
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging the gel-to-sol transition of physically crosslinked thermoresponsive polymer hydrogels to enable reactions induced by lowering temperature†
Romario Lobban a,
Ankan Biswasa,
Kevin J. Ruiz-Márquezb and
Leon M. Bellan*ac
a,
Ankan Biswasa,
Kevin J. Ruiz-Márquezb and
Leon M. Bellan*ac
aDepartment of Mechanical Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: Leon.Bellan@Vanderbilt.edu
bDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA
cDepartment of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
First published on 9th August 2022
Abstract
Much work has been done on the use of heating to trigger reactions via the temperature-dependent removal of a barrier or constraint separating reagents. Far less work, however, has been done on the use of cooling to achieve a similar goal. Numerous applications, such as those involving components or materials susceptible to persistent low temperatures and cases in which energy for heating is not available, would benefit from this inverse approach. Hence, in this study we explore whether physically crosslinked hydrogels can be reliably used as thermoresponsive constraints that allow reagents to react only upon cooling. We achieve this by loading reagents into adjacent blocks of thermoresponsive hydrogel and showing that these reagents can only react with each other after the temperature of the hydrogel falls below its lower critical solution temperature (LCST). Above the LCST, the reagents remain sequestered in separate gels and no reaction occurs; this “OFF” state is stable for extended periods. When the system is allowed to cool, the hydrogels liquify and flow into each other, allowing mixing of the embedded reagents (“ON” state). We tune the hydrogels' LCSTs using NaCl, quantify the NaCl's tuning effect using rheometry, and determine that reactions are triggered reproducibly at temperatures similar to the tuned LCSTs. We also demonstrate generalizability of the concept by exploring situations involving radically different reaction types. This concept therefore constitutes a new approach to autonomous material behavior based on cooling.
Introduction
Stimuli-responsive materials have many potential applications due to their ability to respond dramatically to small environmental changes.1–16 Temperature-responsive materials are particularly attractive as temperature is easy to control and is orthogonal to other relevant environmental conditions.1–16 One possible application of such materials is to enable thermal initiation of mixing of separate payloads. This can be achieved through dramatic temperature-dependent transformation of a constraining material, allowing release from encapsulation,2,6–8,13,14 flow through a blockade or valve,9–12 or the flowing together of previously distinct solid-like gels. This concept can be further applied to initiate chemical reactions by allowing mixing and subsequent interaction of different reagent payloads only upon thermal triggering. Such an approach to thermal initiation of chemical reactions has parameters governed primarily by the thermodynamics of the constraining material; the thermodynamic properties of the reagents themselves are not the controlling factors. Therefore, use of temperature-responsive materials for reaction initiation is attractive as it allows generalization of thermal triggers.There are numerous examples relevant to this concept. Park et al.2 used temperature-dependent degradation of wax capsules to create transient electronic circuits; upon heating, wax capsules degrade and release encapsulated acid that destroys the circuit. Benito-Lopez et al.12 employed chemically crosslinked thermoresponsive hydrogels as temperature-dependent constraints/valves in microfluidic devices; here the hydrogel is swollen and blocks flow at low temperatures, but de-swells and allows flow at high temperatures. Finally, Goertz et al.6 demonstrated an approach using wax capsules to trap payloads. These payloads are sequestered until the capsules are heated sufficiently to melt and release them. As is briefly discussed in the paper, these capsules can contain reagents necessary for a chemical reaction with the surrounding solvent. Again, this demonstrates the utility of temperature-responsive materials in reliably sequestering payloads and releasing them when desired.
All examples of previous work discussed involve thermally triggered transformation and subsequent release due to an increase in temperature. Indeed, this concept is well established in the field of drug delivery where deswelling of chemically crosslinked hydrogels (upon heating) is leveraged to release drugs in a controlled manner.1,8 However, thermoresponsive transformation of a constraining material due to cooling has yet to be fully explored. There has been minimal preliminary work in this area; He and colleagues have used cold responsive nanomaterials to reduce ovarian cancer drug resistance,13 and also employed PNIPAm-B-PLGA-PF127 nanoparticle capsules for cold-triggered release of trehalose (a non-toxic cryoprotectant).14 However, these nanoparticle-based approaches have limited utility at the macroscopic scale. We hypothesized that such cooling-triggered transformations could be made scalable and generalizable to a variety of payloads; this would make macroscopic cooling-triggered reactions possible. Generalizable macroscopic cooling-triggered reactions could enable autonomic material behavior in applications that currently require active circuitry to identify and act on drops in temperature. Such applications include shipment of cold-sensitive produce and even devices for human comfort, like cold-responsive clothing.
To achieve generalizable cooling-triggered reactions, we explore the use of physically crosslinked thermoresponsive hydrogels as the temperature-dependent constraining material. Specifically, we leverage the gel-to-sol transition of physically crosslinked hydrogels to allow flow and subsequent mixing of previously isolated gels (which flow only when below their lower critical solution temperature (LCST)) as shown in Fig. 1. Physically crosslinked hydrogels differ from chemically crosslinked hydrogels in that their chains are not covalently bonded to each other; they are only physically interlocked. Thus, while chemically crosslinked hydrogels form permanent gels and remain gelled when hydrated (below the LCST),1 physically crosslinked hydrogels form reversible (temporary) gels and return to solution when below the LCST.15 Many recent stimuli-responsive drug delivery approaches depend on the deswelling of chemically crosslinked hydrogels at increased temperatures to release drugs sequestered in the hydrated state. However, as physically crosslinked hydrogels are flowing solutions below their LCST (and stationary gels above), their solution state can be used as the “ON” state and the gel state as the “OFF” state for cooling-triggered mixing/reactions.
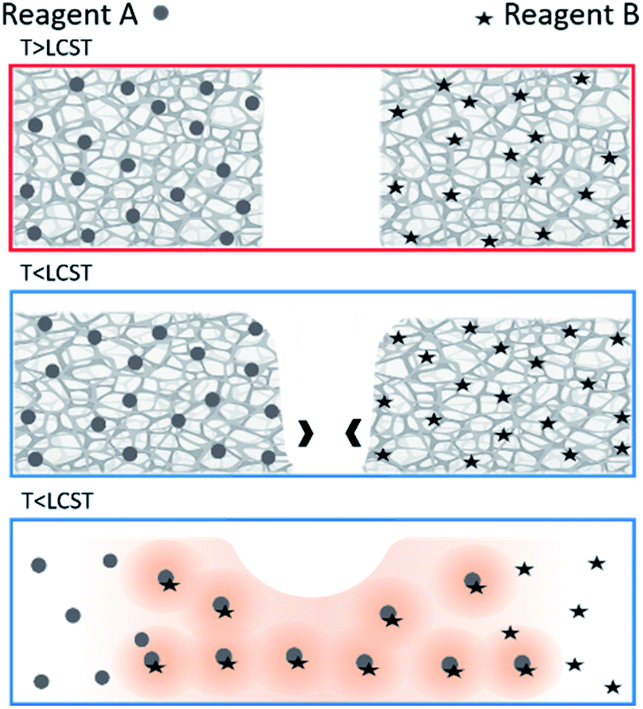 | ||
| Fig. 1 Schematic showing cooling-induced gel-to-sol transition resulting in flow and subsequent mixing and reaction. | ||
There is a wide array of polymeric systems that undergo a gel-to-sol transition upon cooling below their LCST. Jeong et al.17 lists numerous such hydrogels, along with their corresponding LCSTs and other physical properties. Examples include Pluronics/Poloxamers (triblock copolymers of poly (propylene oxide) sandwiched between two blocks of poly (ethylene oxide)); these are known for their utility in drug delivery, tissue engineering, and bioprinting applications.17–19 Another is PNIPAm (poly(N-isopropylacrylamide)), which is known to have a particularly physiologically relevant LCST of ∼32 °C17 and so has been widely applied in tissue engineering, biosensing, and drug delivery.17,20 Finally, methylcellulose (a cellulose derivative in which some hydroxyl groups are substituted by methoxyl groups) has numerous applications ranging from food and cosmetics thickening21 to tissue engineering.22
From the available polymeric systems that undergo gel-to-sol transitions, we choose to use methylcellulose. Thermoreversible gelation of methylcellulose and the various factors affecting its rheological behavior and structural properties have been widely studied.21–26 The dependence of gelation upon methylcellulose concentration is discussed in numerous publications;22–25 Kobayashi et al.23 and Arvidson et al.24 note minimum concentrations necessary for gelation and, along with others, discuss the inverse relationship between concentration and gelation temperature. The relative independence of gelation temperature from molecular weight is also noted.21,22,24,25 Finally, the use of “salting-out” and “salting-in” salts to reduce or increase gelation temperature, respectively22,25–27 is also well-documented. The mechanism involved in the thermogelation of methylcellulose has been described extensively as well. Currently, gelation is attributed to the formation of long, stiff, network-forming fibrils upon heating;21,25,26 this has been observed and quantified using cryo-TEM, SAXS, and SANS. The well-known hysteresis seen between gelation and inverse gelation of methylcellulose21,24,25 is also shown to be linked to the formation and dissolution of fibrils upon heating and cooling, respectively.21,25
In this article, we explore how the well-established sol-to-gel transition of methylcellulose hydrogel can be used to allow reagents to encounter each other only upon cooling. We select a methylcellulose formulation that allows the gel state and solution state to function robustly as the OFF state and ON state, respectively. Next, we demonstrate generalizability of the cooling-triggered reaction concept using two distinctly different reactions. We also demonstrate tunability of the temperature threshold by using NaCl to change the LCST of the methylcellulose and observing the modulation of cooling-triggered behavior. These LCSTs (gel point during heating and solution point during cooling) are corroborated using rheometry.
Experimental section
Preparation of reagent–laden hydrogels
All hydrogels were prepared with 3% w/v concentration of methylcellulose (Sigma-Aldrich; MW ∼ 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; viscosity at 2%, 20 °C ∼ 15 cPs). 12 formulations involving different combinations of NaCl, and reagents were prepared: 0%, 2%, and 3% NaCl combined with 1.67% phenolphthalein (Fisher Chemical), 1.67% NaOH (Fisher Chemical, 50% w/w), 0.05% HRP (Thermo scientific), and 33% TMB substrate (Sigma-Aldrich). All formulations were prepared as follows: 20 ml of PBS solution (distilled water for the NaOH formulations) was heated to 80 °C; the relevant amounts of methylcellulose, NaCl, and reagent (excluding TMB) were then added with vigorous magnetic stirring; the heater was switched off and 40 ml of cold PBS, distilled water, or 50% TMB stock solution added with continued vigorous stirring. All formulations were then chilled at 4 °C for 1 hour, after which they were magnetically stirred for 15 minutes, then stored at 4 °C in closed containers.
000; viscosity at 2%, 20 °C ∼ 15 cPs). 12 formulations involving different combinations of NaCl, and reagents were prepared: 0%, 2%, and 3% NaCl combined with 1.67% phenolphthalein (Fisher Chemical), 1.67% NaOH (Fisher Chemical, 50% w/w), 0.05% HRP (Thermo scientific), and 33% TMB substrate (Sigma-Aldrich). All formulations were prepared as follows: 20 ml of PBS solution (distilled water for the NaOH formulations) was heated to 80 °C; the relevant amounts of methylcellulose, NaCl, and reagent (excluding TMB) were then added with vigorous magnetic stirring; the heater was switched off and 40 ml of cold PBS, distilled water, or 50% TMB stock solution added with continued vigorous stirring. All formulations were then chilled at 4 °C for 1 hour, after which they were magnetically stirred for 15 minutes, then stored at 4 °C in closed containers.
Rheometric characterization of hydrogels
LCSTs (gel and solution points) of the prepared hydrogels were characterized using a TA Instruments Ares G2 rheometer with a 20 mm flat plate geometry. Oscillation temperature ramps were performed between 16 °C and 65 °C at 0.5 °C min−1 (both increasing and decreasing temperature) with a 2-minute period of steady temperature at 65 °C. An oscillation frequency of 1 rad s−1 was used for all formulations. Constant (amplitude) 6 Pa oscillatory stress for HRP–laden and phenolphthalein–laden formulations and 2 Pa oscillatory stress for NaOH–laden and TMB–laden formulations were used. These parameters were chosen to fall within the relevant linear viscous region (LVR) for each formulation. These LVRs were determined by performing isothermal stress sweeps (at 20 °C and 60 °C to characterize LVRs for sol and gel states, respectively) from 0.1 to 20 Pa at 1 rad s−1 for each formulation; this is similar to approaches used elsewhere.28,29 An evaporation shield and silicone oil were used to prevent evaporation during the temperature ramps. The use of silicone oil has been shown to not affect results.29Fabrication of devices for cooling-triggered reactions
An X-shaped mold was printed in ABS using a 3D printer (Stratasys F170). PDMS (Sylgard 184 elastomer base and curing agent) was poured over this mold to create the desired channel with dimensions shown in Fig. 3a. A biopsy punch was then used to create the loading channels (Fig. 3a). Finally, a glass cover slide was plasma bonded (using a Harrick Plasma PDC-001 plasma cleaner) to the PDMS to cover the fabricated channel.Characterization of cooling-triggered reactions
Cooling-triggered reactions were characterized by loading different hydrogel formulations (in gel form) into the device described above and letting them sit for 30 minutes at 65 °C (or 24 hours to demonstrate stability). The devices were then quickly placed into an ice bath. The gel-to-sol transitions and resulting colorimetric reactions upon cooling were captured using a stereoscope (Zeiss Discovery V8 with an Axiocam 105 color camera) set to capture images of the gels in the devices every 5 seconds. Simultaneously, the temperatures of the gels were measured using a calibrated thermistor (Cantherm MF52C1503F3950).Results and discussion
To investigate the use of thermoresponsive polymer hydrogels for cooling-triggered reactions, we prepared formulations of methylcellulose containing different reagent payloads (phenolphthalein, NaOH, horseradish peroxidase (HRP), and tetramethylbenzidine (TMB) substrate). Hydrogels containing phenolphthalein were paired with those containing NaOH; phenolphthalein is a pH indicator that changes from colorless to pink upon mixture with a base, such as NaOH. The color change occurs as the introduction of OH− groups causes phenolphthalein to change into its pink anionic form. Hydrogels containing HRP were paired with those containing TMB substrate; TMB and HRP are both initially colorless, but their mixture results in formation of blue/green to brown coloration. The reaction proceeds as HRP oxidizes the TMB (turning it blue/green to brown) in the substrate while it reduces the hydrogen peroxide also present in the substrate system.30,31 We placed these pairs of hydrogels adjacent to each other to show that no reaction occurs between the reagent payloads stored in them while the hydrogels are kept above their solution point. However, when the hydrogel temperatures fall below their solution points, they liquify and flow into each other, and subsequently a reaction occurs between the sequestered reagents. We also showed that following a reaction, if necessary, reaction products can be separated from the hydrogel for further analysis or reactions using dialysis. Fig. S3 in ESI† shows HPLC data demonstrating the effective separation of reaction products from methylcellulose using a 3.5 kDa MWCO dialysis membrane.Loading reagents into the methylcellulose hydrogels affected their solution points. This was expected as introduction of reagent payloads alters chemical properties of a hydrogel such as ion strength, solvent polarity, and pH; these are known to affect LCST.32 The effect the reagent payloads had on the hydrogels' solution points is shown in Fig. 2 and Table 1. All gel formulations displayed distinct solution points, and the differences in solution point were relatively modest (within 3 °C, except for the NaOH–laden formulation). Additionally, as the solution points of phenolphthalein–laden hydrogels and HRP–laden hydrogels are higher than the solution points of the hydrogels they were paired with, reaction initiation could be characterized using solely their solution points. In other words, these higher solution point “first-flowing gels” undergo gel-to-sol transition and flow into the other gels first during cooling, making their gel-to-sol transition temperatures the critical ones defining reaction initiation. Therefore, to characterize the triggering temperature for a given reaction, the higher solution point among the reagent–laden formulations is taken.
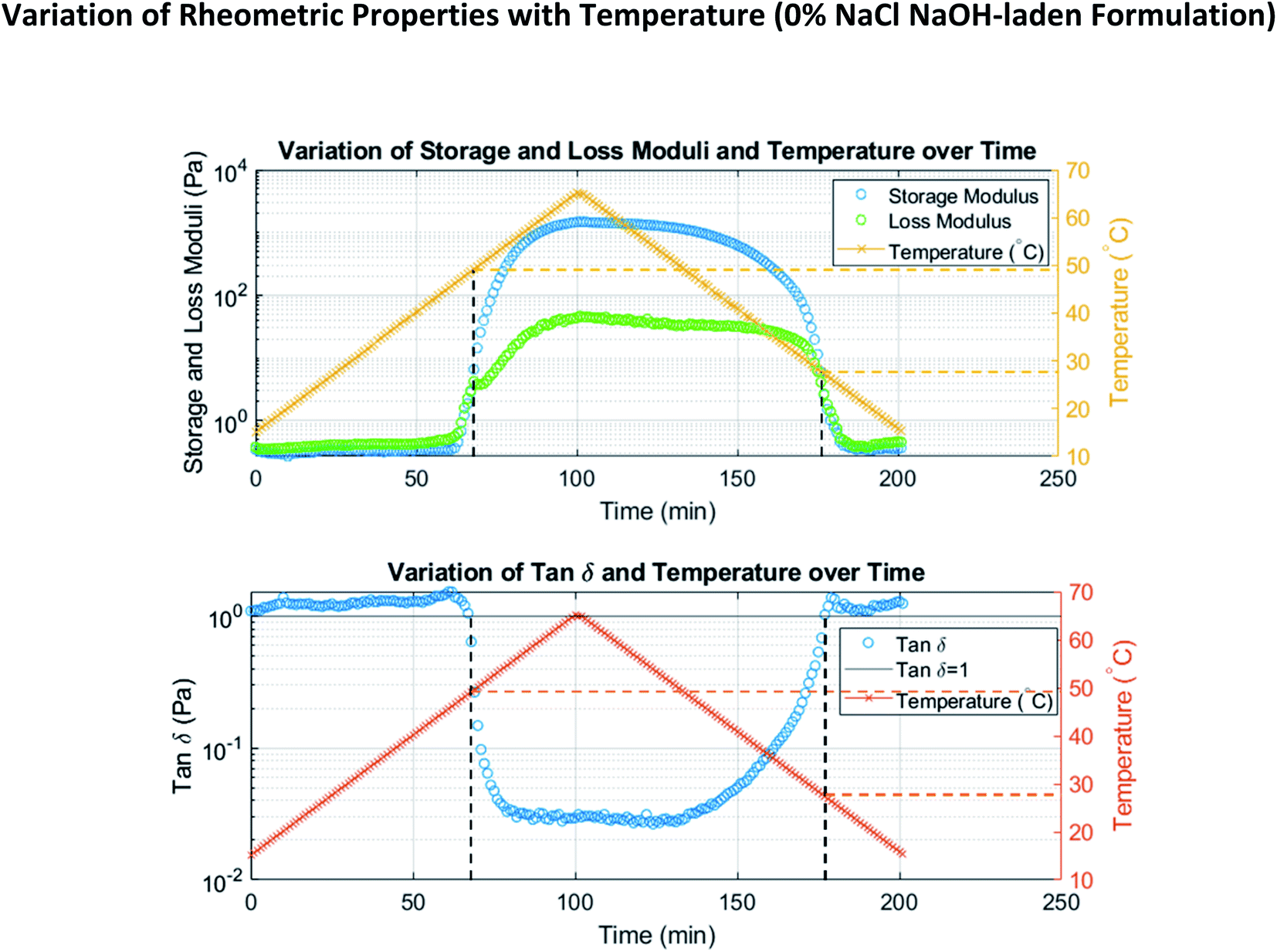 | ||
| Fig. 2 Example of rheometric temperature ramp data. Variation in storage and loss moduli (top) and tanδ (bottom) are shown. The gel point is eventually reached as temperature is increased, while the solution point is eventually reached as temperature is decreased. | ||
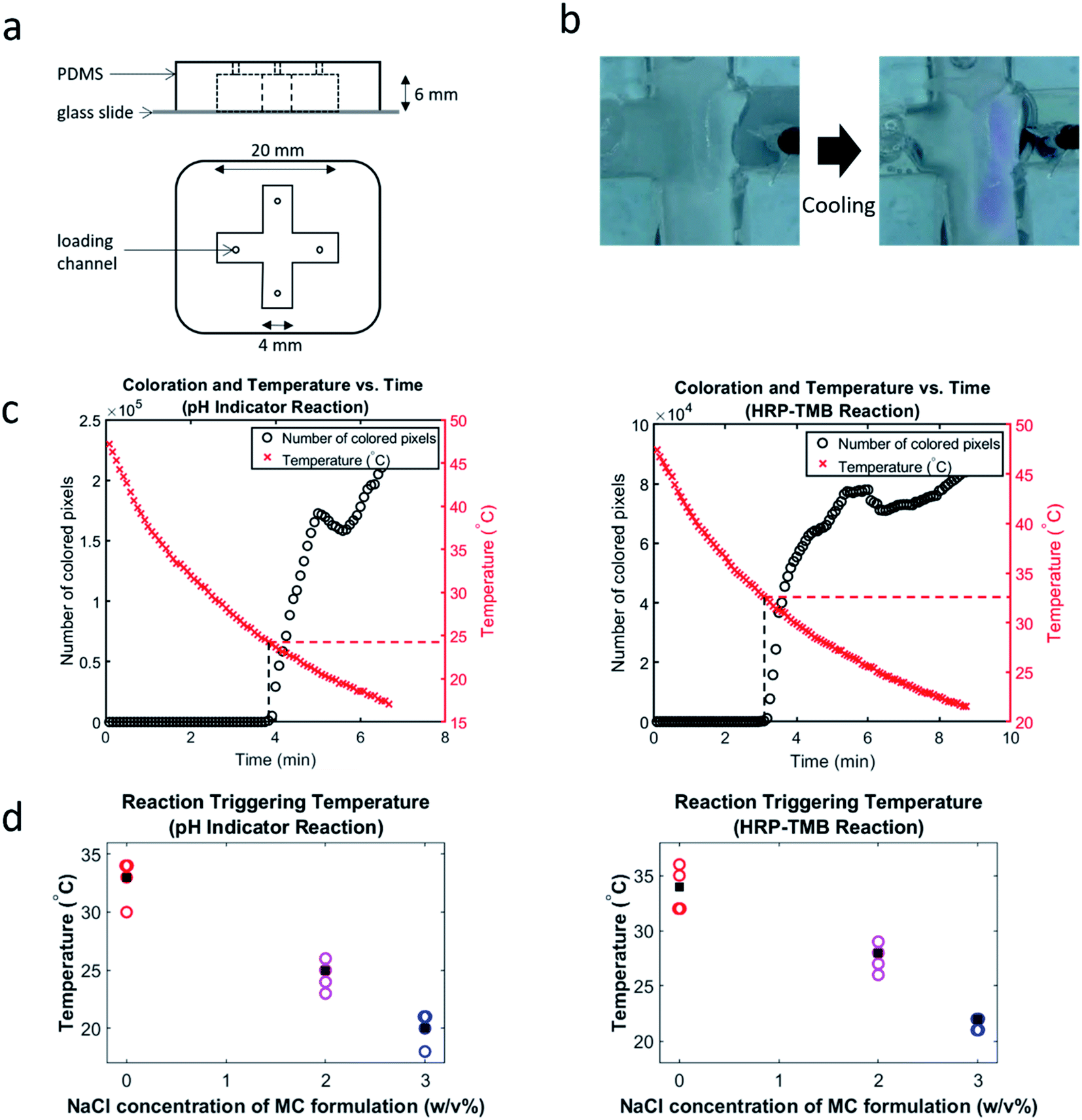 | ||
| Fig. 3 Cooling triggering of chemical reactions. (a) Device used for cooling-triggered reaction experiments. (b) Stereoscope images of a loaded device before (left) and after (right) a cooling-triggered phenolphthalein–NaOH reaction. (c) Coloration and temperature vs. time for a 2% NaCl phenolphthalein–NaOH reaction (left) and a 0% NaCl HRP–TMB reaction (right). (d) Triggering temperature vs. NaCl concentration for phenolphthalein–NaOH reactions (left) and HRP–TMB reactions (right). N = 4 for all formulations. Average triggering temperature for each formulation is represented by a black square. All data points and averages are to the nearest whole number. | ||
| Formulation | Solution point (°C) | Gel point (°C) |
|---|---|---|
| HRP–laden (0% NaCl) | 36 | 55 |
| HRP–laden (2% NaCl) | 27 | 46 |
| HRP–laden (3% NaCl) | 20 | 40 |
| TMB–laden (0% NaCl) | 35 | 53 |
| Phenolphthalein–laden (0% NaCl) | 33 | 51 |
| NaOH–laden (0% NaCl) | 27 | 49 |
To demonstrate tunability among formulations involving the same reagents, NaCl was introduced in various quantities (0%, 2%, and 3%) to all hydrogel formulations. As discussed in the introduction, NaCl and other salts are known to affect the solution point of methylcellulose. It is widely held that adding NaCl to methylcellulose decreases its solution point because dissolving NaCl occupies water molecules (that would otherwise be available to interact with methylcellulose and form a solution) in very strong interactions. A lower temperature is therefore necessary for the methylcellulose chains to compete with the salt ions for water molecules; this results in a lower solution point. The extent to which we observed this effect is shown in Fig. 2 and Table 1. These results demonstrate a robust ability to tune the solution point of methylcellulose using NaCl.
To approximate the gel and solution points shown in Table 1, we examined our temperature ramp data (Fig. 2, for example) to find the temperatures at which storage modulus surpassed loss modulus (tanδ fell below 1) during heating and the temperatures at which storage modulus fell below loss modulus (tanδ rose above 1) during cooling, respectively. This is an approach found in many existing publications.21,24,34 The angular frequency is known to affect the gel and solution points determined using this storage/loss modulus crossover method.24,34 Our use of 1 rad s−1 was therefore informed by the use of this frequency in existing literature;23,26,33–35 this allows for straightforward comparison of results. Similarly, heating/cooling rate is known to affect transition temperatures and the hysteresis between them.24–26,33–35 However, only the gel point is meaningfully affected;24,33 the solution point, which is of greater importance to our results, is not. Again, our use of a rate of 0.5 °C min−1 is informed by its use in previous works23,34,35 and the comparison it therefore enables.
Next, we employed cooling to trigger the two different colorimetric reactions. We fabricated a device (Fig. 3a) to hold hydrogels adjacent to each other such that said gels would not come into contact while remaining in gel form; it allowed contact between liquified gels (and their reactive payloads) (Fig. 3b and Movie S1 in ESI†). We demonstrated that methylcellulose hydrogel can remain in gel form (and thereby prevent reaction initiation) for at least 24 hours in our device kept at 65 °C. We also demonstrated that a reaction could still be triggered after 24 hours at 65 °C without a change in the triggering temperature. Most experiments, however, were performed after allowing 30 minutes for a gel to form in an external container, then a further 30 minutes of incubation within the device. The experiment involving 24 hours of incubation having a triggering profile indistinguishable from those of the other experiments indicates that 24 hours of high-temperature incubation produced negligible changes in the nature of the hydrogel.
Triggering temperatures were determined by simultaneously measuring temperature and coloration every five seconds during cooling (using a thermistor and stereoscope, respectively). Initiation of the colorimetric reactions was determined by visual observation of the image stacks, and by plotting the change in number of “colored” pixels in the images over time. This method of visualizing the reaction's progression allows us to plot both temperature and extent of coloration vs. time (Fig. 3c) and thereby easily note the temperature at which the reaction initiated. Colored pixels were defined by observation of the RGB profile of pixels that emerge only after reaction triggering. This was set as the criterion for counting a pixel as colored. For example, for the phenolphthalein–NaOH reactions, a pixel was counted as colored if its red value was greater than its green value ± an adjustment factor. This adjustment factor was determined for each experiment by observing the relative values of red and green in pixels that are visibly colored pink. The triggering temperatures indicated by this visualization method were corroborated by visual observation of the image stacks; both approaches agreed. We also plotted triggering temperatures vs. salt concentration (Fig. 3d) for both reactions. This visualization of the cooling-triggering data clearly shows that the NaCl concentration had a strong effect on triggering temperature.
Comparing the solution point of each formulation with the corresponding average triggering temperature reveals that reaction initiation generally occurred in the vicinity of the solution point. This shows that reaction initiation was due to the gel-to-sol transition of methylcellulose. Fig. 4 shows average triggering temperatures compared to the corresponding solution points of the HRP–laden hydrogel.
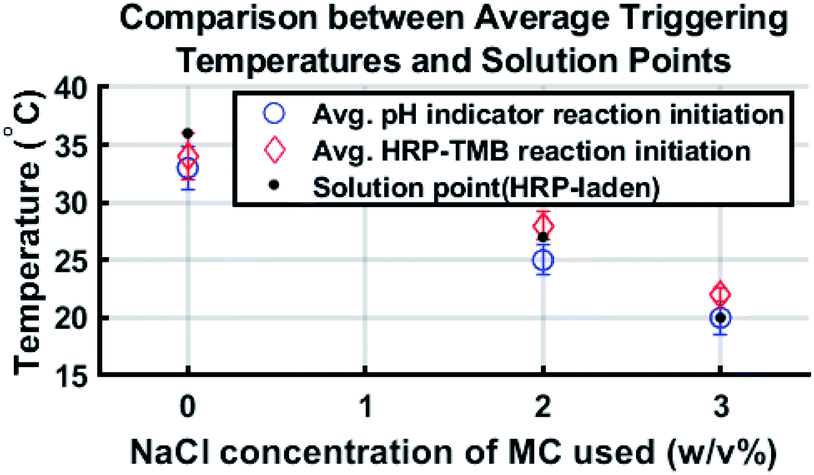 | ||
| Fig. 4 Average triggering temperatures compared to solution points of HRP–laden methylcellulose. | ||
The standard deviation for triggering temperature of a given reaction is at most 2 °C and the average triggering temperature for each HRP–TMB reaction is within 2 °C of the corresponding solution point. As phenolphthalein–laden methylcellulose has a slightly lower solution point, it makes sense that the triggering temperatures for the phenolphthalein–NaOH reactions are consistently lower than those of the HRP–TMB reactions and the solution points of the HRP–laden formulations.
Variation across replicants involving the same formulations can be explained by numerous phenomena. Despite loading 0.2 ml of each hydrogel into the device on all occasions, the separation distance between the two loaded adjacent gels varied from about 2–3.5 mm across replicants. Hence, some replicants displayed triggering more shortly after the sol–gel transition, while others exhibited a longer delay between the transition and triggering (as the solutions took longer to flow into each other). The temperature measurements also possess a margin of error as the thermistors were always inserted in the “second-flowing” gel (to avoid disrupting flow of the “first-flowing gel”). However, given the symmetry of the device and since gels had the same initial temperature and volume and possessed similar specific heat capacity, it was reasonable to assume that at all points in time during cooling, the temperature measured in the second-flowing gel was approximately equal to the temperature in the first-flowing gel. This assumption is corroborated by experimental data showing almost identical (within ∼1 °C) cooling curves as measured at the centers of the two hydrogels (Fig. S1 in ESI†). We also found no evidence that temperature gradients capable of significantly affecting results formed within each individual hydrogel (Fig. S2 in ESI†).
Conclusion
We have demonstrated that methylcellulose, a widely studied example of physically crosslinked thermoresponsive polymer hydrogels, can enable cooling-triggering of mixing and subsequent chemical reactions. Reagent–laden methylcellulose gels reliably remain discrete-preventing any mixing-while above their solution point, but predictably liquify and flow into each other upon cooling. This method is successfully applied to triggering an enzymatic reaction and a pH-indicator reaction, demonstrating generalizability. Additionally, tunability is successfully demonstrated (and quantified with rheometry) by using NaCl to alter the solution points of the hydrogels used. Finally, we also demonstrate that reaction products can be recovered from methylcellulose for further analysis. Confirming the viability of this concept opens a novel approach to autonomous material behavior triggered by cooling. We envision this concept expanding to involve a wide array of other thermoresponsive polymer hydrogels and reagents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medicinal Sciences of the National Institutes of Health under award number R01GM141604. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health.References
- A. S. Hoffman, Applications of Thermally Reversible Polymers and Hydrogels in Therapeutics and Diagnostics, J. Controlled Release, 1987, 6(1), 297–305, DOI:10.1016/0168-3659(87)90083-6.
- C. W. Park, et al., Thermally Triggered Degradation of Transient Electronic Devices, Adv. Mater., 2015, 27(25), 3783–3788, DOI:10.1002/adma.201501180.
- X. Zhang and L. M. Bellan, Composites Formed from Thermoresponsive Polymers and Conductive Nanowires for Transient Electronic Systems, ACS Appl. Mater. Interfaces, 2017, 9(26), 21991–21997, DOI:10.1021/acsami.7b04748.
- X. Zhang, et al., Thermoresponsive Transient Radio Frequency Antennas: Toward Triggered Wireless Transient Circuits, Adv. Mater. Tech., 2019, 4(11), 1900528, DOI:10.1002/admt.201900528.
- M. T. Cook, et al., Polymers Exhibiting Lower Critical Solution Temperatures as a Route to Thermoreversible Gelators for Healthcare, Adv. Funct. Mater., 2021, 31(8), 2008123, DOI:10.1002/adfm.202008123.
- J. P. Goertz, et al., Responsive Capsules That Enable Hermetic Encapsulation of Contents and Their Thermally Triggered Burst-Release, Mater. Horiz., 2019, 6(6), 1238–1243, 10.1039/C9MH00309F.
- S. A. Ryu, et al., Biocompatible Wax-Based Microcapsules with Hermetic Sealing for Thermally Triggered Release of Actives, ACS Appl. Mater. Interfaces, 2021, 13(30), 36380–36387, DOI:10.1021/acsami.1c04652.
- A. P. Esser-Kahn, et al., Triggered Release from Polymer Capsules, Macromolecules, 2011, 44(14), 5539–5553, DOI:10.1021/ma201014n.
- Q. Luo, et al., Monolithic Valves for Microfluidic Chips Based on Thermoresponsive Polymer Gels, Electrophoresis, 2003, 24(21), 3694–3702, DOI:10.1002/elps.200305577.
- S. R. Sershen, et al., Independent Optical Control of Microfluidic Valves Formed from Optomechanically Responsive Nanocomposite Hydrogels, Adv. Mater., 2005, 17(11), 1366–1368, DOI:10.1002/adma.200401239.
- N. Idota, et al., Microfluidic Valves Comprising Nanolayered Thermoresponsive Polymer-Grafted Capillaries, Adv. Mater., 2005, 17(22), 2723–2727, DOI:10.1002/adma.200402068.
- F. Benito-Lopez, et al., Modular Microfluidic Valve Structures Based on Reversible Thermoresponsive Ionogel Actuators, Lab Chip, 2014, 14(18), 3530–3538, 10.1039/C4LC00568F.
- H. Wang, et al., Overcoming Ovarian Cancer Drug Resistance with a Cold Responsive Nanomaterial, ACS Cent. Sci., 2018, 4(5), 567–581, DOI:10.1021/acscentsci.8b00050.
- Y. Zhang, et al., Cold-Responsive Nanoparticle Enables Intracellular Delivery and Rapid Release of Trehalose for Organic-Solvent-Free Cryopreservation, Nano Lett., 2019, 19(12), 9051–9061, DOI:10.1021/acs.nanolett.9b04109.
- H. A. Gaballa, et al., Synthesis and Characterization of Physically Crosslinked N -Vinylcaprolactam, Acrylic Acid, Methacrylic Acid, and N,N -Dimethylacrylamide Hydrogels, J. Polym. Sci., Part B: Polym. Phys., 2013, 51(21), 1555–1564, DOI:10.1002/polb.23369.
- Q. Chai, et al., Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them, Gels, 2017, 3(1), 6, DOI:10.3390/gels3010006.
- B. Jeong, et al., Thermosensitive Sol–Gel Reversible Hydrogels, Adv. Drug Delivery Rev., 2012, 64, 154–162, DOI:10.1016/j.addr.2012.09.012.
- M. S. H. Akash and K. Rehman, Recent Progress in Biomedical Applications of Pluronic (PF127): Pharmaceutical Perspectives, J. Controlled Release, 2015, 209, 120–138, DOI:10.1016/j.jconrel.2015.04.032.
- E. Gioffredi, et al., Pluronic F127 Hydrogel Characterization and Biofabrication in Cellularized Constructs for Tissue Engineering Applications, Procedia CIRP, 2016, 49, 125–132, DOI:10.1016/j.procir.2015.11.001.
- Y. Guan and Y. Zhang, PNIPAM Microgels for Biomedical Applications: From Dispersed Particles to 3D Assemblies, Soft Matter, 2011, 7(14), 6375, 10.1039/c0sm01541e.
- P. Nasatto, et al., Methylcellulose, a Cellulose Derivative with Original Physical Properties and Extended Applications, Polymers, 2015, 7(5), 777–803, DOI:10.3390/polym7050777.
- S. Thirumala, et al., Methylcellulose Based Thermally Reversible Hydrogel System for Tissue Engineering Applications, Cells, 2013, 2(3), 460–475, DOI:10.3390/cells2030460.
- K. Kobayashi, et al., Thermoreversible Gelation of Aqueous Methylcellulose Solutions, Macromolecules, 1999, 32(21), 7070–7077, DOI:10.1021/ma990242n.
- S. A. Arvidson, et al., Interplay of Phase Separation and Thermoreversible Gelation in Aqueous Methylcellulose Solutions, Macromolecules, 2013, 46(1), 300–309, DOI:10.1021/ma3019359.
- McK. L. Coughlin, et al., Methyl Cellulose Solutions and Gels: Fibril Formation and Gelation Properties, Prog. Polym. Sci., 2021, 112, 101324, DOI:10.1016/j.progpolymsci.2020.101324.
- L. Liberman, et al., Salt-Dependent Structure in Methylcellulose Fibrillar Gels, Macromolecules, 2021, 54(5), 2090–2100, DOI:10.1021/acs.macromol.0c02429.
- Y. Xu, et al., Salt-Assisted and Salt-Suppressed Sol−Gel Transitions of Methylcellulose in Water, Langmuir, 2004, 20(3), 646–652, DOI:10.1021/la0356295.
- D. S. Jones, et al., Characterization of the Physicochemical, Antimicrobial, and Drug Release Properties of Thermoresponsive Hydrogel Copolymers Designed for Medical Device Applications, J. Biomed. Mater. Res., Part B, 2008, 85(2), 417–426, DOI:10.1002/jbm.b.30960.
- M. Pakravan, et al., Determination of Phase Behavior of Poly(Ethylene Oxide) and Chitosan Solution Blends Using Rheometry, Macromolecules, 2012, 45(18), 7621–7633, DOI:10.1021/ma301193h.
- L. Gao, et al., Enzyme-Controlled Self-Assembly and Transformation of Nanostructures in a Tetramethylbenzidine/Horseradish Peroxidase/H 2 O 2 System, ACS Nano, 2011, 5(8), 6736–6742, DOI:10.1021/nn2023107.
- P. D. Josephy, et al., The Horseradish Peroxidase-Catalyzed Oxidation of 3,5,3’,5’-Tetramethylbenzidine. Free Radical and Charge-Transfer Complex Intermediates, J. Biol. Chem., 1982, 257(7), 3669–3675, DOI:10.1016/S0021-9258(18)34832-4.
- R. Moreira, et al., Rheological Behaviour of Aqueous Methylcellulose Systems: Effect of Concentration, Temperature and Presence of Tragacanth, LWT, 2017, 84, 764–770, DOI:10.1016/j.lwt.2017.06.050.
- J. R. Lott, et al., Fibrillar Structure of Methylcellulose Hydrogels, Biomacromolecules, 2013, 14(8), 2484–2488, DOI:10.1021/bm400694r.
- L. Li, et al., Gel Network Structure of Methylcellulose in Water, Langmuir, 2001, 17(26), 8062–8068, DOI:10.1021/la010917r.
- L. Su, et al., In Situ Observation of Heat- and Pressure-Induced Gelation of Methylcellulose by Fluorescence Measurement, Int. J. Biol. Macromol., 2014, 64, 409–414, DOI:10.1016/j.ijbiomac.2013.12.021.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02938c |
| This journal is © The Royal Society of Chemistry 2022 |