhibitor of both AChE (IC50 = 0.05 µM) and BChE (IC50 = 0.09 µM) relative to the standard donepezil (IC50 = 0.09 µM for AChE; 0.13 µM for BChE). Moreover, the compounds 311 (IC50 = 0.07 µM) and 312 (IC50 = 0.08 µM) exhibited the highest selective inhibition against AChE as compared to the standard21 (Table 15).
Liu et al. (2020) designed and prepared a series of new flavone derivatives containing 6 or 7-substituted tertiary amine side chain and evaluated those compounds against AChE and BChE inhibition. The results indicated that the alteration of aromatic ring connecting to chromone scaffold markedly changed biological activity. Compared with flavones, the inhibitory activity of 2-naphthyl chromone, 2-anthryl-chromone derivatives against AChE significantly decreased, while that of 2-biphenyl chromone derivatives with 7-substituted tertiary amine side chain is better than relative flavones derivatives. Among the newly synthesized compounds, compound 313 was potent in AChE inhibition (IC50 = 1.29 µmol L−1) with high selectivity for AChE over BChE (selectivity ratio: 27.96)290 (Table 15).
Khan et al. (2021) synthesized a library of quinoline thiosemicarbazones endowed with a piperidine moiety by a microwave-assisted method. The in vitro ChE assay results revealed several compounds as potential inhibitors of AChE (IC50 = 15.8 to 62.3 µM) and BChE (IC50 = 11.59 to 60.02 µM) enzymes. Among all the synthesized compounds, five compounds exhibited IC50 values less than 20 µM. Moreover, compound 314 emerged as the most potent dual inhibitor of AChE and BChE with IC50 values of 9.68 and 11.59 µM, respectively as compared the standard donepezil (IC50 = 2.98 µM for AChE; 7.21 µM for BChE). Moreover, compound 315 appeared to be the selective inhibitor of AChE with an IC50 value of 15.8 µM. Despite several clinically approved drugs and development of anti-Alzheimer's heterocyclic structural leads, the treatment of AD requires safer hybrid therapeutics with characteristic structural and biochemical properties291 (Table 15).
Lomlim et al. (2021) designed and synthesized quinoxaline-based derivatives and evaluated them as novel AChE inhibitors. The results showed that all compounds exhibited potent AChE and BChE inhibitory activities with IC50 values of 0.077 to 50.080 µM and 14.91 to 60.95 µM, respectively. Compound 316 (IC50 = 0.077 µM) displayed the highest AChE inhibitor activity and the compounds 317, IC50 = 40.64 µM and 318, (IC50 = 14.91 µM) resulted in elevated butyrylcholinesterase inhibitory activity as compared to tacrine (IC50 = 0.107 µM for AChE; 0.00066 µM for BChE) and galanthamine (IC50 = 0.59 µM for AChE; 11.55 µM for BChE). Therefore, the quinoxaline analogs could offer the lead for the newly developed candidates as potent AChE inhibitors292 (Table 15).
Munir et al. (2021) synthesized a series of quinoline-thiosemicarbazones and evaluated their AChE inhibitory activity. All the tested hybrid derivatives were found completely selective towards AChE and showed inhibition in the range of 0.12–60.9 µM. In vitro inhibitory results revealed compound 319 as a promising and lead inhibitor with an IC50 value of 0.12 µM, a 5-fold higher potency than standard drug (galanthamine; IC50 = 0.62 µM). The synergistic effect of electron-rich (methoxy) group and ethylmorpholine moiety in quinolinethiosemicarbazone conjugates contributes significantly to improving the inhibition level293 (Table 15).
Bahadur et al. (2021) synthesized a new series of bis-thioureas and screened them for AChE inhibition activity. The results of AChE inhibition assay were found to be active in inhibiting the target enzyme with different IC50 values. The synthesized compounds showed AChE inhibition in the range of IC50 = 0.176 to 25.2063 µM. Among all derivatives, the 4g showed highly potent inhibition potential against AChE enzyme with IC50 value of 0.1761 µM, which is several times better than the reference inhibitor neostigmine methylsulfate IC50 = 2.469 µM. The pharmacokinetic studies guided those compounds possess good lead-like properties with little toxicity and hence be used as a ChE inhibitors in near future for the cure of AD294 (Table 15).
Recently, Cevik et al. (2022) reported some new substituted thiazolylhydrazine analogs and evaluated their inhibitory effects against AChE and BChE enzymes. According to the enzyme inhibition results, the target compounds showed selectivity against BChE. Among all synthesized derivatives, compound 321 was found to be the most potent dual AChE and BChE inhibitor with an IC50 value of 3.63 and 56.01 µM, respectively as compared to the standard donepezil (IC50 = 98.86 µM for AChE; 78.95 µM for BChE). Further alterations in the structural framework of these compounds could be a determining factor to improve their anticholinergic potential which may complement the drug-discovery process against Alzheimer's disease295 (Table 15).
Gulçin et al. (2022) prepared and reported various derivatives of 1,2-aminopropanthiols. These synthesized derivatives were found to be effective inhibitors for the AChE enzyme, with IC50 and Ki values in the range of 25.48 to 60.37 µM and 5.76 to 55.39 µM for AChE, respectively.
Particularly, the evaluation of the inhibitory efficacy of compound 322 (IC50 = 52.50 µM) against AChE enzyme as compared to the standard tacrine (IC50 = 70.87 µM) also indicates the anti-AD potential296 (Table 15).
Jamalis et al. (2022) reported chalcone-based coumarin derivatives and evaluated them as ChE inhibitors for the treatment of AD. The in vitro assessment of the synthesized compounds revealed that all of them showed significant activity (IC50 ranging from 0.42 to 1.296 µM) towards AChE as compared to the standard drug, galantamine (IC50 = 1.142 µM). Amongst the series, compound 323 displayed the most potent inhibitory activity with IC50 value of 0.42 µM. Moreover, bromo substitution on C-5 of thiophene ring in chalcone moiety led to decrease in AChE inhibition activity. The obtained results showed that the linker length connecting chalcone moiety and coumarin core played a vital role in AChE inhibition. Hence, the identified compound may be considered as lead for further study in the search of novel AChE inhibitory agent297 (Table 15).
The SAR studies of the previously mentioned heterocyclic compounds demonstrate that the different derivatives are potent AChE and BChE inhibitors (Fig. 25). All the structural features are performing a significant role in the inhibitory activity, though, a slight variation in the activity of these analogs is due to variability in the nature and positions of substituents on aryl rings. The smaller groups (–CH3, –OCH3, –CF3, –F, –Cl, –NO2, –OH, etc.) attached to the scaffold show higher AChE and BChE inhibitory abilities as compared to the bulky group (benzothiophene, sulfonamide) present on the rings. All the presented analogues so far have shown good to excellent ChE inhibitory abilities with a low risk of toxic side effects. Furthermore, these species are inexpensive and easy to synthesize in the laboratory, making them attractive for viable development and marketing as drugs against cholinesterase.
6 Conclusions and future perspectives
AD is the most common neurodegeneration disease which has a limited number of drug candidates available for its treatment. Currently, anticholinesterase drugs represent the main choice for pharmacotherapy of AD. In this context, the search for new drugs with anticholinesterase activity can be an alternative for treatment and it could be an important form to prolong and improve the life of the patient. This review summarizes the anticholinesterase activity of the most potent N-, O-, S-based heterocyclic compounds. The scope of N-, O-, S-based compounds in medicines is growing daily and their diverse analogs provide a viable and important path for the discovery of drugs with various biological applications. The N-, O-, S-based heterocyclic frameworks offer a high degree of structural diversity that has proven useful for the search of new therapeutic agents with improved pharmacokinetics and other physicochemical features. Research and development of N-, O-, S-based compounds in medicinal chemistry has become a rapidly developing and increasingly active topic. The overwhelming advantages of N-, O- and S-containing drugs in the medicinal field, including easy preparation, low toxicity, less adverse effects, high bioavailability, lower drug resistance, and good biocompatibility, encourage efforts towards further research and development. Although, extensive in vitro testing has been reported on compounds summarized in this review, few have selected for in vivo tests and subsequent steps for development as drug candidates. Thus, further studies should be performed in vivo and more theoretical work should be carried out to predict drug-likeness and drug ability. In addition, we have explored their SAR studies. The SAR studies of the discussed molecules offer a greater understanding of the pattern of substituents on their basic skeleton and appropriate substitutions accountable for their effectiveness and for further exploration of biological efficacy. These significant points confirm the enormous potential of various heterocyclic cores in pharmaceutical applications suggesting a massive scope for these promising moieties because of their diverse molecular targets. We believe that this review article will be valuable for encouraging the structural design and development of sustainable and effective N-, O-, S-based drugs against AD, with minimal side-effects. At last, we hope that this current work could be used as reference to design more potent AChE and BChE inhibitors with excellent inhibitory activity and in the search for new drugs for the treatment of AD.Abbreviations
| AChE | Acetylcholinesterase |
| ACh | Acetylcholine |
| AD | Alzheimer disease |
| BChE | Butyrylcholinesterase |
| ChE | Cholinesterase |
| SAR | Structure–activity relationship |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Deanship of Scientific Research at Umm Al-Qura University, for supporting this work by grant code: 22UQU4320545DSR18. The financial support by the Higher Education Commission of Pakistan (HEC) under Project No. (NRPU-6484 & NRPU-15800) is gratefully acknowledged. Dr Ziad Moussa is grateful to the United Arab Emirates University (UAEU) of Al-Ain and to the Research Office for supporting the research developed in his laboratory (grant no. G00003291).References
- N. N. Makhova, L. I. Belen'kii, G. A. Gazieva, I. L. Dalinger, L. S. Konstantinova, V. V. Kuznetsov, A. N. Kravchenko, M. M. Krayushkin, O. A. Rakitin and A. M. Starosotnikov, Russ. Chem. Rev., 2020, 89, 55 CrossRef CAS.
- P. M. Amisha, M. Pathania and V. K. Rathaur, Fam. Med. Prim. Care Rev., 2019, 8, 2328 CrossRef PubMed.
- J. A. Joule, K. Mills and G. F. Smith, Heterocyclic chemistry, CRC Press, 2020 Search PubMed.
- A. T. Soldatenkov, A. F. Pozharskii and A. R. Katritzky, Heterocycles in life and society: An introduction to heterocyclic chemistry, biochemistry and applications, John Wiley & Sons, 2011 Search PubMed.
- T. Eicher, S. Hauptmann and A. Speicher, The chemistry of heterocycles: structures, reactions, synthesis, and applications, John Wiley & Sons, 2013 Search PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS PubMed.
- H. Zhu, V. Dronamraju, W. Xie and S. S. More, Med. Chem. Res., 2021, 30, 305–352 CrossRef CAS PubMed.
- A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS.
- H. B. Broughton and I. A. Watson, J. Mol. Graphics Modell., 2004, 23, 51–58 CrossRef CAS PubMed.
- A. Mermer, T. Keles and Y. Sirin, Bioorg. Chem., 2021, 114, 105076 CrossRef CAS PubMed.
- M. Gupta, International Journal of Physical, Chemical and Mathematical Sciences, 2015, 4, 21–24 Search PubMed.
- E. U. Mugal, A. Sadiq, J. Ashraf, M. N. Zafar, S. H. Sumrra, R. Tariq, A. Mumtaz, A. Javid, B. A. Khan and A. Ali, Bioorg. Chem., 2019, 91, 103124 CrossRef PubMed.
- E. U. Mughal, M. Ayaz, Z. Hussain, A. Hasan, A. Sadiq, M. Riaz, A. Malik, S. Hussain and M. I. Choudhary, Bioorg. Med. Chem., 2006, 14, 4704–4711 CrossRef PubMed.
- J. Ashraf, E. U. Mughal, A. Sadiq, N. Naeem, S. A. Muhammad, T. Qousain, M. N. Zafar, B. A. Khan and M. Anees, J. Mol. Struct., 2020, 1218, 128458 CrossRef CAS.
- R. J. Obaid, E. U. Mughal, N. Naeem, A. Sadiq, R. I. Alsantali, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 22159–22198 RSC.
- J. Ashraf, E. U. Mughal, R. I. Alsantali, R. J. Obaid, A. Sadiq, N. Naeem, A. Ali, A. Massadaq, Q. Javed and A. Javid, Bioorg. Med. Chem., 2021, 35, 116057 CrossRef CAS PubMed.
- E. U. Mughal, A. Javid, A. Sadiq, S. Murtaza, M. N. Zafar, B. A. Khan, S. H. Sumrra, M. N. Tahir and K. M. Khan, Bioorg. Med. Chem., 2018, 26, 3696–3706 CrossRef CAS PubMed.
- E. U. Mughal, A. Sadiq, S. Murtaza, H. Rafique, M. N. Zafar, T. Riaz, B. A. Khan, A. Hameed and K. M. Khan, Bioorg. Med. Chem., 2017, 25, 100–106 CrossRef CAS PubMed.
- J. Ashraf, E. U. Mughal, A. Sadiq, M. Bibi, N. Naeem, A. Ali, A. Massadaq, N. Fatima, A. Javid and M. N. Zafar, J. Biomol. Struct. Dyn., 2021, 39, 7107–7122 CrossRef CAS PubMed.
- E. U. Mughal, A. Sadiq, M. Ayub, N. Naeem, A. Javid, S. H. Sumrra, M. N. Zafar, B. A. Khan, F. P. Malik and I. Ahmed, J. Biomol. Struct. Dyn., 2021, 39, 6154–6167 CrossRef CAS PubMed.
- R. I. Alsantali, E. U. Mughal, N. Naeem, M. A. Alsharif, A. Sadiq, A. Ali, R. S. Jassas, Q. Javed, A. Javid and S. H. Sumrra, J. Mol. Struct., 2022, 1251, 131933 CrossRef CAS.
- R. J. Obaid, E. U. Mughal, N. Naeem, M. Al Rooqi, A. Sadiq, R. Jassas and S. A. Ahmed, Process Biochem., 2022, 120, 250–259 CrossRef CAS.
- M. A. Alsharif, Q. A. Raja, N. A. Majeed, R. S. Jassas, A. A. Alsimaree, A. Sadiq, N. Naeem, E. U. Mughal, R. I. Alsantali and Z. Moussa, RSC Adv., 2021, 11, 29826–29858 RSC.
- H. Venkatachalam and N. V. A. Kumar, in Heterocycles-Synthesis and Biological Activities, IntechOpen, 2019 Search PubMed.
- P. Kaur, R. Arora and N. Gill, Indo Am. J. Pharm. Res., 2013, 3, 9067–9084 Search PubMed.
- J. Cossy and A. Guerinot, in Advances in Heterocyclic Chemistry, Elsevier, 2016, vol. 119, pp. 107–142 Search PubMed.
- C. Rowlands and R. Farley, Landolt Börnstein, 2008, vol. 26, p. 308 Search PubMed.
- A. U. Hassan, S. H. Sumrra, M. N. Zafar, M. F. Nazar, E. U. Mughal, M. N. Zafar and M. Iqbal, Mol. Diversity, 2022, 26, 51–72 CrossRef CAS PubMed.
- B. Meyer, Chem. Rev., 1976, 76, 367–388 CrossRef CAS.
- R. I. Alsantali, Q. A. Raja, A. Y. Alzahrani, A. Sadiq, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, N. El Guesmi, Z. Moussa and S. A. Ahmed, Dyes Pigm., 2022, 199, 110050 CrossRef CAS.
- E. U. Mughal, R. J. Obaid, A. Sadiq, M. A. Alsharif, N. Naeem, S. Kausar, A. A. Altaf, R. S. Jassas, S. Ahmed and R. I. Alsantali, Dyes Pigm., 2022, 201, 110248 CrossRef CAS.
- R. J. Huxtable, Biochemistry of sulfur, Springer Science & Business Media, 2013 Search PubMed.
- T. H. Maren, Annu. Rev. Pharmacol. Toxicol., 1976, 16, 309–327 CrossRef CAS PubMed.
- M. S. Wilke, A. L. Lovering and N. C. Strynadka, Curr. Opin. Microbiol., 2005, 8, 525–533 CrossRef CAS PubMed.
- R. Kaur, S. Chaudhary, K. Kumar, M. K. Gupta and R. K. Rawal, Eur. J. Med. Chem., 2017, 132, 108–134 CrossRef CAS PubMed.
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- N. Arya, A. Y. Jagdale, T. A. Patil, S. S. Yeramwar, S. S. Holikatti, J. Dwivedi, C. J. Shishoo and K. S. Jain, Eur. J. Med. Chem., 2014, 74, 619–656 CrossRef CAS PubMed.
- Z. Liu, Z. Zhang, W. Zhang and D. Yan, Bioorg. Med. Chem. Lett., 2018, 28, 2454–2458 CrossRef CAS PubMed.
- S. Jain, V. Chandra, P. K. Jain, K. Pathak, D. Pathak and A. Vaidya, Arabian J. Chem., 2019, 12, 4920–4946 CrossRef CAS.
- L. Zhang, X. M. Peng, G. L. Damu, R. X. Geng and C. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed.
- J. Zhang, S. Wang, Y. Ba and Z. Xu, Eur. J. Med. Chem., 2019, 174, 1–8 CrossRef CAS PubMed.
- R. Gupta, Int. J. Comput. Appl., 2015, 975, 8887 Search PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- V. Polshettiwar and R. S. Varma, Curr. Opin. Drug Discovery Dev., 2007, 10, 723–737 CAS.
- A. Padwa and S. K. Bur, Tetrahedron, 2007, 63, 5341 CrossRef CAS PubMed.
- D. M. D'Souza and T. J. Mueller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC.
- G. Eren, S. Ünlü, M.-T. Nuñez, L. Labeaga, F. Ledo, A. Entrena, E. Banoğlu, G. Costantino and M. F. Şahin, Bioorg. Med. Chem., 2010, 18, 6367–6376 CrossRef CAS PubMed.
- J. Hepworth and A. Boulton, Comprehensive Heterocyclic Chemistry, ed. M. G. McKillop, Pergamon Press, Oxford, 1984, vol. 3 Search PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- R. DeSimone, K. Currie, S. Mitchell, J. Darrow and D. Pippin, Comb. Chem. High Throughput Screening, 2004, 7, 473–493 CrossRef CAS PubMed.
- S. Andreescu and J.-L. Marty, Biomol. Eng., 2006, 23, 1–15 CrossRef CAS PubMed.
- N. Ben Oujji, I. Bakas, G. Istamboulié, I. Ait-Ichou, E. Ait-Addi, R. Rouillon and T. Noguer, Sensors, 2012, 12, 7893–7904 CrossRef CAS PubMed.
- N. A. Hosea, H. A. Berman and P. Taylor, Biochemistry, 1995, 34, 11528–11536 CrossRef CAS PubMed.
- G. S. Nunes, T. Montesinos, P. B. O. Marques, D. Fournier and J. L. Marty, Anal. Chim. Acta, 2001, 434, 1–8 CrossRef CAS.
- G. D. Stanciu, A. Luca, R. N. Rusu, V. Bild, S. I. Beschea Chiriac, C. Solcan, W. Bild and D. C. Ababei, Biomolecules, 2020, 10, 40 CrossRef CAS PubMed.
- M. A. DeTure and D. W. Dickson, Mol. Neurodegener., 2019, 14, 1–18 CrossRef PubMed.
- A. Martinez and A. Castro, Expert Opin. Invest. Drugs, 2006, 15, 1–12 CrossRef CAS PubMed.
- M. Mehta, A. Adem and M. Sabbagh, J. Alzheimer’s Dis., 2012, 2012, 1–8 CrossRef PubMed.
- P. J. Ghumatkar, S. P. Patil, P. D. Jain, R. M. Tambe and S. Sathaye, Pharmacol., Biochem. Behav., 2015, 135, 182–191 CrossRef CAS PubMed.
- Y. Sun, M. S. Lai, C. J. Lu and R. C. Chen, Eur. J. Paediatr. Neurol., 2008, 15, 278–283 CrossRef CAS PubMed.
- M. Seto, R. L. Weiner, L. Dumitrescu and T. J. Hohman, Mol. Neurodegener., 2021, 16, 1–16 CrossRef PubMed.
- A. S. Association, Alzheimers. Dement., 2020, 16, 391–460 CrossRef PubMed.
- H. Ahmad, S. Ahmad, M. Ali, A. Latif, S. A. A. Shah, H. Naz, N. ur Rahman, F. Shaheen, A. Wadood and H. U. Khan, Bioorg. Chem., 2018, 78, 427–435 CrossRef CAS PubMed.
- J. D. Ulrich and D. M. Holtzman, ACS Chem. Neurosci., 2016, 7, 420–427 CrossRef CAS PubMed.
- A. M. Palmer, Trends Pharmacol. Sci., 2011, 32, 141–147 CrossRef CAS PubMed.
- J.-C. Li, J. Zhang, M. C. Rodrigues, D.-J. Ding, J. P. F. Longo, R. B. Azevedo, L. A. Muehlmann and C.-S. Jiang, Bioorg. Med. Chem. Lett., 2016, 26, 3881–3885 CrossRef CAS PubMed.
- A. Rahman, M. T. Ali, M. M. A. K. Shawan, M. G. Sarwar, M. A. Khan and M. A. Halim, SpringerPlus, 2016, 5, 1–14 CrossRef PubMed.
- N. Bulut, U. M. Kocyigit, I. H. Gecibesler, T. Dastan, H. Karci, P. Taslimi, S. Durna Dastan, I. Gulcin and A. Cetin, J. Biochem. Mol. Toxicol., 2018, 32, 22006 CrossRef PubMed.
- M. Xu, Y. Peng, L. Zhu, S. Wang, J. Ji and K. Rakesh, Eur. J. Med. Chem., 2019, 180, 656–672 CrossRef CAS PubMed.
- D. K. Lahiri, M. R. Farlow, N. H. Greig and K. Sambamurti, Drug Dev. Res., 2002, 56, 267–281 CrossRef CAS.
- Z. Najafi, M. Mahdavi, M. Saeedi, E. Karimpour-Razkenari, R. Asatouri, F. Vafadarnejad, F. H. Moghadam, M. Khanavi, M. Sharifzadeh and T. Akbarzadeh, Eur. J. Med. Chem., 2017, 125, 1200–1212 CrossRef CAS PubMed.
- P. Anand and B. Singh, Arch. Pharmacal Res., 2013, 36, 375–399 CrossRef CAS PubMed.
- R. M. Lane, S. G. Potkin and A. Enz, Int. J. Neuropsychopharmacol., 2006, 9, 101–124 CrossRef CAS PubMed.
- N. H. Greig, T. Utsuki, Q.-s. Yu, X. Zhu, H. W. Holloway, T. Perry, B. Lee, D. K. Ingram and D. K. Lahiri, Curr. Med. Res. Opin., 2001, 17, 159–165 CrossRef CAS PubMed.
- A. Nordberg, C. Ballard, R. Bullock, T. Darreh-Shori and M. Somogyi, The primary care companion for CNS disorders, 2013, vol. 15 Search PubMed.
- P. B. Watkins, H. J. Zimmerman, M. J. Knapp, S. I. Gracon and K. W. Lewis, JAMA, 1994, 271, 992–998 CrossRef CAS PubMed.
- H. Khan, S. Amin, M. A. Kamal and S. Patel, Biomed. Pharmacother., 2018, 101, 860–870 CrossRef CAS PubMed.
- B. Ahmad, S. Mukarram Shah, H. Khan and S. Hassan Shah, J. Enzyme Inhib. Med. Chem., 2007, 22, 730–732 CrossRef CAS PubMed.
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef CAS PubMed.
- E. C. Ballinger, M. Ananth, D. A. Talmage and L. W. Role, Neuron, 2016, 91, 1199–1218 CrossRef CAS PubMed.
- H. Khan, M. Ali Khan and I. Hussan, J. Enzyme Inhib. Med. Chem., 2007, 22, 722–725 CrossRef CAS PubMed.
- V. P. Nair and J. M. Hunter, Critical Care and Pain, 2004, 4, 164–168 Search PubMed.
- M. Yuksel, K. Biberoglu, S. Onder, K. G. Akbulut and O. Tacal, Biochimie, 2018, 146, 105–112 CrossRef CAS PubMed.
- H. Khan, M. Saeed, M. A. Khan, N. Muhammad, A. Khan and A. Ullah, J. Chem. Soc. Pak., 2014, 36, 865–869 CAS.
- S. Zacks and J. Blumberg, J. Histochem. Cytochem., 1961, 9, 317–324 CrossRef CAS PubMed.
- S. Kuwabara, Landmark Papers in Neurology 2015, vol. 12, p. 429 Search PubMed.
- O. Lykhmus, L. Koval, D. y. Pastuhova, M. Zouridakis, S. Tzartos, S. Komisarenko and M. Skok, Immunobiology, 2016, 221, 1355–1361 CrossRef CAS PubMed.
- K. Vrolix, J. Fraussen, M. Losen, J. Stevens, K. Lazaridis, P. C. Molenaar, V. Somers, M. A. Bracho, R. Le Panse and P. Stinissen, J. Autoimmun., 2014, 52, 101–112 CrossRef CAS PubMed.
- M. Almasieh, Y. Zhou, M. Kelly, C. Casanova and A. Di Polo, Cell Death Dis., 2010, 1, 27–29 CrossRef PubMed.
- S. H. Song, S. M. Choi, J. E. Kim, J. E. Sung, H. A. Lee, Y. H. Choi, C. J. Bae, Y. W. Choi and D. Y. Hwang, Neurosci. Lett., 2017, 638, 121–128 CrossRef CAS PubMed.
- N. D. Belyaev, K. A. Kellett, C. Beckett, N. Z. Makova, T. J. Revett, N. N. Nalivaeva, N. M. Hooper and A. J. Turner, J. Biol. Chem., 2010, 285, 41443–41454 CrossRef CAS PubMed.
- H. Ashton, Curr. Opin. Psychiatry, 2005, 18, 249–255 CrossRef PubMed.
- Y. Dgachi, O. Sokolov, V. Luzet, J. Godyń, D. Panek, A. Bonet, H. Martin, I. Iriepa, I. Moraleda and C. García-Iriepa, Eur. J. Med. Chem., 2017, 126, 576–589 CrossRef CAS PubMed.
- Q. Li, S. He, Y. Chen, F. Feng, W. Qu and H. Sun, Eur. J. Med. Chem., 2018, 158, 463–477 CrossRef CAS PubMed.
- H. Zakut, J. Lieman‐Hurwitz, R. Zamir, L. Sindell, D. Ginzberg and H. Soreq, Prenatal Diagn., 1991, 11, 597–607 CrossRef CAS PubMed.
- M. Mesulam, A. Guillozet, P. Shaw and B. Quinn, Neurobiol. Dis., 2002, 9, 88–93 CrossRef CAS PubMed.
- M.-M. Mesulam, A. Guillozet, P. Shaw, A. Levey, E. Duysen and O. Lockridge, Neuroscience, 2002, 110, 627–639 CrossRef CAS PubMed.
- A. Chatonnet and O. Lockridge, Biochem. J., 1989, 260, 625–634 CrossRef CAS PubMed.
- A. Mack and A. Robitzki, Prog. Neurobiol., 2000, 60, 607–628 CrossRef CAS PubMed.
- O. Lockridge, Structure of human serum cholinesterase, Bioessays, 1988, 9, 125–128 CrossRef CAS PubMed.
- C. G. Carolan, G. P. Dillon, D. Khan, S. A. Ryder, J. M. Gaynor, S. Reidy, J. F. Marquez, M. Jones, V. Holland and J. F. Gilmer, J. Med. Chem., 2010, 53, 1190–1199 CrossRef CAS PubMed.
- B. Li, E. G. Duysen, M. Carlson and O. Lockridge, J. Pharmacol. Exp. Ther., 2008, 324, 1146–1154 CrossRef CAS PubMed.
- I. Manoharan, R. Boopathy, S. Darvesh and O. Lockridge, Clin. Chim. Acta, 2007, 378, 128–135 CrossRef CAS PubMed.
- N. H. Greig, T. Utsuki, D. K. Ingram, Y. Wang, G. Pepeu, C. Scali, Q.-S. Yu, J. Mamczarz, H. W. Holloway and T. Giordano, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17213–17218 CrossRef CAS PubMed.
- G. Mushtaq, N. H. Greig, J. A. Khan and M. A. Kamal, CNS and Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS and Neurological Disorders), 2014, vol. 13, pp. 1432–1439 Search PubMed.
- W. Xie, J. A. Stribley, A. Chatonnet, P. J. Wilder, A. Rizzino, R. D. McComb, P. Taylor, S. H. Hinrichs and O. Lockridge, J. Pharmacol. Exp. Ther., 2000, 293, 896–902 CAS.
- Q. Li, H. Yang, Y. Chen and H. Sun, Eur. J. Med. Chem., 2017, 132, 294–309 CrossRef CAS PubMed.
- B. Brus, U. Kosak, S. Turk, A. Pislar, N. Coquelle, J. Kos, J. Stojan, J.-P. Colletier and S. Gobec, J. Med. Chem., 2014, 57, 8167–8179 CrossRef CAS PubMed.
- D. Knez, B. Brus, N. Coquelle, I. Sosič, R. Šink, X. Brazzolotto, J. Mravljak, J.-P. Colletier and S. Gobec, Bioorg. Med. Chem., 2015, 23, 4442–4452 CrossRef CAS PubMed.
- S. N. Dighe, G. S. Deora, E. De la Mora, F. Nachon, S. Chan, M.-O. Parat, X. Brazzolotto and B. P. Ross, J. Med. Chem., 2016, 59, 7683–7689 CrossRef CAS PubMed.
- A. Nordberg, C. Ballard, R. Bullock, T. Darreh-Shori and M. Somogyi, The Primary Care Companion for CNS Disorders, 2013, vol. 15, p. 26731 Search PubMed.
- Y. Oda and I. Nakanishi, The distribution of cholinergic neurons in the human central nervous system, 2000 Search PubMed.
- M. K. Tripathi, P. Sharma, A. Tripathi, P. N. Tripathi, P. Srivastava, A. Seth and S. K. Shrivastava, J. Comput.-Aided Mol. Des., 2020, 34, 983–1002 CrossRef CAS PubMed.
- M. Reale, E. Costantini, M. Di Nicola, C. D'Angelo, S. Franchi, M. D'Aurora, M. Di Bari, V. Orlando, S. Galizia and S. Ruggieri, Sci. Rep., 2018, 8, 1–9 Search PubMed.
- D. J. Selkoe, Cell, 1989, 58, 611–612 CrossRef CAS PubMed.
- S. S. Sisodia and D. L. Price, FASEB J., 1995, 9, 366–370 CrossRef CAS PubMed.
- A. A. Geronikaki, J. C. Dearden, D. Filimonov, I. Galaeva, T. L. Garibova, T. Gloriozova, V. Krajneva, A. Lagunin, F. Z. Macaev and G. Molodavkin, J. Med. Chem., 2004, 47, 2870–2876 CrossRef CAS PubMed.
- P. Piplani and C. C. Danta, Bioorg. Chem., 2015, 60, 64–73 CrossRef CAS PubMed.
- P. N. Tripathi, P. Srivastava, P. Sharma, A. Seth and S. K. Shrivastava, Bioorg. Med. Chem., 2019, 27, 1327–1340 CrossRef CAS PubMed.
- A. L. Guillozet, S. Weintraub, D. C. Mash and M. M. Mesulam, Arch. Neurol., 2003, 60, 729–736 CrossRef PubMed.
- C. R. Tyler and A. M. Allan, Curr. Environ. Health Rep., 2014, 1, 132–147 CrossRef PubMed.
- P. Srivastava, P. N. Tripathi, P. Sharma, S. N. Rai, S. P. Singh, R. K. Srivastava, S. Shankar and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 163, 116–135 CrossRef CAS PubMed.
- A. Blokland, Brain Res. Rev., 1995, 21, 285–300 CrossRef CAS PubMed.
- J. Pagonabarraga and J. Kulisevsky, Neurobiol. Dis., 2012, 46, 590–596 CrossRef PubMed.
- M.-C. Buhot, Curr. Opin. Neurobiol., 1997, 7, 243–254 CrossRef CAS PubMed.
- A. Easton, V. Douchamps, M. Eacott and C. Lever, Neuropsychologia, 2012, 50, 3156–3168 CrossRef PubMed.
- G. Johnson and S. Moore, Curr. Pharm. Des., 2006, 12, 217–225 CrossRef CAS PubMed.
- G. V. De Ferrari, M. A. Canales, I. Shin, L. M. Weiner, I. Silman and N. C. Inestrosa, Biochemistry, 2001, 40, 10447–10457 CrossRef CAS PubMed.
- K. Jomova, D. Vondrakova, M. Lawson and M. Valko, Mol. Cell. Biochem., 2010, 345, 91–104 CrossRef CAS PubMed.
- S. Melov, N. Wolf, D. Strozyk, S. R. Doctrow and A. I. Bush, Free Radical Biol. Med., 2005, 38, 258–261 CrossRef CAS PubMed.
- W. R. Markesbery, Free Radical Biol. Med., 1997, 23, 134–147 CrossRef CAS PubMed.
- L. Peauger, R. Azzouz, V. Gembus, M.-L. Tintas, J. Sopková-de Oliveira Santos, P. Bohn, C. Papamicael and V. Levacher, J. Med. Chem., 2017, 60, 5909–5926 CrossRef CAS PubMed.
- S. Thompson, K. L. Lanctôt and N. Herrmann, Expert Opin. Drug Saf., 2004, 3, 425–440 CrossRef CAS PubMed.
- T. B. Ali, T. R. Schleret, B. M. Reilly, W. Y. Chen and R. Abagyan, PLoS One, 2015, 10, 0144337 Search PubMed.
- A. Tripathi, P. K. Choubey, P. Sharma, A. Seth, P. N. Tripathi, M. K. Tripathi, S. K. Prajapati, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 183, 111707 CrossRef PubMed.
- E. Mikiciuk-Olasik, P. Szymański and E. Żurek, Diagnostics and therapy of Alzheimer's disease, 2007 Search PubMed.
- A. Jain and P. Piplani, Bioorg. Chem., 2020, 103, 104151 CrossRef CAS PubMed.
- M. S. Malik, B. H. Asghar, R. Syed, R. I. Alsantali, M. Morad, H. M. Altass, Z. Moussa, I. I. Althagafi, R. S. Jassas and S. A. Ahmed, Front. Chem., 2021, 9, 666573–666584 CrossRef CAS PubMed.
- M. S. Malik, R. A. Alsantali, A. Y. Alzahrani, Q. M. S. Jamal, E. M. Hussein, K. A. Alfaidi, M. M. Al-Rooqi, R. J. Obaid, M. A. Alsharif and S. F. Adil, J. Saudi Chem. Soc., 2022, 26, 101449 CrossRef CAS.
- M. S. Malik, S. Farooq Adil, Z. Moussa, H. M. Altass, I. I. Althagafi, M. Morad, M. A. Ansari, Q. M. Sajid Jamal, R. J. Obaid and A. A. Al-Warthan, Front. Chem., 2021, 21, 630357–630369 CrossRef PubMed.
- R. Mehmood, A. Sadiq, R. I. Alsantali, E. U. Mughal, M. A. Alsharif, N. Naeem, A. Javid, M. M. Al-Rooqi, G.-e.-S. Chaudhry and S. A. Ahmed, ACS Omega, 2022, 7, 17444–17461 CrossRef PubMed.
- M. S. Malik, R. I. Alsantali, Q. M. S. Jamal, Z. S. Seddigi, M. Morad, M. A. Alsharif, E. M. Hussein, R. S. Jassas, M. M. Al-Rooqi and Z. Abduljaleel, Front. Chem., 2021, 9, 808556–808566 CrossRef CAS PubMed.
- M. S. Malik, R. I. Alsantali, M. A. Alsharif, S. I. Aljayzani, M. Morad, R. S. Jassas, M. M. Al-Rooqi, A. A. Alsimaree, H. M. Altass and B. H. Asghar, Arabian J. Chem., 2022, 15, 103560 CrossRef CAS.
- J. Ashraf, E. U. Mughal, R. I. Alsantali, A. Sadiq, R. S. Jassas, N. Naeem, Z. Ashraf, Y. Nazir, M. N. Zafar and A. Mumtaz, RSC Adv., 2021, 11, 35077–35092 RSC.
- E. M. Hussein, M. S. Malik, R. I. Alsantali, B. H. Asghar, M. Morad, M. A. Ansari, Q. M. S. Jamal, A. A. Alsimaree, A. N. Abdalla and A. S. Algarni, J. Mol. Struct., 2021, 1246, 131232 CrossRef CAS.
- E. M. Hussein, M. M. Al-Rooqi, A. A. Elkhawaga and S. A. Ahmed, Arabian J. Chem., 2020, 13, 5345–5362 CrossRef CAS.
- E. M. Hussein, M. M. Al-Rooqi, S. M. Abd El-Galil and S. A. Ahmed, BMC Chem., 2019, 13, 1–18 CrossRef CAS PubMed.
- S. Faazil, M. S. Malik, S. A. Ahmed, R. I. Alsantali, P. Yedla, M. A. Alsharif, I. N. Shaikh and A. Kamal, Bioorg. Chem., 2022, 105869 CrossRef CAS PubMed.
- S. Gauthier, Can. Med. Assoc. J., 2002, 166, 616–623 Search PubMed.
- J. L. McGaugh and L. F. Petrinovich, Int. Rev. Neurobiol., 1965, 8, 139–196 CrossRef CAS PubMed.
- A. Weissman, Int. Rev. Neurobiol., 1967, 10, 167–198 CrossRef CAS PubMed.
- J. J. Sramek, J. Hourani, S. S. Jhee and N. R. Cutler, Life Sci., 1999, 64, 1215–1221 CrossRef CAS PubMed.
- M. M. Koola, S. K. Praharaj and A. Pillai, Current Behavioral Neuroscience Reports, 2019, vol. 6, pp. 37–50 Search PubMed.
- P. Bacalhau, A. A. San Juan, A. Goth, A. T. Caldeira, R. Martins and A. J. Burke, Bioorg. Chem., 2016, 67, 105–109 CrossRef CAS PubMed.
- A. Fallah, F. Mohanazadeh and M. Safavi, Med. Chem. Res., 2020, 29, 341–355 CrossRef CAS.
- M. Danish, M. A. Raza, U. Anwar, U. Rashid and Z. Ahmed, J. Chin. Chem. Soc., 2019, 66, 1408–1415 CrossRef CAS.
- P. P. Roy, P. Banjare, S. Verma and J. Singh, Mol. Inf., 2019, 38, 1800151 CrossRef CAS PubMed.
- R. Shamsimeymandi, Y. Pourshojaei, K. Eskandari, M. Mohammadi‐Khanaposhtani, A. Abiri, A. Khodadadi, A. Langarizadeh, F. Sharififar, B. Amirheidari and T. Akbarzadeh, Arch. Pharm., 2019, 352, 1800352 CrossRef PubMed.
- M. Bajda, K. Łątka, M. Hebda, J. Jończyk and B. Malawska, Bioorg. Chem., 2018, 78, 29–38 CrossRef CAS PubMed.
- G. Makhaeva, N. Kovaleva, S. Lushchekina, E. Rudakova, N. Boltneva, A. Proshin, B. Lednev, I. Serkov and S. Bachurin, in Doklady Biochemistry and Biophysics, Springer, 2018, pp. 369–373 Search PubMed.
- N. Nunes, G. P. Rosa, S. Ferraz, M. C. Barreto and M. de Carvalho, J. Appl. Phycol., 2020, 32, 759–771 CrossRef CAS.
- D.-H. Shi, X.-D. Ma, Y.-W. Liu, W. Min, F.-J. Yin, Z.-M. Tang, M.-Q. Song, C. Lu, X.-K. Song and W.-W. Liu, J. Chem. Res., 2018, 42, 366–370 CrossRef CAS.
- F. Turkan, A. Cetin, P. Taslimi and İ. Gulçin, Arch. Pharm., 2018, 351, 1800200 CrossRef PubMed.
- Y. Xu, Z. Zhang, X. Jiang, X. Chen, Z. Wang, H. Alsulami, H.-L. Qin and W. Tang, Eur. J. Med. Chem., 2019, 181, 111598 CrossRef CAS PubMed.
- Taslimi, F. Türkan, A. Cetin, H. Burhan, M. Karaman, I. Bildirici, I. Gulcin and F. Şen, Bioorg. Chem., 2019, 92, 103213–103221 CrossRef CAS PubMed.
- G. Gutti, D. Kumar, P. Paliwal, A. Ganeshpurkar, K. Lahre, A. Kumar, S. Krishnamurthy and S. K. Singh, Bioorg. Chem., 2019, 90, 103080 CrossRef CAS PubMed.
- S. Shaikh, P. Dhavan, G. Pavale, M. Ramana and B. Jadhav, Bioorg. Chem., 2020, 96, 103589 CrossRef CAS PubMed.
- R. M. Ghalib, R. Hashim, S. F. Alshahateet, S. H. Mehdi, O. Sulaiman, K.-L. Chan, V. Murugaiyah and A. Jawad, J. Chem. Crystallogr., 2012, 42, 783–789 CrossRef CAS.
- Y. K. Yoon, M. A. Ali, A. C. Wei, T. S. Choon, K.-Y. Khaw, V. Murugaiyah, H. Osman and V. H. Masand, Bioorg. Chem., 2013, 49, 33–39 CrossRef CAS PubMed.
- F. Ahmad, M. J. Alam, M. Alam, S. Azaz, M. Parveen, S. Park and S. Ahmad, J. Mol. Struct., 2018, 1151, 327–342 CrossRef CAS.
- Y. Xu, H. Wang, X. Li, S. Dong, W. Liu, Q. Gong, Y. Tang, J. Zhu, J. Li and H. Zhang, Eur. J. Med. Chem., 2018, 143, 33–47 CrossRef CAS PubMed.
- B. Kuzu, M. Tan, P. Taslimi, İ. Gülçin, M. Taşpınar and N. Menges, Bioorg. Chem., 2019, 86, 187–196 CrossRef CAS PubMed.
- A. I. Almansour, N. Arumugam, R. S. Kumar, D. Kotresha, T. S. Manohar and S. Venketesh, Bioorg. Med. Chem. Lett., 2020, 30, 126789 CrossRef CAS PubMed.
- I. A. Khodja, H. Boulebd, C. Bensouici and A. Belfaitah, J. Mol. Struct., 2020, 1218, 128527 CrossRef.
- B. Barut, S. Sari, S. Sabuncuoğlu and A. Özel, J. Mol. Struct., 2021, 1235, 130268 CrossRef CAS.
- M. A. Abbasi, A. Akhtar, K. Nafeesa, S. Z. Siddiqui, K. M. Khan, M. Ashraf and S. A. Ejaz, J. Chil. Chem. Soc., 2013, 58, 2186–2190 CrossRef CAS.
- A. Kamal, A. B. Shaik, G. N. Reddy, C. G. Kumar, J. Joseph, G. B. Kumar, U. Purushotham and G. N. Sastry, Med. Chem. Res., 2014, 23, 2080–2092 CrossRef CAS.
- M. Mohammadi‐Khanaposhtani, M. Mahdavi, M. Saeedi, R. Sabourian, M. Safavi, M. Khanavi, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Chem. Biol. Drug Des., 2015, 86, 1425–1432 CrossRef PubMed.
- S. Z. Siddiqui, M. A. Abbasi, M. Ashraf, B. Mirza and H. Ismail, Pak. J. Pharm. Sci., 2017, 30, 1743–1751 CAS.
- A. Ibrar, A. Khan, M. Ali, R. Sarwar, S. Mehsud, U. Farooq, S. Halimi, I. Khan and A. Al-Harrasi, Front. Chem., 2018, 6, 61 CrossRef PubMed.
- A. Rehman, A. Fatima, N. Abbas, M. A. Abbasi, K. M. Khan, M. Ashraf, I. Ahmad and S. A. Ejaz, Pak. J. Pharm. Sci., 2013, 26, 345–352 Search PubMed.
- A. Rehman, J. Iqbal, M. A. Abbasi, S. Z. Siddiqui, H. Khalid, S. Jhaumeer Laulloo, N. Akhtar Virk, S. Rasool and S. A. A. Shah, Cogent Chem., 2018, 4, 1441597 CrossRef.
- A. Rehman, K. Nafeesa, M. A. Abbasi, S. Z. Siddiqui, S. Rasool, S. A. A. Shah and M. Ashraf, Cogent Chem., 2018, 4, 1472197 CrossRef.
- J. Zhang, J.-C. Li, J.-L. Song, Z.-Q. Cheng, J.-Z. Sun and C.-S. Jiang, J. Asian Nat. Prod. Res., 2018, 1090–1103 Search PubMed.
- P. N. Tripathi, P. Srivastava, P. Sharma, A. Seth and S. K. Shrivastava, Bioorg. Med. Chem., 2019, 27, 1327–1340 CrossRef CAS PubMed.
- P. Mishra, P. Sharma, P. N. Tripathi, S. K. Gupta, P. Srivastava, A. Seth, A. Tripathi, S. Krishnamurthy and S. K. Shrivastava, Bioorg. Chem., 2019, 89, 103025 CrossRef CAS PubMed.
- A. Tripathi, P. K. Choubey, P. Sharma, A. Seth, P. N. Tripathi, M. K. Tripathi, S. K. Prajapati, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 183, 111707 CrossRef PubMed.
- P. Sharma, A. Tripathi, P. N. Tripathi, S. S. Singh, S. P. Singh and S. K. Shrivastava, ACS Chem. Neurosci., 2019, 10, 4361–4384 CrossRef CAS PubMed.
- X. Yu, Y.-F. Zhao, G.-J. Huang and Y.-F. Chen, J. Asian Nat. Prod. Res., 2021, 23, 866–876 CrossRef CAS PubMed.
- A. Fallah, F. Mohanazadeh and M. Safavi, Med. Chem. Res., 2020, 29, 341–355 CrossRef CAS.
- P. K. Choubey, A. Tripathi, M. K. Tripathi, A. Seth and S. K. Shrivastava, Bioorg. Chem., 2021, 111, 104922 CrossRef CAS PubMed.
- A. Tripathi, P. K. Choubey, P. Sharma, A. Seth, P. Saraf and S. K. Shrivastava, Bioorg. Chem., 2020, 95, 103506 CrossRef CAS PubMed.
- G. Ucar, N. Gokhan, A. Yesilada and A. A. Bilgin, Neurosci. Lett., 2005, 382, 327–331 CrossRef CAS PubMed.
- V. Jayaprakash, S. Yabanoglu, B. Sinha and G. Ucar, Turk. J. Biochem., 2010, 35, 91–98 CAS.
- M. D. Altintop, A. Özdemir, Z. A. Kaplancikli, G. Turan‐Zitouni, H. E. Temel and G. A. Çiftçi, Arch. Pharm., 2013, 346, 189–199 CrossRef CAS PubMed.
- S. Chigurupati, M. Selvaraj, V. Mani, K. K. Selvarajan, J. I. Mohammad, B. Kaveti, H. Bera, V. R. Palanimuthu, L. K. Teh and M. Z. Salleh, Bioorg. Chem., 2016, 67, 9–17 CrossRef CAS PubMed.
- M. S. Shah, S. U. Khan, S. A. Ejaz, S. Afridi, S. U. F. Rizvi, M. Najam-ul-Haq and J. Iqbal, Biochem. Biophys. Res. Commun., 2017, 482, 615–624 CrossRef CAS PubMed.
- H. E. Temel, M. D. Altintop and A. Özdemir, Turk. J. Pharm. Sci., 2018, 15, 333 CAS.
- F. Turkan, A. Cetin, P. Taslimi, H. S. Karaman and I. Gulcin, Arch. Pharm., 2019, 352, 1800359 CrossRef PubMed.
- A. Mumtaz, A. Majeed, S. Zaib, S. U. Rahman, S. Hameed, A. Saeed, H. Rafique, E. Mughal, A. Maalik and I. Hussain, Bioorg. Chem., 2019, 90, 103036 CrossRef CAS PubMed.
- C. Yamali, H. I. Gul, C. Kazaz, S. Levent and I. Gulcin, Bioorg. Chem., 2020, 96, 103627 CrossRef CAS PubMed.
- B. Sever, C. Türkeş, M. D. Altıntop, Y. Demir and Ş. Beydemir, Int. J. Biol. Macromol., 2020, 163, 1970–1988 CrossRef CAS PubMed.
- M. Tuğrak, H. İ. Gül and İ. Gülçin, J. Res. Pharm., 2020, 24, 464–471 Search PubMed.
- M. Mohammadi-Khanaposhtani, M. Saeedi, N. S. Zafarghandi, M. Mahdavi, R. Sabourian, E. K. Razkenari, H. Alinezhad, M. Khanavi, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2015, 92, 799–806 CrossRef CAS PubMed.
- M. A. Munawar, F. A. Chattha, S. Kousar, J. Munir, T. Ismail, M. Ashraf and M. A. Khan, Bioorg. Med. Chem., 2015, 23, 6014–6024 CrossRef PubMed.
- S. P. Mantoani, T. P. Chierrito, A. F. Vilela, C. L. Cardoso, A. Martínez and I. Carvalho, Molecules, 2016, 21, 193 CrossRef PubMed.
- J.-Y. Park, S. Shin, K. C. Park, E. Jeong and J. H. Park, J. Korean Chem. Soc., 2016, 60, 125–130 CrossRef CAS.
- G. Wu, Y. Gao, D. Kang, B. Huang, Z. Huo, H. Liu, V. Poongavanam, P. Zhan and X. Liu, MedChemComm, 2018, 9, 149–159 RSC.
- L. Yin, L. Wang, X.-J. Liu, F.-C. Cheng, D.-H. Shi, Z.-L. Cao and W.-W. Liu, Heterocycl. Commun., 2017, 23, 231–236 CAS.
- Z. Najafi, M. Mahdavi, M. Saeedi, E. Karimpour-Razkenari, R. Asatouri, F. Vafadarnejad, F. H. Moghadam, M. Khanavi, M. Sharifzadeh and T. Akbarzadeh, Eur. J. Med. Chem., 2017, 125, 1200–1212 CrossRef CAS PubMed.
- Z. Najafi, M. Mahdavi, M. Saeedi, E. Karimpour-Razkenari, N. Edraki, M. Sharifzadeh, M. Khanavi and T. Akbarzadeh, Bioorg. Chem., 2019, 89, 303–316 CrossRef PubMed.
- M. Son, H. Lee, C. Jeon, Y. Kang, C. Park, K. W. Lee and J. H. Park, Bull. Korean Chem. Soc., 2019, 40, 544–553 CrossRef CAS.
- M. Özil, H. T. Balaydın and M. Şentürk, Bioorg. Chem., 2019, 86, 705–713 CrossRef PubMed.
- A. Rastegari, H. Nadri, M. Mahdavi, A. Moradi, S. S. Mirfazli, N. Edraki, F. H. Moghadam, B. Larijani, T. Akbarzadeh and M. Saeedi, Bioorg. Chem., 2019, 83, 391–401 CrossRef CAS PubMed.
- A. Singh, S. Sharma, S. Arora, S. Attri, P. Kaur, H. K. Gulati, K. Bhagat, N. Kumar, H. Singh and J. V. Singh, Bioorg. Med. Chem. Lett., 2020, 30, 127477 CrossRef CAS PubMed.
- M. Yazdani, N. Edraki, R. Badri, M. Khoshneviszadeh, A. Iraji and O. Firuzi, Mol. Diversity, 2020, 24, 641–654 CrossRef CAS PubMed.
- M. Mehrazar, M. Hassankalhori, M. Toolabi, F. Goli, S. Moghimi, H. Nadri, S. N. A. Bukhari, L. Firoozpour and A. Foroumadi, Mol. Diversity, 2020, 24, 997–1013 CrossRef CAS PubMed.
- G. Le-Nhat-Thuy, N. N. Thi, H. Pham-The, T. A. D. Thi, H. N. Thi, T. H. N. Thi, S. N. Hoang and T. Van Nguyen, Bioorg. Med. Chem. Lett., 2020, 30, 127404 CrossRef CAS PubMed.
- N. Riaz, M. Iftikhar, M. Saleem, S. Hussain, F. Rehmat, Z. Afzal, S. Khawar, M. Ashraf and M. Al-Rashida, Results Chem., 2020, 2, 100041 CrossRef CAS.
- M. de Freitas Silva, E. Tardelli Lima, L. Pruccoli, N. G. Castro, M. J. R. Guimarães, F. M. da Silva, N. Fonseca Nadur, L. L. de Azevedo, A. E. Kümmerle and I. A. Guedes, Molecules, 2020, 25, 3165 CrossRef CAS PubMed.
- H. K. Askarani, A. Iraji, A. Rastegari, S. N. A. Bukhari, O. Firuzi, T. Akbarzadeh and M. Saeedi, BMC Chem., 2020, 14, 1–13 CrossRef PubMed.
- A. Rani, A. Singh, J. Kaur, G. Singh, R. Bhatti, N. Gumede, P. Kisten, P. Singh and V. Kumar, Bioorg. Chem., 2021, 105053 CrossRef CAS PubMed.
- M. Saeedi, A. Maleki, A. Iraji, R. Hariri, T. Akbarzadeh, N. Edraki, O. Firuzi and S. S. Mirfazli, J. Mol. Struct., 2021, 1229, 129828 CrossRef CAS.
- I. Khan, M. Hanif, M. T. Hussain, A. A. Khan, M. A. S. Aslam, N. H. Rama and J. Iqbal, Aust. J. Chem., 2012, 65, 1413–1419 CrossRef CAS.
- A. Skrzypek, J. Matysiak, M. M. Karpińska and A. Niewiadomy, J. Enzyme Inhib. Med. Chem., 2013, 28, 816–823 CrossRef CAS PubMed.
- M. Rafiq, M. Saleem, M. Hanif, M. R. Maqsood, N. H. Rama, K.-H. Lee and S.-Y. Seo, Bull. Korean Chem. Soc., 2012, 33, 3943–3949 CrossRef CAS.
- A. Skrzypek, J. Matysiak, A. Niewiadomy, M. Bajda and P. Szymański, Eur. J. Med. Chem., 2013, 62, 311–319 CrossRef CAS PubMed.
- I. Khan, S. Zaib, A. Ibrar, N. H. Rama, J. Simpson and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 167–177 CrossRef CAS PubMed.
- W.-W. Liu, Q.-X. Li and D.-H. Shi, Heterocycles, 2015, 91, 275–286 CrossRef CAS.
- X.-J. Liu, L. Wang, L. Yin, F.-C. Cheng, H.-M. Sun, W.-W. Liu, D.-H. Shi and Z.-L. Cao, J. Chem. Res., 2017, 41, 571–575 CrossRef CAS.
- D.-H. Shi, H.-L. Zhu, Y.-W. Liu, Z.-M. Tang, C. Lu, X.-D. Ma, X.-K. Song, W.-W. Liu, T. Dong and M.-Q. Song, J. Chem. Res., 2017, 41, 664–667 CrossRef CAS.
- R. Ujan, A. Saeed, P. A. Channar, F. A. Larik, Q. Abbas, M. F. Alajmi, H. R. El-Seedi, M. A. Rind, M. Hassan and H. Raza, Molecules, 2019, 24, 860 CrossRef PubMed.
- S. Lotfi, T. Rahmani, M. Hatami, B. Pouramiri, E. T. Kermani, E. Rezvannejad, M. Mortazavi, S. F. Hafshejani, N. Askari and N. Pourjamali, Bioorg. Chem., 2020, 105, 104457 CrossRef CAS PubMed.
- N. Aggarwal, S. Jain and N. Chopra, Hybrids of thiazolidin-4-ones and 1, 3, 4-thiadiazole: synthesis and biological screening of a potential new class of acetylcholinesterae inhibitors, Biointerface Res. Appl. Chem., 2021, 12(3), 2800–2812 Search PubMed.
- A. Skrzypek, J. Matysiak, M. Karpińska, K. Czarnecka, P. Kręcisz, D. Stary, J. Kukułowicz, B. Paw, M. Bajda and P. Szymański, Bioorg. Chem., 2021, 107, 104617 CrossRef CAS PubMed.
- Y.-J. Lee, Y.-R. Han, W. Park, S.-H. Nam, K.-B. Oh, H.-S. Lee, J. Das, R. V. Moquin, A. J. Dyckman and T. Li, Bioorg. Med. Chem. Lett., 2010, 20, 6865–6881 CrossRef.
- A. I. Almansour, R. S. Kumar, N. Arumugam, A. Basiri, Y. Kia, M. A. Ali, M. Farooq and V. Murugaiyah, Molecules, 2015, 20, 2296–2309 CrossRef PubMed.
- A. Basiri, B. M. Abd Razik, M. O. Ezzat, Y. Kia, R. S. Kumar, A. I. Almansour, N. Arumugam and V. Murugaiyah, Bioorg. Chem., 2017, 75, 210–216 CrossRef CAS PubMed.
- L. W. Mohamed, S. M. Abuel-Maaty, W. A. Mohammed and M. A. Galal, Bioorg. Chem., 2018, 76, 210–217 CrossRef CAS PubMed.
- A. M. Srour, D. H. Dawood, M. N. Khalil and Z. M. Nofal, Bioorg. Chem., 2019, 83, 226–234 CrossRef CAS PubMed.
- M. A. Youssef, S. S. Panda, R. A. El-Shiekh, E. M. Shalaby, D. R. Aboshouk, W. Fayad, N. G. Fawzy and A. S. Girgis, RSC Adv., 2020, 10, 21830–21838 RSC.
- H. Girisha, J. N. S. Chandra, S. Boppana, M. Malviya, C. Sadashiva and K. S. Rangappa, Eur. J. Med. Chem., 2009, 44, 4057–4062 CrossRef CAS PubMed.
- H. Khalid, A. U. Rehman, M. A. Abbasi, R. Hussain, K. M. Khan, M. Ashraf, S. A. Ejaz and M. Q. Fatmi, Turk. J. Chem., 2014, 38, 189–201 CrossRef CAS.
- G. Brahmachari, C. Choo, P. Ambure and K. Roy, Bioorg. Med. Chem., 2015, 23, 4567–4575 CrossRef CAS PubMed.
- P. Meena, V. Nemaysh, M. Khatri, A. Manral, P. M. Luthra and M. Tiwari, Bioorg. Med. Chem., 2015, 23, 1135–1148 CrossRef CAS PubMed.
- K. S. S. Kumar, C. D. Mohan, K. S. R. Swamy Jagadish, A. Hanumappa and K. S. R. Basappa, Synthesis, 2015, 8, 142–148 CAS.
- N. Karaman, Y. Sıcak, T. Taşkın-Tok, M. Öztürk, A. Karaküçük-İyidoğan, M. Dikmen, B. Koçyiğit-Kaymakçıoğlu and E. E. Oruç-Emre, Eur. J. Med. Chem., 2016, 124, 270–283 CrossRef CAS PubMed.
- L. Yurttaş, Z. A. Kaplancıklı and Y. Özkay, J. Enzyme Inhib. Med. Chem., 2013, 28, 1040–1047 CrossRef PubMed.
- S. Hamulakova, J. Imrich, L. Janovec, P. Kristian, I. Danihel, O. Holas, M. Pohanka, S. Böhm, M. Kozurkova and K. Kuca, Int. J. Biol. Macromol., 2014, 70, 435–439 CrossRef CAS PubMed.
- B. Kaya, Y. Özkay, H. E. Temel and Z. A. Kaplancıklı, J. Chem., 2016, 2016, 1–7 CrossRef.
- W. Hussein, B. N. Sağlık, S. Levent, B. Korkut, S. Ilgın, Y. Özkay and Z. A. Kaplancıklı, Molecules, 2018, 23, 2033 CrossRef PubMed.
- M. Saeedi, D. Mohtadi‐Haghighi, S. S. Mirfazli, M. Mahdavi, R. Hariri, H. Lotfian, N. Edraki, A. Iraji, O. Firuzi and T. Akbarzadeh, Chem. Biodiversity, 2019, 16, 1800433 CrossRef PubMed.
- P. N. Tripathi, P. Srivastava, P. Sharma, M. K. Tripathi, A. Seth, A. Tripathi, S. N. Rai, S. P. Singh and S. K. Shrivastava, Bioorg. Chem., 2019, 85, 82–96 CrossRef CAS PubMed.
- Ş. Demirayak, Z. Şahin, M. Ertaş, E. F. Bülbül, C. Bender, S. N. Biltekin, B. Berk, B. N. Sağlık, S. Levent and L. Yurttaş, J. Heterocycl. Chem., 2019, 56, 3370–3386 CrossRef.
- M. Abbasi, M. Irshad, S. Siddiqui, H. Junaid, S. Shah and M. Ashraf, Pharm. Chem. J., 2020, 54, 596–603 CrossRef CAS.
- P. Szymanski, M. Markowicz, M. Bajda, B. Malawska and E. Mikiciuk-Olasik, Lett. Drug Des. Discovery, 2012, 9, 645–654 CrossRef CAS.
- Z. Najafi, M. Saeedi, M. Mahdavi, R. Sabourian, M. Khanavi, M. B. Tehrani, F. H. Moghadam, N. Edraki, E. Karimpor-Razkenari and M. Sharifzadeh, Bioorg. Chem., 2016, 67, 84–94 CrossRef CAS PubMed.
- Z. Sang, W. Pan, K. Wang, Q. Ma, L. Yu and W. Liu, Bioorg. Med. Chem., 2017, 25, 3006–3017 CrossRef CAS PubMed.
- M. Eghtedari, Y. Sarrafi, H. Nadri, M. Mahdavi, A. Moradi, F. H. Moghadam, S. Emami, L. Firoozpour, A. Asadipour and O. Sabzevari, Eur. J. Med. Chem., 2017, 128, 237–246 CrossRef CAS PubMed.
- N. A. Khan, I. Khan, S. Abid, S. Zaib, A. Ibrar, H. Andleeb, S. Hameed and J. Iqbal, Med. Chem., 2018, 14, 74–85 CAS.
- M. S. Shah, M. Najam-ul-Haq, H. S. Shah, S. U. F. Rizvi and J. Iqbal, Comput. Biol. Chem., 2018, 76, 310–317 CrossRef CAS PubMed.
- J. Mo, H. Yang, T. Chen, Q. Li, H. Lin, F. Feng, W. Liu, W. Qu, Q. Guo and H. Chi, Bioorg. Chem., 2019, 93, 103310 CrossRef PubMed.
- J. de Oliveira C Brum, D. C. F. Neto, J. S. F. de Almeida, J. A. Lima, K. Kuca, T. C. C. França and J. D. Figueroa-Villar, Int. J. Mol. Sci., 2019, 20, 3944 CrossRef PubMed.
- D. N. Rosado-Solano, M. A. Barón-Rodríguez, P. L. Sanabria Florez, L. K. Luna-Parada, C. E. Puerto-Galvis, A. s. F. Zorro-González, V. V. Kouznetsov and L. Y. Vargas-Méndez, J. Agric. Food Chem., 2019, 67, 9210–9219 CrossRef CAS PubMed.
- I. Bazine, Z. Cheraiet, R. Bensegueni, C. Bensouici and A. Boukhari, J. Heterocycl. Chem., 2020, 57, 2139–2149 CrossRef CAS.
- B. Macha, R. Kulkarni, C. Bagul, A. K. Garige, R. Akkinepally and A. Garlapati, Med. Chem. Res., 2021, 30, 685–701 CrossRef CAS.
- B. Macha, R. Kulkarni, A. K. Garige, S. Pola, R. Akkinepally and A. Garlapati, ChemistrySelect, 2021, 6, 1947–1957 CrossRef CAS.
- T. Mohamed, X. Zhao, L. K. Habib, J. Yang and P. P. Rao, Bioorg. Med. Chem., 2011, 19, 2269–2281 CrossRef CAS PubMed.
- S. Ahmad, F. Iftikhar, F. Ullah, A. Sadiq and U. Rashid, Bioorg. Chem., 2016, 69, 91–101 CrossRef CAS PubMed.
- T. U. Rehman, I. U. Khan, M. Ashraf, H. Tarazi, S. Riaz and M. Yar, Arch. Pharm., 2017, 350, 1600304 CrossRef PubMed.
- V. Kumar, B. Kumar, A. Ranjan Dwivedi, D. Mehta, N. Kumar, B. Bajaj, T. Arora, V. Prashar, J. Parkash and V. Kumar, ChemistrySelect, 2020, 5, 8021–8032 CrossRef CAS.
- S. Manzoor, S. K. Prajapati, S. Majumdar, M. K. Raza, M. T. Gabr, S. Kumar, K. Pal, H. Rashid, S. Kumar and S. Krishnamurthy, Eur. J. Med. Chem., 2021, 215, 113224 CrossRef CAS PubMed.
- M. Maqbool, A. Manral, E. Jameel, J. Kumar, V. Saini, A. Shandilya, M. Tiwari, N. Hoda and B. Jayaram, Bioorg. Med. Chem., 2016, 24, 2777–2788 CrossRef CAS PubMed.
- S. Akocak, B. Mehmet, N. Lolak, M. Tuneg and R. K. Sanku, J. Turk. Chem. Soc., Sect. A, 2019, 6, 63–70 CrossRef CAS.
- N. Lolak, M. Boga, M. Tuneg, G. Karakoc, S. Akocak and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2020, 35, 424–431 CrossRef CAS PubMed.
- S. Parlar, G. Bayraktar, A. H. Tarikogullari, V. Alptüzün and E. Erciyas, Chem. Pharm. Bull., 2016, 64, 1281–1287 CrossRef CAS PubMed.
- F. Pandolfi, D. De Vita, M. Bortolami, A. Coluccia, R. Di Santo, R. Costi, V. Andrisano, F. Alabiso, C. Bergamini and R. Fato, Eur. J. Med. Chem., 2017, 141, 197–210 CrossRef CAS PubMed.
- R. Ghobadian, M. Mahdavi, H. Nadri, A. Moradi, N. Edraki, T. Akbarzadeh, M. Sharifzadeh, S. N. A. Bukhari and M. Amini, Eur. J. Med. Chem., 2018, 155, 49–60 CrossRef CAS PubMed.
- S. K. Singh, S. K. Sinha and M. K. Shirsat, Indian J. Pharm. Educ. Res., 2018, 52, 644–654 CrossRef CAS.
- N. Sultana, M. Sarfraz, S. T. Tanoli, M. S. Akram, A. Sadiq, U. Rashid and M. I. Tariq, Bioorg. Chem., 2017, 72, 256–267 CrossRef CAS PubMed.
- T. Mohamed and P. P. Rao, Eur. J. Med. Chem., 2017, 126, 823–843 CrossRef CAS PubMed.
- M. Sarfraz, N. Sultana, M. Jamil and M. I. Tariq, Rev. Roum. Chim., 2018, 63, 227–234 Search PubMed.
- U. M. Kocyigit, Y. Budak, M. B. Gürdere, F. Ertürk, B. Yencilek, P. Taslimi, İ. Gülçin and M. Ceylan, Arch. Physiol. Biochem., 2018, 124, 61–68 CrossRef CAS PubMed.
- T.-S. Tran, M.-T. Le, T.-C.-V. Nguyen, T.-H. Tran, T.-D. Tran and K.-M. Thai, Molecules, 2020, 25, 3916 CrossRef CAS PubMed.
- S. M. Sanad and A. E. Mekky, J. Iran. Chem. Soc., 2021, 18, 213–224 CrossRef CAS.
- P. Suwanhom, T. Nualnoi, P. Khongkow, V. Sanghiran Lee and L. Lomlim, Med. Chem. Res., 2020, 29, 564–574 CrossRef CAS.
- S. Shaikh, P. Dhavan, M. Ramana and B. Jadhav, Mol. Diversity, 2021, 25, 811–825 CrossRef CAS PubMed.
- Q. Q. Lu, Y. M. Chen, H. R. Liu, J. Y. Yan, P. W. Cui, Q. F. Zhang, X. H. Gao, X. Feng and Y. Z. Liu, Drug Dev. Res., 2020, 81, 1037–1047 CrossRef CAS PubMed.
- R. Munir, M. Zia-ur-Rehman, S. Murtaza, S. Zaib, N. Javid, S. J. Awan, K. Iftikhar, M. M. Athar and I. Khan, Molecules, 2021, 26, 656 CrossRef CAS PubMed.
- P. Suwanhom, J. Saetang, P. Khongkow, T. Nualnoi, V. Tipmanee and L. Lomlim, Molecules, 2021, 26, 4895 CrossRef CAS PubMed.
- S. Zaib, R. Munir, M. T. Younas, N. Kausar, A. Ibrar, S. Aqsa, N. Shahid, T. T. Asif, H. O. Alsaab and I. Khan, Molecules, 2021, 26, 6573 CrossRef CAS PubMed.
- R. Ujan, P. A. Channar, A. Bahadur, Q. Abbas, M. Shah, S. Rashid, S. Iqbal, A. Saeed, H. S. Abd-Rabboh and H. Raza, J. Mol. Struct., 2021, 1246, 131136 CrossRef CAS.
- A. Işık, U. Acar Çevik, A. Karayel, I. Celik, T. Erçetin, A. Koçak, Y. Özkay and Z. Kaplancıklı, SAR QSAR Environ. Res., 2022, 33, 193–214 CrossRef PubMed.
- A. Huseynova, R. Kaya, P. Taslimi, V. Farzaliyev, X. Mammadyarova, A. Sujayev, B. Tüzün, U. M. Kocyigit, S. Alwasel and İ. Gulçin, J. Biomol. Struct. Dyn., 2022, 40, 236–248 CrossRef CAS PubMed.
- A. H. Hasan, S. Murugesan, S. I. Amran, S. Chander, M. M. Alanazi, T. B. Hadda, S. Shakya, M. R. F. Pratama, B. Das and S. Biswas, Bioorg. Chem., 2022, 119, 105572 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |