DOI:
10.1039/D2RA02848D
(Paper)
RSC Adv., 2022,
12, 20991-21003
Profiling of in vivo, in vitro and reactive zorifertinib metabolites using liquid chromatography ion trap mass spectrometry†
Received
5th May 2022
, Accepted 18th July 2022
First published on 21st July 2022
Abstract
Zorifertinib (AZD-3759; ZFB) is a potent, novel, oral, small molecule used for the treatment of non-small cell lung cancer (NSCLC). ZFB is Epidermal Growth Factor Receptor (EGFR) inhibitor that is characterized by good permeability of the blood–brain barrier for (NSCLC) patients with EGFR mutations. The present research reports the profiling of in vitro, in vivo and reactive metabolites of ZFB. Prediction of vulnerable metabolic sites and reactivity pathways (cyanide and GSH) of ZFB were performed by WhichP450™ module (StarDrop software package) and XenoSite reactivity model (XenoSite Web Predictor-Home), respectively. ZFB in vitro metabolites were done by incubation with isolated perfused rat liver hepatocytes and rat liver microsomes (RLMs). Extraction of ZFB and its related metabolites from the incubation matrix was done by protein precipitation. In vivo metabolism was performed by giving ZFB (10 mg kg−1) through oral gavage to Sprague Dawley rats that were housed in metabolic cages. Urine was collected at specific time intervals (0, 6, 12, 18, 24, 48, 72, 96 and 120 h) from ZFB dosing. The collected urine samples were filtered then stored at −70 °C. N-Methyl piperazine ring of ZFB undergoes phase I metabolism forming iminium intermediates that were stabilized using potassium cyanide as a trapping agent. Incubation of ZFB with RLMs were performed in the presence of 1.0 mM KCN and 1.0 mM glutathione to check reactive intermediates as it is may be responsible for toxicities associated with ZFB usage. For in vitro metabolites there were six in vitro phase I metabolites, three in vitro phase II metabolites, seven reactive intermediates (four GSH conjugates and three cyano adducts) of ZFB were detected by LC-IT-MS. For in vivo metabolites there were six in vivo phase I and three in vivo phase II metabolites of ZFB were detected by LC-IT-MS. In vitro and in vivo phase I metabolic pathways were N-demethylation, O-demethylation, hydroxylation, reduction, defluorination and dechlorination. In vivo phase II metabolic reaction was direct sulphate and glucuronic acid conjugation with ZFB.
1. Introduction
Non-small-cell lung carcinoma (NSCLC) is any type of epithelial lung cancer other than small cell lung carcinoma (SCLC). NSCLC accounts for about 85% of all lung cancers.1 NSCLC treatment had a breakthrough in the past 15 years with tyrosine kinase inhibitors, specifically with the first generation gefitinib and erlotinib and, currently, with the second generation afatinib. These agents are referred to as “targeted” therapy since they target epidermal growth factor (EGF) mutations in lung cancer adenocarcinoma patients.2–4 EGF is a single polypeptide of fifty-three amino acid residues which is contributed in the cell proliferation regulation. EGF and EGFR play a crucial role in wound healing through stimulating dermal and epidermal regeneration.
The new therapies development for improving wound healing has involved the usage of EGF. In addition, EGFR inhibitors have become a way for the cancer treatment. Thus, therapies targeting EGF/EGFR are suitable for the treatment of cancer and cutaneous wounds.5 The first agent developed as a target cancer therapy was EGFR inhibitor. There are two classes of EGFR inhibitors are in current use: the monoclonal antibodies (cetuximab, panitumumab, and matuzumab) that target the extracellular ligand-binding domain and small-molecule tyrosine kinase inhibitors such as ZFB which target intracellular domain.6,7
ZFB is a novel small molecule used for NSCLC treatment through inhibition of EGFR and characterized by good permeability of the blood–brain barrier (BBB) for (NSCLC) patients with EGFR mutations.8 ZFB can penetrate the BBB and was confirmed to be effective in both clinical and preclinical settings, and showed promising activity in patients with central nervous system (CNS) metastasis as monotherapy.8,9 ZFB is currently at a phase II/III clinical trials.10 Side effects of ZFB was skin and gastrointestinal disorders of occurred in 92% and 76% patients respectively. Other disorders included hepatobiliary and renal disorders (13%), asthenia (7%), infections and infestations (7%), and metabolism and nutrition disorders at 4% and 7%, respectively.11
ZFB chemical name is 4-((3-chloro-2-fluorophenyl)amino)-7-methoxyquinazolin-6-yl (R)-2,4-dimethylpiperazine-1-carboxylate. ZFB chemical structure contains N-methyl piperazine and phenyl amine rings that could undergo metabolic bioactivation generating iminium and iminoquinone reactive intermediate, respectively that can be captured by nucleophile such as potassium cyanide (KCN) and GSH, respectively forming cyano adducts and GSH conjugates, respectively that are stable and could be identified by liquid chromatography ion trap mass spectrometry (LC-IT-MS). Extraction, identification, separation and characterization of these stabilized adducts and conjugates were performed using LC-IT-MS.12–14 These reactive metabolites helped us to predict toxicities associated with ZFB.
Our research team previously established LC-MS/MS analytical method for ZFB quantification with application to assess its metabolic stability which revealed the moderate extraction ratio and the good bioavailability of ZFB.15,16 This research will focus on the profiling of ZFB metabolites either in vitro or in vivo using LC-IT-MS. Metabolic bioactivation of a drug to reactive intermediates that can covalently modify proteins leading to drug-induced organ toxicities. Hence, we will check the reactive intermediates that will aid in predicting the reasons for toxicities associated with these drugs. Reactive intermediates cannot be checked in vivo as a result of as once they are formed, they will bind to endogenous materials as proteins or DNA that prevents the detection by LC-IT-MS.17
2. Chemicals and methods
2.1. Chemicals and instruments
Zorifertinib was purchased from Med Chem Express LLC company (Princeton, NJ, USA). Reference powders (analytical grade) and solvents (HPLC grade) were used. Sprague Dawley rats were obtained from Health Research Center of Prince Naif bin Abdul Aziz at King Saud University (Riyadh, KSA). Rat liver microsomes (RLMs) were prepared in our laboratory following a published protocol using Sprague Dawley rats.18,19 Acetonitrile (ACN), nicotinamide adenine dinucleotide phosphate (NADPH), formic acid (HCOOH), potassium cyanide (KCN) and glutathione reductase (GSH) were purchased from Sigma-Aldrich company (St. Louis, MO, USA). Water (HPLC grade) was supplied by Milli-Q plus purification system (Billerica, MA, USA) that is available in-house. Tween 80 were obtained from Eurostar Scientific Ltd. (Liverpool, UK). Ethical approval for the Animal experiments of were obtained from the Animal Ethics Committee at King Saud University (No. KSU-SE-19-52). The separation of ZFB and its related metabolites and adducts was done on an Agilent HPLC 1200 series that consists of G1367B HIP-ALS auto sampler, G1322A degasser, G1316 thermostated column compartment and G1311A binary pump that was connected using an electrospray ionization (ESI) to an Agilent 6320 Ion Trap (Agilent Technologies, Palo Alto, CA, USA). Mass Hunter software (Agilent Technologies, Palo Alto, CA, USA) was used to regulate the data acquisition and instruments.
2.2. Chromatographic conditions
The analytical parameters that were used for separation of ZFB and its related metabolites are listed are mentioned in Table 1. Agilent eclipse plus C18 analytical column (150 mm × 2.1 mm, 3.5 μm particle size) (Agilent Technologies, Palo Alto, CA, USA). Column temperature was kept constant at 23 ± 2 °C. The most appropriate chromatographic conditions were attained at a flow rate of 0.25 mL min−1 with a gradient system for 65 min. Mobile phase composed of 0.1% HCOOH (pH: 3.2) and ACN. The post time was 15 min. Sample injection volume was 10 μL. Mass parameters were optimized for ZFB. Dissociation for ZFB and related metabolites was done using collision-induced dissociation (CID) in the collision cell.
Table 1 Adjusted parameters of the proposed LC-MS experiment
| Liquid chromatographic parameters |
Mass spectrometric parameters |
| HPLC |
Agilent 1200 |
Mass spectrometer |
Agilent 6320 ion trap |
| Gradient mobile phase |
A: 1% HCOOH in H2O |
Ionization source |
Positive ESI |
| B: ACN |
Drying gas: N2 gas |
| Flow rate: 0.25 mL min−1 |
Flow rate (10 L min−1) |
| Run time: 65 min |
Pressure (60 psi) |
| Agilent eclipse plus C18 column |
Length |
150 mm |
ESI temperature: 350 °C |
| Internal diameter |
2.1 mm |
Capillary voltage: 4000 V |
| Particle size |
3.5 μm |
Collision gas |
High purity N2 |
| Temperature |
23 ± 2 °C |
Modes |
Mass scan and MS2 |
| Gradient system |
Time |
% B |
Analytes |
ZFB and its metabolites |
| 0 |
5 |
|
| 5 |
5 |
Mass parameters |
|
| 30 |
30 |
Fragmentor voltage: 145 V |
| 40 |
40 |
Amplitude: 1.25 V |
| 50 |
80 |
|
| 60 |
90 |
|
| 65 |
5 |
|
2.3. In silico ZFB metabolism prediction using WhichP450™ (StarDrop software)
Our target is to recognize the susceptibility of metabolism key sites, as shown by the low value of the site lability that was revealed by the composite site lability (CSL) and also the proposed major isoforms regioselectivity that was supposed to be responsible for metabolism. The results from the WhichP450™ software, displayed by the pie chart used for indication of most probably cyp450 isoform that has the major role in ZFB metabolism. These predictions were used as a director during profiling of ZFB in vivo and in vitro metabolites.20,21
2.4. In silico of ZFB reactive intermediates and toxicity prediction using XenoSite reactivity model and DEREK software
XenoSite reactivity model is freely available online at https://swami.wustl.edu/xenosite that was used to detect the labile sites that could produce reactive intermediates that could form complex with GSH, cyanide, DNA and protein stable adducts.22–24 This in silico model is based on neural networks of around 680 molecules.25 This free online software characterized by short running time depending on the molecule size.21 SMILES format (CC(C)Oc1ccc(cc1)NC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)N2CCN(CC2)c3c4cc(c(cc4ncn3)OCCCN5CCCCC5)OC) of ZFB was uploaded to XenoSite reactivity model that is an online website. The outcomes were used in searching and identifying reactive metabolites of ZFB.
O)N2CCN(CC2)c3c4cc(c(cc4ncn3)OCCCN5CCCCC5)OC) of ZFB was uploaded to XenoSite reactivity model that is an online website. The outcomes were used in searching and identifying reactive metabolites of ZFB.
DEREK module (StarDrop software package) was utilized for screening for ZFB and its related metabolites structural alerts and to verify our bioactivation pathway proposal. DEREK software can be used to propose structural modification at the bioactive sites that may stop the initiation of toxicity sequence. The chemical structure of ZFB and its related metabolites (SMILES format) was uploaded on the program software for the toxicity prediction.
2.5. In vitro metabolism
2.5.1. RLMs incubations. RLMs were incubated with ZFB following a previously published procedure.26,27 Thirty μM ZFB in DMSO was added to 0.08 M phosphate buffer (pH 7.4) with RLMs (1 mg mL−1). The incubation mixture was first kept in a shaking water bath for 5 min at 37 °C. Incubation was initiated by NADPH (1 mM) solution. The final volume of incubation mixture is 1 mL and the total time of incubation is 90 min. Controls were prepared either by replacing RLMs or NAPDH with buffer. The metabolic reaction was stopped adding 2 mL ice-cold acetonitrile then centrifugation at 9000g for 10 min for protein precipitation. The supernatant was removed to clean vials for evaporation using a stream of nitrogen as inert gas. The residue was solubilized in a mixture of 0.1% HCOOH and ACN in a ratio similar to mobile phase and transferred to HPLC vial for analysis. To trap reactive intermediates, the same experiment was repeated using 1.0 mM KCN or 1.0 mM GSH. Similarly, negative controls were done using the above steps. Phenytoin (2 μM) was used to evaluate the activity of RLMs as the positive control.
2.5.2. Liver hepatocytes incubation. Rat liver hepatocytes (RLHs) will be done following the established steps.28 RLHs incubation experiment with ZFB (30 μM) was performed in a rotary evaporation instrument fitted with a custom made 4-way adaptor to permit the connections of four 100 mL round bottom flasks. The RLHs incubations were performed at 37 °C for 2 hours. One mL aliquots (at 0, 15, 30, 60, 90, and 120 minutes were taken from each flask, mixed with 2 mL of ACN and immediately frozen at −70 °C refrigerator). Cell viability was retested using trypan blue at the end of the experiment. Stored frozen samples were thawed and then centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Supernatants were transferred to new vials and evaporated under a stream of inert gas nitrogen. The residue was reconstituted in 300 μl of the mobile phase in HPLC vials for LC-MS analysis. Negative control experiments were done following the above steps.
000 rpm. Supernatants were transferred to new vials and evaporated under a stream of inert gas nitrogen. The residue was reconstituted in 300 μl of the mobile phase in HPLC vials for LC-MS analysis. Negative control experiments were done following the above steps.
2.6. In vivo metabolism
Four male one month Sprague-Dawley rats of average weight (400 g) were housed individually in a metabolism cage that was kept in animal care facility in a 12 h light/dark cycle (7:00–19:00). Rats had free access to standard animal food and water. Rats were kept in metabolism cages for 72 h before the start of the experiment. ZFB was solubilized in a special solution (4% DMSO, 5% Tween 80 and 30% PEG 300, in HPLC H2O) to allow ZFB dispersion and to maintain the drug solubility. The ZFB dose for rats was calculated by conversion from reported human dose to rat dose.29 Each rat received a specific dose of ZFB, depending on its weight. The ZFB dose for rat was 10 mg per kg per day.
3. Results and discussion
3.1. In silico prediction of ZFB metabolites
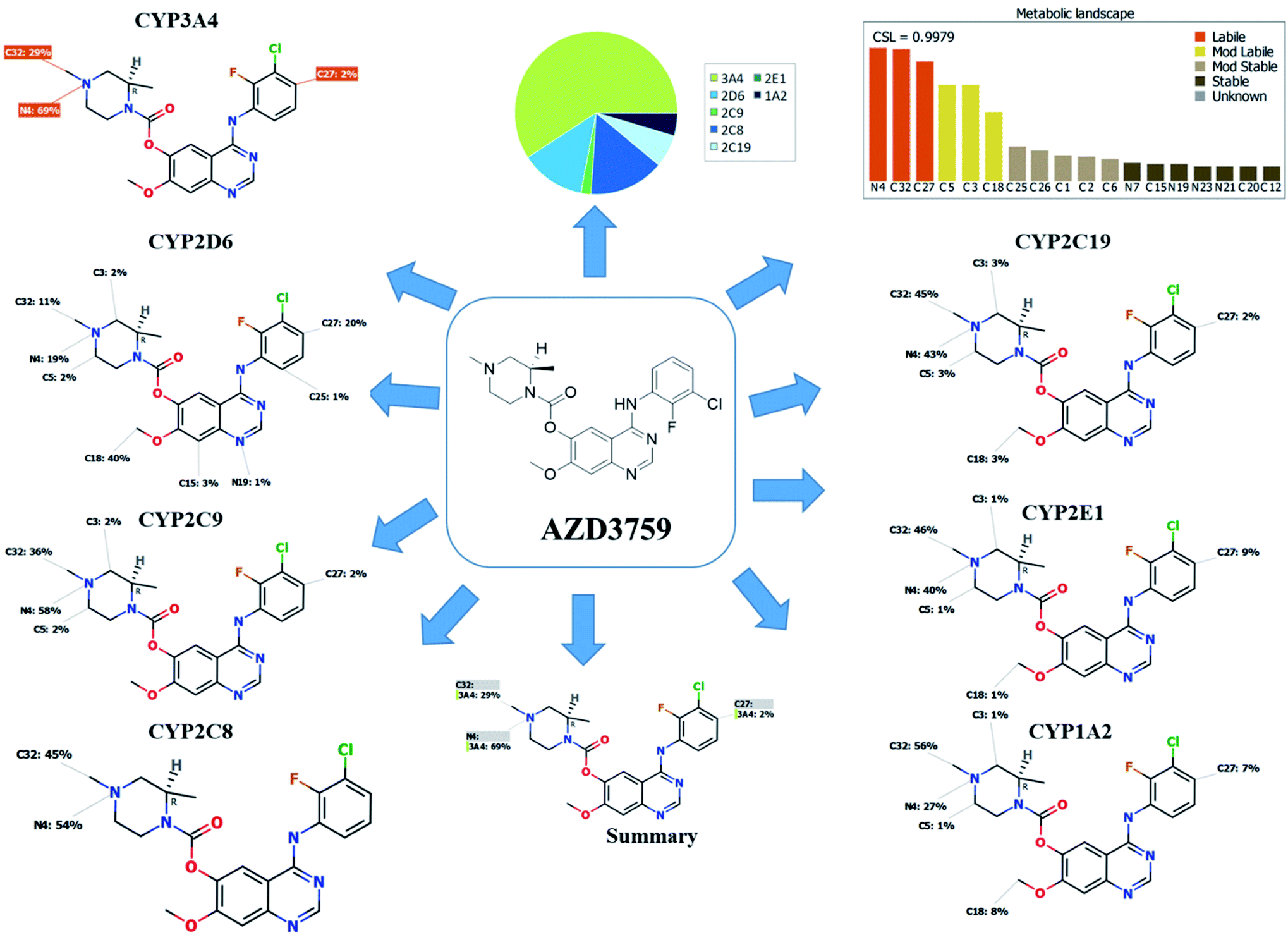
The composite site lability for ZFB is 0.9979 that indicates the high lability of ZFB to metabolism. The metabolic Landscape indicates that atoms of C32 and N4 of N-methyl piperazine ring and C27 of phenyl group are the most labile atoms to metabolism, while C3 and C5 (the α carbons) of the N-methyl piperazine ring and C18 of methoxy group are moderate labile atoms to metabolism that matched with the current experiments (Fig. 1). Cyp3A4 had the main role in ZFB metabolism. Consecutively, depending on the literature knowledge and in silico postulations, a list of predicted reactive intermediates and metabolites was prepared and used as a guide during practical experiments.
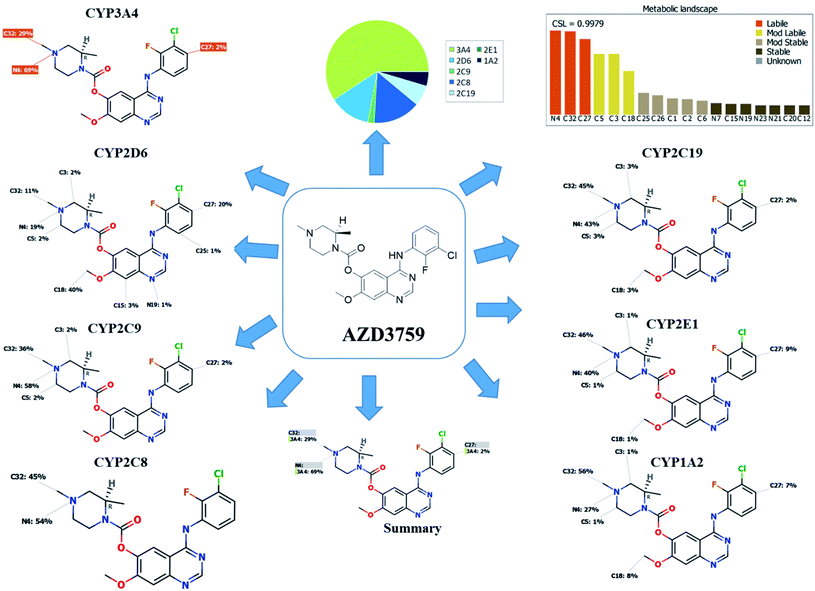 |
| | Fig. 1 Predicted atomic sites of metabolism for zorifertinib by XenoSite web predictor. | |
3.2. In silico ZFB bioactivity and toxicity prediction
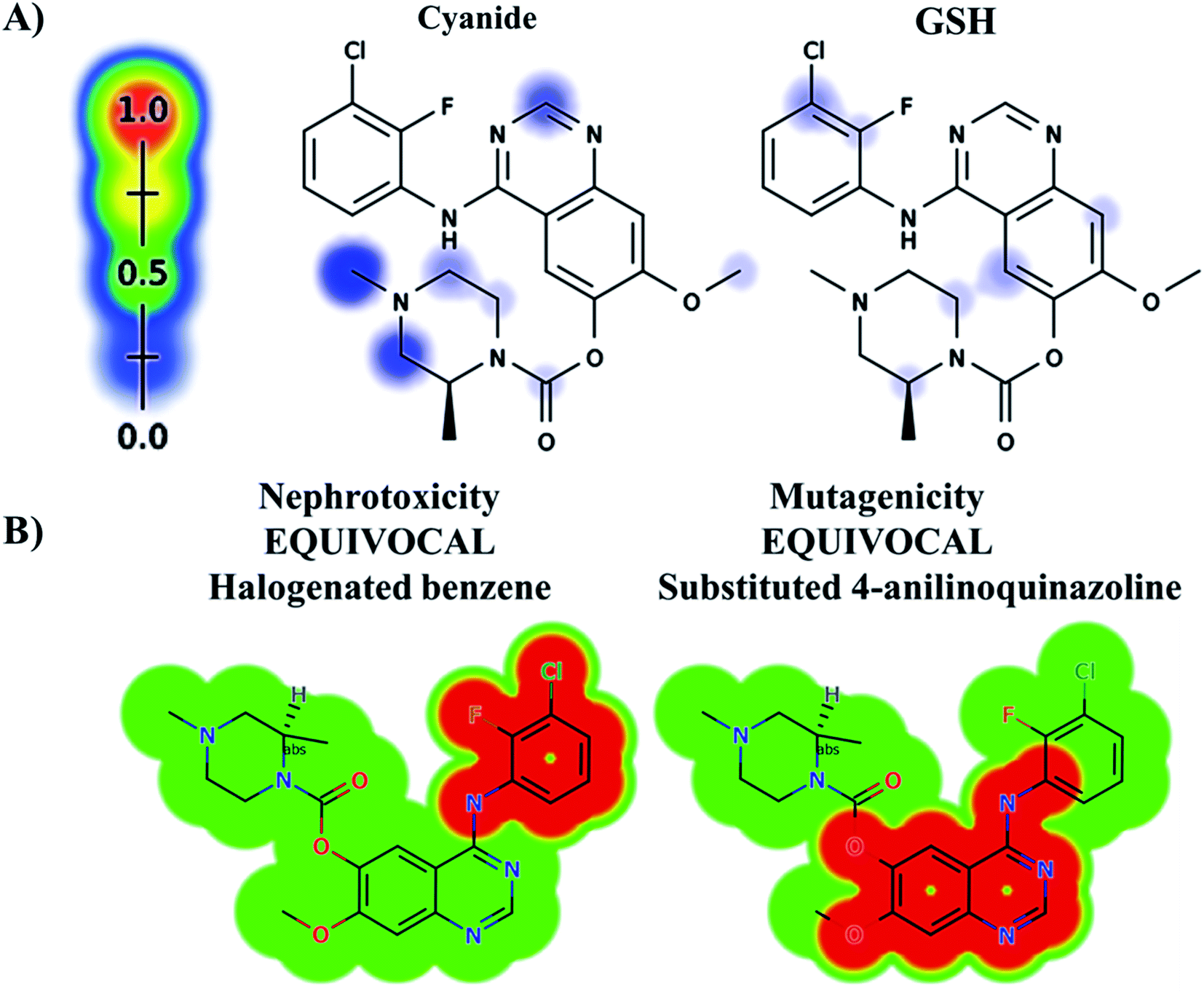
The ZFB atomic sites were proposed for generation of electrophiles that could be trapped by cyanide and GSH. The results were displayed as gradient color scale bar where the white color and red color indicates no or highest possibility of reactive intermediate formation at the atomic site, respectively (Fig. 2). GSH model reveals the possibility of reactive intermediate generation at phenyl group that could be trapped with GSH. KCN model reveals the possibility of reactive iminium ion intermediate generation at the piperazine ring that could be captured by cyanide forming cyano adduct that was confirmed by in vitro experiments. The outcomes of the predictions are shown in Fig. 2A.
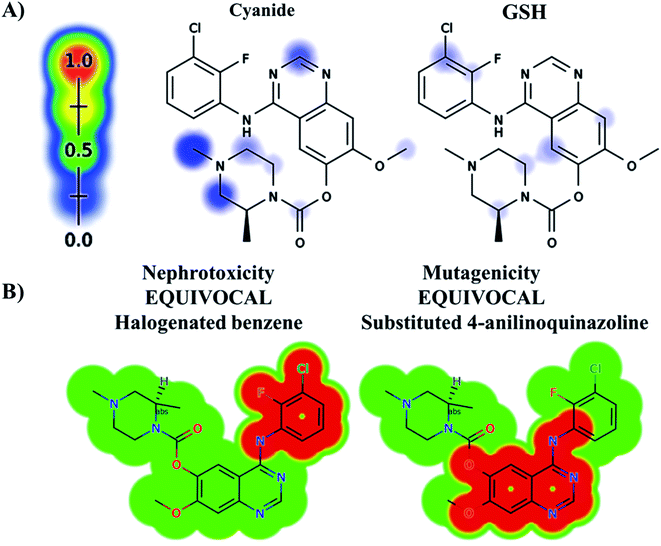 |
| | Fig. 2 Predicted bioactive sites of zorifertinib by XenoSite web predictor including GSH and cyano bioactive centers (A), outcomes of in silico toxicity studies of zorifertinib using DEREK toxicity module of StarDrop software. Red color indicates structural alerts (B). | |
In silico toxicity assessment of ZFB metabolites was done using DEREK module (StarDrop software package) (Fig. 2B). ZFB and its metabolites show equivocal nephrotoxicity and equivocal mutagenicity due to halogenated benzene and substituted 4-anilinoquinazoline, respectively (Fig. 2B). M4 shows plausible HERG channel inhibition due to 2-aminophenol derivative. Table 2 shows a complete list of in vitro and in vivo ZFB metabolites with DEREK outcomes for supposed toxicity profile.
Table 2 DEREK results for ZFB and its in vitro and in vivo metabolites
| ZFB and its metabolites |
Nephrotoxicity |
Mutagenicity |
HERG channel inhibition |
Teratogenicity and other toxicological parameters |
| Structural alert |
Halogenated benzene |
Substituted 4-anilinoquinazoline |
2-Aminophenol derivative |
para-Phenylenediamine |
| AZD 3759 |
Equivocal |
Equivocal |
Na |
Na |
| M1 |
Equivocal |
Equivocal |
Na |
Na |
| M2 |
Equivocal |
Equivocal |
Na |
Na |
| M3 |
Equivocal |
Equivocal |
Na |
Na |
| M4 |
Equivocal |
Equivocal |
Plausible |
Na |
| M5 |
Equivocal |
Equivocal |
Na |
Na |
| M6 |
Equivocal |
Equivocal |
Na |
Na |
| M7 |
Equivocal |
Equivocal |
Na |
Na |
| M8 |
Equivocal |
Equivocal |
Na |
Na |
| M9 |
Equivocal |
Equivocal |
Na |
Na |
| M1 |
Equivocal |
Equivocal |
Na |
Na |
| M10 |
Equivocal |
Equivocal |
Na |
Na |
| M11 |
Equivocal |
Equivocal |
Na |
Na |
| M12 |
Equivocal |
Equivocal |
Na |
Na |
| M13 |
Equivocal |
Equivocal |
Na |
Na |
| M14 |
Equivocal |
Equivocal |
Na |
Na |
| M15 |
Equivocal |
Equivocal |
Na |
Na |
3.3. MS/MS fragmentation behavior of ZFB
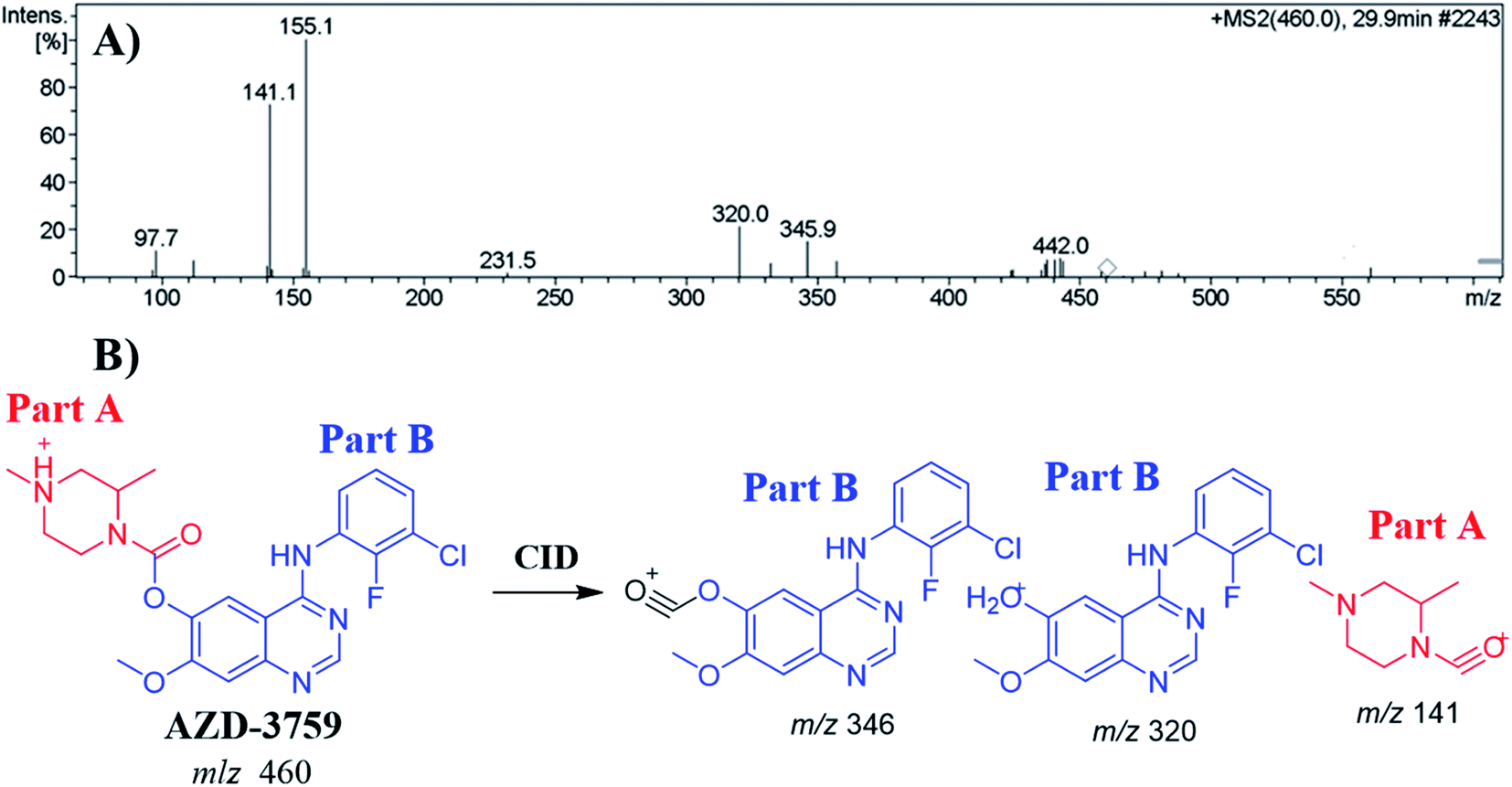
The ZFB precursor ion peak (PIP) appeared at 29.9 min in the total ion chromatogram (TIC). Dissociation of precursor ion (PI) at m/z 460 resulted in three characteristic fragment ions at m/z 346, m/z 320 and m/z 141 (Fig. 3A). The dissociation of ZFB targets the weak carboxylate link between the piperazine and quinazoline moieties inside its chemical structure. Fragment ions at m/z 320 and m/z 141 represent the two building blocks of ZFB. Fragment ion at m/z 141 was used for confirmation of the metabolic reaction at piperazine ring while m/z 320 for the remaining chemical structure (Fig. 3B).30
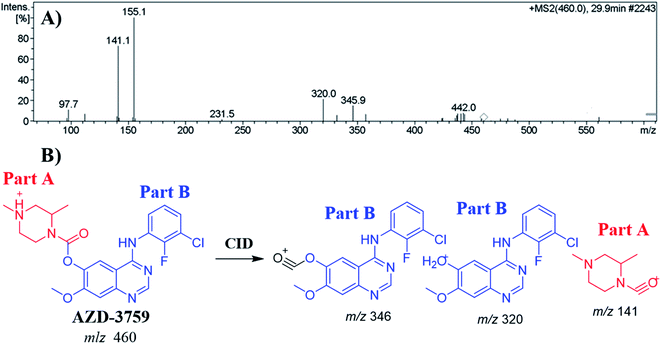 |
| | Fig. 3 MS2 mass spectrum of zorifertinib (A). Proposed zorifertinib structure and its corresponding fragment ions (B). | |
3.4. Identification of ZFB in vitro metabolites
Purified extracts recovered from RLMs incubates were injected into LC-IT-MS. Six phase I metabolic reactions produced by N-demethylation, O-demethylation, hydroxylation, reduction, α-oxidation and dechlorination. One sulphate conjugate and two glucuronic acid conjugates were characterized after incubation of ZFB with isolated perfused RLHs as phase II metabolite (Table 3). In vitro phase I metabolites (M2, M3, M4, M5 and M6) figures and schemes have been moved to a ESI (Fig. S1–S5†). In vitro phase II metabolites (M9) figures and schemes have been moved to a ESI (Fig. S6†).
Table 3 In vitro phase I and metabolites of ZFB
| |
MS scan |
Major fragment ions |
tR (min) |
Proposed metabolic reaction |
| ZFB |
460 |
320, 285, 127, 141 |
29.9 |
ZFB |
|
| In vitro phase I metabolites |
| M1 |
446 |
320, 428, 285, 100 |
27.6 |
N-Demethylation at piperazine ring |
| M2 |
446 |
306, 428 |
45.3 |
O-Demethylation |
| M3 |
476 |
320, 458 |
27.7 |
Hydroxylation |
| M4 |
434 |
306, 416, 287 |
34.7 |
N-Demethylation, O-demethylation and reduction |
| M5 |
456 |
438, 173 |
36.8 |
α-Oxidation, dechlorination, defluorination then hydroxylation |
| M6 |
430 |
412, 335, 161 |
44.2 |
N-Demethylation, O-demethylation, hydroxylation, and dechlorination |
|
| In vitro phase I metabolites |
| Sulphate conjugates |
| M7 |
526 |
446, 508 |
44.1 |
N-Demethylation and conjugation of sulphate group |
| Glucuronide conjugates |
| M8 |
622 |
430, 446, 605 |
26.7 |
O-Demethylation, and glucuronide conjugates |
| M9 |
648 |
472, 438, 630 |
29.1 |
Hydroxylation, α-oxidation, dechlorination then hydroxylation and, glucuronide conjugates |
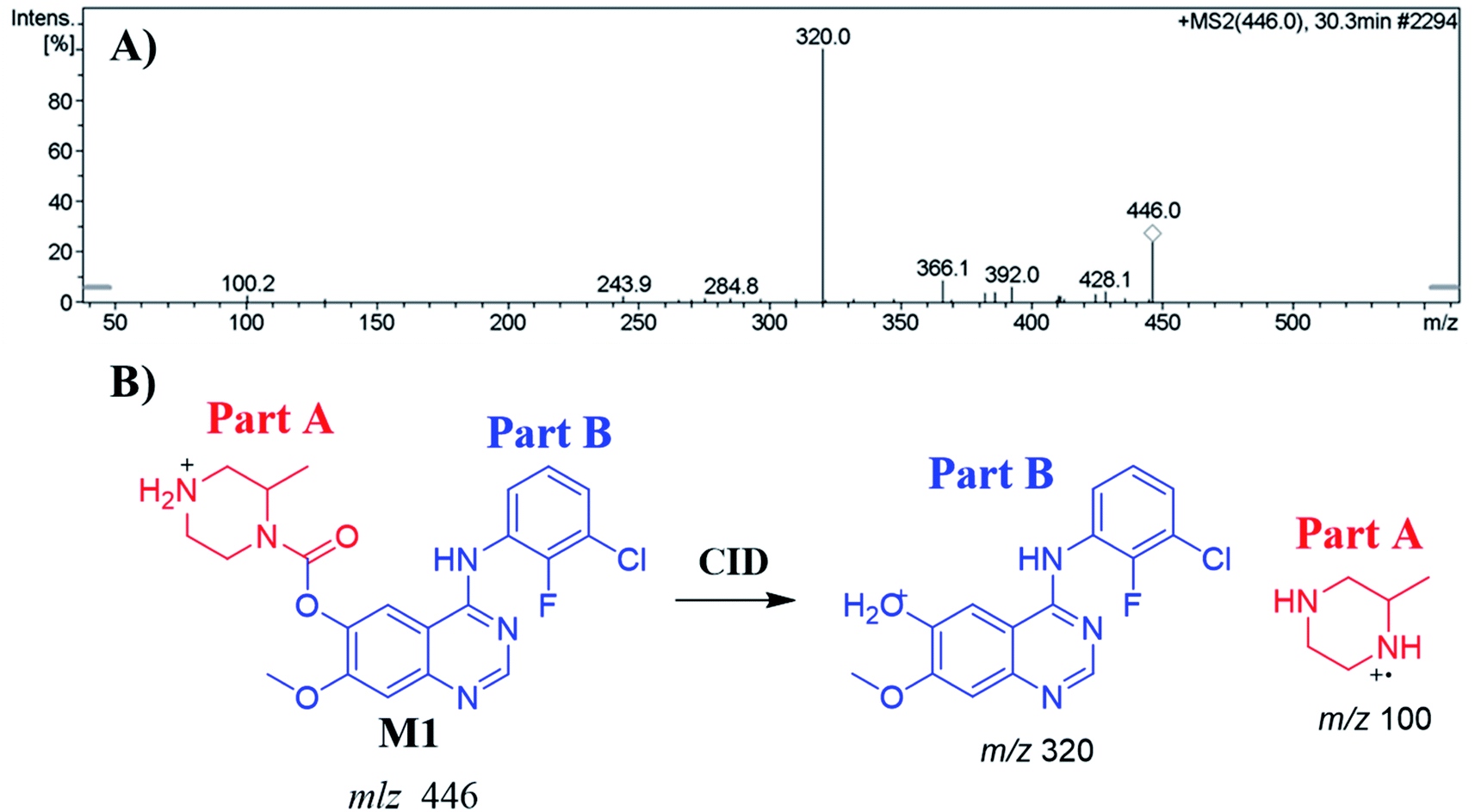
3.4.1. Identification of the M1 metabolite of ZFB. The M1 PIP appeared at 30.3 min in the TIC. Dissociation of PI at m/z 446 resulted in two characteristic fragment ions at m/z 320 and m/z 100 (Fig. 4A). Comparing to the dissociation of ZFB, fragment ion at m/z 320 indicates that no metabolic reaction in part B while part A shows a 14 m/z less that indicates the N-demethylation of piperazine ring (Fig. 4B). Radical cation (m/z 100) was formed in small intensity due to collision induced dissociation (CID) in LC-IT-MS.31
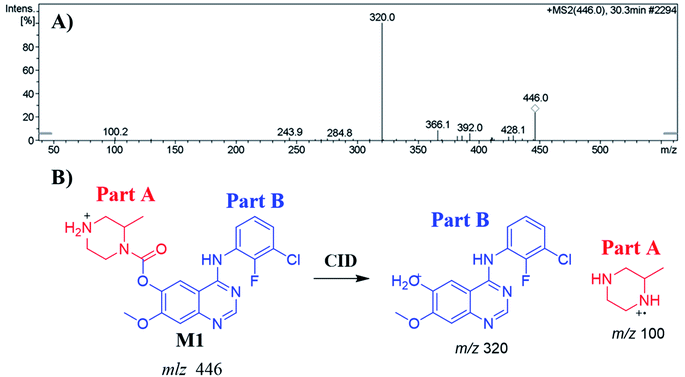 |
| | Fig. 4 MS2 mass spectrum of M1 (A). Proposed M1 structure and its corresponding fragment ions (B). | |
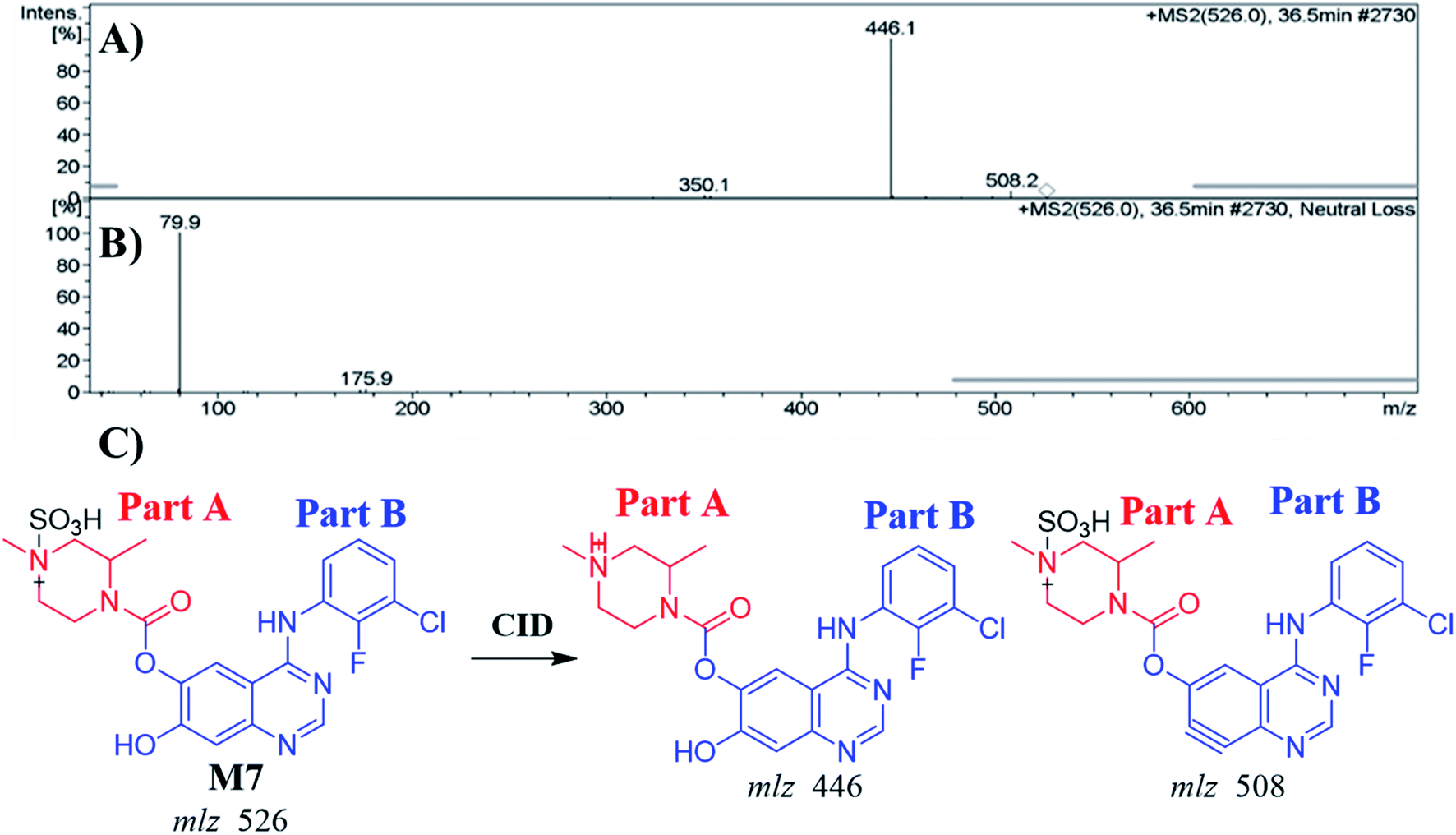
3.4.2. Identification of the M7 metabolite of ZFB. The M7 PIP appeared at 36.5 min (m/z 526) in the TIC. PIP exhibits 80 m/z higher than that of the M1 (precursor metabolite) with m/z 446 that was predicted to be sulfate conjugation of M1. Dissociation of PI at m/z 526 resulted in two characteristic fragment ions at m/z 508 and m/z 446 (Fig. 5A). Comparing to the dissociation of ZFB,32 fragment ion at m/z 446 indicates that the metabolic reaction in part B (14 m/z less) while part A shows no metabolic reaction that indicates the O-demethylation of methoxy group attached to quinazoline moiety. Fragment ion at m/z 508 (18 m/z less) that indicates water loss and confirm the O-demethylation metabolic reaction. M7 was proposed to be a direct conjugation of M1 with sulfate ion (Fig. 5B). Another LC-IT-MS screening for sulfate adduct was done by neutral loss scan of ions that lose 80 Da (Fig. 5C).33
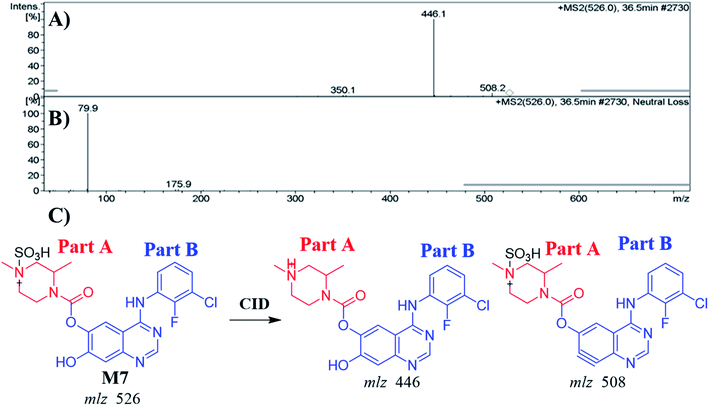 |
| | Fig. 5 MS2 mass spectrum of M7 (A). Constant neutral loss scan of M7 (B). Proposed M7 structure and its corresponding fragment ions (C). | |
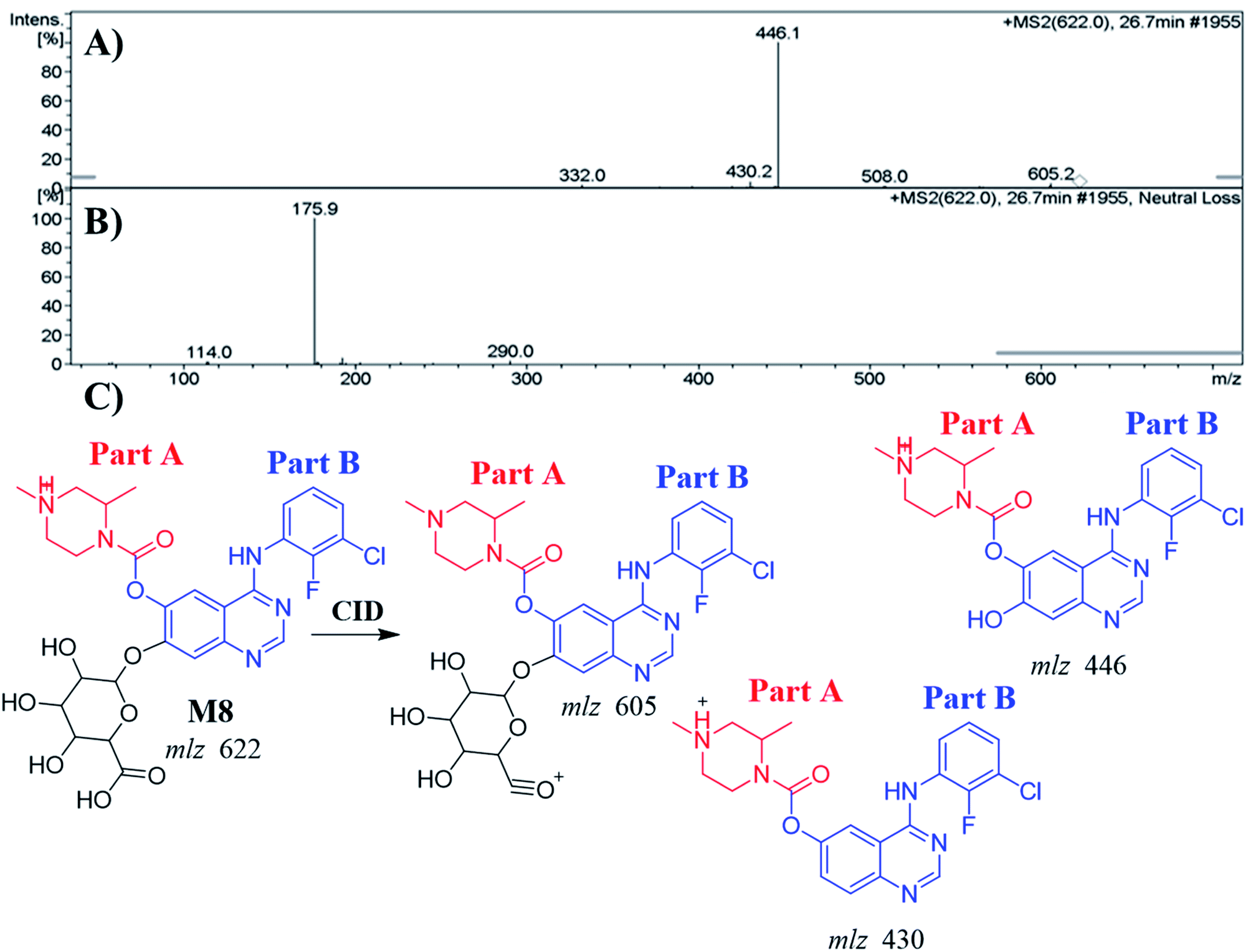
3.4.3. Identification of the M8 metabolite of ZFB. The M8 PPI appeared at 26.7 min in the TIC. Dissociation of PI at m/z 622 resulted in three characteristic fragment ions at m/z 605, m/z 446 and m/z 430 (Fig. 6A). It showed 176 m/z higher than that of PI with m/z 446 (M2), which proposed to be a glucuronic acid conjugate of ZFB metabolite. Fragment ion at m/z 446 that showed neutral loss of glucuronic acid (loss of 176 m/z units) that matched with the other fragment ions at m/z 430 and m/z 605 (Fig. 6B and C). Constant neutral loss scan for M8 metabolite conjugates are more specific (due to the characteristic loss of 176 mass units), and provide much cleaner mass spectral data.32
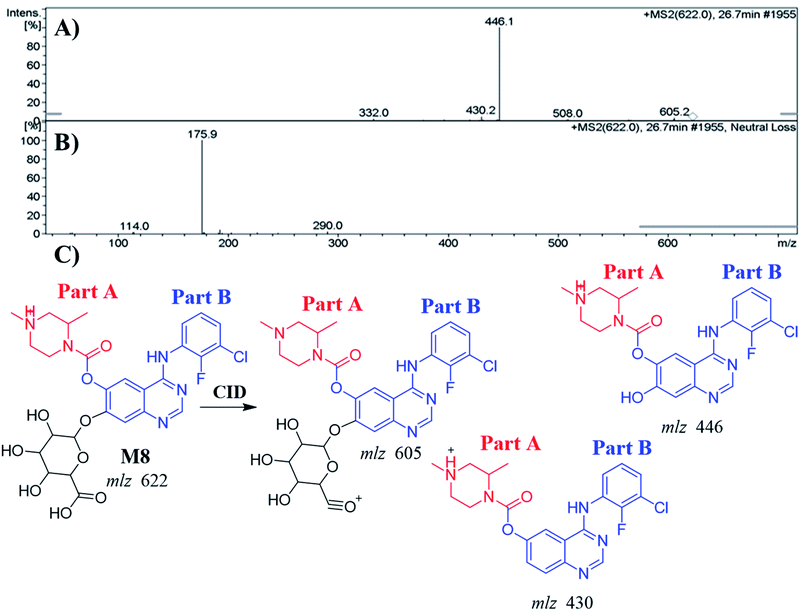 |
| | Fig. 6 MS2 mass spectrum of M8 (A). Constant neutral loss scan of M8 (B). Proposed M8 structure and its corresponding fragment ions (C). | |
3.5. Identification of ZFB in vivo metabolites
PIP mass spectra comparison between urine extracts with control urine samples as well as PIP comparison of ZFB proposed metabolites (Table 4) allowed the profiling of six in vivo phase I and three in vivo phase II metabolites. Metabolic pathways for in vivo phase I metabolites were supposed to be N-demethylation, hydroxylation, reduction, dechlorination and α oxidation, while for phase II metabolites were the result of two glucuronic acid conjugation and one sulphate (Table 4). M1 metabolites is previously mentioned in in vitro ZFB phase I metabolism. M7 and M9 metabolites are previously mentioned in in vivo ZFB phase II metabolism. In vivo phase I metabolites (M10, M11, M12, M13 and M14) figures and schemes have been moved to a ESI (Fig. S7–S10†).
Table 4 In vivo phase I and phase II metabolites of ZFB using MS scan and fragment ions
| |
MS scan |
Major fragment ions |
tR (min) |
Proposed metabolic reaction |
| In vivo phase I metabolites |
| M1 |
446 |
320, 285, 428, 100 |
30.3 |
N-Demethylation at piperazine ring |
| M10 |
476 |
336, 141 |
26.4 |
Hydroxylation |
| M11 |
476 |
320, 458 |
34.3 |
Hydroxylation |
| M12 |
476 |
320, 458 |
35.4 |
Hydroxylation |
| M13 |
462 |
320, 143, 443 |
37.1 |
Reduction |
| M14 |
488 |
320, 169 |
38.9 |
α Oxidation |
|
| In vivo phase II metabolites |
| Sulphate conjugate |
| M7 |
526 |
446, 508 |
36.5 |
N-Demethylation and conjugation of sulphate group |
| Glucuronide conjugates |
| M9 |
648 |
472, 438, 630 |
29.1 |
Hydroxylation, α oxidation, dechlorination then hydroxylation and, glucuronide conjugates |
| M15 |
638 |
462, 141 |
26.8 |
Hydroxylation, O-demethylation of benzene group and conjugation of glucuronic acid |
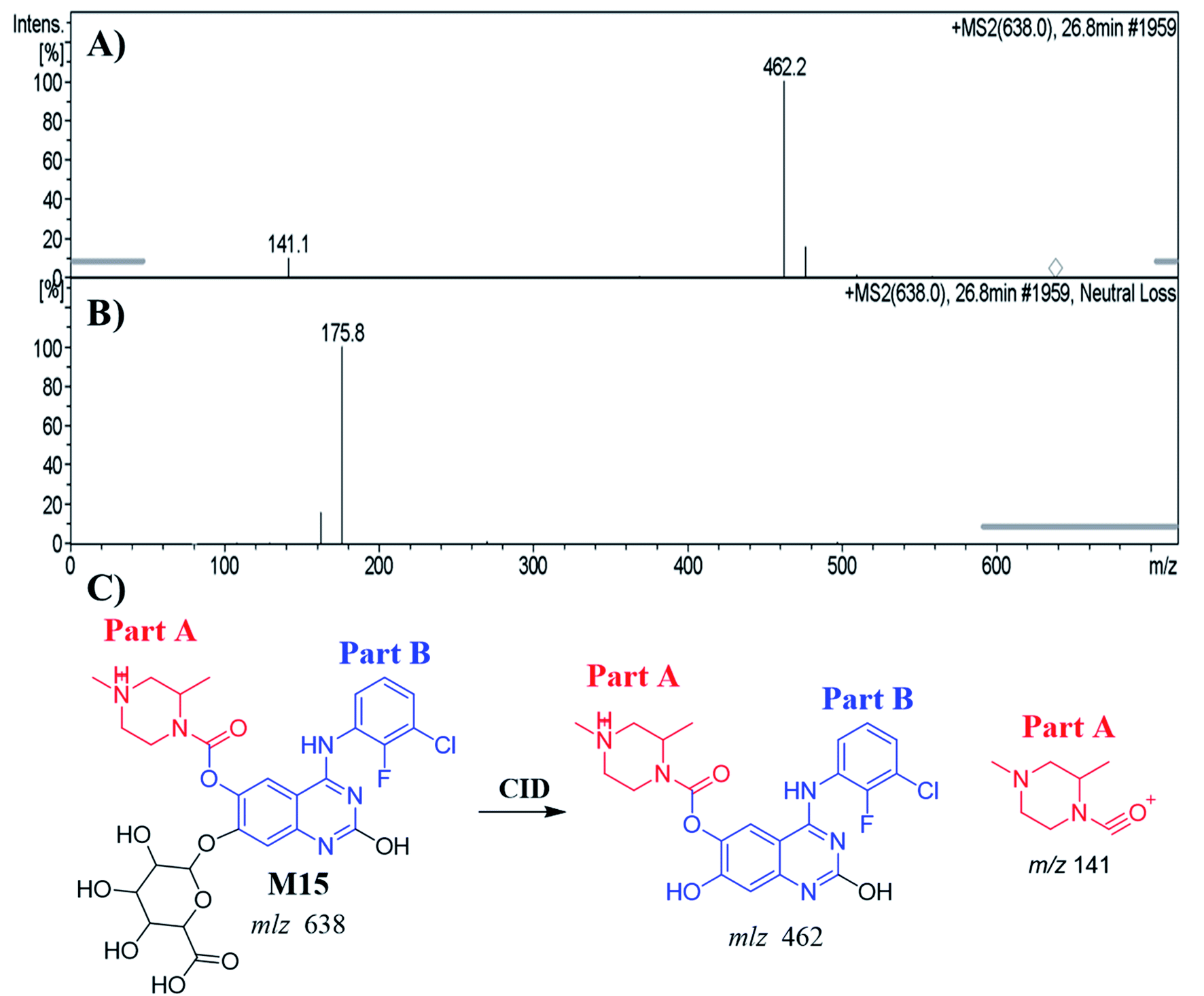
3.5.1. Identification of the M15 metabolite of ZFB. The M15 PIP appeared at 26.8 min in the TIC. Dissociation of PI at m/z 638 resulted in two characteristic fragment ions at m/z 462 and m/z 141 (Fig. 7A). Comparing to the dissociation of ZFB, fragment ion at m/z 141 indicates that no metabolic reaction in part A. Fragment ion at 462 indicates the neutral loss of glucuronic acid (loss of 176 m/z) that indicates O-glucuronidation after O-demethylation of methoxy group. The proposed metabolic reactions were hydroxylation reaction at quinazoline moiety, O-demethylation and O-glucuronidation (Fig. 7B and C). Glucuronidation was confirmed by running neutral loss scan mode that shows the precursor ion at m/z 638.32
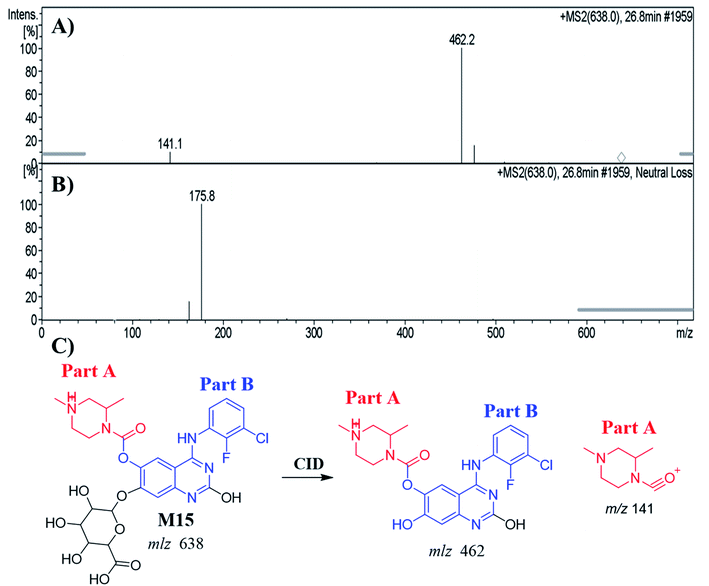 |
| | Fig. 7 MS2 mass spectrum of M15 (A). Constant neutral loss scan of M15 (B). Proposed M15 structure and its corresponding fragment ions (C). | |
3.6. Identification of in vitro reactive metabolites of ZFB
The same metabolic incubation (ZFB with RLMs) was done in the presence of 1.0 mM KCN and 1.0 mM GSH to trap iminium and electro deficient conjugated system intermediates, respectively. Four GSH conjugates and three cyanide adducts were identified (Table 5), revealing that the N-phenyl amine group and methyl piperazine ring in ZFB can become bioactivated and then trapped by the nucleophile GSH and cyanide ion.34 ZFB cyano adducts (AZDCN515, AZDCN501) and ZFB GSH conjugates (AZDGSH761, AZDGSH763, AZDGSH717) figures and schemes have been moved to a ESI (Fig. S11–S15†).
Table 5 Reactive metabolites of ZFB
| |
MS scan |
Most abundant fragment ions |
Rt. (min) |
Metabolic reaction |
| Cyanide adducts |
| AZDCN485 |
485 |
148, 458 |
46.6 |
Attack of KCN at bioactivated piperazine ring of parent drug |
| AZDCN515 |
515 |
371, 488, 197 |
43.2 |
Hydroxylation, α-oxidation and attack of KCN at bioactivated piperazine ring |
| AZDCN501 |
501 |
294, 327, 474 |
45.5 |
Hydroxylation at piperazine ring and attack of KCN at bioactivated piperazine ring |
|
| GSH conjugates |
| AZDGSH715 |
715 |
586, 398 |
42.4 |
N-Demethylation, dechlorination, defluorination then hydroxylation and GSH conjugate at benzene ring |
| AZDGSH761 |
761 |
632, 731 |
43.1 |
Double hydroxylation at piperazine ring, dechlorination, defluorination then hydroxylation and conjugation of GSH at benzene ring |
| AZDGSH763 |
763 |
634, 458 |
36.4 |
O-Demethylation, double hydroxylation at piperazine ring, dechlorination, defluorination then hydroxylation and conjugation of GSH at benzene ring |
| AZDGSH717 |
717 |
588, 307 |
31 |
N-Demethylation, reduction, dechlorination, defluorination then hydroxylation and conjugation of GSH at benzene ring |
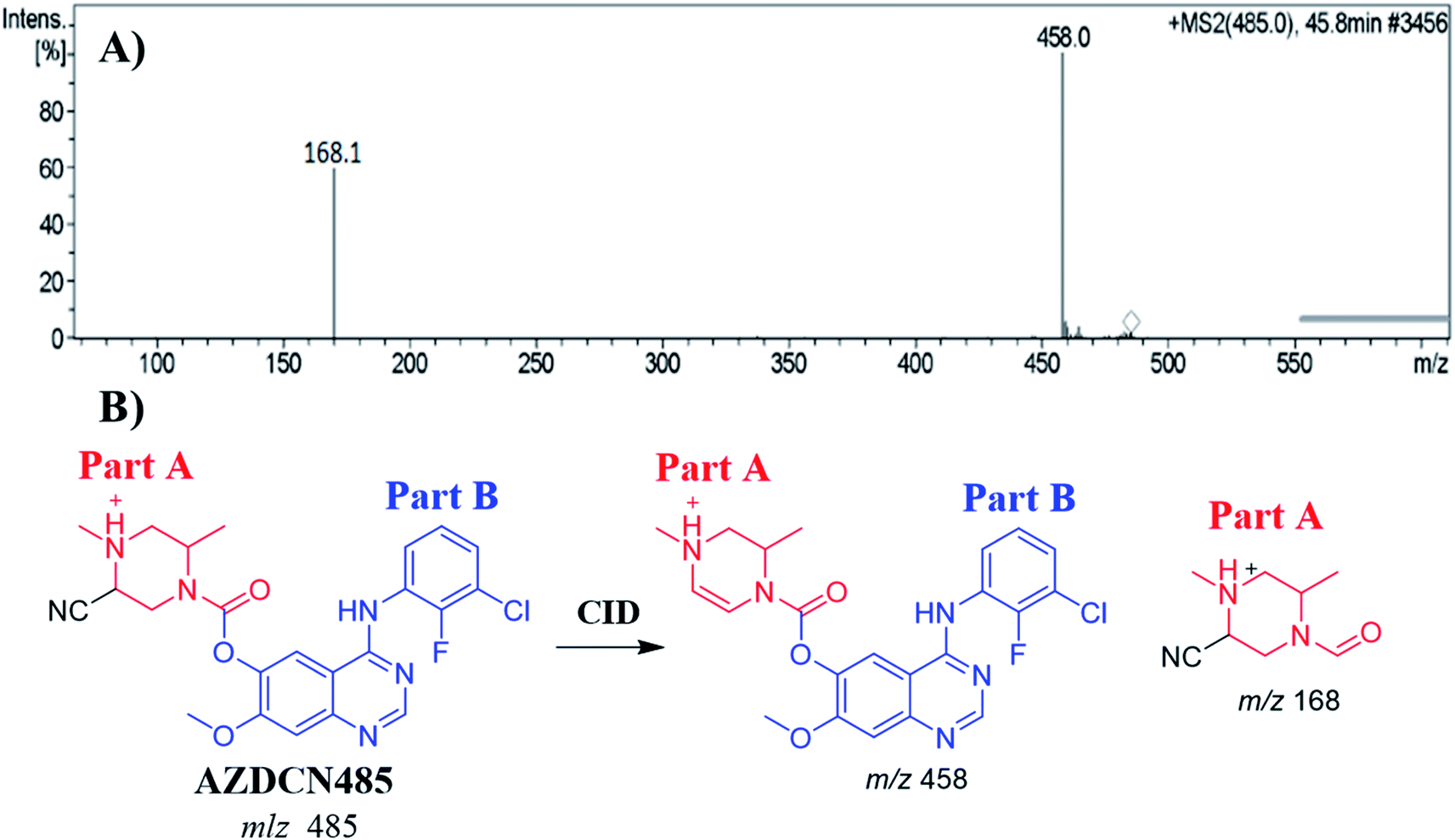
3.6.1. Identification of AZDCN485 cyano adduct. The AZDCN485 PIP appeared at 46.6 min in the total ion chromatogram (TIC). Dissociation of PI at m/z 485 resulted in two characteristic fragment ions at m/z 458 and m/z 168 (Fig. 8A). Fragment ion at m/z 458 (27 m/z less) that indicates HCN loss and confirms the cyano adduct formation. Comparing to the dissociation of ZFB, fragment ion at m/z 168 indicates that cyano nucleophile trapped the bioactivated piperazine ring (part A) forming cyano adduct (Fig. 8B).
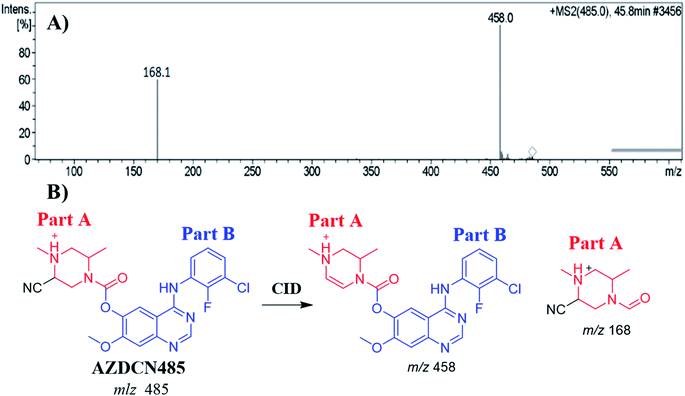 |
| | Fig. 8 MS2 mass spectrum of AZDCN485 (A). Proposed AZDCN485 structure and its corresponding fragment ions (B). | |
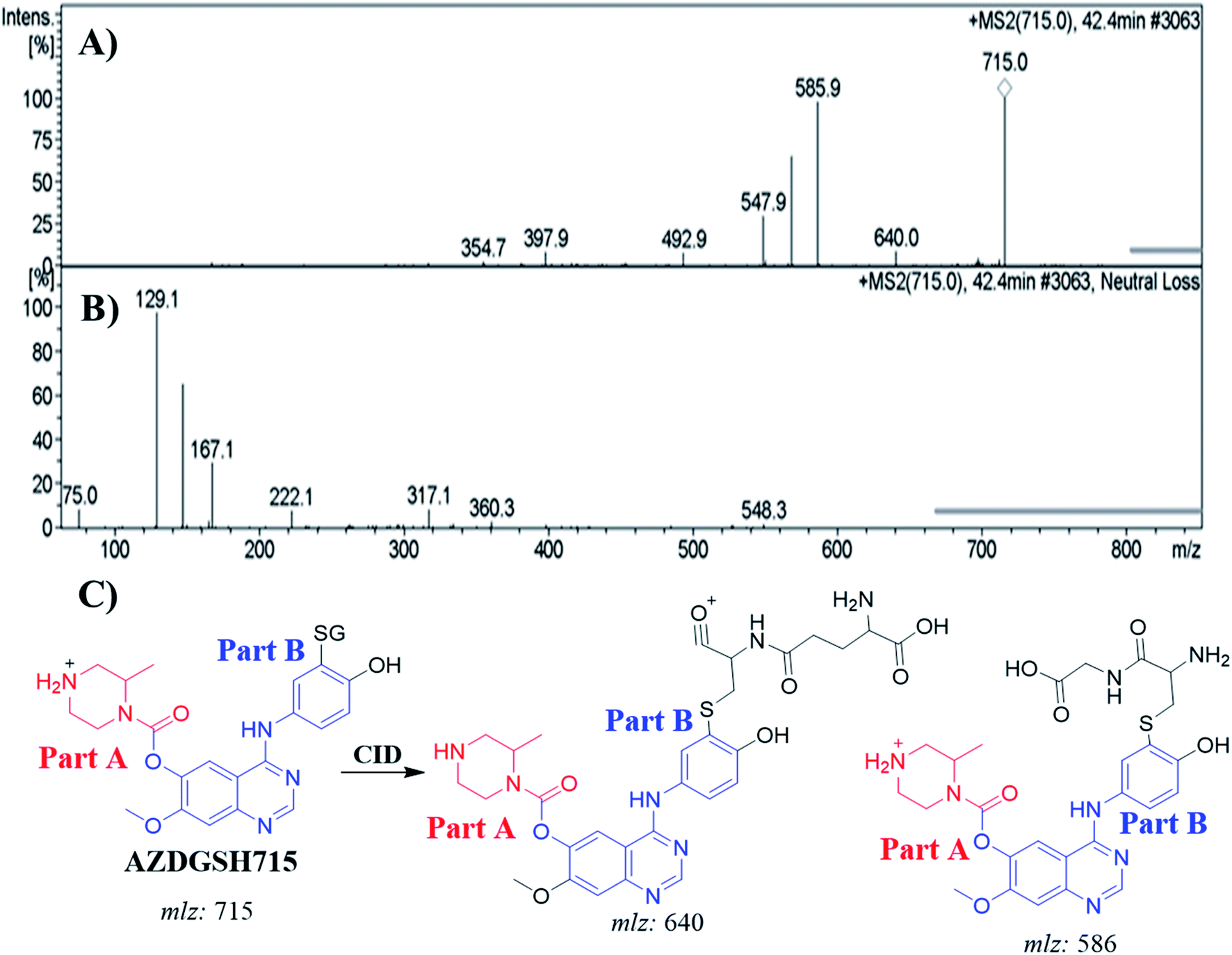
3.6.2. Identification of AZDGSH715 GSH conjugates. The AZDGSH715 PIP appeared at 42.4 min in the TIC. Dissociation of PI at m/z 715 resulted in two characteristic fragment ions at m/z 640 and m/z 586 (Fig. 9A). Fragment ion at m/z 586 (129 m/z less) that indicates anhydroglutamic acid moiety and confirms the formation of GSH conjugate. Constant neutral loss scan for screening for GSH adduct was done by monitoring of ions that lose 129 Da (Fig. 9B).32 AZDGSH715 formation indicated the phenyl ring bioactivation in in vitro metabolism of ZFB. The metabolic reactions that occurred in AZDGSH715 were proposed dechlorination, N-demethylation, defluorination then hydroxylation and GSH conjugation at bioactivated phenyl ring (iminoquinone derivative) (Fig. 9C).
 |
| | Fig. 9 MS2 mass spectrum of AZDGSH715 (A). Constant neutral loss scan of AZDGSH715 (B). Proposed AZDGSH715 structure and its corresponding fragment ions (C). | |
3.7. Proposed pathways of bioactivation of ZFB
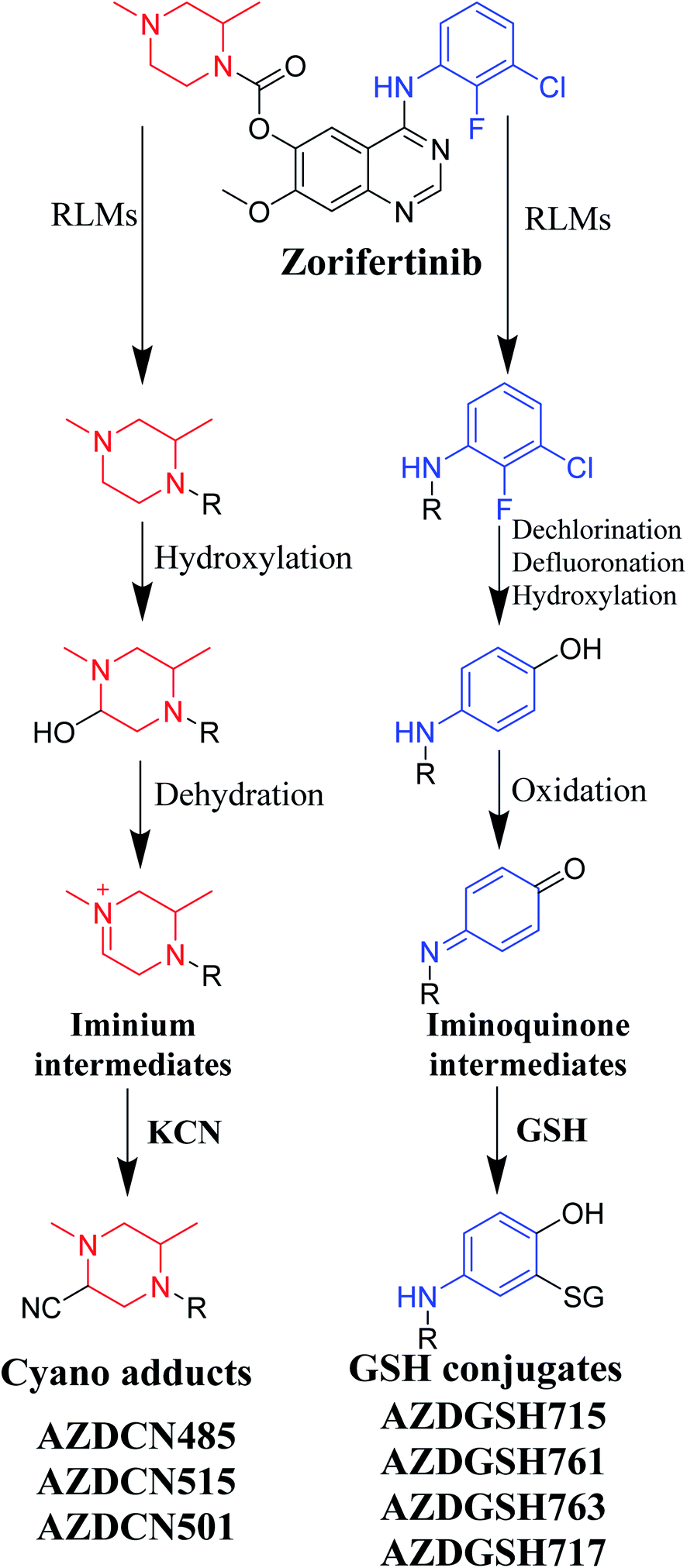
Bioactivation pathways of ZFB are proposed in Fig. 10. AZDCN485, AZDCN515 and AZDCN501 cyanide adducts revealed the metabolic formation of reactive iminium intermediates at N-methyl piperazine ring in ZFB metabolism. The bioactivation pathway was predicted as hydroxylation metabolic reaction at N-methyl piperazine ring in ZFB followed by loss of H2O (dehydration) that resulted in iminium ions intermediates (reactive hard nucleophile) that were trapped using cyanide electrophile (KCN) forming stable cyanide adducts that were characterized and identified using LC-IT-MS. This bioactivation pathway that involves iminium intermediates formation is previously reported with other drugs containing cyclic tertiary amine ring.35,36 The formation of AZDGSH715, AZDGSH761, AZDGSH763 and AZDGSH717 supposed the formation of iminoquinone reactive intermediates in the metabolism of ZFB that were formed by dechlorination, defluorination and hydroxylation of phenyl group and then oxidation34 generating reactive intermediates that were trapped using GSH leading to the formation of stable conjugates that were characterized and identified using LC-IT-MS (Fig. 10).37
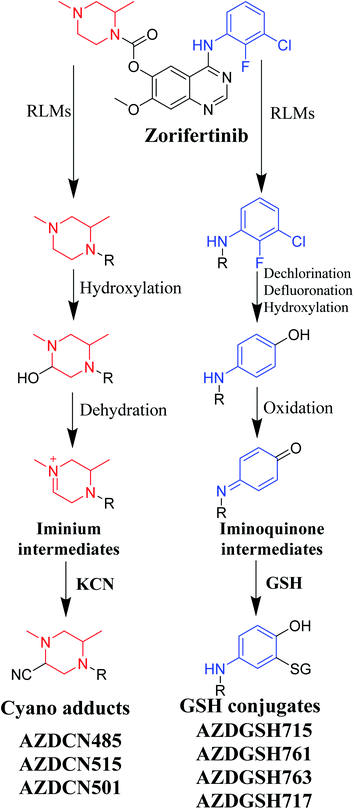 |
| | Fig. 10 Proposed pathways for zorifertinib bioactivation for cyano adducts and GSH conjugates. | |
4. Conclusions
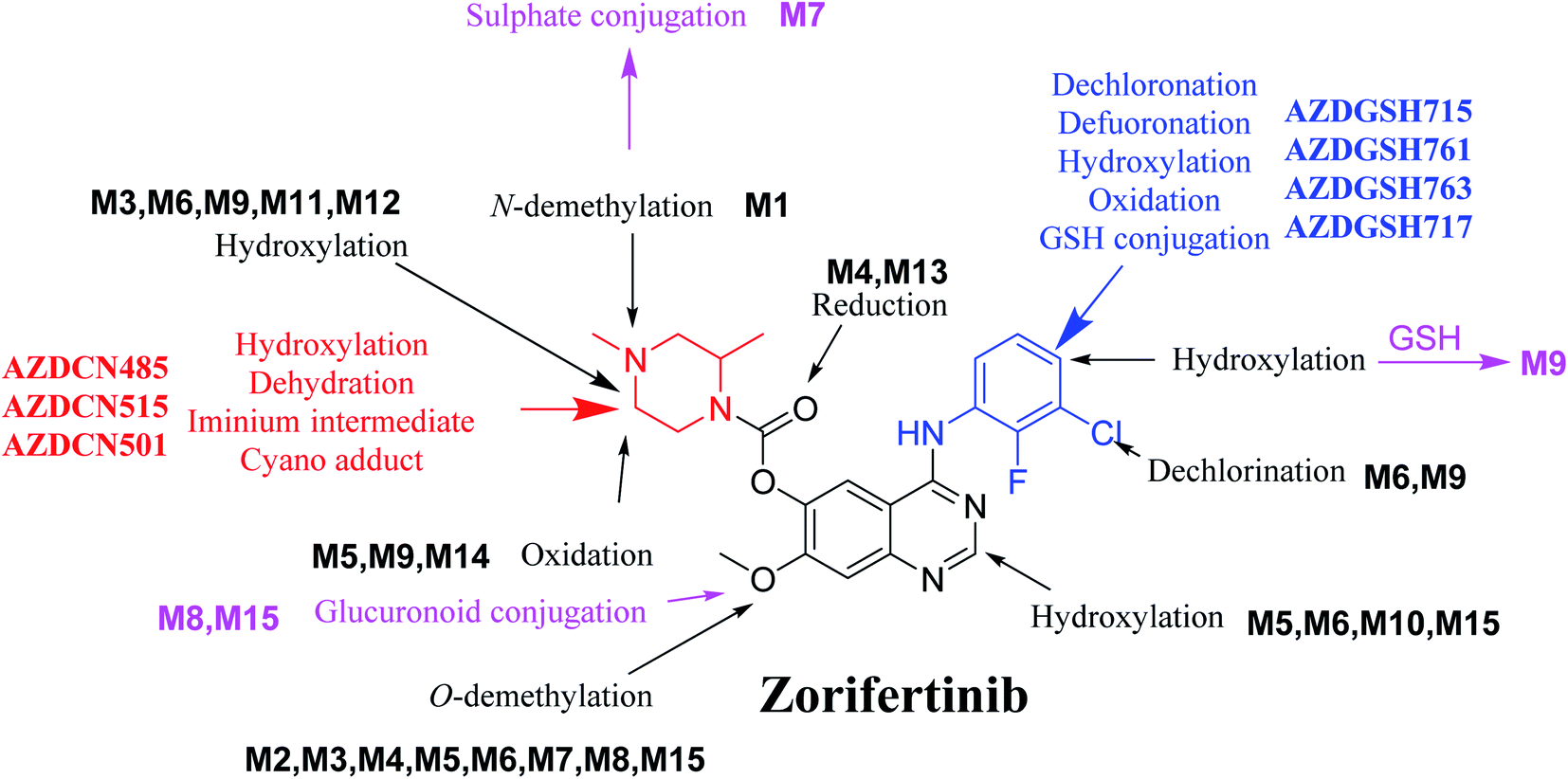
In silico software (WhichP450™ module, DEREK and XenoSite reactivity model) has proposed the most labile atoms in the ZFB structure that revealed to be the same as seen in the detected metabolites in phase I metabolism and the same groups were bioactivated as expected. Three in vitro phase II metabolites (sulphate and glucuronic acid conjugates) and six in vitro phase I (N-demethylation, O-demethylation, hydroxylation, reduction, and oxidation) and were found for ZFB. Three in vivo phase II metabolites (one sulphate and two glucuronic acid conjugates) and six in vivo phase I metabolites (N-demethylation, α-oxidation, hydroxylation, reduction, and dechlorination) of ZFB were identified (Fig. 11). Seven reactive intermediates (four GSH conjugates and three cyano adducts) were characterized and the mechanisms of their formation were proposed (Fig. 11) that may illuminate the way for figuring out the causes behind ZFB toxic side effects.
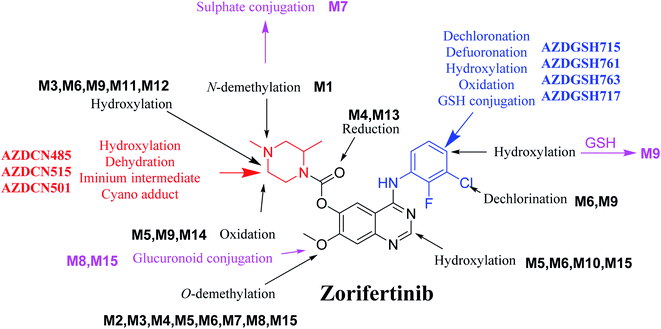 |
| | Fig. 11 Chemical structure of zorifertinib showing bioactivation pathways including iminium, and electro deficient conjugated system. | |
Ethics approval
Ethical approval for the Animal experiments of were obtained from the Animal Ethics Committee of King Saud University (No. KSU-SE-19-52).
Author contributions
Nasser S. Al-Shakliah: conceptualization, methodology, visualization, software. Mohamed Attwa: data curation, writing – original draft preparation. Adnan Kadi: supervision, investigation, software, validation. Haitham AlRabiah: supervision, writing – reviewing and editing. Haya Aljohar: writing – reviewing and editing.
Conflicts of interest
The authors declare no conflicts of interest for the current work.
Abbreviations
| LC-IT-MS | Liquid chromatography ion trap mass spectrometry |
| ACN | Acetonitrile |
| ESI | Electrospray ionization |
| RLMs | Rat liver microsomes |
| TKIs | Tyrosine kinase inhibitors |
| PIP | Precursor ion peak |
| TIC | Total ion chromatogram |
| CID | Collision-induced dissociation |
Acknowledgements
The authors are grateful to King Saud University, Riyadh, Saudi Arabia for funding the work through the Researcher Supporting Project (No. RSP2022R501).
References
- J. R. Molina, P. Yang, S. D. Cassivi, S. E. Schild and A. A. Adjei, Mayo Clin. Proc., 2008, 83, 584–594 CrossRef PubMed.
- E. Papadopoulou, N. Tsoulos, A. Tsirigoti, A. Apessos, K. Agiannitopoulos, V. Metaxa-Mariatou, K. Zarogoulidis, P. Zarogoulidis, D. Kasarakis, S. Kakolyris, J. Dahabreh, F. Vlastos, C. Zoublios, A. Rapti, N. G. Papageorgiou, D. Veldekis, M. Gaga, G. Aravantinos, V. Karavasilis, N. Karagiannidis and G. Nasioulas, Oncol. Lett., 2015, 10, 2176–2184 CrossRef CAS PubMed.
- A. Milovancev, V. Stojsic, B. Zaric, T. Kovacevic, T. Sarcev, B. Perin, K. Zarogoulidis, K. Tsirgogianni, L. Freitag and K. Darwiche, OncoTargets Ther., 2015, 8, 2915 CAS.
- S. Lampaki, G. Lazaridis, K. Zarogoulidis, I. Kioumis, A. Papaiwannou, K. Tsirgogianni, A. Karavergou, T. Tsiouda, V. Karavasilis and L. Yarmus, J. Cancer, 2015, 6, 568 CrossRef PubMed.
- R. J. Bodnar, Adv. Wound Care, 2013, 2, 24–29 CrossRef PubMed.
- L. Tanoue, Year Book of Pulmonary Disease, 2009, vol. 2009, pp. 177–179 Search PubMed.
- P. M. Harari, G. W. Allen and J. A. Bonner, J. Clin. Oncol., 2007, 25, 4057–4065 CrossRef CAS PubMed.
- Z. Yang, Q. Guo, Y. Wang, K. Chen, L. Zhang, Z. Cheng, Y. Xu, X. Yin, Y. Bai and S. Rabbie, Sci. Transl. Med., 2016, 8, 368ra172 Search PubMed.
- Q. Zeng, J. Wang, Z. Cheng, K. Chen, P. Johnstrom, K. Varnas, D. Y. Li, Z. F. Yang and X. Zhang, J. Med. Chem., 2015, 58, 8200–8215 CrossRef CAS PubMed.
- N. Li, Z. Bian, M. Cong and Y. Liu, Front. Oncol., 2021, 11, 723562 CrossRef PubMed.
- M.-J. Ahn, D.-W. Kim, B. C. Cho, S.-W. Kim, J. S. Lee, J.-S. Ahn, T. M. Kim, C.-C. Lin, H. R. Kim and T. John, Lancet Respir. Med., 2017, 5, 891–902 CrossRef CAS.
- S. Ma and M. Zhu, Chem.-Biol. Interact., 2009, 179, 25–37 CrossRef CAS PubMed.
- A. F. Stepan, D. P. Walker, J. Bauman, D. A. Price, T. A. Baillie, A. S. Kalgutkar and M. D. Aleo, Chem. Res. Toxicol., 2011, 24, 1345–1410 Search PubMed.
- A. Tolonen, M. Turpeinen and O. Pelkonen, Drug Discovery Today, 2009, 14, 120–133 CrossRef CAS PubMed.
- G. Evans, A handbook of bioanalysis and drug metabolism, CRC press, 2004 Search PubMed.
- M. W. Attwa, N. S. Al-Shakliah, H. AlRabiah, A. A. Kadi and A. S. Abdelhameed, J. Pharm. Biomed. Anal., 2022, 211, 114626 CrossRef CAS PubMed.
- F. Li, J. Lu and X. Ma, Chem. Res. Toxicol., 2011, 24, 744–751 Search PubMed.
- J. Vrbanac and R. Slauter, in A Comprehensive Guide to Toxicology in Preclinical Drug Development, ed. A. S. Faqi, Academic Press, 2013, pp. 3–30, DOI:10.1016/B978-0-12-387815-1.00002-2.
- A. A. Kadi, M. W. Attwa and H. W. Darwish, RSC Adv., 2018, 8, 1182–1190 RSC.
- P. A. Hunt, M. D. Segall and J. D. Tyzack, J. Comput.-Aided Mol. Des., 2018, 32, 537–546 CrossRef CAS PubMed.
- J. D. Tyzack and J. Kirchmair, Chem. Biol. Drug Des., 2019, 93, 377–386 CrossRef CAS PubMed.
- M. W. Attwa, A. A. Kadi and A. S. Abdelhameed, RSC Adv., 2020, 10, 5412–5427 RSC.
- T. B. Hughes, G. P. Miller and S. J. Swamidass, Chem. Res. Toxicol., 2015, 28, 797–809 Search PubMed.
- T. B. Hughes, N. L. Dang, G. P. Miller and S. J. Swamidass, ACS Cent. Sci., 2016, 2, 529–537 Search PubMed.
- M. W. Attwa, A. A. Kadi, A. S. Abdelhameed and H. A. Alhazmi, Drug Des., Dev. Ther., 2020, 14, 783–793 Search PubMed.
- M. W. Attwa, A. A. Kadi, H. W. Darwish, S. M. Amer and N. S. Al-Shakliah, Chem. Cent. J., 2018, 12, 99 Search PubMed.
- A. A. Kadi, S. M. Amer, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7, 36279–36287 RSC.
- D. R. Mudra and A. Parkinson, Curr. Protoc. Toxicol., 2001, ch. 14, Unit 14.12 Search PubMed.
- Q. Wu, A. Hua, Y. Sun, C. Ma, W. Tian, C. Huang, H. Yu, P. Jiao, S. Wang and H. Tong, Thorac. Cancer, 2018, 9, 1383–1389 CrossRef CAS PubMed.
- D. P. Demarque, A. E. Crotti, R. Vessecchi, J. L. Lopes and N. P. Lopes, Nat. Prod. Rep., 2016, 33, 432–455 RSC.
- M. Schäfer, M. Drayß, A. Springer, P. Zacharias and K. Meerholz, Eur. J. Org. Chem., 2007, 2007, 5162–5174 CrossRef.
- K. Levsen, H.-M. Schiebel, B. Behnke, R. Dötzer, W. Dreher, M. Elend and H. Thiele, J. Chromatogr. A, 2005, 1067, 55–72 CrossRef CAS PubMed.
- A. Lafaye, C. Junot, B. Ramounet Le Gall, P. Fritsch, E. Ezan and J. C. Tabet, J. Mass Spectrom., 2004, 39, 655–664 CrossRef CAS PubMed.
- M. W. Attwa, A. A. Kadi and A. S. Abdelhameed, J. Pharm. Biomed. Anal., 2019, 164, 659–667 CrossRef CAS PubMed.
- A. A. Kadi, H. W. Darwish, M. W. Attwa and S. M. Amer, RSC Adv., 2016, 6, 72575–72585 RSC.
- S. M. Amer, A. A. Kadi, H. W. Darwish and M. W. Attwa, Chem. Cent. J., 2017, 11, 1–7 CrossRef PubMed.
- M. W. Attwa, A. A. Kadi, H. Alrabiah and H. W. Darwish, J. Pharm. Biomed. Anal., 2018, 160, 19–30 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Haya I. Aljohar
,
Haya I. Aljohar