
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFly ash derived calcium silicate hydrate as a highly efficient and fast adsorbent for Cu(II) ions: role of copolymer functionalization†
Shengrui Sun ab,
Ya Tangac,
Jiayi Lia,
Jiahui Kou*c and
Yangqiao Liu*ab
ab,
Ya Tangac,
Jiayi Lia,
Jiahui Kou*c and
Yangqiao Liu*ab
aShanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: yqliu@mail.sic.ac.cn
bJiangsu Research Institute of Advanced Inorganic Materials, Taicang 215488, China
cCollege of Materials Science and Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: koujiahui@163.com
First published on 15th August 2022
Abstract
The environmental issues caused by heavy metal accumulation from polluted water are becoming serious and threaten human health and the ecosystem. The adsorption technology represented by calcium silicate hydrate has attracted much attention, but suffers from high manufacturing costs and poor stability bottlenecks. Here, we have proposed a “trash-to-treasure” conversion strategy to prepare a thin sheet calcium silicate hydrate material (ACSH) using solid waste fly ash as silicon source and a small amount of Acumer2000 as modifier. The obtained materials showed fast adsorption rates, superior adsorption capacities and remarkable long-term stability for Cu(II) removal. Under the conditions of 0.5 g L−1 adsorbent concentration and 100 mL Cu(II) solution with a concentration of 100 mg L−1, ACSH can adsorb 95.6% Cu(II) within 5 min. The adsorption isotherms conformed to Langmuir models and the maximum adsorption capacity was 532 mg g−1. Using X-ray diffraction, scanning electron microscopy, X-ray photoelectron spectroscopy, Fourier transform infrared spectroscopy, specific surface area and pore structure analysis, it was found that the excellent adsorption performance could be attributed to the ultrahigh surface area (356 m2 g−1), abundant pores and multiple active sites induced by Acumer2000 modification. Moreover, the encapsulation effect from carboxylate and long carbon chains in Acumer2000 endowed modified samples with strong corrosion resistance to CO2, which effectively inhibited the formation of by-product CaCO3 and retained the remarkable adsorption performance for more than 100 days. Interestingly enough, the advantages of ACSH in economy and performance could been maintained in ACSH based adsorptive membranes. This work is of great significance for solid waste utilization as well as the preparation of high quality, cost-effective and long-term stability calcium silicate hydrate materials.
1 Introduction
Cu(II) ions, as one of the most popular heavy metal ions, can reach water bodies through the electronics industry, mining industry, cable industry, metal smelting, metal rinse processes, corroded plumbing systems and so on. Cu(II) accumulation from water has become a serious problem threatening human health and the ecosystem.1 It has spawned many techniques for water pollutant treatment, including adsorption, coagulation, microbial treatment and advanced oxidation processes.2–4 Among these techniques, adsorption has been considered a promising approach because of its simplicity, energy saving and low cost.5At present, the most common adsorption material is activated carbon.6 Recently, new kinds of adsorbents such as silica,7 resins,8 metal organic frameworks9 and layered double hydroxides,10 have also been investigated. However, for the heavy metal ion adsorption, the excellent performance of calcium silicate hydrate material is superior to other materials, resulting from its large specific surface area, high porosity and controllable structure. Shao et al. prepared flowerlike calcium silicate hydrate with a specific surface area of 77 m2 g−1, showing higher adsorption capacity for metal ions Cu(II), Zn(II) and Pb(II), than that of activated carbon with a specific surface area of 921 m2 g−1.11 Qi et al. analyzed the adsorption mechanism of calcium silicate hydrate to cobalt ions and found that ion exchange played an important role, accounting for about 76.7%, and the complexation of hydroxyl groups on the surface of calcium silicate hydrate was weak.12 So far a variety of calcium silicate hydrate and calcium silicate hydrate based-composite materials have been reported, such as ultra-thin calcium silicate hydrate nanosheets, magnetic based composite, doped calcium silicate hydrate materials, etc., which has been investigated as adsorbents not only for metal ions, but also for drugs, phosphorus, proteins, etc.11–16
Co-precipitation is a simple method to fabricate calcium silicate hydrate, with calcium salt or calcium hydroxide as calcium source and sodium silicate or tetraethyl orthosilicate as silicon source. The cost of silicon source accounts for a large part of the material cost. In order to meet the requirement of low cost in industrial scale application, a few researchers have committed to the preparation of calcium silicate hydrate with solid wastes which are rich in silicon. For instance, Qi et al. prepared calcium silicate hydrate using fly ash desilication liquid as raw material, and the adsorption capacity to Co(II) reached 109.6–154.8 mg g−1.12 Fly ash is one of the most productive solid wastes all over the world, obtained from coal combustion in power plants, where the SiO2 content could exceed 40%. It endows the fly ash with great potential as a cheap silicon source that can be extracted by alkali reaction. Accordingly, the desilication liquid owns high alkalinity, in which high levels of carbon dioxide (CO2) are unavoidable. So the formation of by-product calcium carbonate (CaCO3) was hard to avoid in the fabrication of calcium silicate hydrate using solid waste desilication liquid.11,12 Furthermore, the amount of CaCO3 would constantly increase through the reaction between atmospheric CO2 and calcium in adsorbent as long as calcium silicate hydrate material is exposed to air. This reaction can continue for several weeks until the adsorption properties of the material are completely destroyed.17 The drawback of calcium silicate hydrate in long-term stability greatly hinders its large scale application. Although there are many studies on calcium silicate hydrate, the advances in preventing the corrosion of calcium silicate hydrate by CO2 are scarce.
In this work, fly ash-derived calcium silicate hydrate with high purity and excellent adsorption performance was synthesized with an effective modifier—Acumer2000. Previous studies have proved that modifying the surface of calcium silicate hydrate with functional groups, such as sulfhydryl group and amino group, could effectively improve the capacity and adsorption rate by complexation effect.18,19 However, the synthesis strategies usually need a lot of organic solvents, which may result in cytotoxicity for subsequent applications. Acumer2000 is a water-soluble dispersant composed of strong acid (sulfonic group) and weak acid (carboxyl group) groups, which have strong attraction with Ca(II). During the nucleation and the growth of calcium silicate hydrate, Acumer2000 could bridge onto the surface of calcium silicate hydrate through chelate effect. On one hand, the functional groups can improve the surface complexation during the process of adsorption. On the other hand, the functional groups, especially the carboxyl groups, can effectively inhibit the generation of CaCO3 by encapsulating Ca atoms and interfering with the normal lattice arrangement.20,21 Overall, the application of Acumer2000 in the synthesis of calcium silicate hydrate is expected to improve the adsorption performance by inhibiting the formation of CaCO3 and enhancing the surface complexation. No relevant studies have been reported so far.
In this work, the impact of Acumer2000 for calcium silicate hydrate was elucidated by X-ray diffraction (XRD), Fourier infrared spectrophotometer (FTIR), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and N2 adsorption–desorption test. Using Cu(II) as adsorbate, the adsorption capacity and stability of modified calcium silicate hydrate on pollutants were studied and the adsorption mechanism deduced from the above results was also discussed. We anticipate this “trash-to-treasure” conversion strategy to enrich the understanding and pathway of cost-effective and high-quality adsorbent fabrication.
Moreover, the potential of modified calcium silicate hydrate in the field of membrane was also explored. In previous studies,11–16 most of calcium silicate hydrates achieved strong adsorption performance in the state of dispersed powder, suffering from the difficulty of recovery and the potential threat of secondary pollution. It can be solved by assembling adsorbent to porous membrane, with the advantages of fast adsorption rate, easy recovery and industrial amplification. While, it has very high requirements for the force between pollutant and adsorbent, which challenges the performance of adsorbent. The application of calcium silicate hydrate in adsorption membrane area has not been reported. In our work, a simple and universal method was used to prepare the calcium silicate hydrate based adsorption film, of which the adsorption efficiency for Cu(II) uptake from water were investigated. We expect this attempt can broaden the materials range in the membrane field.
2 Material and methods
2.1 Material
Sodium hydroxide (NaOH), calcium hydroxide (Ca(OH)2), copper chloride dishydrate (CuCl2·2H2O), ethanol, hydrochloric acid are all analytical grade, purchased from Sinopharm Chemical Reagent Co., Ltd. Acumer2000 solution (Mw = 4500, solid content 43%, density 1.21 g mL−1, pH = 4) was purchased from Rohm & Hass company. The raw fly ash was obtained as waste from a power plant in Jiaxing city, Zhejiang province, China. All chemicals have been used without further purification. The water used in this work is deionized water.2.2 Calcium silicate hydrate preparation
Silicon component in fly ash was extracted by high-temperature alkali activation method: 5 g fly ash and 8 g NaOH were mixed and grinded, then heated at 600 °C for 2 h. When cooled to room temperature, appropriate amount of deionized water was added into the mixture and stirred for 1 h. After standing overnight, fly ash slags (named as desilication fly ash) were filtered out and obtain desilication solution (SiO2 = 6 mg mL−1).100 mL desilication solution mixed with 0.1 g Acumer2000 heated up to 60 °C, and 1.5 wt% Ca(OH)2 suspension was added drop by drop until the mole ratio of Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Si was about 1. Then, the temperature was increased to 80 °C and maintained for 2 h. Finally, the products were obtained by filtering, washing with distilled water and ethanol in turn, and drying in a vacuum oven at 60 °C for 12 h, which were denoted as ACSH. In addition, sample was prepared in the absence of Acumer2000, denoted as CSH.
:Si was about 1. Then, the temperature was increased to 80 °C and maintained for 2 h. Finally, the products were obtained by filtering, washing with distilled water and ethanol in turn, and drying in a vacuum oven at 60 °C for 12 h, which were denoted as ACSH. In addition, sample was prepared in the absence of Acumer2000, denoted as CSH.
In addition, 100 mg ACSH was dispersed in ethanol solution, then the suspension was filtered through the polypropylene substrate to form membrane with radius of 2 cm. The membrane was named as ACSH-M. CSH-M was prepared in the same operation but with CSH powder.
2.3 Characterization
The composition of raw fly ash and desilication fly ash were determined by X-ray fluorescence spectrometer (XRF, Axios, Holland). Inductively coupled plasma mass spectrometer (ICP, 725 ICP-OES, Agilent) was used to determine the ion concentration in desilication solution. XRD patterns were obtained on a Bruker Advance D8 diffractometer (Germany) using a filtered Cu Kα radiate source (λ = 1.54178 Å), scanned from 20 to 50° with 1°/min. Field emission scanning electron microscopy (FESEM, Magellan 400) equipped with an energy-dispersive X-ray spectrometer (EDS) were used to investigate the morphology and composition of the sample. The specific surface area of catalysts was investigated by N2 adsorption at 77 K, using the multipoint Brunauer–Emmett–Teller (BET) method (Micromeritics ASAP2020). Prior to the measurements, the samples were dehydrated in vacuum at 120 °C for 24 h. The pore size distributions, average pore width and volume of pores were calculated from adsorption branches of the isotherms by the Barrett–Joyner–Halenda (BJH) method. Mid-IR spectra of samples were recorded on a FTIR Spectrometer (Bruker EQUINOX55) using the conventional KBr pellet method. The elemental and chemical valence information on the surface of material was determined by XPS (Thermo Scientific K-Alpha) using X-ray source Al Kα radiation (hv = 1486.6 eV) and all binding energies were calibrated to the neutral C 1s peak at 284.8 eV.2.4 Batch adsorption experiments
A series of adsorption experiments on Cu(II) were carried out at 20 °C with the synthesized powder samples as adsorbents, including the adsorption capacity under different time and pH and the adsorption isotherms. Aqueous solutions containing heavy metal Cu(II) ions were prepared by dissolving appropriate amount of CuCl2·2H2O in deionized water to prepare 100–1000 mg L−1 Cu(II) solution.The adsorption capacity on samples under different time were obtained by dispersing 50 mg sample into 100 mL aqueous solution (100 mg L−1, initial pH = 6). The suspensions were in a shaking table (250 rpm) for a certain time. In the experiment, samples were taken at regular intervals, and the clear liquid was filtered by needle filter, and the concentration of copper ion in the clear liquid was analyzed by HACH2800 water quality analyzer. Tests were conducted in triplicate and the average value was used for calculation. Adsorption capacity (q, mg g−1) and residue percentage (η, %) were calculated according to the following equations (eqn (1) and (2)), respectively:
| q = (C0 − C) × V m−1 | (1) |
| η = (C/C0) × 100% | (2) |
To examine the influence of initial pH, experiments were performed in Cu(II) solution of 300 mg L−1. The initial pH of the solution varied from 2.0 ± 0.3 to 6.0 ± 0.3, adjusted with 0.1 M HCl or 0.1 M NaOH. The sorbent dosage was 0.5 g L−1. For the adsorption isotherms studies, the initial concentration of Cu(II) solution varied from 100 to 1000 mg L−1 and the initial pH were adjusted to pH = 5.0 ± 0.3. The sorbent dosage was 0.5 g L−1, too. While all other operation were the same as that in the previous experiment.
In addition, 20 mg L−1 Cu(II) solution was passed through adsorption membrane at a flow rate of 3 mL min−1. The concentration of copper ions in the filtrate was investigate by HACH2800 water quality analyzer. Copper ion removal rate (R, %) were calculated according to eqn (3).
| R = [(C0 − C)/C0] × 100% | (3) |
3 Results and discussion
3.1 Component characterization of fly ash and desilication solution
Table S1† presents XRF results of raw fly ash and desilication fly ash. It can be seen that the main components of raw fly ash are SiO2 and Al2O3, containing trace Fe, Ca, Mg and other components. It is well known that the components of fly ash will be changed because of the variance in raw materials and combustion methods. All experiments in our study were performed using the same batch of fly ash. Through high-temperature alkali melting reaction (eqn (4)), a certain amount of silicon element was removed into water to form desilication solution. ICP results (in Table S2†) of the desilication solution shows that 38.3% silicon in raw fly ash have been leached and used for further samples synthesis. The other silicon might exist in a refractory state, or be wrapped into the fly ash particles, hardly reacted with alkali. The desilication solution also contained a great deal of Na arising from alkali, most of which are not involved in the reaction (eqn (4)) but dissolve into the solution. The desilication solution had a high alkalinity and would readily absorb CO2 from the air. And as shown in Table S1,† desilication fly ash comprised of significant amount of various metals, and after alkali melting reaction these metals were likely to be exposed on the surface. Desilication fly ash can be viewed as a potential source for cost-effective catalysts or catalyst supports to removal pollution from water, which will be one of the research directions for our team in future.| SiO2(S) + 2NaOH(S) → Na2SiO3(S) + H2O(g) → Na2SiO3(aq) | (4) |
3.2 Characterization of sample
XRD patterns of CSH and ACSH were presented in Fig. 1a. For sample ACSH, characteristic diffraction peaks appeared at 29.5°, 32.1°, 34.1° and 43.3°, corresponding to (111), (200), (015) and (009) of calcium silicate hydrate (JCPDS 29-0329), respectively. The peaks widened seriously, implying poor crystallinity due to lower synthesized temperature. For CSH, a series of sharp diffraction peaks appeared at 23.1°, 29.4°, 36.0°, 39.4° and 43.2°, corresponding to CaCO3 (JCPDS 47-1743). The formation of calcium silicate hydrate was usually accompanied by CaCO3 as a by-product. Similar results have been reported in other literatures.11,12 Because of these sharp and strong peaks for CaCO3, the peaks with poor crystallinity for calcium silicate hydrate were hard to observe. XRD results confirm that calcium silicate hydrate with poor crystallinity can be obtained using fly ash as raw material and the formation of impurity CaCO3 is unavoidable in the absence of Acumer2000. While, the introduction of Acumer2000 can greatly inhibit the generation of CaCO3. The purity of samples can be improved accordingly.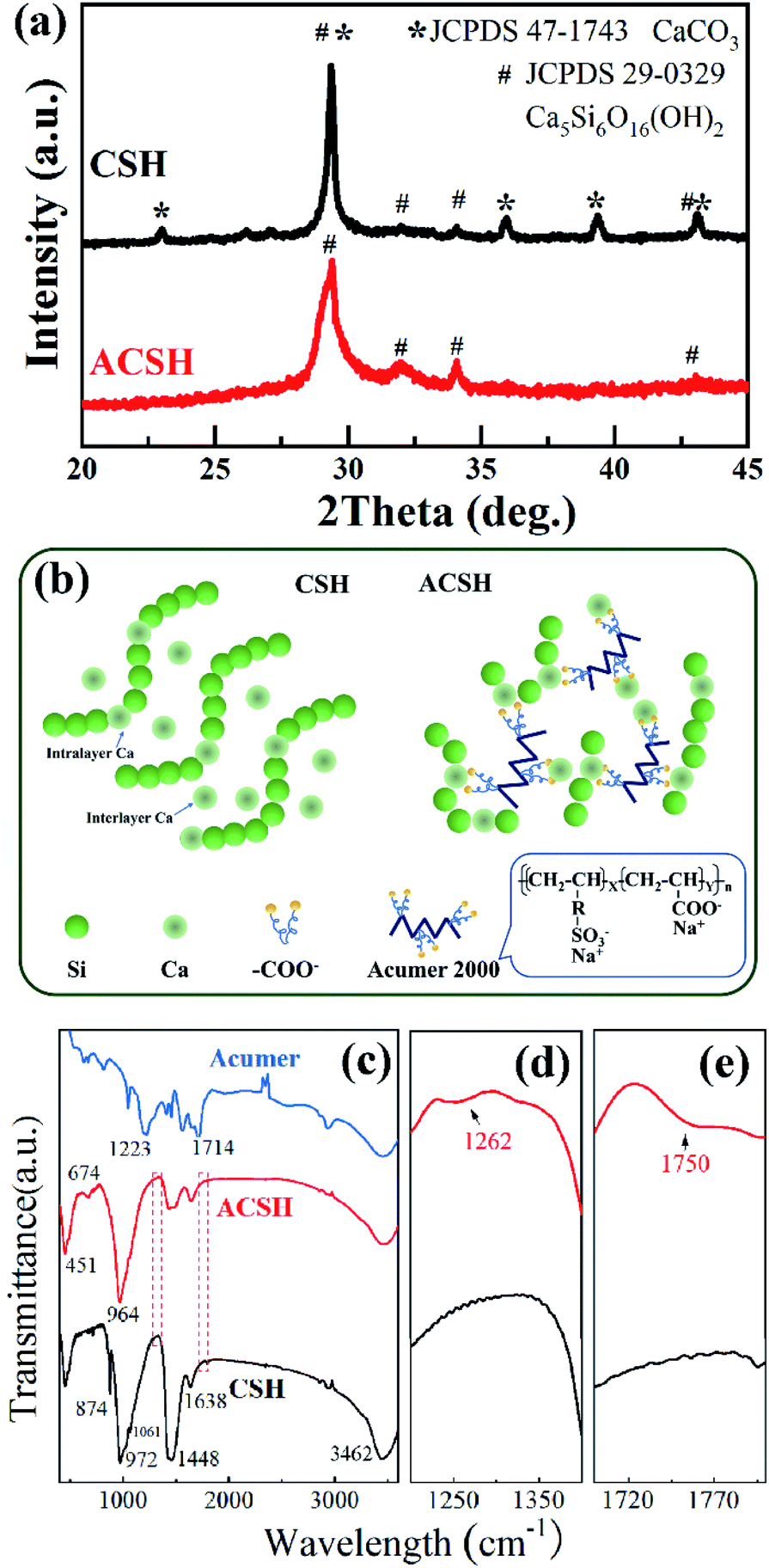 | ||
| Fig. 1 (a) XRD patterns, (b) schematic illustration of nanostructure and (c–e) FTIR spectra of CSH and ACSH. | ||
Copolymer Acumer2000 is made up by sulfonic group, carboxyl group and water-compatible carbon chains (see in Fig. 1b). Although the inhibition mechanism to CaCO3 has not been fully understood, it is believed that the encapsulation to Ca(II) from carboxylate plays an important role. As illustrated in Fig. 1b, the CSH has a layer-stacked structure composed of intralayer Ca and wollastonite type Si chains.22,23 When the molar Ca/Si ratio is 1, there are some distorted interlayer Ca atoms between layers,23 which prefer to react with CO2 in desilication solution and form by-product CaCO3.17 While, in the presence of Acumer2000, the carboxyl groups can recognize and chelate to Ca(II) ions, and its carbon chains with sulfonic groups can give rise to steric and electrostatic repulsion. Both intralayer Ca and interlayer Ca are in high dispersion and encapsulate state, which effectively inhibit the generation of CaCO3. Similar inhibitition mechanism were reported in Alendronic Acid and MLn systems.21,24 More interestingly, without the intervention of copolymer, CSH nuclei would grow into layer structure with a large size. Under the help of Acumer2000, the chelation of carboxylate with Ca(II) ions as well as the repulsion between crystal nuclei arising from sulfonic groups and the long carbon chains would slow down the nucleation and crystal growth rate, which can induce the layer structure of ACSH with much small size but high dispersancy.
Due to poor crystallinity of calcium silicate hydrate, it is difficult to obtain comprehensive information through XRD analysis. Therefore, FTIR was conducted. As shown in Fig. 1c, both CSH and ACSH presented characteristic peaks of calcium silicate hydrate at 451 cm−1,674 cm−1 and 1061 cm−1, which are attributed to Si–O–Si bending vibration, Si–O asymmetric stretching vibration and Si–O–Si asymmetric stretching vibration, respectively. It demonstrates that calcium silicate hydrate existed in CSH and ACSH. While, the peak at 972 cm−1 in CSH was red shifted to 964 cm−1 in ACSH, indicating that Si–O–Ca in calcium silicate hydrate was affected by the modification of Acumer2000.25,26 Meanwhile, weak signals of sulfonic acid group and carboxylic acid group could be detected in ACSH shown in Fig. 1d and e. However, compared with Acumer2000, the peak of sulfonic acid group in ACSH blue shifted from 1223 cm−1 to 1262 cm−1, and that of carboxylic acid group blue shifted from 1714 cm−1 to 1750 cm−1. It confirmed that Acumer2000 bridged onto the surface of calcium silicate hydrate through interaction with Si–O–Ca. In addition, the peak at 1448 cm−1 assigned to C–O antisymmetric stretching vibration was also observed in ACSH, which was due to the influence of CO2 in air during the test. For CSH, the peak at 1448 cm−1 was significantly enhanced, and the out-of-plane deformation vibration peak attributed to CO32− appeared at 874 cm−1, which was far beyond the influence of CO2 in air, reflecting the existence of CaCO3 materials in CSH.
Low amount of Acumer2000 in ACSH (<4 wt%) leads to weak signal observed in FTIR spectrum. In order to further analyze the influence of Acumer2000, XPS was used to characterize the samples and the results are presented in Fig. 2. Fig. 2a demonstrates that the surfaces of ACSH and CSH were composed of Ca, Si, O and C. For ACSH, the characteristic peak attributed to S 2p also was observed in the range of 150–175 eV, arising from the sulfonic acid group of Acumer2000. The high resolution XPS spectrum and fitting parameters for Ca 2p are shown in Fig. 2b and Table S3.† For CSH, the peaks of Ca 2p3/2 and Ca 2p1/2 in calcium silicate hydrate were located at 347.1 eV and 350.7 eV, respectively, while that in CaCO3 were located at 346.7 eV and 350.3 eV, respectively.27,28 According to the calculation of integral area, the mole ratio of Ca atoms in CaCO3 and calcium silicate hydrate was 3:7. For ACSH, the peaks of Ca 2p3/2 and Ca 2p1/2 were located at 346.8 eV and 350.4 eV, respectively. The shift to low binding energy indicated the increase of electron cloud density, which was likely that the sulfonate groups and carboxylic groups in Acumer2000 bonded on the surface of ACSH and reduced the binding energy of calcium atom. Similar phenomena have been reported in other systems.29,30 The peaks at 284.8 eV, 286.6 eV and 288.5 eV in Fig. 2c belonged to C–C, C–R and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C derived from polluted carbon in the test. For CSH, a characteristic peak of CO32− appeared at 289.3 eV, which belonged with CaCO3 in sample. That peak did not appear in ASCH, but a new peak appeared at 288.8 eV, which was attributed to the carboxylic acid group. Combined with XRD and FTIR data, it indicated that both calcium silicate hydrate and CaCO3 existed in CSH, further confirming that the purity of ACSH was higher than that of CSH.
C derived from polluted carbon in the test. For CSH, a characteristic peak of CO32− appeared at 289.3 eV, which belonged with CaCO3 in sample. That peak did not appear in ASCH, but a new peak appeared at 288.8 eV, which was attributed to the carboxylic acid group. Combined with XRD and FTIR data, it indicated that both calcium silicate hydrate and CaCO3 existed in CSH, further confirming that the purity of ACSH was higher than that of CSH.
 | ||
| Fig. 2 XPS spectra of (a) survey spectrum, (b) Ca 2p high-resolution spectrum and (c) C 1s high-resolution spectrum for samples. | ||
Fig. S1† shows SEM and EDS results of samples. Fig. S1a† indicated that there were two kinds of materials with different morphologies in CSH. Combing with EDS results (Fig. S1b and c†) and above XRD, FTIR and XPS analysis, the block material (region 1 in Fig. S1a†) with a size of about 2 μm and dense surface, containing a lot of calcium and trace silicon, was CaCO3. The flake material (region 2 in Fig. S1a†) mainly composed of calcium, silicon and oxygen, was calcium silicate hydrate. Obviously, CSH sample was made up by flake calcium silicate hydrate and a small amount of block CaCO3. And there were many slit holes formed by flake stacking. Based on the results from Fig. S1d–f,† the morphology and composition distribution of ACSH were more uniform. It mainly comprised lamellar calcium silicate hydrate particles. Because of the intervention of Acumer2000, the size and thickness of the ACSH sheets were smaller in contrast to that of CSH, but the dispersancy was higher. Similarly, a large number of slit holes also appeared in ACSH due to the stacking of lamellar. While, the size of these slits was smaller and the number was more abundant than that of CSH.
Pore structure and surface area are important factors for the adsorption performance. Therefore, N2 adsorption and desorption test was conducted. The isotherm of CSH and ACSH (Fig. 3a) could be classified as a type-IV with H3 hysteresis loop, meaning the existence of slit-shaped pores, which was consistent with SEM observation.31 For ACSH sample, the broad hysteresis loop in the relative pressures from 0.4 to 1.0 respected that there were a few macropores and plenty of mesopores. The pore size distribution (Fig. 3b) proved that most of the mesopores for ACSH existed in with diameter from 2 to 10 nm. While for CSH, the pore size was concentrated at about 19 nm. Calculated from the adsorption branch of the isotherm with BJH method, average diameter (DBJH) and cumulative pore volume (VBJH) were determined with the pore size in the range of 2–50 nm, which was benefit for the adsorption of Cu(II). The specific surface area (SBET), DBJH and VBJH were shown in the inset of Fig. 3a. Obviously, Acumer2000 brought a high mesoporosity and the small pores for ACSH, displaying the number of mesopores in sample ACSH was extremely high, which contributed greatly to the specific surface area of 356 m2 g−1. In addition, the hysteresis loop of ACSH on the nitrogen adsorption/desorption isotherms was more pronounced than that of CSH, implying that the surface of ACSH had a stronger force on adsorbents.32,33 A large specific surface area, abundant pores and an effective interaction with the adsorbate constituted an ideal condition for Cu(II) adsorption, which was expected to exhibit excellent performance.
 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution curves of samples. The inset shows the specific surface area and the average pore width and volume of pores determined by the BJH method using adsorption isotherms. | ||
3.3 Adsorption performance and mechanism
Based on the above analysis, ACSH has high purity, small size, rich mesopores, large specific surface area and strong adsorption force, owning obvious characteristics for adsorption. In order to confirm its superiority, Cu(II) was taken as target pollutant. The experiment was performed at 20 °C for 2 h, the initial concentration of Cu(II) was 100 mg L−1, the pH was 6 ± 0.3 and the dosage of adsorbent was 0.5 g L−1. Fig. 4a and b presents the curve of sorption capacity and residue percentage of pollutants with different adsorption time. For ACSH, the adsorption capacity was as high as 191.1 mg g−1 at 5 min, and only 4.4% Cu(II) was left in the solution. With the increase of time, the performance was maintained without Cu(II) desorption. For CSH, the sorption capacity was 74.2 mg g−1 at 5 min, when 62.9% Cu(II) remained in the solution. With the increase of time, the sorption capacity increased gradually, reaching 135.4 mg g−1 at 2 h, and still 32.3% Cu(II) failed to adsorb. Fig. 4c shows the sorption capacities for aged CSH and ACSH samples. By exposed in air for 100 days, the sorption capacity of ACSH kept as high as that of fresh sample. However, aged CSH completely lost adsorption ability in this condition, likely due to the formation of CaCO3 in large quantities during aging period. Overall, the modified calcium silicate hydrate not only had a high sorption capacity but also displayed a long-term stability. | ||
| Fig. 4 Cu(II) residue percentage and sorption capacity varied with different adsorption time of (a) ACSH and (b) CSH; (c) Cu(II) sorption capacity at 5 and 30 min for fresh samples and aged samples; Sorption capacity of (d) ACSH varied with different pH and (e) different samples; (f) Linearized fitting curves of Langmuir model for isothermal adsorption of ACSH and CSH. | ||
The pH effect on ACSH sorption capacity was investigated (Fig. 4d). The experimental data was performed at 20 °C for 2 h, the initial concentration of Cu(II) was 300 mg L−1 and the dosage of adsorbent was 0.5 g L−1. Due to the appearance of white turbidity caused by Cu(OH)2 precipitation when pH > 6, the range of pH was 2–6 in this experiment. In general, the adsorption capacity of ACSH for Cu(II) increased gradually with the raise of pH. There were a certain amount of hydroxyl groups, sulfonic groups and carboxylic acid groups on surface of ACSH. When the pH was 2–4, active groups reacted with hydrogen ions under acidic conditions. Adsorbent was difficult to coordinate with metal ions and the adsorption performance mainly depended on the high specific surface area of ACSH. The adsorption capacity was relatively low. When the range of pH was 5–6, the functional groups on the surface of ACSH were deprotonated and the coordination effect increased. Accordingly, the sorption capacity increased significantly. In summary, the high sorption capacity of ACSH for Cu(II) stems from the synergistic effect of abundant surface active groups and high specific surface area.
Fig. 4e presents the sorption capacities of ACSH and commercial activated carbon. The experimental data was performed at 20 °C for 2 h, the initial concentration of Cu(II) was 300 and 500 mg L−1, without adjusting pH, and the dosage of adsorbent was 0.5 g L−1. The specific surface area of commercial activated carbon was 658 m2 g−1, much higher than that of ACSH (356 m2 g−1). In the 300 mg L−1 Cu(II) solution, the sorption capacity of activated carbon was only 42 mg g−1, which was 14.3% of that of ACSH. When the initial concentration of Cu(II) solution was raised to 500 mg L−1, the sorption capacity of ACSH was further increased to 374 mg g−1, which was 11.7 times as large as that of activated carbon.
In order to explore the adsorption mechanism of ACSH, the sorption capacities and process were evaluated by adsorption isotherm. Langmuir (eqn (5)) model and Freundlich (eqn (6)) model, which are most commonly used to analyze the adsorption process, were adopted:
 | (5) |
 | (6) |
Fig. S2† and 4f shows the adsorption isotherm and fitting curve of different models. The experiment was performed at 20 °C for 4 h, the initial concentration of Cu(II) varied from 100 to 1000 mg L−1, the initial pH was adjusted to 5.0 ± 0.3 and the dosage of adsorbent was 0.5 g L−1. Related parameters are provided in Table 1. Obviously, the correlation coefficients R2 of Langmuir isothermal model was higher than that of Freundlich model for both ACSH and CSH, meaning Cu(II) adsorption process was carried out on the homogeneous surface and the adsorption was monolayer adsorption. In Langmuir model, the maximum adsorption capacities of ACSH and CSH were 526.3 mg g−1 and 294.1 mg g−1, respectively, close to the experimental value 532 mg g−1 and 278 mg g−1. For ACSH, the high sorption capacity was due to rich pore structure, high specific surface area and resistance to corrosion of CO2, which significantly improved the number of active adsorption sites, the purity of materials and their bonding ability to pollutants. In addition, the KL for ACSH adsorbent was only 0.0826, indicating that the adsorption process was highly favorable. Table S4† presents the adsorption performances for Cu(II) using different adsorbents reported previously. In contrast to these results, ACSH showed strong competitiveness for heavy metal adsorption.
| Sample | Freundlich | Langmuir | ||||
|---|---|---|---|---|---|---|
| KF | n | R2 | KL | qmax | R2 | |
| ACSH | 209 | 6.78 | 0.8767 | 0.0826 | 526.3 | 0.9974 |
| CSH | 54 | 3.81 | 0.8892 | 0.0200 | 294.1 | 0.9954 |
The saturated adsorbent precipitations during this work were filtered by vacuum, washed and dried to obtain CSH-Cu and ACSH-Cu samples, which were analyzed by XRD and XPS. Fig. 5a presents XRD pattern. The phase compositions of the samples after adsorption were obviously different from that before adsorption, as shown in Fig. 1a. After Cu(II) adsorption, no diffraction peak of calcium silicate hydrate was observed for ACSH-Cu but new characteristic peaks were observed at 16.2°, 31.6°, 32.2° and 39.4°. They were attributed to copper salt (JCPDS 77-0116). The peaks were strong and the width was narrow, indicating a relatively good crystallinity. Therefore, the characteristic diffraction peaks of calcium silicate hydrate with poor crystallinity were difficult to observe. Similar phenomena have been reported in other literatures related to calcium silicate hydrate.34 For CSH-Cu, the diffraction peaks attributed to copper salt widened, resulting from a smaller adsorption capacity to Cu(II) and negative condition for the nucleation and growth of copper salt. And for CSH-Cu, the characteristic peak (29.4°) corresponding to CaCO3 material (JCPDS 47-1743) still existed.
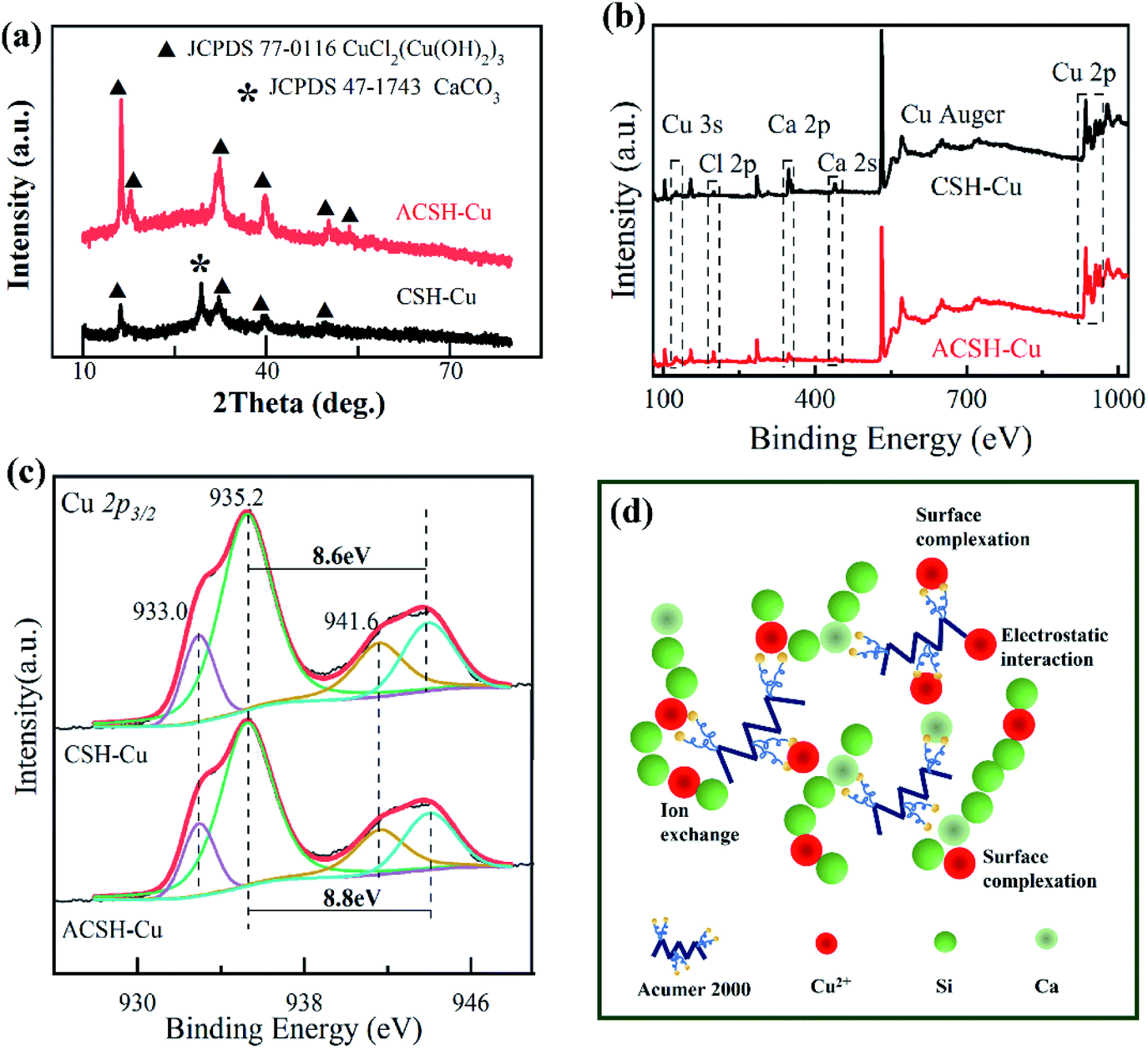 | ||
| Fig. 5 (a) XRD patterns of samples CSH-Cu and ACSH-Cu; XPS spectra of (b) survey spectrum and (c) Cu 2p3/2 high-resolution spectrum for samples; (d) schematic illustration of the possible adsorption mechanism for ACSH. | ||
Fig. 5b shows the XPS spectra of samples after adsorption. Compared with Fig. 2a and 5b demonstrated that there were Cu and Cl elements on the surface of CSH-Cu and ACSH-Cu samples, besides the original Ca, Si, O and C elements. The signal of Ca atom decreased significantly, which confirmed that ion exchange was still an important way in the adsorption process for CSH and ACSH. Because CSH contained a certain amount of CaCO3, which was difficult to participate in the ion exchange and kept in sample CSH-Cu. Compared with CSH-Cu, the signal of Ca atoms in ACSH-Cu samples was much weaker. These results are consistent with the XRD data in Fig. 5a. Fig. 5c presents the Cu 2p3/2 spectrum of the samples. The peak located at 933.0 eV was attributed to Cu–OH and the satellite peak was located at 941.6 eV. For CSH–Cu, the peak located at 935.2 eV belonged to Cu–Cl, and the satellite peak was located at 943.8 eV. The separation between the main peak and the satellite peak was 8.6 eV. While, for ACSH-Cu, the peak located at 935.2 eV moved to a higher binding energy, which was 944.0 eV. The separation increased to 8.8 eV. The satellite peak depends on the hybridized degree between 3d orbital of copper atom and 2p orbital of O atom. According to theoretical calculation,35 the increase of separation meant the functional groups on the surface of ACSH working like electron donor for Cu(II), which would induce the strong intervention between surface of modified calcium silicate hydrate and copper atom. It is consistent with the results of Fig. 3a. Similar phenomena have been reported in other literatures.36,37
Combining the XRD and XPS analysis for the samples before and after adsorption with the results of adsorption isotherm fitting, although the adsorption mechanism for ACSH is not yet fully understood, we believe that the synergistic effect of ion exchange and surface complexation is an important factor for the remarkable performance. As illustrated in Fig. 5d, ACSH modified by Acumer2000 is composed of a large number of ultra-thin calcium silicate hydrate sheets without impurities, which can form aggregates with plenty of slit channels and pores, providing the possibility of physical adsorption between ACSH and Cu(II) and benefiting the further ion exchange between Ca(II) on the thin sheet and Cu(II) in the surrounding solution. Furthermore, the surface of ACSH contains multiple sites for Cu(II), such as hydroxyl groups, carboxyl groups and sulfonic groups, exhibiting excellent chelating ability. Through strong complexation, electrostatic interaction, ion exchange or a combination of the above, high sorption capacity could achieve in short time. Finally, due to the corrosion resistance of ACSH to CO2, the excellent adsorption performance can remain at a high level for a long time.
3.4 Performance of adsorption membrane
ACSH powder material synthesized based on fly ash desilication liquid has significant adsorption advantages. In order to investigate its practical application potential, this work prepared calcium silicate hydrate based adsorption film (CSH-M and ACSH-M) by a simple filtration technology and tested the performance of adsorption films using the device shown in Fig. 6a. A peristaltic pump was used to push Cu(II) solution through the adsorption membrane at a flow rate of 3 mL min−1. Considering that the concentration of metal ions in actual waste water is low, 20 mg L−1 Cu(II) solution was taken. As shown in Fig. 6b, part of Cu(II) in the solution will interact with active sites on the surface of adsorbents while the others will pass through filtration membrane in the process of experiment. The removal rates of Cu(II) can be calculated by eqn (3) and results are shown in Fig. 6c. When flux was 30 mL, the removal rates for CSH-M and ACSH-M maintained at ∼94% and ∼95%, respectively. When flux increased to 120 mL, the removal rate of ACSH-M membrane decreased slightly, but was as high as 94%. However, the performance of CSH-M decreased to ∼87%. In general, ACSH-M had better adsorption efficiency and stability, showing potential for practical application. In comparison to previously reported work listed in Table S5,† both the water flux and rejection of Cu(II) obtained by ACSH-M were higher. Most importantly, the fabrication of the membrane materials reported previously either required complex CVD treatment, or high-temperature hydrothermal processes, or a large number of organic reagents, which are not conducive for large-scale production. Compared with the above materials, the modified calcium silicate hydrate in our work, which was synthesized using solid waste fly ash as silicon source and a small amount of Acumer2000 as modifier by a simple co-precipitation process without other organic solvent, exhibited remarkable economic advantages. | ||
| Fig. 6 (a) Photograph and (b) schematic illustration of the experimental setup, (c) Cu(II) removal performance of ACSH-M and CSH-M. | ||
4 Conclusions
In summary, we have proposed a “trash-to-treasure” strategy to develop a modified calcium silicate hydrate materials from fly ash for highly efficient Cu(II) removal applications. The obtained ACSH samples not only owned a high purity but also possessed ideal structure for adsorption, resulting from the modifier Acumer2000's inhibitition to by-product CaCO3 and modulation to micromorphology. Through strong complexation, electrostatic interaction, ion exchange mechanism, ACSH achieved 95.6% Cu(II) removal rate within 5 min, under the conditions of 0.5 g L−1 adsorbent concentration and 100 mL Cu(II) solution with concentration of 100 mg L−1. The adsorption isotherms conformed to Langmuir models and the maximum adsorption capacity was 532 mg g−1. Moreover, the excellent performance could be maintained for 100 days, displaying excellent long-term stability. When applied to the membrane adsorption process, the adsorbent still showed a high removal rate of Cu(II) under a high water flux, ensuring the promising application potential. Our work solves the bottleneck problems in cost and stability of calcium silicate hydrate, explores a new area for fly ash utilization and is of great significance for practical applications. In this study, about 60% silicon in fly ash have not been developed, and much desilication fly ash has been produced during the experiment. Future research needs to develop treatment methods with higher silicon-lift rates in order to fully exploit the silicon resources in fly ash. At the same time, the study on application of desilication fly ash would be carried out.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51878647, 61574148) and Taicang Basic Research Program of China (TC2020JC08).References
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4, 36 CrossRef CAS.
- G. T. Tee, X. Y. Gok and W. F. Yong, Environ. Res., 2022, 212, 113248 CrossRef CAS PubMed.
- J. Liu, Y. Huang, H. Li and H. Duan, Chemosphere, 2022, 287, 132021 CrossRef CAS PubMed.
- Z. Xu, T. Wu, Y. Cao, C. Chen, S. Ke, X. Zeng and P. Lin, Chem. Eng. J., 2020, 392, 123639 CrossRef CAS.
- S. Rajendran, A. K. Priya, P. S. Kumar, T. K. A. Hoang, K. Sekar, K. Y. Chong, K. S. Khoo, H. S. Ng and P. L. Show, Chemosphere, 2022, 303, 135146 CrossRef CAS PubMed.
- A. T. Hoang, S. Kumar, E. Lichtfouse, C. K. Cheng, R. S. Varma, N. Senthilkumar, P. Q. P. Nguyen and X. P. Nguyen, Chemosphere, 2022, 303, 134825 CrossRef PubMed.
- M. Santhamoorthy, K. Thirupathi, T. Periyasamy, D. Thirumalai, V. Ramkumar, S. P. Asrafali and S.-C. Kim, Surf. Interfaces, 2022, 29, 101808 CrossRef CAS.
- W. Fu, G. Ji, H. Chen, S. Yang, H. Yang, B. Guo and Z. Huang, J. Environ. Chem. Eng., 2020, 8, 104072 CrossRef CAS.
- Y. Yan, Y. Chu, M. A. Khan, M. Xia, M. Shi, S. Zhu, W. Lei and F. Wang, Sci. Total Environ., 2022, 806, 150652 CrossRef CAS PubMed.
- G. Rathee, S. Kohli, A. Awasthi, N. Singh and R. Chandra, RSC Adv., 2020, 10, 19371 RSC.
- N. Shao, S. Tang, Z. Liu, L. Li, L. Yan, S. Li and Z. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 14926–14935 CrossRef CAS.
- G. Qi, X. Lei, L. Li, C. Yuan, Y. Sun, J. Chen, J. Chen, Y. Wang and J. Hao, Chem. Eng. J., 2015, 279, 777–787 CrossRef CAS.
- J. Wu, Y. J. Zhu and F. Chen, Small, 2013, 9, 2911–2925 CrossRef CAS PubMed.
- T. Taweekarn, W. Wongniramaikul and A. Choodum, J. Environ. Manage., 2022, 301, 113923 CrossRef CAS PubMed.
- F. Valenzuela, G. Quintana, A. Briso, V. Ide, C. Basualto, J. Gaete and G. Montes, J. Water Process. Eng., 2021, 40, 101951 CrossRef.
- L. Zeng and L. Yang, J. Chem., 2015, 131050 Search PubMed.
- T. F. Sevelsted and J. Skibsted, Cem. Concr. Res., 2015, 71, 56–65 CrossRef CAS.
- L. Liu, T. Li, G. Yang, Y. Wang, A. Tang and Y. Ling, J. Environ. Chem. Eng., 2017, 5, 6201–6215 CrossRef CAS.
- L. Liu, J. Liu, T. Li, G. Yang, A. Tang and Y. Ling, Desalin. Water Treat., 2018, 107, 165–181 CrossRef CAS.
- L. C. Wang, W. D. Zhao, Y. Che, R. K. Gou, H. B. Liu, C. G. Zhu, L. B. Wang and Z. Guan, J. Appl. Polym. Sci., 2020, 137, 1–4 Search PubMed.
- M. F. Mady, R. Ortega and M. A. Kelland, Energy Fuels, 2022, 36, 1863–1873 CrossRef CAS.
- J. J. Beaudoin, H. Dramé, L. Raki and R. Alizadeh, J. Mater. Res., 2008, 23, 2804–2815 CrossRef CAS.
- J. Li, W. Zhang and P. J. M. Monteiro, ACS Sustainable Chem. Eng., 2020, 8, 12453–12461 CrossRef CAS.
- G. Liu, M. Xue, Q. Liu, H. Yang, J. Yang and Y. Zhou, RSC Adv., 2017, 7, 24723–24729 RSC.
- A. Meiszterics, L. Rosta, H. Peterlik, J. Rohonczy, S. Kubuki, P. Henits and K. Sinkó, J. Phys. Chem. A, 2010, 114, 10403–10411 CrossRef CAS PubMed.
- Y. Kuwahara, S. Tamagawa, T. Fujitani and H. Yamashita, J. Mater. Chem. A, 2013, 1, 7199–7210 RSC.
- L. Black, K. Garbev, G. Beuchle, P. Stemmermann and D. Schild, Cem. Concr. Res., 2006, 36, 1023–1031 CrossRef CAS.
- C. S. Gopinath, S. G. Hegde, A. V. Ramaswamy and S. Mahapatra, Mater. Res. Bull., 2002, 37, 1323–1332 CrossRef CAS.
- J. Laisney, A. Rosset, V. Bartolomei, D. Predoi, D. Truffier-Boutry, S. Artous, V. Bergé, G. Brochard and I. MichaudSoret, Environ. Sci.: Nano, 2021, 8, 297–310 RSC.
- Y. Chu, X. Zhang, W. Chen, F. Wu, P. Wang, Y. Yang, S. Tao and X. Wang, Sci. Total Environ., 2019, 681, 124–132 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemiemiewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- Y. Kuwahara, S. Tamagawa, T. Fujitani and H. Yamashita, Materials, 2021, 14, 3914 CrossRef PubMed.
- Y. K. Tovbin and A. B. Rabinovich, Russ. Chem. Bull., 2009, 58, 1627–1636 CrossRef CAS.
- L. Chen, X. Wang, Y. Chen, Z. Zhuang, F. Chen, Y. Zhu and Y. Yu, Chem. Eng. J., 2020, 402, 125922 CrossRef CAS.
- G. Moretti and H. P. Beck, Surf. Interface Anal., 2022, 54, 803–812 CrossRef.
- N. Masciocchi, E. Corradi, A. Sironi, G. Moretti, G. Minelli and P. Porta, J. Solid State Chem., 1997, 31, 252–262 CrossRef.
- L. Chang, Y. Pu, P. Jing, Y. Cui, G. Zhang, S. Xu, B. Cao, J. Guo, F. Chen and C. Qiao, Appl. Surf. Sci., 2021, 541, 148400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03007a |
| This journal is © The Royal Society of Chemistry 2022 |