
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUiO-66-based metal–organic framework for dispersive solid-phase extraction of vanillylmandelic acid from urine before analysis by capillary electrophoresis
Roghayeh Hassanpour-Sabeta,
Behruoz Seyfinejadbc,
Elnaz Marzi Khosrowshahi a,
Mahboob Nematia,
Mohammad Reza Afshar Mogaddam*ab and
Abolghasem Jouyban*bd
a,
Mahboob Nematia,
Mohammad Reza Afshar Mogaddam*ab and
Abolghasem Jouyban*bd
aFood and Drug Safety Research Center, Tabriz University of Medical Sciences, Tabriz, Iran. E-mail: mr.afsharmogaddam@yahoo.com; afsharmogaddam@tbzmed.ac.ir; Fax: +98 41 31773333; Tel: +98 41 33372250
bPharmaceutical Analysis Research Center and Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. E-mail: ajouyban@hotmail.com
cStudent Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
dFaculty of Pharmacy, Near East University, PO BOX: 99138 Nicosia, Mersin 10, North Cyprus, Turkey
First published on 10th October 2022
Abstract
Dispersive solid-phase extraction (DSPE) was developed for the extraction of vanillylmandelic acid (VMA) in urine samples prior to capillary electrophoresis with diode array detection (CE-DAD). Extraction of VMA by DSPE was carried out by direct addition of 7.5 mg of synthesized amino-functionalized UiO-66 (Zr) metal–organic framework adsorbent into the 5 mL sample solution (pH 4.0), followed by sonication and centrifugation. The supernatant layer was discarded, then the sedimented adsorbent was eluted using borate buffer (75 mM, pH 10). Effective extraction parameters including the amount of adsorbent, sample pH, adsorption and desorption time, type, volume and pH of eluent, and type of adsorbent dispersion method were systematically investigated. Under the optimized conditions, linearity of the method was from 40 to 2000 μg L−1 with a correlation coefficient over 0.9948. The method detection and quantification limits were 12 and 40 μg L−1, respectively. The relative standard deviations for intra-and inter-day precision were 2.4 and 2.8% (n = 5), respectively. The extraction recovery and enrichment factor values were 90% and 9.0 respectively.
1. Introduction
Vanillylmandelic acid (VMA) is the end product of epinephrine and norepinephrine metabolism in the human body.1 The concentration of VMA increases during some neurological or metabolic diseases such as autism,2 pheochromocytoma,3 neuroblastoma,4 and tumors of the neural crest.5 Therefore, its determination from body fluids might be useful in assessing these diseases. Various analytical techniques such as high-performance liquid chromatography (HPLC),6–21 gas chromatography,22–25 immunoassay techniques,26,27 and electrochemical methods28–30 have been reported for the determination of VMA. HPLC used for quantification of VMA has the drawbacks of slow separation, low resolving power, and large sample volume.31 GC offers high resolution, but the time-consuming derivatization step is required for the analyte to become volatile and stable.32 Capillary electrophoresis (CE) has been developed to determine VMA in human urine.33–52 CE has attracted increased interest because of its high separation efficiency, short analysis time, small sample volume, and low reagent consumption which makes it an attractive alternative to HPLC.34 Commercial CE instruments are equipped with UV absorbance detectors due to their low cost and simplicity of instrumentation. As previously reported, direct injection of urine samples into CE is possible.33,51 However, the presence of a large number of different endogenous compounds can impair resolution. In addition, under such conditions, when solutes are monitored using a UV detector, peak identification can be difficult, especially in complex matrices such as plasma and urine samples. On the other hand, urine samples are often diluted for direct injection, which reduces the concentration in real samples. For example, Shirao et al.46 described the method of CE with UV detection to determine VMA, but the developed method can not quantify the content of VMA in the urine of healthy individuals due to the high limit of detection (LOD). Due to the poor sensitivity of the CE-UV as the result of short optical path length and low injection volume, preconcentration should also be considered. Consequently, before CE-UV analysis, the sample preparation step is usually needed for the selective extraction of analytes of interest from biological samples and to clean up the sample matrix and concentrate the analyte. Solid-phase extraction (SPE)52,53 and liquid–liquid extraction (LLE)31,39,54 methods are usually used to treat urine samples prior to the analysis of VMA. Straightforward methods such as dilution,42 protein precipitation,37,49 and filtration have also been reported to clean up urine samples prior to injection to CE,35,38,40 but interfering compounds hamper the identification of the analyte of interest and also reduce the selectivity of the method. Besides, separation column performance diminishes under the influence of unremoved compounds. Dispersive-solid phase extraction (DSPE) as a miniaturized sample preparation technique is a modified arrangement of SPE in which the sorbent particles are dispersed into the sample solution instead of being packed in a cartridge. In this case, due to the high contact surface between the analyte and the sorbent, the sorption kinetics increase, which increases the extraction efficiency and reduces the extraction time.55 The use of an appropriate adsorbent for the efficient extraction of analytes is crucial in DSPE. Metal–organic frameworks (MOFs) with a large surface area and excellent adsorption capacity have received growing attention as novel solid adsorbents in DSPE. MOFs are crystalline porous materials composed of positively charged metal ions or clusters coordinated to organic linkers. However, some MOFs are hydrolyzed in the presence of water, which destroys the porous structure and hinders their use in aqueous samples.56 UiO-66 is a MOF made up of [Zr6O4(OH)4] clusters with 1,4-benzodicarboxylic acid linkers. UiO-66 exhibits good chemical resistance against water because of the strong coordination bonds between the Zr(IV) atoms and carboxylate oxygens.57 Modification of organic ligands with different functional groups is a common way to improve extraction performance. The amino-functionalized UiO-66-NH2 offers satisfactory selectivity to acidic analytes.58Therefore, the main goal of the present study was to utilize the amino-functionalized UiO-66-NH2 as a sorbent of DSPE for selective extraction of VMA from urine samples and then followed by CE separation and UV detection by diode array detector (DAD). For this purpose, the effects of various experimental parameters, such as the amount of adsorbent, the type and volume of the eluent solvent, adsorption and desorption time, pH and salt effect, and type of sample agitation were investigated.
2. Experimental
2.1. Chemicals
VMA (purity ≥ 98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium hydroxide (NaOH), sodium chloride (NaCl), sodium tetraborate decahydrate (borax), acetonitrile (ACN), ethanol, and methanol (MeOH), zirconium nitrate (ZrNO3), 2-aminoterephthalate (H2ATA), and N,N-dimethylformamide (DMF) were purchased from Merck (Darmstadt, Germany). All chemicals were of analytical grade and used without further purification. Deionized (DI) water was supplied by Shahid Ghazi Pharmaceutical Co. (Tabriz, Iran).2.2. Standard solutions and biological matrices
The stock solution of VMA (100 mg mL−1) was prepared in ethanol and used for the preparation of calibration standards. All required working standard solutions were prepared daily by dilution of the stock solution in the water. Urine samples were collected from healthy individuals and were extracted by the developed DSPE method under optimized conditions (see section DSPE procedure).2.3. Preparation of amino-functionalized UiO-66(Zr)
The amino-functionalized UiO-66(Zr) was prepared by the procedures reported in the literature.27 In brief, 0.186 g of H2ATA (1.029 mmol) and 0.349 g of Zr(NO3)4 (1.029 mmol) were mixed in 60 mL of anhydrous DMF. After the addition of 0.19 mL of deionized water, the mixture was stirred for 5 min (24 × g) at room temperature and then transferred to a Teflon liner stainless steel autoclaves (100 mL) and reacted at 120 °C for 24 h in an oven. Finally, the resulting product was harvested by centrifugation (9000 × g for 10 min) and washed with DMF and methanol three times (activation), respectively. The powder was then soaked in methanol for two days. Finally, yellow powder of UiO-66(Zr)-NH2 was obtained after thoroughly drying at 100 °C under a vacuum overnight.2.4. DSPE procedure
Firstly, 7.5 mg UiO-66(Zr)-NH2 powder was added into a 5 mL aqueous solution containing 1 mg L−1 of VMA adjusted at pH 4 using acetate buffer (c = 0.5 M). The mixture was then sonicated for 1 min for analyte sorption. Then the adsorbent was separated from the solution by centrifugation at 1075 × g for 5 min. Subsequently, the supernatant was discarded and 500 μL borate buffer (75 mM, pH 10; elution solvent) was added to the centrifuge tube and was ultrasonicated for 3 min to desorb the analyte from the adsorbent particles. After centrifugation for 5 min at 1075 × g, an aliquot of 50 μL of elution solvent was collected using a microsyringe and transferred to a CE microvial to further analysis by CE.2.5. Instrumentation
CE analyses were carried out using a HP 3D CE device (Hewlett–Packard, Palo Alto, CA, USA), equipped with a DAD detector. Instrumental control and data acquisition were performed using ChemStation (Agilent Technologies, Waldbronn, Germany). The capillary used was from Agilent Technology (Waldbronn, Germany) 50 μm i.d. × 40 cm (31.5 cm to the detection window). The new capillary was pretreated with 1 M NaOH for 30 min, deionized water for 30 min, and the background electrolyte (BGE) solution for 30 min. The rinsing pressure was 950 mbar in all preconditioning steps. Before each run, the capillary was flushed with 0.1 M NaOH for 2 min, deionized water for 2 min, and BGE solution for 4 min. The optimized conditions of electrophoretic separation were: the BGE solution was 100 mM borate buffer at pH 9.3. The sample was injected by hydrodynamic pressure at 50 mBar for 10 s. The separation voltage was +25 kV and the capillary was thermostatted at 25 °C. The detection wavelength was 200 nm. BGE solution was sonicated and filtered through a 0.20 μm pore size PTFE syringe filter (Chromafil, Germany). Brunauer–Emmett–Teller (BET) analysis based on nitrogen adsorption/desorption method at a temperature of 77 K was used to access information about the surface properties of the sorbent such as specific surface area and average pores diameter. In the current survey, BET analysis was performed using BELSORP mini II analyzer (BEL, Japan). Infrared spectra were recorded using a Fourier transform infrared (FT-IR) spectrophotometer (Bruker, Billerica, USA). A Cary-100 UV-Vis Spectrophotometer was used for recording the UV-Vis spectrum. A D500 diffractometer (Siemens, Karlsruhe, Germany) was used for obtaining X-ray diffraction (XRD) pattern operating at a voltage of 35 kV at room temperature.3. Results and discussion
3.1. Characterization of sorbent
The SEM images were recorded for the morphology investigation of MOF structures and the particle sizes of the MOF were ranged under 500 nm, which is sufficiently small for these particles to be used in extraction.27 BET analysis was performed through measuring the nitrogen adsorption isotherms (Fig. 1a). Considering the data, the specific surface area and mean pores diameter (d) of the utilized MOF were 60.15 m2 g−1 and 73.9 nm, respectively. According to the fact that d is between 2 and 50 nm, the particles are mesopore in nature. The UV-Vis absorption spectrum of the MOF was depicted in Fig. 1b. The absorption peaks at 250 and 325 nm are related to the light absorption of Zr6 clusters, the amine group of the MOF ligand, respectively. The XRD pattern (Fig. 1c) represents typical Bragg peaks positioned at 2θ = 7.4°, 8.4°, 26.7°, and 35.9° related to 111, 002, 006, and 116 spacing of the MOF, respectively. These peaks confirm successful synthesis of MOF. Based on FTIR spectrum (Fig. 1d), the wide band on 3429 cm−1 can be attributed to the stretch vibration of the single bond NH2 group. The presence of the single bond NH2 group is corroborated by the N–H bending vibration, observed at 1680 cm−1 in the MOF. The bands at 1571 and 1430 cm−1 are corresponded to the asymmetric and symmetric stretching vibrations of the carboxylate groups from terephthalic ligand. The vibration peaks around 664 and 491 cm−1 are related to the stretching vibration of Zr–O of the Zr6 cluster. All of the data are compatible with the results reported in literature.27 | ||
| Fig. 1 (a) Nitrogen adsorption/desorption isotherm (b) UV-vis spectrum, (c) XRD pattern, and (d) FTIR spectrum of the MOF. | ||
3.2. Optimization of the DSPE procedure
Sample pH plays a vital role in the extraction of ionizable analytes because the interaction between adsorbent and analyte has a close relationship with the existing form of the analyte. The effect of sample pH on extraction efficiency was investigated in the range of 2.0–10.0. It can be seen from Fig. 2A, the method efficiency increases by increasing the sample solution pH to 4 and then decreases. Based on the pKa value of VMA (3.1), decreasing the method efficiency can be related to the formation of carboxylate ions of VMA and increasing its solubility in the sample solution. It must be noted that decreasing the method performance at acidic pHs (pH = 2.0) can be related to the ionization of –NH2 groups of the MOF and its instability in the solution. So, the sample solution pH was adjusted at 4.0 using acetate buffer (0.5 M). | ||
| Fig. 2 The effect of affective variables in the adsorption step on DSPE of VMA; effects of sample pH, (A) adsorbent amount, (B) sample agitation type, (C) adsorption time, (D) and salt addition. (E) The error bars indicate minimum and maximum values of three independent determinations. | ||
To obtain maximum extraction recovery, the amount of adsorbent was studied in the range of 2.5–12.5 mg. As can be seen from Fig. 2B, there was an increase in extraction recovery with the increase of adsorbent amount from 2.5 to 7.5. At high values, the recovery was reduced, which was probably due to the insufficiency of the elution solvent for eluting the whole analyte.27 In addition, analysis of the sample solution after the sorption step showed that 7.5 mg is enough to isolate whole the analyte. So, 7.5 mg adsorbent was selected for further studies.
In DSPE, the adsorption and desorption of analyte to the adsorbent is accelerated by vortex or ultrasonic. The effect of these dispersion methods on the efficiency of adsorption and desorption steps was investigated. The results (Fig. 2C and 3A) showed that at the same time, the performance of sonication was better than vortex in both adsorption and desorption steps. Therefore, the rest of the experiments were performed using ultrasonication.
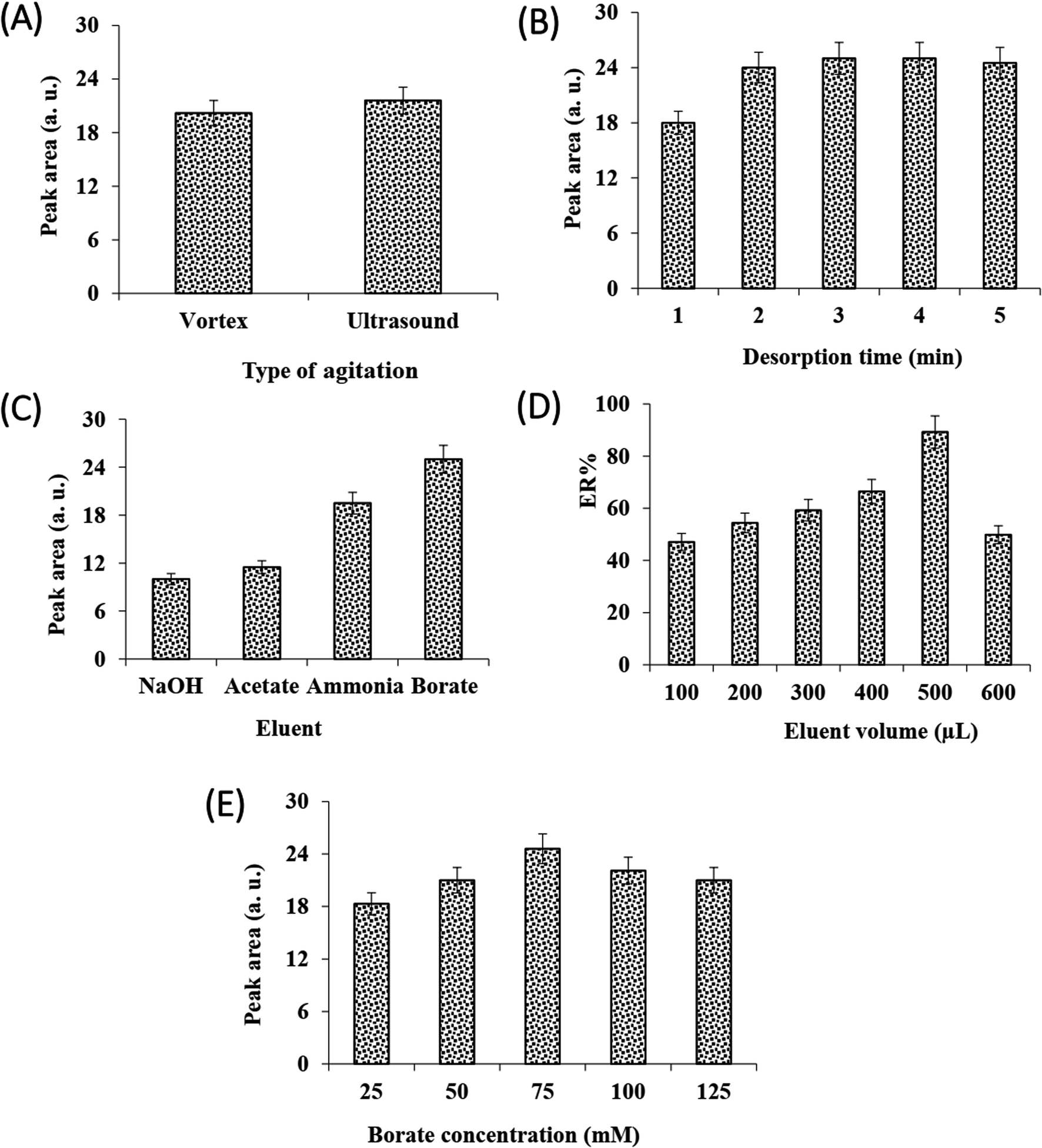 | ||
| Fig. 3 The effect of affective variables in the desorption step on DSPE of VMA; effects of sample agitation type, (A) adsorption time, (B) eluent type, (C) eluent volume, (D) and eluent concentration. (E) The error bars indicate minimum and maximum values of three independent determinations. | ||
The influence of adsorption and desorption times was investigated within 0–7 min. According to the results, the 1 min of adsorption time was sufficient for high extraction recovery, however, at longer times the amount of recovery is reduced, which can be due to back extraction (Fig. 2D). In the case of desorption time, as can be seen from Fig. 3B, extraction recovery rises up to 3.0 min and then remains almost constant. Subsequently, 1 and 3 min were considered as the optimum times for adsorption and desorption, respectively.
The solubility of analytes alters with the change in ionic strength of the solution. Adding salt can be the most straightforward way to increase the ionic strength of the solution, which reduces the solubility of the analyte by the salting-out effect, thus increasing the analyte extraction efficiency. To study the effect of salt addition on extraction recovery, different concentrations of NaCl were used (0–12.5% w/v). Results showed (Fig. 2E) that there was a slight increase in extraction recovery with the increase of NaCl concentration from 0 to 7.5% w/v. No change was observed at high salt concentrations, so 7.5% was selected as the optimal value.
They kind of elution solvent for desorption of the analytes from the MOF is key effect on the method efficiency. The preliminary tests confirmed that nearly 100% of the analyte was adsorbed onto the sorbent by analyzing the sample solution after extraction of the analytes with the MOF particles. Thus, the analyte must be eluted effectively from the sorbent surface. To select the proper elution solvent, a series of experiments were done with different organic solvents consist of ethanol, ACN, chloroform, and n-hexane, independently. For this purpose, 100 μL of each pure solvent was added onto the separated MOF (after centrifugation and discarding the supernatant) to desorb the analyte. After evaporating and reconstituting, the obtained solution was injected into the CE, no peak was observed for any of the solvents, indicating the inability of the solvents to desorb the analyte. In the following, due to acidic nature of VMA, its elution from the sorbent surface was tested by using different buffers including borate, ammonia, and acetate and sodium hydroxide solutions adjusted at pH = 9.0. The elution solvent concentration was 100 mM. According to the data (Fig. 3C), the highest extraction recovery was obtained using borate buffer. It is well established that borate can form complexes with polyol compounds,59,60 so borate was used in the subsequent steps.
The effect of elution solvent (buffer solution) volume, concentration, and pH on the method efficiency was investigated. The results showed (Fig. 3D) that with increasing the volume of borate buffer up to 500 μL, recovery increases but decreased with further increment in borate buffer volume. It indicated 500 μL borate buffer could effectively desorb the VMA with the desired enrichment factor.
Therefore, 500 μL was selected for the next experiments. From Fig. 3E, it can be seen that with increasing buffer concentration up to 75 mM recovery increases but after that, it decreased. As the concentration of buffer increases up to 75 mM, the recovery increases due to the complex formation between the borate and the VMA, but at high concentrations of buffer due to the salting-out effect, the VMA may be eluted into the buffer in smaller amounts. Therefore, the 75 mM buffer was chosen for further studies. Finally, the effect of buffer borate pH on desorption was studied within 9.0–11.0. It was found that pH 10 is more suitable for desorption (data not shown).
Based on the findings of the optimization of the DSPE method, the mechanism of adsorption and desorption of the VMA with the UiO-66(Zr)-NH2 can be explained as follows. In the adsorption step under basic conditions, the negatively charged VMA is electrostatically interacted with the positive charge of the adsorbent surface and is extracted using the ion-pair mechanism. In the desorption step under basic conditions, the negatively charged analyte is exchanged by negative ions of the elution buffer, consequently desorbed from the adsorbent surface by the ion exchange mechanism.
3.3. Method validation and analytical parameters
To investigate the analytical performance of the proposed method, several main parameters were determined including LOD, the limit of quantification (LOQ), linear range, intra-and inter-day precision, recovery, enrichment factor (EF), and extraction recovery. An eight-point calibration curve was used to check the linearity of response for standards of VMA at concentrations ranging from 40 to 2000 μg L−1. The linearity was assessed by plotting the peak area of VMA versus the corresponding concentrations. The obtained calibration curve equation was y = 32.178x + 0.55 and good linearity was achieved for VMA over the studied concentration range with a correlation coefficient higher than 0.994. The LOD and LOQ were determined based on signal-to-noise ratios of 3 and 10, respectively. The LOD and LOQ were 12 and 40 μg L−1. According to literature61 the normal concentration range of VMA in urine is 11.6 to 28.7 μM (or 2.3 to 5.7 μg mL−1), thus the analyzed urine samples must be diluted before performing the established method on the samples. The repeatability of the method was carried out by performing the method on aqueous solutions containing the analyte at the concentrations of 100, 500 and 1000 μg L−1 in a same day (n = 5) and different days (n = 3). The repeatability was expressed as relative standard deviation (RSD). RSDs for intra-and inter-day precisions were 2.4 and 2.8% in all studied concentrations, respectively. The recovery experiments were accomplished to investigate the accuracy of the method. Recovery (trueness) of VMA was measured by a standard addition method in the real urine samples that were spiked at three separate concentrations (Table 1). VMA concentrations in spiked urines were measured by the developed method and recovery was estimated by comparing the values found, with those of the standards. The recovery results are summarized in Table 1. The average recoveries were within the ranges of 87.6–101.6%. It can be seen that the obtained values are in the acceptable range (between 80 and 120%). The EF value was calculated using the ratio of the VMA concentration in the elution solvent to the initial sample concentration. ER and EF for the VMA were 90% and 9.0 respectively. The high surface area of the UiO-66-NH2 and its functionalized surface lead to extraction with high recovery and selectivity.| Sample | Added (μg L−1) | Found (μg L−1) | Mean relative recovery ± standard deviation (n = 3, %) |
|---|---|---|---|
| Urine 1 | 50 | 44.5 | 89 ± 5.1 |
| 100 | 93.0 | 93 ± 4.6 | |
| 250 | 254.1 | 101.6 ± 3.5 | |
| Urine 2 | 50 | 43.8 | 87.6 ± 3.8 |
| 100 | 91.6 | 91.6 ± 4.2 | |
| 250 | 249.2 | 99.7 ± 4.8 |
3.4. Real samples analysis
Under optimized conditions, the proposed DSPE-CE-DAD method was used to determine VMA in the urine samples from healthy volunteers. Since the normal concentration of the VMA in healthy individuals is higher than the linear range of the method, urine samples were extracted after 10-fold dilutions. VMA concentrations in real samples were found 13 and 8.2 mg L−1. The sample donors signed a consent form registered by the Tabriz University of Medical Sciences Ethical Committee (approval code, IR. TBZMED.VCR.REC.1401.162). A typical electropherogram of a urine sample is shown in Fig. 4. It is observed that due to dilution, the concentration of other endogenous compounds decreases, which leads to clean electropherograms. In the following, to acquire data about matrix effect of the investigated samples, added–found method was utilized. For this purpose, two urine samples were spiked with the analyte (50, 100, and 250 μg L−1) and then quantified using the suggested approach. The initial concentration in the spiked samples were divided to the found concentration and multiplied by 100. The results are reported as mean relative recoveries in Table 1. Based on outcomes, the matrices of the samples have a negligible effect on the applicability of the developed approach.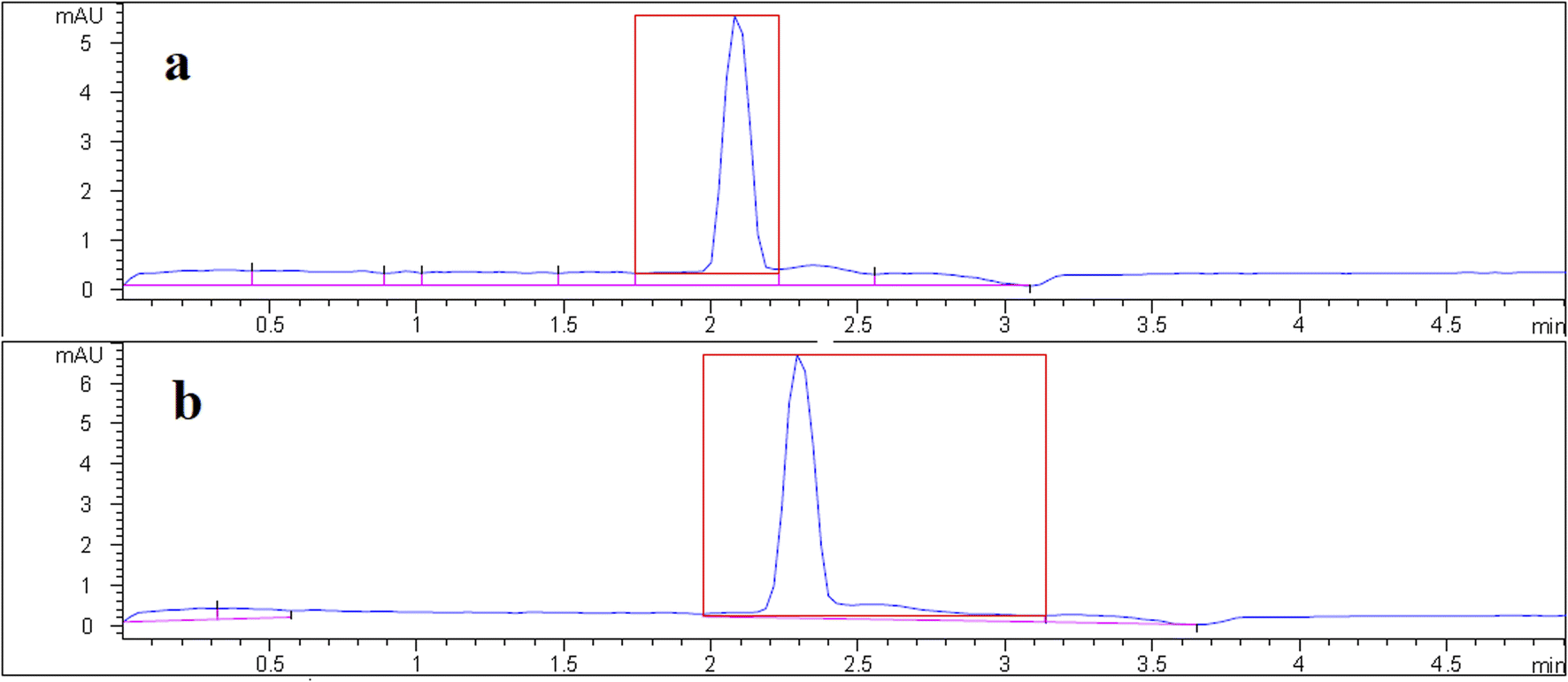 | ||
| Fig. 4 Typical electropherogram of urine #1 sample (a) before and (b) after spike with 100 μg L−1 of VMA. | ||
3.5. Comparison with other reported methods
The performance of the developed DSPE-CE-DAD method for the extraction and determination of VMA was compared with other reported CE-UV-based methods. The methods were compared from the viewpoint of LOD, LOQ, linearity, and RSD. The comparison results are summarized in Table 2. It can be seen that the proposed method is more precise than other methods. Also, the wide linear range is an advantage of this method over other methods. However, the LOD and LOQ of some methods are better than the proposed method.| Analysis method | Sample treatment/reagent | LOD (μg mL−1) | LOQ (μg mL−1) | LR (μg mL−1) | Method imprecision (RSD, %) | Ref. |
|---|---|---|---|---|---|---|
| a ECD – electrochemical detection, UV – ultraviolet, DAD – diode array detection, CE – capillary electrophoresis, LLE – liquid–liquid extraction, SPE – solid-phase extraction, PP – protein precipitation, LR – linear range, LOD – limit of detection, LOQ – limit of quantification. DSPE – dispersive solid-phase extraction. | ||||||
| CE-DAD | Centrifugation and filtration | 0.001 | 0.025 | — | — | 35 |
| CE-ECD | LLE/ether; agitation by vortex | 0.16 | 1.0 | 1.0–100 | 4.9 | 31 |
| CE-UV | PP/1 mM formic acid containing 20% ACN and dilution (10 times) | 0.062 | — | — | 3.1 | 37 |
| CE-ECD | Centrifugation, filtration and dilution | 0.005 | 0.01 | 0.01–5.0 | 3.4 | 38 |
| CE-DAD | LLE/diethyl ether; agitation by vortex | 0.10 | 0.50 | 0.50–50 | ≤9.1 | 39 |
| CE-UV | Centrifugation and dilution (1 time) | 0.006 | 0.60 | 0.60–19.5 | 7.0 | 33 |
| CE-DAD | DSPE/UiO-66-NH2 MOF | 0.012 | 0.04 | 0.04–2.0 | 2.8 | This work |
4. Conclusions
In conclusion, the amino-functionalized UiO-66 was firstly used as a DSPE adsorbent for the extraction and enrichment of VMA. The developed method of DSPE-CE-DAD offers sound analytical performances with good sensitivity, high extraction recovery, and short separation time. The results reveal that the adsorption and desorption mechanisms in this method are ion-pair and ion exchange, respectively. Under optimal conditions, these two mechanisms lead to selective and clean extraction of the VMA from the urine sample. Since no organic solvent was used in the VMA desorption step and also the separations performed by CE did not use any organic solvent, this method is a green and environmentally friendly method. The developed method would have a significant application potential for the isolation of other endogenous compounds in different biological fluids.Abbreviations
| VMA | Vanillylmandelic acid |
| DSPE | Dispersive solid phase extraction |
| EF | Enrichment factor |
| ER | Extraction recovery |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| RSD | Relative standard deviation |
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors thank the Research Council of the Tabriz University of Medical Science for financial support under grant number 68927.References
- C. Schulz, G. Eisenhofer and H. Lehnert, Principles of catecholamine biosynthesis, metabolism and release, in Pheochromocytoma: Pathophysiology and Clinical Management, ed. H. Lehnert, Front. Horm. Res. Basel, 2004, pp. 1–25 Search PubMed.
- J. Kaluzna-Czaplinska, E. Socha and J. Rynkowski, Determination of homovanillic acid and vanillylmandelic acid in urine of autistic children by gas chromatography/mass spectrometry, Med. Sci. Monit., 2010, 16, 445–450 Search PubMed.
- J. G. Boyle, D. F. Davidson, C. G. Perry and J. M. Connell, Comparison of diagnostic accuracy of urinary free metanephrines, vanillyl mandelic acid, and catecholamines and plasma catecholamines for diagnosis of pheochromocytoma, J. Clin. Endocrinol. Metab., 2007, 92, 4602–4608 CrossRef.
- S. Barco, I. Gennai, G. Reggiardo, B. Galleni, L. Barbagallo, A. Maffia, E. Viscardi, F. De Leonardis, V. Cecinati and S. Sorrentino, Urinary homovanillic and vanillylmandelic acid in the diagnosis of neuroblastoma: report from the Italian cooperative group for neuroblastoma, Clin. Biochem., 2014, 47, 848–852 CrossRef.
- E. Pussard, M. Neveux and N. Guigueno, Reference intervals for urinary catecholamines and metabolites from birth to adulthood, Clin. Biochem., 2009, 42, 536–539 CrossRef CAS PubMed.
- J. Mika, J. Barek, J. Zima, J. C. Moreira and H. Dejmková, Simultaneous determination of homovanillic and vanillylmandelic acid by HPLC using a coulometric detector with renewable glassy carbon microbeads based working electrode, Electroanalysis, 2018, 30, 1455–1460 CrossRef CAS.
- A. Makrlikova, H. Dejmkova, T. Navratil, J. Barek and V. Vyskocil, HPLC-ED/UV for determination of vanillylmandelic acid in human urine after solid phase extraction, Proceedings of the 14th ISC modern analytical chemistry, Prague, 2018, 82–86 Search PubMed.
- P. Moleman and J. Borstrok, Determination of urinary vanillylmandelic acid by liquid chromatography with electrochemical detection, Clin. Chem., 1983, 29, 878–881 CrossRef CAS.
- Y. Sato, J. Hanai and T. Takasugi Ntakeda, Determination of urinary vanillylmandelic acid and homovanillic acid by high performance liquid chromatography for mass screening of neuroblastoma, Tohoku J. Exp. Med., 1986, 150, 169–174 CrossRef PubMed.
- K. Yamada, E. Kayama, Y. Aizawa, K. Oka and S. Hara, Determination of vanillylmandelic acid in urine by pre-column dansylation using micro high-performance liquid chromatography with fluorescence detection, J. Chromatogr. B, 1981, 223, 176–178 CrossRef.
- A. Gironi, G. Seghieri, M. Niccolai and P. Mammini, Simultaneous liquid-chromatographic determination of urinary vanillylmandelic acid, homovanillic acid, and 5-hydroxyindoleacetic acid, Clin. Chem., 1988, 34, 2504–2506 CrossRef.
- G. M. Anderson, F. C. Feibel and D. J. Cohen, Liquid-chromatographic determination of vanillylmandelic acid in urine, Clin. Chem., 1985, 31, 819–821 CrossRef.
- M. Valko-Rokytovská, P. Očenáš, A. Salayová and Z. Kostecká, New developed UHPLC method for selected urine metabolites, J. Chromatogr. Sep. Tech., 2018, 9, 2–8 Search PubMed.
- X. Xiong and Y. Zhang, A new approach for urinary vanillylmandelic acid determination using eVol microextraction by packed sorbent coupled to liquid chromatography-tandem mass spectrometry, J. Anal. Sci. Technol., 2020, 11, 1–14 CrossRef.
- Z. Xie, P. Lorkiewicz, D. W. Riggs, A. Bhatnagar and S. Srivastava, Comprehensive, robust, and sensitive UPLC-MS/MS analysis of free biogenic monoamines and their metabolites in urine, J. Chromatogr. B, 2018, 1099, 83–91 CrossRef PubMed.
- Y. Shen, J. Lu, Q. Tang, Q. Guan, Z. Sun, H. Li and L. Cheng, Rapid, easy analysis of urinary vanillylmandelic acid for diagnostic testing of pheochromocytoma by liquid chromatography tandem mass spectrometry, J. Chromatogr. B, 2015, 1002, 92–97 CrossRef PubMed.
- Z. D. Clark, J. M. Cutler, I. Y. Pavlov, F. G. Strathmann and E. L. Frank, Simple dilute-and-shoot method for urinary vanillylmandelic acid and homovanillic acid by liquid chromatography tandem mass spectrometry, Clin. Chim. Acta, 2017, 468, 201–208 CrossRef CAS PubMed.
- K. Sadilkova, K. Dugaw, D. Benjamin and R. M. Jack, Analysis of vanillylmandelic acid and homovanillic acid by UPLC–MS/MS in serum for diagnostic testing for neuroblastoma, Clin. Chim. Acta, 2013, 424, 253–257 CrossRef CAS.
- L. Lionetto, A. M. Lostia, A. Stigliano, P. Cardelli and M. Simmaco, HPLC–mass spectrometry method for quantitative detection of neuroendocrine tumor markers: vanillylmandelic acid, homovanillic acid and 5-hydroxyindoleacetic acid, Clin. Chim. Acta, 2008, 398, 53–56 CrossRef CAS.
- L. Fang, Y. Lv, X. Sheng and S. Yao, Sensitive, rapid and easy analysis of three catecholamine metabolites in human urine and serum by liquid chromatography tandem mass spectrometry, J. Chromatogr. Sci., 2012, 50, 450–456 CAS.
- L. Konieczna, A. Roszkowska, M. Niedźwiecki and T. Bączek, Hydrophilic interaction chromatography combined with dispersive liquid–liquid microextraction as a preconcentration tool for the simultaneous determination of the panel of underivatized neurotransmitters in human urine samples, J. Chromatogr. A, 2016, 1431, 111–121 CrossRef.
- R. Allenbrand and U. Garg, Quantitation of homovanillic acid (HVA) and vanillylmandelic acid (VMA) in urine using gas chromatography-mass spectrometry (GC/MS), in Clinical Applications of Mass Spectrometry. Methods in Molecular Biology (Methods and Protocols), ed. U. Garg and C. Hammett-Stabler, Humana Press, 2010, pp. 261–269 Search PubMed.
- M. T. C. Tran, J. Baglin, T. T. T. Tran, K. T. Hoang, L. T. Phung, A. Read and R. F. Greaves, Development of a new biochemical test to diagnose and monitor neuroblastoma in Vietnam: Homovanillic and vanillylmandelic acid by gas chromatography–mass spectrometry, Clin. Biochem., 2014, 47, 206–215 CrossRef PubMed.
- G. Fauler, H. Leis, E. Huber, C. Schellauf, R. Kerbl, C. Urban and H. Gleispach, Determination of homovanillic acid and vanillylmandelic acid in neuroblastoma screening by stable isotope dilution GC-MS, J. Mass Spectrom., 1997, 32, 507–514 CrossRef.
- M. Tuchman, P. Crippin and W. Krivit, Capillary gas-chromatographic determination of urinary homovanillic acid and vanillylmandelic acid, Clin. Chem., 1983, 29, 828–831 CrossRef.
- F. Taran, H. Bernard, A. Valleix, C. Créminon, J. Grassi, D. Olichon, J. R. Deverre and P. Pradelles, Competitive enzyme immunoassay for urinary vanillylmandelic acid, Clin. Chim. Acta, 1997, 264, 177–192 CrossRef.
- A. Abaslizadeh, S. M. Sorouraddin, M. A. Farajzadeh, M. Nemati and M. R. Afshar Mogaddam, Dispersive solid phase extraction of several pesticides from fruit juices using a hydrophobic metal organic framework prior to HPLC-MS/MS determination, J. Food Compost. Anal., 2022, 114, 104788 CrossRef.
- S. Baluchová, J. Barek, L. I. Tomé, C. M. Brett and K. Schwarzová-Pecková, Vanillylmandelic and homovanillic acid: Electroanalysis at non-modified and polymer-modified carbon-based electrodes, J. Electroanal. Chem., 2018, 821, 22–32 CrossRef.
- B. Fu, H. Chen, Z. Yan, Z. Zhang, J. Chen, T. Liu and K. Li, A simple ultrasensitive electrochemical sensor for simultaneous determination of homovanillic acid and vanillylmandelic acid in human urine based on MWCNTs-Pt nanoparticles as peroxidase mimics, J. Electroanal. Chem., 2020, 866, 114165 CrossRef CAS.
- M. Libansky, J. Zima, J. Barek and H. Dejmkova, Voltammetric determination of homovanillic acid and vanillylmandelic acid on a disposable electrochemical measuring cell system with integrated carbon composite film electrodes, Monatsh. Chem., 2016, 147(1), 89–96 CrossRef CAS.
- X. Li, W. Jin and Q. Weng, Separation and determination of homovanillic acid and vanillylmandelic acid by capillary electrophoresis with electrochemical detection, Anal. Chim. Acta, 2002, 461, 123–130 CrossRef CAS.
- W. H. de Jong, E. G. de Vries and I. P. Kema, Current status and future developments of LC-MS/MS in clinical chemistry for quantification of biogenic amines, Clin. Biochem., 2011, 44, 95–103 CrossRef CAS PubMed.
- A. Garcıa, M. Heinänen, L. Jiménez and C. Barbas, Direct measurement of homovanillic, vanillylmandelic and 5-hydroxyindoleacetic acids in urine by capillary electrophoresis, J. Chromatogr. A, 2000, 871, 341–350 CrossRef.
- J. S. Toraño, R. Ramautar and G. de Jong, Advances in capillary electrophoresis for the life sciences, J. Chromatogr. B, 2019, 1118, 116–136 CrossRef.
- H. Sirén and U. Karjalainen, Study of catecholamines in patient urine samples by capillary electrophoresis, J. Chromatogr. A, 1999, 853, 527–533 CrossRef.
- N. Miękus, I. Olędzka, A. Plenis, P. Kowalski, E. Bień, A. Miękus, M. A. Krawczyk, E. Adamkiewicz-Drożyńska and T. Bączek, Determination of urinary biogenic amines' biomarker profile in neuroblastoma and pheochromocytoma patients by MEKC method with preceding dispersive liquid–liquid microextraction, J. Chromatogr. B, 2016, 1036, 114–123 CrossRef.
- M. M. Hsieh, E. P. Lin and S. W. Huang, On-line concentration and separation of cationic and anionic neurochemicals by capillary electrophoresis with UV absorption detection, Talanta, 2012, 88, 638–645 CrossRef.
- Z. H. Zhang, Z. Li, J. Zhang, Y. Zhang, J. Ye, Q. Chu and M. Zhang, Simultaneous determination of catecholamines and related metabolites by capillary electrophoresis with amperometric detection, Chem. Res. Chin. Univ., 2013, 29, 850–853 CrossRef.
- N. Miękus, P. Kowalski, I. Olędzka, A. Plenis, E. Bień, A. Miękus, M. Krawczyk, E. Adamkiewicz-Drożyńska and T. Bączek, Cyclodextrin-modified MEKC method for quantification of selected acidic metabolites of catecholamines in the presence of various biogenic amines. Application to diagnosis of neuroblastoma, J. Chromatogr. B, 2015, 1003, 27–34 CrossRef.
- P. Tůma, E. Samcová and K. Štulík, Determination of the spectrum of low molecular mass organic acids in urine by capillary electrophoresis with contactless conductivity and ultraviolet photometric detection-An efficient tool for monitoring of inborn metabolic disorders, Anal. Chim. Acta, 2011, 685, 84–90 CrossRef PubMed.
- W. Jin, L. Jin, G. Shi and J. Ye, Determination of monoamine transmitters and their metabolites by capillary electrophoresis with electrochemical detection, Anal. Chim. Acta, 1999, 382, 33–37 CrossRef.
- L. Zhou, J. D. Glennon and J. H. Luong, Electrophoretic analysis of biomarkers using capillary modification with gold nanoparticles embedded in a polycation and boron doped diamond electrode, Anal. Chem., 2010, 82, 6895–6903 CrossRef CAS PubMed.
- X. Li, W. Jin and Q. Weng, Separation and determination of homovanillic acid and vanillylmandelic acid by capillary electrophoresis with electrochemical detection, Anal. Chim. Acta, 2002, 461, 123–130 CrossRef CAS.
- J. Caslavska, E. Gassmann and W. Thormann, Modification of a tunable UV-visible capillary electrophoresis detector for simultaneous absorbance and fluorescence detection: profiling of body fluids for drugs and endogenous compounds, J. Chromatogr. A, 1995, 709, 147–156 CrossRef CAS.
- M. M. Hsieh and H. T. Chang, Impact of halides on the simultaneous separation of aromatic amines and their acidic metabolites by capillary electrophoresis with laser-induced native fluorescence detection under acidic conditions, J. Chromatogr. A, 2006, 1102, 302–308 CrossRef CAS.
- M. K. Shirao, S. Suzuki, J. Kobayashi, H. Nakazawa and E. Mochizuki, Analysis of creatinine, vanilmandelic acid, homovanillic acid and uric acid in urine by micellar electrokinetic chromatography, J. Chromatogr. B Biomed. Appl., 1997, 693, 463–467 CrossRef.
- S. Ma, Z. Xu and J. Ren, Analysis of neurochemicals by capillary electrophoresis in athletes' urine and a pilot study of their changes responding to sport fatigue, Anal. Methods, 2019, 11, 2712–2719 RSC.
- H. J. Issaq, K. Delviks, G. M. Janini and G. M. Muschik, Capillary zone electrophoretic separation of homovanillic and vanillylmandelic acids, J. Liq. Chromatogr. Relat. Technol., 1992, 15, 3193–3201 CrossRef.
- W. L. Tseng, S. M. Chen, C. Y. Hsu and M. M. Hsieh, On-line concentration and separation of indolamines, catecholamines, and metanephrines in capillary electrophoresis using high concentration of poly (diallyldimethylammonium chloride), Anal. Chim. Acta, 2008, 613, 108–115 CrossRef CAS.
- H. Sirén, M. Mielonen and M. Herlevi, Capillary electrophoresis in the determination of anionic catecholamine metabolites from patients' urine, J. Chromatogr. A, 2004, 1032, 289–297 CrossRef.
- D. Flottmann, J. Hins, C. Rettenmaier, N. Schnell, Z. Kuçi, G. Merkel, G. Seitz and G. Bruchelt, Two-dimensional isotachophoresis for the analysis of homovanillic acid and vanillylmandelic acid in urine for cancer therapy monitoring, Microchim. Acta, 2006, 154, 49–53 CrossRef CAS.
- M. Moriarty, A. Lee, B. O'Connell, A. Kelleher, H. Keeley and A. Furey, Development of an LC-MS/MS method for the analysis of serotonin and related compounds in urine and the identification of a potential biomarker for attention deficit hyperactivity/hyperkinetic disorder, Anal. Bioanal. Chem., 2011, 401, 2481–2493 CrossRef CAS PubMed.
- N. Gan, T. Li, L. Wang and Q. Jiang, Determination of seven biogenic amines in fish using micellar electrokinetic capillary chromatography, Chin. J. Anal. Chem., 2007, 25, 934–938 CAS.
- J. Płonka, Methods of biological fluids sample preparation–biogenic amines, methylxanthines, water-soluble vitamins, J. Chromatogr. B Biomed. Appl., 2015, 29, 1–20 Search PubMed.
- M. Nemati, M. Tuzen, M. A. Farazajdeh, S. Kaya and M. R. Afshar Mogaddam, Development of dispersive solid-liquid extraction method based on organic polymers followed by deep eutectic solvents elution; application in extraction of some pesticides from milk samples prior to their determination by HPLC-MS/MS, Anal. Chim. Acta, 2022, 1199, 339570 CrossRef CAS.
- Z. Hasan and S. H. Jhung, Removal of hazardous organics from water using metal-organic frameworks (MOFs): plausible mechanisms for selective adsorptions, J. Hazard. Mater., 2015, 283, 329–339 CrossRef CAS PubMed.
- X. Mao, W. Xiao, Y. Wan, Z. Li, D. Luo and H. Yang, Dispersive solid-phase extraction using microporous metal-organic framework UiO-66: Improving the matrix compounds removal for assaying pesticide residues in organic and conventional vegetables, Food Chem., 2021, 345, 128807 CrossRef CAS.
- F. Qu, L. Xia, C. Wu, L. Liu, G. Li and J. You, Sensitive and accurate determination of sialic acids in serum with the aid of dispersive solid-phase extraction using the zirconium-based MOF of UiO-66-NH2 as sorbent, RSC Adv., 2016, 6, 64895–64901 RSC.
- Y. Tsuzuki, The nature of the complex formation between boric acid and organic polyoxy compounds, Bull. Chem. Soc. Jpn., 1941, 16, 23–31 CrossRef.
- S. W. Sinton, Complexation chemistry of sodium borate with poly (vinyl alcohol) and small diols: a boron-11 NMR study, Macromolecules, 1987, 20, 2430–2441 CrossRef CAS.
- A. Garcıa, M. Heinänen, L. Jiménez and C. Barbas, Direct measurement of homovanillic, vanillylmandelic and 5-hydroxyindoleacetic acids in urine by capillary electrophoresis, J. Chromatogr. A, 2000, 871, 341–350 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |