
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study on heterogeneous OH oxidation of 3-methyltetraol sulfate in the atmosphere under high NO conditions†
Chuanen Guoa,
Luyao Xub and
Chenxi Zhang *c
*c
aJudicial Expertise Center, Shandong University of Political Science and Law, Jinan 250014, P. R. China
bEnvironment Research Institute, Shandong University, Qingdao 266200, P. R. China
cJia Si-xie Agricultural College, Weifang University of Science and Technology, Weifang 262700, P. R. China. E-mail: sdzhangcx@163.com
First published on 1st August 2022
Abstract
Organosulfates (OSs), also known as organic sulfate esters, are ubiquitous in atmospheric particles and used as secondary organic aerosol (SOA) markers. However, the chemical transformation mechanism of these OSs remains unclear. Therefore, we investigated the heterogeneous OH oxidation of 3-methyltetraol sulfate (3-MTS), which is one of the most abundant particulate organosulfates, by using quantum chemical and kinetic calculations. 3-MTS can easily undergo abstraction reaction with OH radicals, and the reaction rate constant is about 7.87 × 10−12 cm3 per molecule per s. The generated HCOOH, CH3COOH, HCHO, CH3CHO and 2-methyl-2,3-dihydroxypropionic acid are low-volatility species with increased water solubility, which are the main components of SOA. In addition, the OH radicals obtained from the reaction can continue to promote the oxidation reaction. The results of this study provide insights into the heterogeneous OH reactivity of other organosulfates in atmospheric aerosols, and it also provides a new understanding of the conversion of sulfur (S) between its organic and inorganic forms during the heterogeneous OH oxidation of organic sulfates.
Introduction
OSs generally refer to sulfate-containing ester compounds and their derivatives, which are an important class of SOA, accounting for about 5–30% of the total mass fraction of organic matter in PM10.1–3 OSs have been observed in atmospheric particles collected in rural, urban, ocean, forest and arctic regions.4–11 Due to the hydrophilic and hydrophobic functional groups the OSs molecules, they can contribute to climate change by affecting the hygroscopicity and light absorption of aerosols.12,13 Given this, it is imperative to understand the origin, formation and transformation of OSs species in the atmosphere.OSs can be formed from the heterogeneous reaction of biogenic volatile organic compounds (BVOCs) such as isoprene, monoterpenes, sesquiterpenes, oxidized derivatives, and some chlorophyll alcohols with acidic sulfates in the atmosphere.14–19 In addition to natural sources, anthropogenic alkanes, polycyclic aromatic hydrocarbons, unsaturated fatty acids, and diesel fuels can also serve as precursors for OSs.20–24 These organic precursors can be further oxidized and then react with sulfur-containing nucleophiles to form OS.
The most abundant OSs in ambient aerosols, isoprene-derived organosulfates, are formed from the reaction of sulfates with isoprene oxidation products in the particulate phase.25–31 Isoprene can undergo atmospheric oxidation reaction with hydroxyl radicals (OH), and the generated OH-isoprene will be oxidized to form isoprene hydroperoxide (ISOPOOH) under low nitrogen oxide (NOx) conditions.32,33 ISOPOOH can further react with OH radicals to form isomeric isoprene epoxydiols (IEPOX). Under acid-catalyzed conditions, it is partitioned into sulfate aerosols through a ring-opening reaction. At the surface of the aerosol, sulfate attacks IEPOX to form a large amount of isoprene-derived OSs.25,34–37 Of these, the most abundant OS is methyltetraol sulfate (MTS). The data shows that, in the PM2.5 of downtown Atlanta and Look Rock in the United States, the proportion of organic carbon of MTS accounts for as high as 13%.38,39
Although the formation mechanism of OSs has been extensively studied, their chemical transformation remains unclear.24,40–42 These low-volatility OSs preferentially exist in the particulate phase, where they are oxidized at the aerosol surface by gas-phase oxidants, such as OH radicals, O3 and NO3 radicals.18,43–47 The most reactive atmospheric gas-phase radicals, especially OH radicals, readily facilitate this chemical reaction through surface interactions. For MTS, Lam et al. investigated the heterogeneous OH oxidation of potassium 3-MTS (C5H11SO7K) at 70.8% RH by using an aerosol flow tube reactor.32 The effective rate constant for the heterogeneous reaction is 4.74 ± 0.2 × 10−13 cm3 per molecule per s, corresponding to an atmospheric lifetime of 16.2 ± 0.3 day. Chen et al. investigated the oxidative aging of 2-MTS aerosols by gas-phase OH radicals at 61 ± 1% relative humidity.48 Kinetic measurements reveal that the effective heterogeneous OH rate constant is 4.9 ± 0.6 × 10−13 cm3 per molecule per s, with an atmospheric lifetime against OH oxidation of 16 ± 2 day, which is close to the results of 3-MTS. These suggest that MTS, as 3-MTS or 2-MTS, can exist for a longer periods in the atmosphere. In terms of the reaction mechanism, using aerosol mass spectrometry, Lam et al. found that OH oxidation of 3-MTS only resulted in an increase in hydrogen sulfate ions (HSO4−), while no other oxidation products were detected.32 Analyzing the reason, it is likely that other products are volatile and redistribute to the gas phase. Combined with aerosol phase reactions reported in previous literature, four reaction pathways with different initial hydrogen extraction points are proposed.44,46 Chen et al. paid more concerned to how 2-MTS and OH radicals were converted into OSs monitored by HILIC/ESI-HR-QTOFMS.48 Thus, the specific oxidation reaction mechanism between MTS and OH radicals is still unclear.
In this article, the degradation processes of the 3-MTS with OH radicals were investigated via the quantum chemical calculation approaches. Our results provide a pathway for atmospheric transformation of isoprene-derived organosulfates, which can better understand their potential effect on air quality and climate change.
Computational methods
The Gaussian 09 package was chosen to perform configuration optimization and energy calculations.49 The M06-2X density functional method is one of the best functionals for calculating chemical thermodynamics and non-covalent interactions of main group elements.50 The optimal configurations of reactants, transition states, intermediates and products were obtained at the level of the 6-311++G(d,p) basis set. Frequency analysis was performed at the same level. Stable reactants, intermediates and products are free of imaginary frequencies. The transition state has one and only one imaginary frequency, and is further determined as the transition state corresponding to the reactants and products by the calculation of intrinsic reaction coordinates (IRC).51 All DLPNO-CCSD(T) single point energy calculations were carried out using the ORCA program (version 5.0) in conjunction with the cc-pVTZ basis set.52,53 In addition, the polarized continuum model (PCM) within a self-consistent reaction field (SCRF) theory was used as the model of the continuum solvent effects.54,55 The PCM has been proven to be flexible and accurate, in particularly, when the solute is accommodated in a cavity of realistic molecular shape and has been widely used for the study of many chemical processes.56Rate constants were calculated by the KiSTelP program over a temperature range of 298 K and a pressure of 1.0 bar.57 The procedure is mainly based on transition state theory (TST) and Wigner tunneling correction.58
Results and discussion
It is more favorable for the OH to be partially solvated at the surface rather than fully solvated in bulk water.59 Therefore, the first oxidation step is the abstraction reaction of 3-MTS, which exists in the droplet in its ionic form, with gas-phase OH radical on the surface of the aerosol.Initial reactions with OH radical
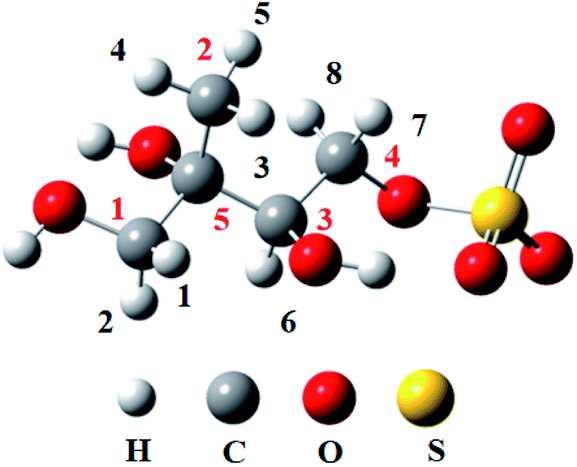 | ||
| Fig. 1 The labeled numbers in the structure of 3-MTS. | ||
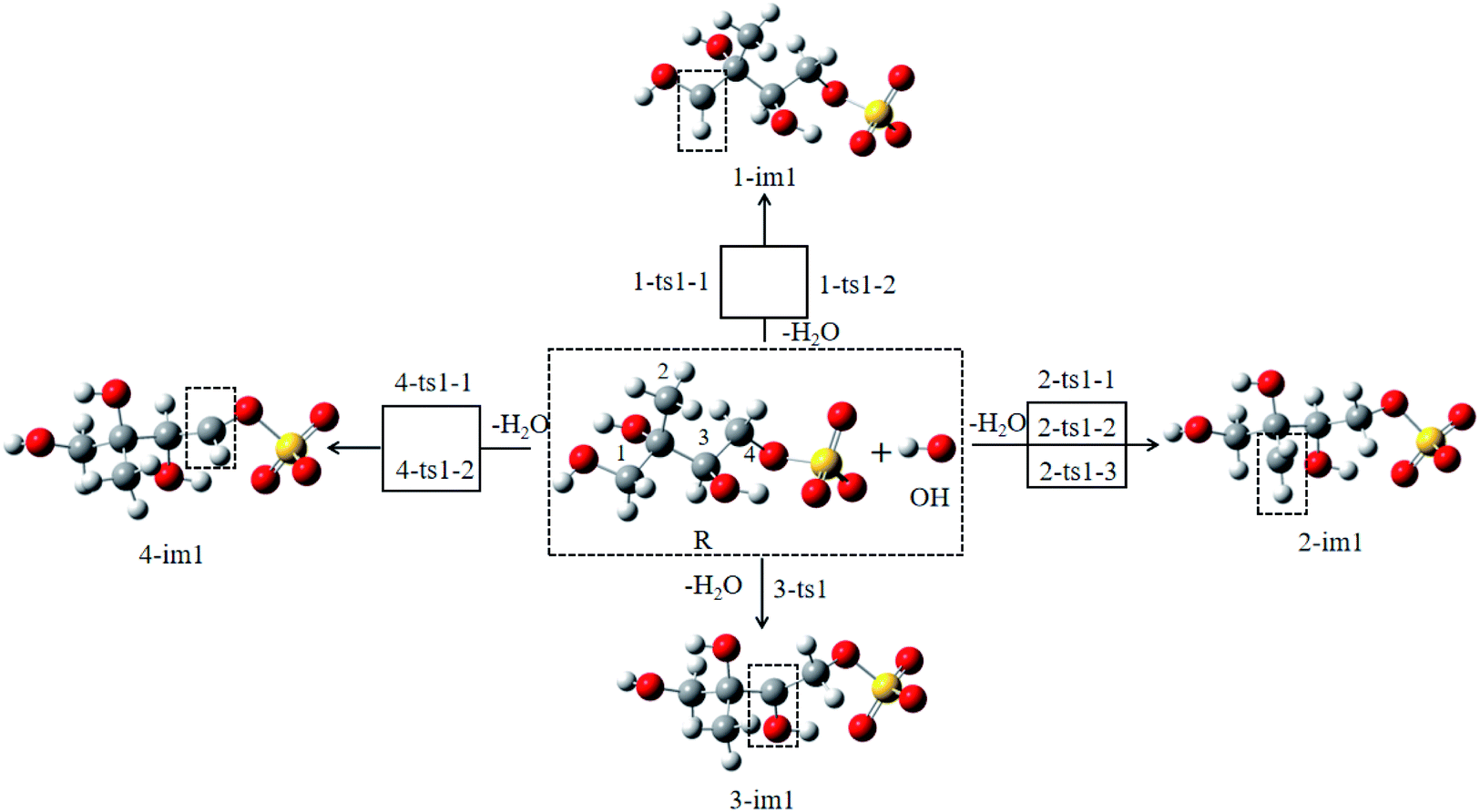 | ||
| Fig. 2 The possible reactions of 3-MTS with OH radicals. | ||
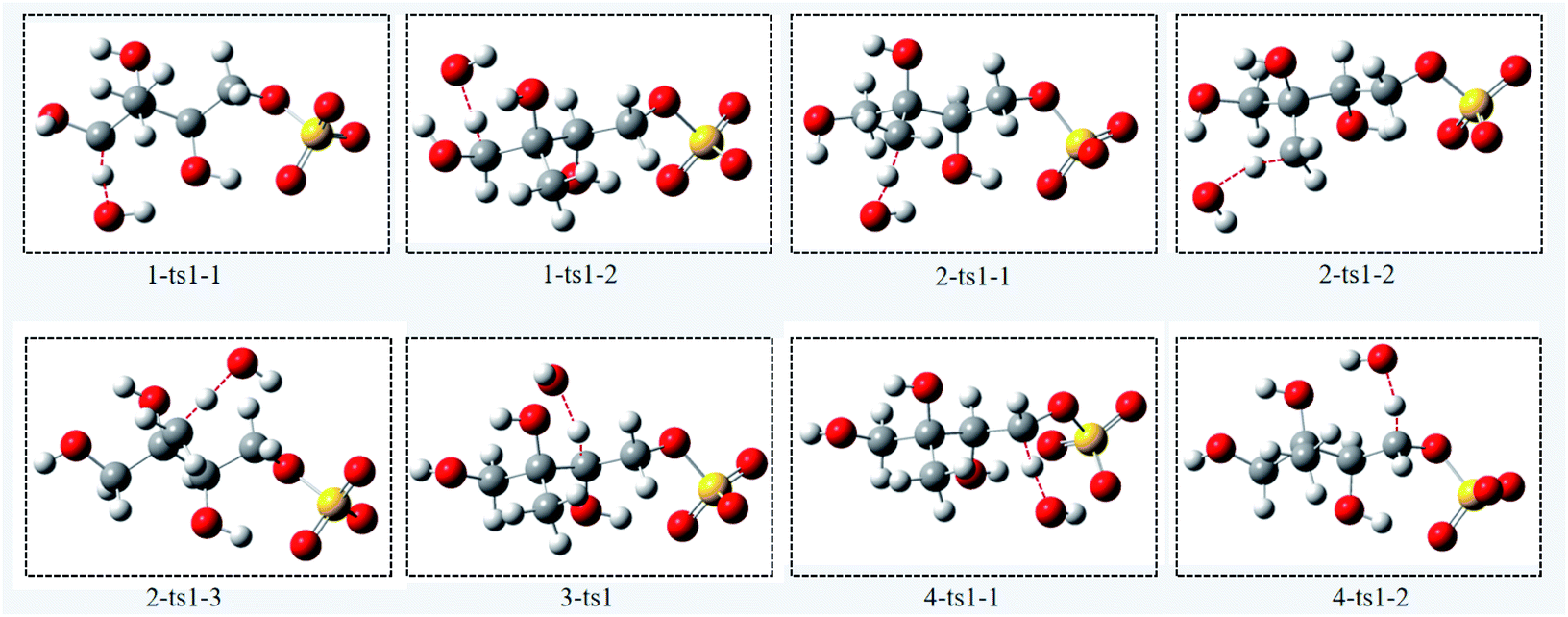 | ||
| Fig. 3 The optimized transition state structures of 3-MTS with OH radicals. | ||
| Reaction | k268 K | k278 K | k288 K | k298 K | k308 K | k318 K | ΔGts–R(298 K) | ΔGim–R(298 K) | R298 K |
|---|---|---|---|---|---|---|---|---|---|
| R + OH → 1-ts1-1 → 1-im1 + H2O | 5.13 × 10−12 | 4.54 × 10−12 | 4.06 × 10−12 | 3.67 × 10−12 | 3.34 × 10−12 | 3.07 × 10−12 | 4.61 | −22.96 | 52.67% |
| R + OH → 1-ts1-2 → 1-im1 + H2O | 5.60 × 10−13 | 5.28 × 10−13 | 5.01 × 10−13 | 4.77 × 10−13 | 4.57 × 10−13 | 4.40 × 10−13 | 5.99 | ||
| R + OH → 1-ts1-1 → 2-im1 + H2O | 1.50 × 10−13 | 1.45 × 10−13 | 1.41 × 10−13 | 1.37 × 10−13 | 1.34 × 10−13 | 1.31 × 10−13 | 6.63 | −16.22 | 2.05% |
| R + OH → 2-ts1-2 → 2-im1 + H2O | 2.11 × 10−14 | 2.17 × 10−14 | 2.23 × 10−14 | 2.29 × 10−14 | 2.35 × 10−14 | 2.41 × 10−14 | 7.24 | ||
| R + OH → 2-ts1-3 → 2-im1 + H2O | 1.68 × 10−15 | 1.93 × 10−15 | 2.21 × 10−15 | 2.50 × 10−15 | 2.82 × 10−15 | 3.16 × 10−15 | 11.27 | ||
| R + OH → 3-ts1 → 3-im1 + H2O | 4.78 × 10−12 | 4.22 × 10−12 | 3.77 × 10−12 | 3.39 × 10−12 | 3.08 × 10−12 | 2.83 × 10−12 | 3.09 | −25.44 | 43.06% |
| R + OH → 4-ts1-1 → 4-im1 + H2O | 1.04 × 10−13 | 1.03 × 10−13 | 1.03 × 10−13 | 1.02 × 10−13 | 1.02 × 10−13 | 1.02 × 10−13 | 5.89 | −21.22 | 2.22% |
| R + OH → 4-ts1-2 → 4-im1 + H2O | 7.13 × 10−14 | 7.21 × 10−14 | 7.29 × 10−14 | 7.38 × 10−14 | 7.47 × 10−14 | 7.58 × 10−14 | 5.67 | ||
| Total (cm3 per molecule per s) | 1.08 × 10−11 | 9.63 × 10−12 | 8.67 × 10−12 | 7.87 × 10−12 | 7.21 × 10−12 | 6.68 × 10−12 |
As shown in Fig. 1, there are 8 different H atoms in 3-MTS that can undergo abstraction reactions. We are mainly divided into four types of hydrogen atoms, namely H1(H2) attached to C1, H3 (H4, H5) attached to C2, H6 attached to C3 and H7 (H8) attached to C4, and then we will discuss these four cases separately.
For the C1 site, OH radical can abstract two different H atoms to form the same product. Through geometry optimization and energy calculation, two transition states (1-ts1-1 and 1-ts1-2) were found in the abstraction reactions with the Gibbs energy barrier of 4.61 and 5.99 kcal mol−1, respectively. During the abstraction process, the H1 and H2 atom on C1 site start to transfer to the O atom on the OH radical, and finally the C1–H1 and C1–H2 bonds are broken with the formation of the O–H1 and O–H2 bonds, and then the 1-im1 is formed, and a H2O molecule is removed. The reaction releases 22.96 kcal mol−1 of Gibbs energy.
For the C2 site, H atom abstraction from the methyl group. After three different transition states (2-ts1-1, 2-ts2-2 and 2-ts2-3), the alkyl radical 2-im1 is formed. The potential Gibbs free energy barriers for the three reaction processes are 6.63, 7.24 and 11.27 kcal mol−1, respectively. H atom abstraction reaction from C2 site is exothermic by 16.22 kcal mol−1.
As for the C3 and C4 sites, their reaction mechanism is similar to the C1 and C2 sites. The whole H atom abstraction reactions form the C3 and C4 sites are strongly exothermic by 25.44 and 21.22 kcal mol−1.
The above results indicate that the H-abstraction reactions are all exothermic reactions with lower reaction Gibbs free energy barriers, which are expected to occur easily and may play an important role in the conversion of 3-MTS in the atmosphere.
As the results shown in Table 1, the rate constants of C1 site, C2 site, C3 site and C4 site are 4.15 × 10−12, 1.62 × 10−13, 3.39 × 10−12 and 1.76 × 10−13 cm3 per molecule per s at 298 K and 1 atm pressure, respectively. Thus the ktotal is 7.87 × 10−12 cm3 per molecule per s, which is an order of magnitude higher than the experimental data 4.74 ± 0.2 × 10−13 cm3 per molecule per s.32 This may be related to the fact that DFT can generally reduce transition state energy.
The branching ratio (R) can be more intuitively express the contribution of each pathway. It is obvious that pathway C1-abs and C3-abs are dominant, whose R is around 52.67% and 43.06%. Thus, we will focus on the fate of 1-im1 and 3-im1. These two intermediates have unpaired electrons, which are highly reactive and will react rapidly with oxygen molecules to form peroxy radicals.
Reactions of 1-im1
1-im1 can be further oxidized by the ubiquitous O2 in the atmosphere, which is a barrier-free reaction process. As shown in Fig. 4, it generates intermediate 1-im2, releasing Gibbs free energy of 20.34 kcal mol−1. Then the peroxy radical adduct 1-im2 can undergo further reaction via reaction with NO to form 1-im3. This process continues to release 5.70 kcal mol−1 of Gibbs free energy. Next, it will go through the transition state 1-ts2 and take off NO2. In this reaction, while the O–O bond of O2 is broken, the C1–C5 bond is also broken at the same time, and finally HCOOH and 1-im4 are generated. This reaction needs to cross a very high Gibbs free energy barrier, about 25.32 kcal mol−1, and continues to release 34.57 kcal mol−1 of Gibbs free energy. It is the rate determining mining step in the reaction path.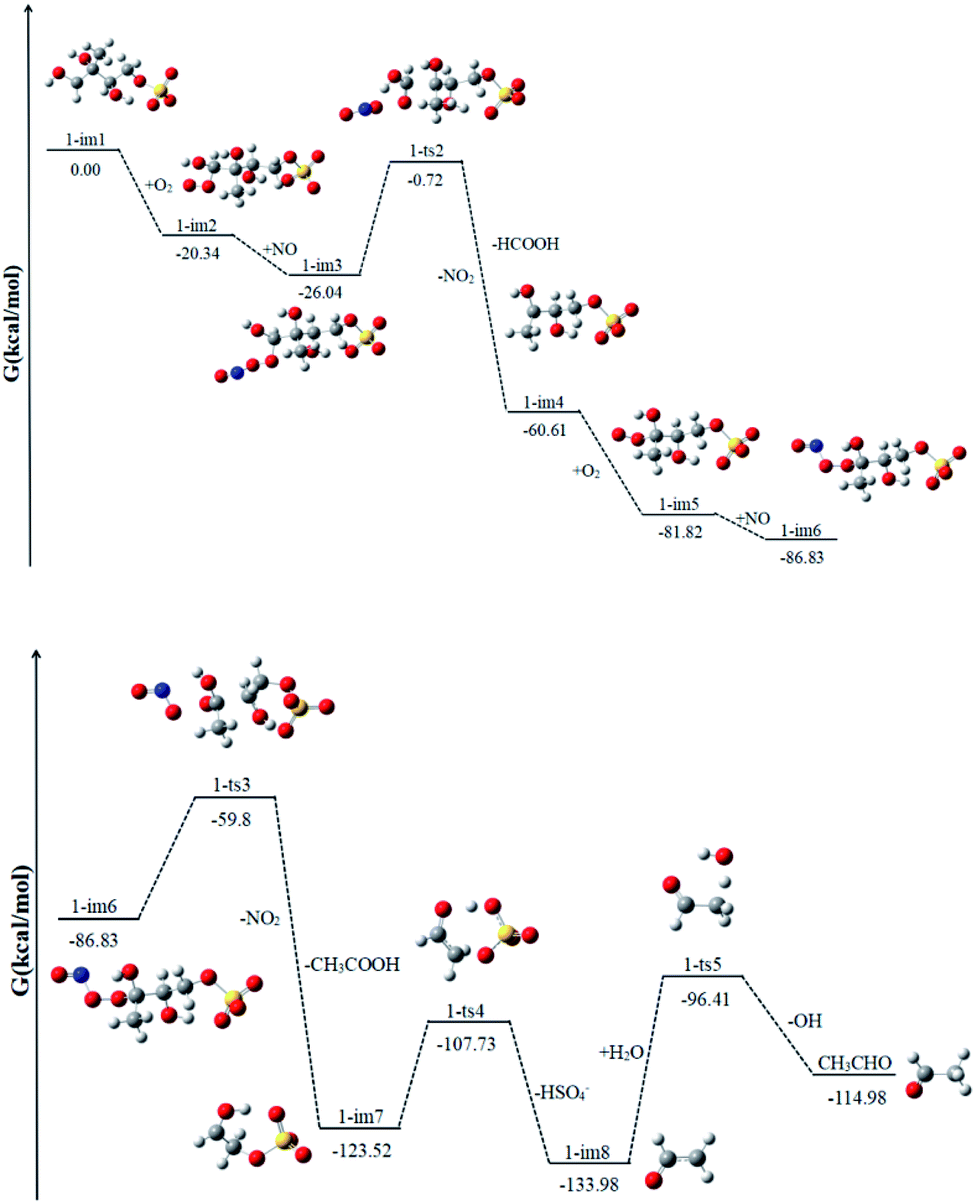 | ||
| Fig. 4 The profile of the potential energy surface for the reaction of 1-im1 at the DLPNO-CCSD(T)/cc-pVTZ//M06-2X/6-311++G(d,p) level of theory. | ||
Since 1-im4 has unpaired electrons, it will continue to react with O2 and NO to form 1-im6, releasing 26.22 kcal mol−1 of Gibbs free energy. After crossing the Gibbs free energy barrier of 27.03 kcal mol−1, NO2 will be removed, and with the cleavage of the C5–C3 bond, acetic acid and 1-im7 will be obtained. SO4− in im7 can extract a H atom from the adjacent O–H bond, while the C4–O cleavage occurs to form bisulfate ion (HSO4−) and ˙CH2CHO radicals (1-im8). 1-im8 can extract H atoms from H2O to generate CH3CHO with regenerating OH radicals. The regenerated OH radicals will initiate a new round of reactions.
The difference between our findings and the route proposed by Lam et al. is that in the case of high NO content, the RO2 radical can react with NO and the removal of NO2 will be accompanied by the cleavage of the C–C bond.32 The process can produce less volatile and more water soluble species, such as HCOOH, CH3CHO and CH3COOH, which can form SOA by nucleation, condensation, and/or partitioning between the condensed and gas phases. The production of HCOOH was also detected during the reaction of 2-MTS with OH.48 In addition, the produced HSO4− has been detected by aerosol mass spectrometry.32 And experiments show that the HSO4− content increases obviously with the increase of time.
Reactions of 3-im1
Similar to 1-im1, 3-im1 can undergo three elementary reactions: O2 addition, NO addition, NO2 elimination (Fig. 5). It should be pointed out that O2 addition and NO addition are barrier-free combination, resulting in an energy-rich intermediate (3-im3) that can be further reacted through unimolecular decomposition. The NO2 elimination reaction has a high potential Gibbs free energy barrier of 31.43 kcal mol−1. When the NO2 is removed, the C3–C4 bond will also be broken. This process will form the intermediate 3-im4 and P1 (2-methyl-2,3-dihydroxypropionic acid). The 3-im4 can undergo the O2 addition, NO addition and NO2 elimination to yield 3-im7. Then the 3-im7 undergo fragmentation to yield a HSO4− and a formyl radical (CHO), which is also mentioned in the mechanism of the heterogeneous OH oxidation reaction of sodium methosulfate.60 The subsequent reactions of CHO radical can react with H2O to yield CO2 and OH radical. CO2 is volatile and can be redistributed back into the gas phase.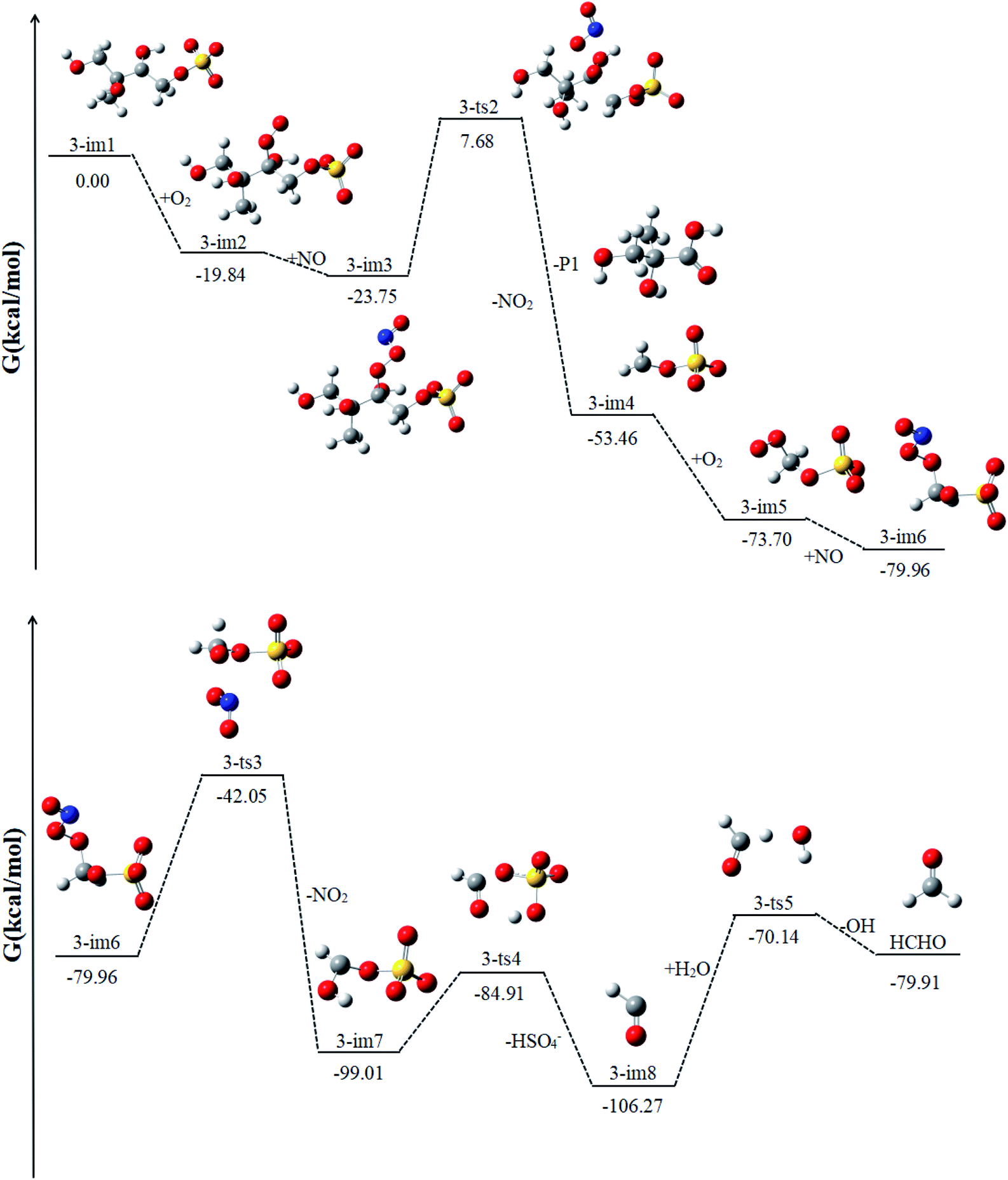 | ||
| Fig. 5 The profile of the potential energy surface for the reaction of 3-im1 at the DLPNO-CCSD(T)/cc-pVTZ//M06-2X/6-311++G(d,p) level of theory. | ||
Conclusions
In this paper, we applied the method of quantum chemical calculation to study the heterogeneous reaction mechanism of 3-MTS with OH radicals in the atmosphere under high NOx conditions, and calculated the rate constants for the reaction of 3-MTS with OH radicals. The study developed a model describing the kinetics of oxidation and the formation of inorganic sulfur species. Through the research, the following meaningful conclusions have been obtained.(1) 3-MTS can easily undergo abstraction reaction with OH radicals in the atmosphere, and its total reaction rate constant is 7.87 × 10−12 cm3 per molecule per s. In 3-MTS, there are eight C-linked H atoms that can be extracted by OH radicals to generate four intermediates. Among them, H1 connected to C1 and H6 connected to C3 are the most easily extracted.
(2) The alkyl radicals obtained by the abstraction reaction can continue to react with oxygen in the air to generate alkoxy radicals. Then under NOx-rich conditions, NO addition and NO2 removal reactions occur. In the process of NO2 removal, it is often accompanied by the breaking of C–C bonds. The generated HCOOH, CH3COOH, HCHO, CH3CHO, and 2-methyl-2,3-dihydroxypropionic acid are the main components of SOA.
(3) The CHO radical can react with H2O to yield CO2 and OH radicals. CO2 can be redistributed back into the gas phase. The OH radicals obtained by the reaction can continue to react with 3-MTS, thereby contributing to the occurrence of the oxidation reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (21607011), Key Research and Development Project of Shandong Province (2019GSF109021).References
- K. M. Shakya and R. E. Peltier, Environ. Sci. Technol., 2013, 47, 9332–9338 CrossRef CAS PubMed.
- K. M. Shakya and R. E. Peltier, Atmos. Environ., 2015, 100, 159–166 CrossRef CAS PubMed.
- M. Brüggemann, R. Xu, A. Tilgner, K. C. Kwong, A. Mutzel, H. Y. Poon, T. Otto, T. Schaefer, L. Poulain, M. N. Chan and H. Herrmann, Environ. Sci. Technol., 2020, 54, 3767–3782 CrossRef PubMed.
- S. F. Maria, L. M. Russell, B. J. Turpin, R. J. Porcja, T. L. Campos, R. J. Weber and B. J. Huebert, J. Geophys. Res., 2003, 108, 8637 CrossRef.
- A. A. Frossard, P. M. Shaw, L. M. Russell, J. H. Kroll, M. R. Canagaratna, D. R. Worsnop, P. K. Quinn and T. S. Bates, J. Geophys. Res., 2011, 116, D05205 CrossRef.
- K. Kristensen and M. Glasius, Atmos. Environ., 2011, 45, 4546–4556 CrossRef CAS.
- L. R. Mazzoleni, P. Saranjampour, M. M. Dalbec, V. Samburova, A. G. Hallar, B. Zielinska, D. H. Lowenthal and S. Kohl, Environ. Chem., 2012, 9, 285–297 CrossRef CAS.
- E. A. Stone, L. Yang, L. E. Yu and M. Rupakheti, Atmos. Environ., 2012, 47, 323–329 CrossRef CAS.
- A. M. K. Hansen, K. Kristensen, Q. T. Nguyen, A. Zare, F. Cozzi, J. K. Nojgaard, H. Skov, J. Brandt, J. H. Christensen, J. Strom, P. Tunved, R. Krejci and M. Glasius, Atmos. Chem. Phys., 2014, 14, 7807–7823 CrossRef.
- Y. Ma, X. Xu, W. Song, F. Geng and L. Wang, Atmos. Environ., 2014, 85, 152–160 CrossRef CAS.
- Q. M. Pei, E. Saikawa, S. Kaspari, D. Widory, C. F. Zhao, G. M. Wu, M. Loewen, X. Wan, S. C. Kang, X. P. Wang, Y. L. Zhang and Z. Y. Cong, Earth-Sci. Rev., 2021, 220, 103753 CrossRef CAS.
- C. Song, M. Gyawali, R. A. Zaveri, J. E. Shilling and W. P. Arnott, J. Geophys. Res.: Atmos., 2013, 118, 11741–11749 CrossRef.
- N. T. Tsona and L. Du, Atmos. Environ., 2019, 216, 116921 CrossRef CAS.
- J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2007, 41, 517–527 CrossRef CAS PubMed.
- M. S. Shalamzari, A. Kahnt, R. Vermeylen, T. E. Kleindienst, M. Lewandowski, F. Cuyckens, W. Maenhaut and M. Claeys, Environ. Sci. Technol., 2014, 48, 12671–12678 CrossRef CAS PubMed.
- L. E. Mael, M. I. Jacobs and M. J. Elrod, J. Phys. Chem. A, 2015, 119, 4464–4472 CrossRef CAS PubMed.
- M. S. Shalamzari, R. Vermeylen, F. Blockhuys, T. E. Kleindienst, M. Lewandowski, R. Szmigielski, K. J. Rudzinski, G. Spólnik, W. Danikiewicz, W. Maenhaut and M. Claeys, Atmos. Chem. Phys., 2016, 16, 7135–7148 CrossRef CAS.
- R. S. Xu, Y. Ge, K. C. Kwong, H. Y. Poon, K. R. Wilson, J. Z. Yu and M. N. Chan, ACS Earth Space Chem., 2020, 4, 2041–2049 CrossRef CAS.
- K. Gao and T. Zhu, Sci. Total Environ., 2021, 784, 147244 CrossRef CAS PubMed.
- M. Riva, S. Tomaz, T. Cui, Y. H. Lin, E. Perraudin, A. Gold, E. A. Stone, E. Villenave and J. D. Surratt, Environ. Sci. Technol., 2015, 49, 6654–6664 CrossRef CAS PubMed.
- M. Passananti, L. Kong, J. Shang, Y. Dupart, S. Perrier, J. Chen, D. J. Donaldson and C. George, Angew. Chem., Int. Ed., 2016, 55, 10336–10339 CrossRef CAS PubMed.
- J. Shang, M. Passananti, Y. Dupart, R. Ciuraru, L. Tinel, S. Rossignol, S. Perrier, T. Zhu and C. George, Environ. Sci. Technol. Lett., 2016, 3, 67–72 CrossRef CAS.
- S. L. Blair, A. C. MacMillan, G. T. Drozd, A. H. Goldstein, R. K. Chu, L. Pasa-Tolic, J. B. Shaw, N. Tolic, P. Lin, J. Laskin, A. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2017, 51, 119–127 CrossRef CAS PubMed.
- W. Fan, T. Chen, Z. Zhu, H. Zhang, Y. Qiu and D. Yin, J. Hazard. Mater., 2022, 430, 128406 CrossRef CAS PubMed.
- K. D. Froyd, S. M. Murphy, D. M. Murphy, J. A. de Gouw, N. C. Eddingsaas and P. O. Wennberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21360–21365 CrossRef CAS PubMed.
- L. E. Hatch, J. M. Creamean, A. P. Ault, J. D. Surratt, M. N. Chan, J. H. Seinfeld, E. S. Edgerton, Y. Su and K. A. Prather, Environ. Sci. Technol., 2011, 45, 5105–5111 CrossRef CAS PubMed.
- Y. H. Lin, E. M. Knipping, E. S. Edgerton, S. L. Shaw and J. D. Surratt, Atmos. Chem. Phys., 2013, 13, 8457–8470 CrossRef.
- Q. F. He, X. Ding, X. M. Wang, J. Z. Yu, X. X. Fu, T. Y. Liu, Z. Zhang, J. Xue, D. H. Chen, L. J. Zhong and N. M. Donahue, Environ. Sci. Technol., 2014, 48, 9236–9245 CrossRef CAS PubMed.
- J. Liao, K. D. Froyd, D. M. Murphy, F. N. Keutsch, G. Yu, P. O. Wennberg, J. M. St Clair, J. D. Crounse, A. Wisthaler, T. Mikoviny, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. W. Hu, T. B. Ryerson, I. B. Pollack, J. Peischl, B. E. Anderson, L. D. Ziemba, D. R. Blake, S. Meinardi and G. Diskin, J. Geophys. Res.: Atmos., 2015, 120, 2990–3005 CrossRef CAS PubMed.
- S. H. Budisulistiorini, X. Li, S. T. Bairai, J. Renfro, Y. Liu, Y. J. Liu, K. A. McKinney, S. T. Martin, V. F. McNeill, H. O. T. Pye, A. Nenes, M. E. Neff, E. A. Stone, S. Mueller, C. Knote, S. L. Shaw, Z. Zhang, A. Gold and J. D. Surratt, Atmos. Chem. Phys., 2015, 15, 8871–8888 CrossRef CAS.
- M. Riva, Y. Z. Chen, Y. Zhang, Z. Y. Lei, N. E. Olson, H. C. Boyer, S. Narayan, L. D. Yee, H. S. Green, T. Q. Cui, Z. F. Zhang, K. Baumann, M. Fort, E. Edgerton, S. H. Budisulistiorini, C. A. Rose, I. O. Ribeiro, R. L. e Oliveira, E. O. dos Santos, C. M. D. Machado, S. Szopa, Y. Zhao, E. G. Alves, S. S. de Sá, W. W. Hu, E. M. Knipping, S. L. Shaw, S. D. Junior, R. A. F. de Souza, B. B. Palm, J.-L. Jimenez, M. Glasius, A. H. Goldstein, H. O. T. Pye, A. Gold, B. J. Turpin, W. Vizuete, S. T. Martin, J. A. Thornton, C. S. Dutcher, A. P. Ault and J. D. Surratt, Environ. Sci. Technol., 2019, 53, 8682–8694 CrossRef CAS PubMed.
- H. K. Lam, K. C. Kwong, H. Y. Poon, J. F. Davies, Z. Zhang, A. Gold, J. D. Surratt and M. N. Chan, Atmos. Chem. Phys., 2019, 19, 2433–2440 CrossRef CAS.
- Y. Wang, M. Hu, Y. C. Wang, X. Li, X. Fang, R. Tang, S. H. Lu, Y. S. Wu, S. Guo, Z. J. Wu, M. Hallquist and J. Z. Yu, Environ. Sci. Technol. Lett., 2020, 7, 787–794 CrossRef CAS.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, Atmos. Chem. Phys., 2009, 9, 4987–5005 CrossRef CAS.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. S. Clair, J. H. Seinfeld and P. O. Wennberg, Science, 2009, 325, 730–733 CrossRef PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- Y. H. Lin, Z. Zhang, K. S. Docherty, H. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton, T. E. Kleindienst, A. Gold and J. D. Surratt, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS PubMed.
- A. P. S. Hettiyadura, E. A. Stone, S. Kundu, Z. Baker, E. Geddes, K. Richards and T. Humphry, Atmos. Meas. Tech., 2015, 8, 2347–2358 CrossRef CAS.
- T. Cui, Z. Zeng, E. O. dos Santos, Z. Zhang, Y. Chen, Y. Zhang, C. A. Rose, S. H. Budisulistiorini, L. B. Collins, W. M. Bodnar, R. A. F. Souza, S. T. de Martin, C. M. D. Machado, B. J. Turpin, A. Gold, A. P. Ault and J. D. Surratt, Environ. Sci.: Processes Impacts, 2018, 20, 1524–1536 RSC.
- E. C. Minerath, M. P. Schultz and M. J. Elrod, Environ. Sci. Technol., 2009, 43, 8133–8139 CrossRef CAS PubMed.
- B. Nozière, S. Ekström, T. Alsberg and S. Holmström, Geophys. Res. Lett., 2010, 37, L05806 CrossRef.
- T. B. Nguyen, M. M. Coggon, K. H. Bates, X. Zhang, R. H. Schwantes, K. A. Schilling, C. L. Loza, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Atmos. Chem. Phys., 2014, 14, 3497–3510 CrossRef.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- I. J. George and J. P. D. Abbatt, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- D. D. Huang, Y. J. Li, B. P. Lee and C. K. Chan, Environ. Sci. Technol., 2015, 49, 3672–3679 CrossRef CAS PubMed.
- J. H. Kroll, C. Y. Lim, S. H. Kessler and K. R. Wilson, J. Phys. Chem. A, 2015, 119, 10767–10783 CrossRef CAS PubMed.
- A. D. Estillore, A. P. S. Hettiyadura, Z. Qin, E. Leckrone, B. Wombacher, T. Humphry, E. A. Stone and V. H. Grassian, Environ. Sci. Technol., 2016, 50, 4259–4268 CrossRef CAS PubMed.
- Y. Chen, Y. Zhang, A. T. Lambe, R. Xu, Z. Lei, N. E. Olson, Z. F. Zhang, T. Szalkowski, T. Q. Cui, W. Vizuete, A. Gold, B. J. Turpin, A. P. Ault, M. N. Chan and J. D. Surratt, Environ. Sci. Technol. Lett., 2020, 7, 460–468 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 808–821 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, e1606 Search PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- O. Tapia and O. Goscinski, Mol. Phys., 1975, 29, 1653–1661 CrossRef CAS.
- J. Tomasi, R. Cammi and B. Mennucci, Int. J. Quantum Chem., 1999, 75, 783–803 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- M. Roeselová, J. Vieceli, L. X. Dang, B. C. Garrett and D. J. Tobias, J. Am. Chem. Soc., 2004, 126, 16308–16309 CrossRef PubMed.
- K. C. Kwong, M. M. Chim, J. F. Davies, K. R. Wilson and M. N. Chan, Atmos. Chem. Phys., 2018, 18, 2809–2820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02958h |
| This journal is © The Royal Society of Chemistry 2022 |