
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of chiral 1,2-oxazinane and hexahydropyridazin spirocyclic scaffolds by organocatalytic [4 + 2] cycloaddition†
Heng-Zhi Tiana,
Qing-Gang Tanga,
Guo-Qiang Linab and
Xing-Wen Sun *ab
*ab
aDepartment of Chemistry, Fudan University, Shanghai 200433, China. E-mail: sunxingwen@fudan.edu.cn
bKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 24th May 2022
Abstract
A novel approach to synthesize chiral 1,2-oxazinane spirocyclic scaffolds by organocatalytic [4 + 2] cycloaddition reaction between methyleneindolinones and γ-aminooxy-α,β-unsaturated ester has been disclosed. Furthermore, a hydrazide 1,4-synthon is designed and synthesized to construct chiral hexahydropyridazin spirocyclic scaffolds with methyleneindolinones via [4 + 2] cycloaddition reaction. Both reactions give corresponding products in good to excellent yield, excellent diastereoselectivity and good enantioselectivity.
Spiro-oxindole scaffolds bearing a quaternary carbon stereocenter at the 3-position, especially with a heterocycle, are privileged and wide-spread among natural products and medicinal compounds.1 Among the vast array of spiro-oxindole scaffolds, these structures which contain two heteroatoms at the spiro ring are of great importance due to their remarkable bioactivites (Fig. 1). For example, compound I and compound II possess antineoplastic activities.2 Compound III and compound IV are used as a CRTH2 antagonist or p53/Hdm2 interaction inhibitor, respectively.3 And the spiro-oxindoles fused with the ring containing two heteroatoms at the C3-position, have recently become one of the most important synthetic subjects because of significance for drug design.4 In addition, 1,2-oxazinane5 and hexahydropyridazin6 constitute two other groups of bioactive compounds owing to the presence of two heteroatoms (Fig. 1). FR900482 (V) and FK317 (VI) are antibiotics with exceptionally potent antitumor activity. Piperazimycin A1 (VII) is a kind of cytotoxic hexadepsipeptides. 1-Azafagomine (VIII) is considered as an inhibitor of glycosyl-cleaving enzyme which offers the opportunity to treat AIDS, diabetes, and tumor metastasis. Given the relevance of spiro-oxindole scaffolds and 1,2-oxazinane or hexahydropyridazin, it is anticipated that incorporation of both pharmaceutical and biological structural motifs into one molecule might lead to the creation of new compounds with potentially interesting properties. Therefore, it is significant for drug design to introduce 1,2-oxazinane and hexahydropyridazin into spiro-oxindole scaffolds at the C3-position.
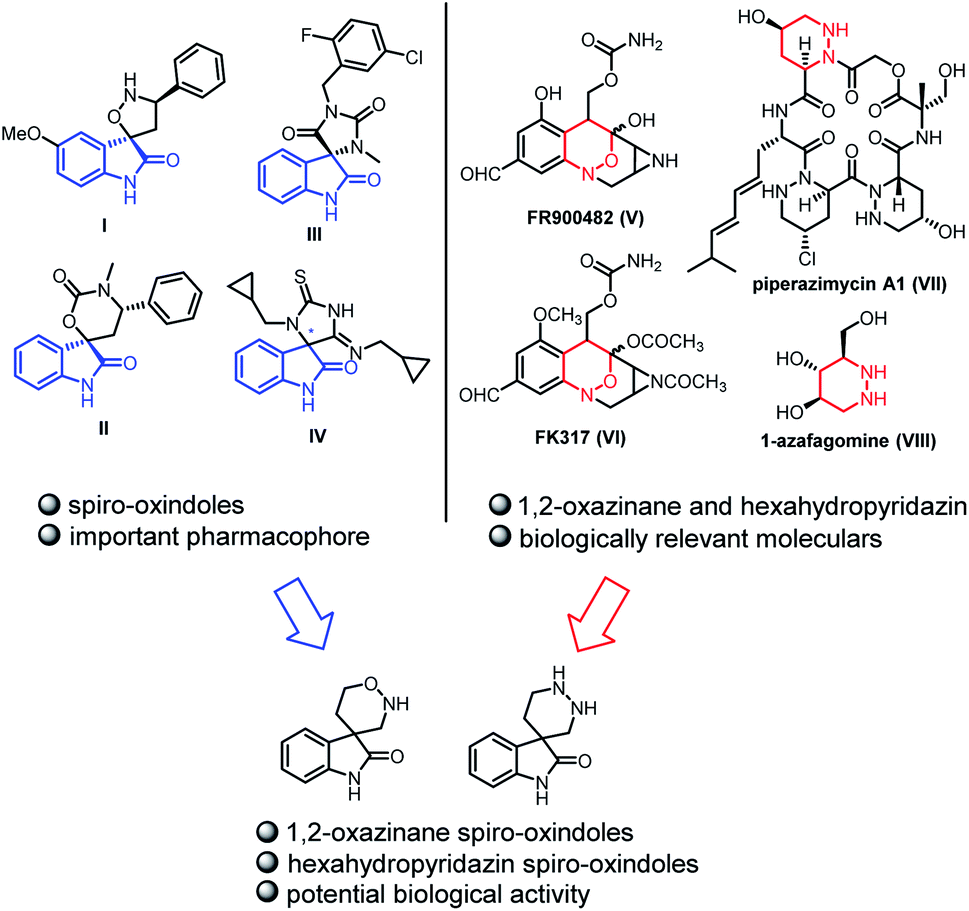 | ||
| Fig. 1 Examples of natural compounds including 1,2-oxazinane, hexahydropyridazin and spiro-oxindole skeletons. | ||
Although, a few accesses chiral 1,2-oxazinane spiro-oxindoles are disclosed recently,7 metal-free methods to construct this structure are still rare. As for chiral hexahydropyridazin spiro-oxindoles, some methods for the construction of other chiral pyridazin skeletons have been reported before. Gong8 has found chiral gold(I) complex-catalyzed can highly regio- and enantioselectively catalyze azo hetero-Diels–Alder reaction between 2,4-hexadiene and urea-based diazene dienophiles to synthesize chiral hexahydropyridazin. Wang9 has utilized α-chloro N-benzoyl hydrazine to form several kinds of chiral parallel hexahydropyridazin with Cu complex as the catalysts. The asymmetric synthesis of spiro-hexahydropyridazin has not been reported until 2021. Deng10 and co-workers design the asymmetric annulation between isatin N,N′-cyclic azomethine imine and cinnamaldehyde in the presence of secondary amine catalyst to construct chiral spiro-hexahydropyridazin. Even so, there is no effective method to form that structure we exhibit in Fig. 1. Developing novel and metal-free synthetic methods to construct chiral 1,2-oxazinane spiro-oxindoles and hexahydropyridazin spiro-oxindoles, are still in demand as ever.
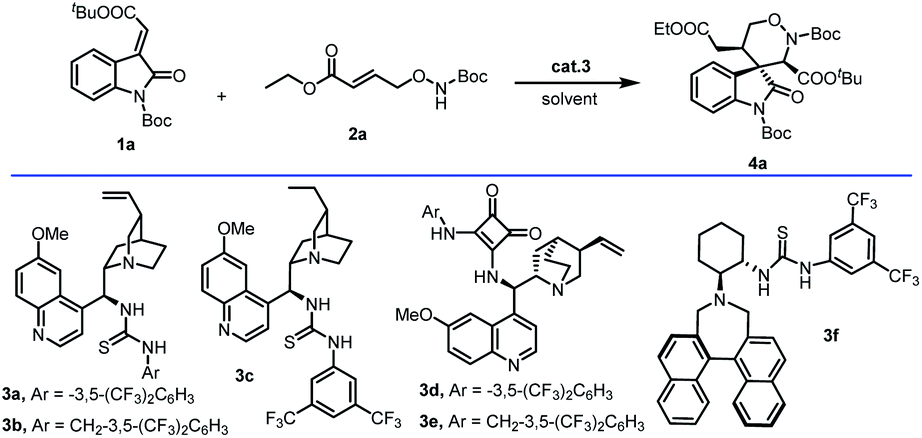
Our studies were initiated with the reaction between methyleneindolinones (1a) and γ-aminooxy-α,β-unsaturated ester (2a) in DCM with 5 mol% organocatalyst 3a. The desired 1,2-oxazinane spiro-oxindole 4a (derivative compound 8, CCDC 2153695) was generated with 80% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 95% ee. Optimization studies were performed using methyleneindolinones (1a) and γ-aminooxy-α,β-unsaturated ester (2a) as model substrates (Table 1). Preliminary catalyst screening (Table 1, entries 2–6) showed that the desired reaction profile could be accessed when squaramide or thiourea as the hinge of catalyst 3, and catalyst 3c gave the best result (87% yield, dr > 20:1, 96% ee). But in the same condition, when 3f with larger steric hindrance was used, this process hardly worked (yield < 20%). Subsequent solvent screening (Table 1, entries 7–10) enabled us to identify DCM as the most suitable solvent for the devised reaction.
:1 dr and 95% ee. Optimization studies were performed using methyleneindolinones (1a) and γ-aminooxy-α,β-unsaturated ester (2a) as model substrates (Table 1). Preliminary catalyst screening (Table 1, entries 2–6) showed that the desired reaction profile could be accessed when squaramide or thiourea as the hinge of catalyst 3, and catalyst 3c gave the best result (87% yield, dr > 20:1, 96% ee). But in the same condition, when 3f with larger steric hindrance was used, this process hardly worked (yield < 20%). Subsequent solvent screening (Table 1, entries 7–10) enabled us to identify DCM as the most suitable solvent for the devised reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | drb | Yieldb | cee |
| a Typical reaction conditions: 1a (0.11 mmol), 2a (0.10 mmol), 5 mol% catalyst 3 in DCM (0.2 mL), at 25 °C for 48 h.b Determined by 1H NMR analysis of the crude reaction mixture before purification.c Determined by HPLC analysis. | |||||
| 1 | 3a | DCM | >20:1 |
80 | 95 |
| 2 | 3b | DCM | >20:1 |
83 | 94 |
| 3 | 3c | DCM | >20:1 |
87 | 96 |
| 4 | 3d | DCM | >20:1 |
53 | 97 |
| 5 | 3e | DCM | >20:1 |
85 | 94 |
| 6 | 3f | DCM | nd | <20 | nd |
| 7 | 3c | CHCl3 | >20:1 |
90 | 95 |
| 8 | 3c | THF | >20:1 |
53 | 94 |
| 9 | 3c | Toluene | >20:1 |
92 | 94 |
| 10 | 3c | DCE | >20:1 |
90 | 93 |
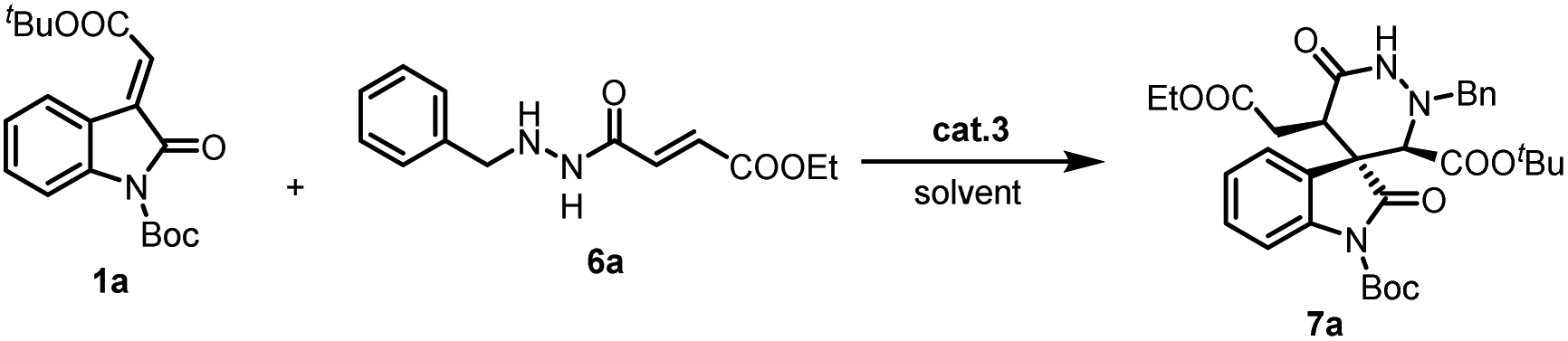
With the optimized reaction conditions in hand, the scope of the methodology was investigated (Scheme 1). Initially, different methyleneindolinones 1 were reacted with γ-aminooxy-α,β-unsaturated ester 2a. It was found that the protective group on the methyleneindolinones R1 was crucial. It influenced this reaction in electron withdrawing, catalyst binding and steric hindrance. When R1 was Ac or Et, the reaction did not work. But similar Cbz group was compatible. Importantly, the introduction of functional groups in the phenyl ring in 1 also proved to be possible. The influence of the electron properties and position of substituents on the C-5 of the methyleneindolinones 1 was investigated. The use of methyleneindolinones bearing halogen atoms, Me, MeO or nitro afforded corresponding products 4 in good yield and stereoselectivity. Similarly, on the C-6 and C-7 position of the methyleneindolinones 1, the introduction of halogen atoms was also compatible. Subsequently, the ester group on methyleneindolinones 1 (R3) was studied. It was found that different ester groups had no major influence on the reaction outcome both in terms of yield and stereoselectivity as demonstrated in the synthesis of 4l, 4m and 4n. Ultimately, the introduction of different groups in γ-aminooxy-α,β-unsaturated ester 2 was investigated. The result show that Ac group was proved to be possible, but Bz group afforded product 4p with deteriorated enantioselectivity. Notably, in all of the cases the diastereoselectivity was excellent (>20:1).
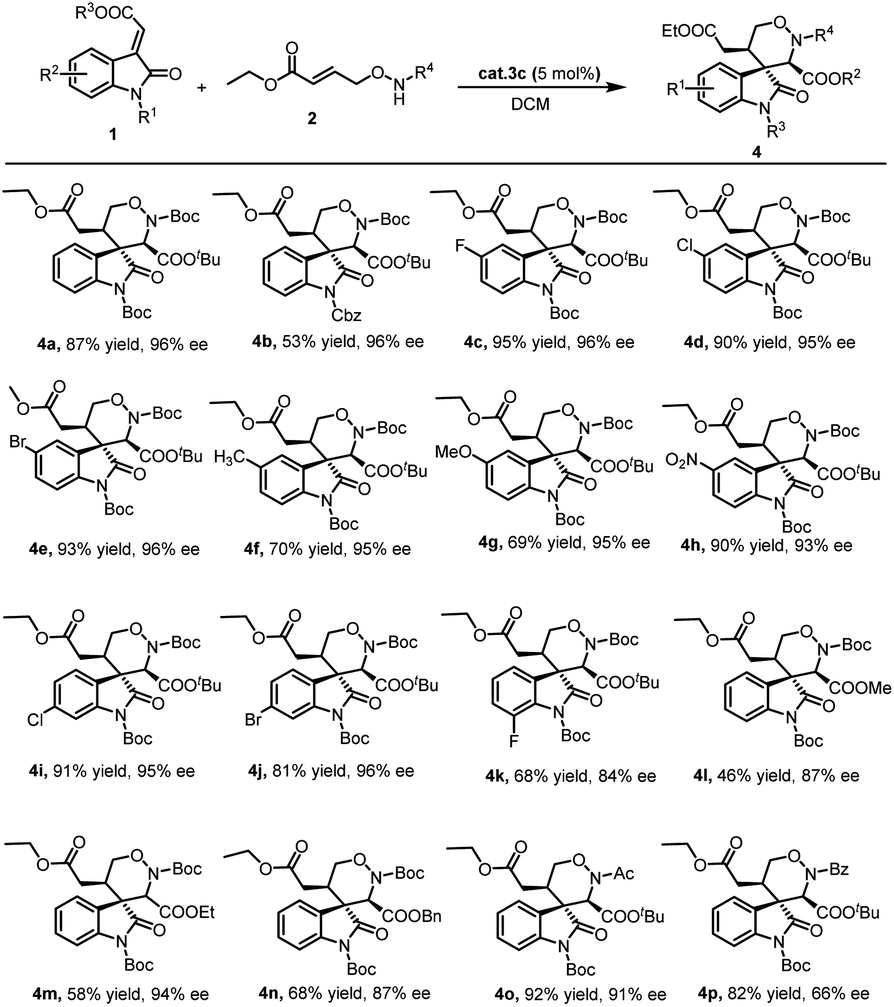 | ||
| Scheme 1 Substrate scope examination of product 4. | ||
Having established the scope of the desired 1,2-oxazinane spiro-oxindole 4, the possibility to synthesize the skeleton of hexahydropyridazin spiro-oxindoles by analogic methodology was put on agenda. Based on the above research, a 1,4-synthon containing N and O atoms as used to construct the 1,2-oxazinane spiro-oxindole 4. It was automatic to consider constructing hexahydropyridazin spiro-oxindoles by similar 1,4-synthon containing double N atoms. Although, Deng (Fig. 2a) developed the methodology to form hexahydropyridazin spiro-oxindoles by isatin N,N′-cyclic azomethine imine, there is still no known 1,4-synthon containing double N atoms disclosed, which was able to form our desired chiral hexahydropyridazin spiro-oxindoles. In the face of new area, we intended to design and synthesize new 1,4-synthon for synthesis of hexahydropyridazin spiro-oxindoles. Initially, a transformed synthon 5 which substituted N–O fragment in γ-aminooxy-α,β-unsaturated ester 2 with N–N fragment was taken into consideration. However, it was easy to figure out from the analysis of charge distributions that designed synthon 5 was supposed to initiate reaction from another N atom, and tetrahydropyrrole spiro-oxindoles rather than hexahydropyridazin spiro-oxindoles would be obtained when synthon 5 was utilized to reacted with methyleneindolinones 1. Therefore, a 1,4-synthon 6 that possessed reversed charge distribution with synthon 5 was designed and synthesized to construct desired hexahydropyridazin spiro-oxindoles.
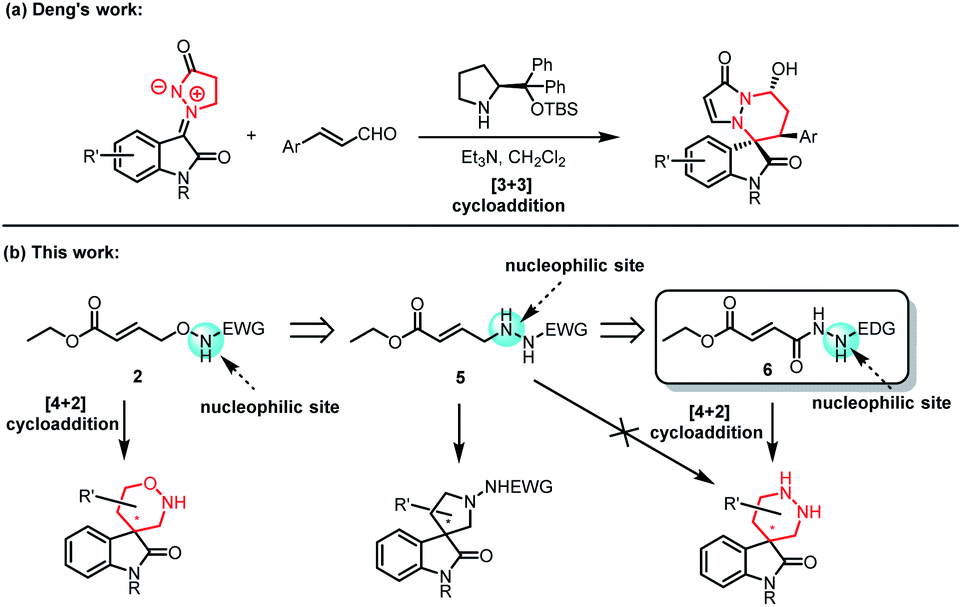 | ||
| Fig. 2 The design of new 1,4-synthon 6. | ||
Fortunately, a novel group of hexahydropyridazin spiro-oxindoles 7a (CCDC 2153627) was synthesized from methyleneindolinones 1a and new 1,4-synthon 6a catalyzed by organocatalyst 3a, with 77% yield, >20:1 dr and 49% ee (Table 2, entry 1). Nonetheless, it was anticipated that a proper choice of the organocatalyst was able to increase enantioselectivity and retain diastereoselectivity and yield on this reaction simultaneously.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T(°C) | Yieldb | eec |
| a Typical reaction conditions: 1a (0.15 mmol), 6a (0.10 mmol), 10 mol% catalyst 3 in DCM (0.2 mL), at 25 °C for 72 h. All reactions in this table, dr values >20:1.b Determined by 1H NMR analysis of the crude reaction mixture before purification.c Determined by HPLC analysis.d Reacted for 7 days.e 15 mol% catalyst was used.f Reaction concentration was 1.0 mol L−1.g Reacted for 10 days. |
|||||
| 1 | 3a | DCM | 25 | 77 | 49 |
| 2 | 3b | DCM | 25 | 63 | 39 |
| 3 | 3c | DCM | 25 | 70 | 40 |
| 4 | 3d | DCM | 25 | 75 | 25 |
| 5 | 3e | DCM | 25 | 40 | 45 |
| 6 | 3f | DCM | 25 | 80 | 78 |
| 7 | 3f | CHCl3 | 25 | 76 | 78 |
| 8 | 3f | DCE | 25 | 77 | 77 |
| 9 | 3f | Toluene | 25 | 55 | 73 |
| 10 | 3f | CH3CN | 25 | 80 | 73 |
| 11d | 3f | DCM | 0 | 70 | 83 |
| 12d | 3f | DCM | −20 | 61 | 87 |
| 13d | 3f | DCM | −30 | 49 | 89 |
| 14d | 3f | DCM | −35 | 41 | 92 |
| 15d,e | 3f | DCM | −35 | 58 | 92 |
| 16d,e,f | 3f | DCM | −35 | 63 | 91 |
| 17e,f,g | 3f | DCM | −35 | 74 | 91 |
Optimization studies started from the catalysts screening with methyleneindolinones 1a and fumaric acid monoester monoamide 6a as model substrates (Table 2). It was different from the reaction of 1,2-oxazinane spiro-oxindole 4, analogous squaramide or thiourea catalyst 3b–3e gave the desired hexahydropyridazin spiro-oxindoles 7a with poor enantioselectivity (Table 2, entries 2–5). However, the product 7a was formed with 78% ee value under the catalysis of larger steric hindrance catalyst 3f (Table 2, entry 6), which was unable to catalyze the previous convention of 1,2-oxazinane spiro-oxindole 4. Subsequent solvent screening (Table 2, entries 7–10) enabled us to identify DCM as the most suitable solvent for the devised reaction. With the best suited solvent being established, the optimization efforts were directed towards the reaction temperature (Table 2, entries 11–14). As the falling of reaction temperature, remarkable increasing of enantioselectivity and reducing of yield was observed. The desired product 7a was obtained in 92% ee value and 41% yield under the condition of −35 °C. Finally, through the adjustment of catalyst amount, reaction concentration and reaction time, the optimized reaction conditions were found: 15 mol% 3f as the catalyst in DCM solvent, 1.0 mol L−1 1,4-synthon 6a and 1.5 mol L−1 methyleneindolinones 1a reacted for 10 days under the temperature of −35 °C and desired hexahydropyridazin spiro-oxindoles 7a was obtained in 74% yield, dr > 20:1, 91% ee.
With the optimized reaction conditions in hand, the substrate scope and limitations of this new cascade reaction were explored. As summarized in Scheme 2, the Boc protective group on the methyleneindolinones 1 was irreplaceable. Other protective groups (such as Ac or Et), restrained this cascade reaction even though similar Cbz group. Subsequently, an array of methyleneindolinones 1 bearing different functional substitutions (R1) on C-5, C-6 and C-7 were investigated in the reactions with fumaric acid monoester monoamide 6a. It was found that halogen atoms, Me, MeO on C-5 were well tolerated (7b–7f). However, C-6 and C-7 position was proved general compatibility. Except F atom, other substitutions deteriorated enantioselectivity in different degree. Ultimately, the ester group on methyleneindolinones 1 (R2) and fumaric acid monoester monoamide 6 (R3) was studied. It was found that different ester groups had no major influence on the reaction outcome both in terms of yield and stereoselectivity as demonstrated in the synthesis of 7n–7r.
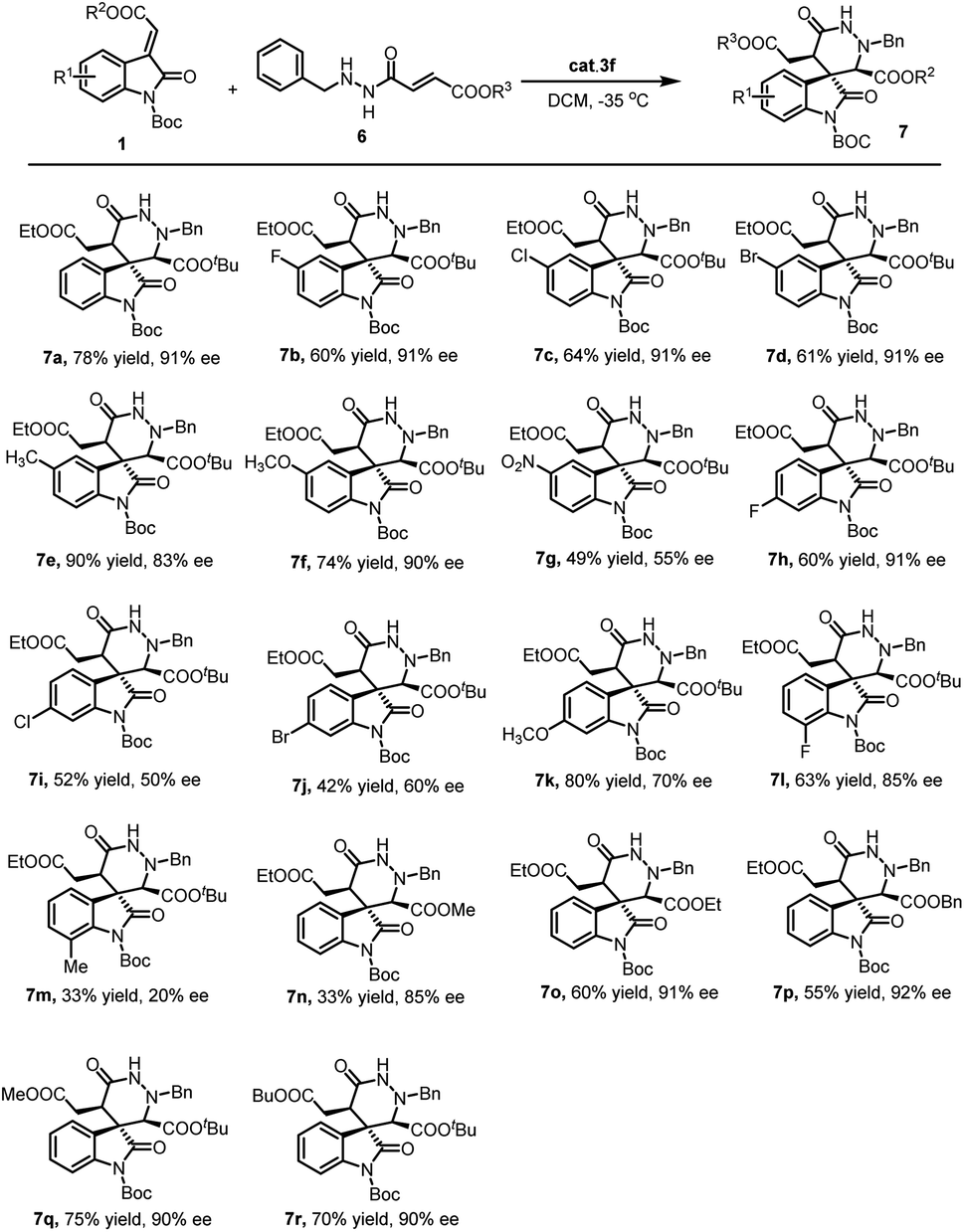 | ||
| Scheme 2 Substrate scope examination of product 7. | ||
To illustrate the potential application of this reaction, several transformations of 1,2-oxazinane spiro-oxindole 4a and hexahydropyridazin spiro-oxindoles 7a were carried out. As illustrated in Scheme 3, two Boc groups in 4a could be eliminated completely with TFA at the same. But in the condition of SmI2 (2.5 eq.), only the Boc group on indole ring was removed selectively. Both transformations retain the enantioselectivity of products. Unexpectedly, hexahydropyridazin spiro-oxindoles 7a eliminated the Boc group on indole ring and the carboxyl group in the condition of TFA. This transformation optimized the structure of product 7a for more applications.
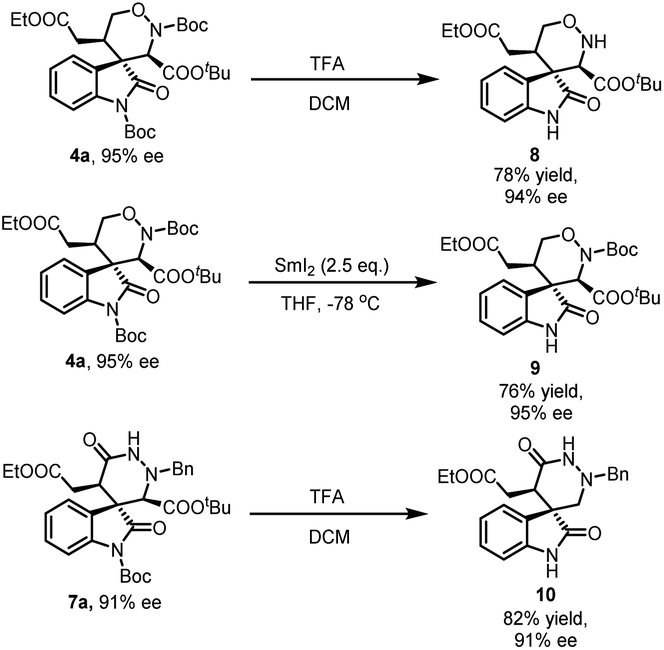 | ||
| Scheme 3 Transformation of 4a and 7a. | ||
In conclusion, two novel asymmetric organocatalytic reactions for the preparation of chiral 1,2-oxazinane spiro-oxindole and chiral hexahydropyridazin spiro-oxindole has been presented. Further investigations on expanding the scope of these two reactions prove wide range tolerance of other methyleneindolinones. Furthermore, in order to construct the undiscovered chiral hexahydropyridazin spiro-oxindoles, a new fumaric acid monoester monoamide 1,4-synthon is designed and synthesized. The developed 1,4-synthon possibly offers a facile, efficient and general entry to other heterocyclic skeletons containing two N atoms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Natural Science Foundation of Shanghai (20ZR1405500).Notes and references
- (a) Y. Ding, S. Gruschow, T. J. Greshock, J. M. Finefield, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2008, 71, 1574 CrossRef CAS PubMed; (b) B. H. Lee, G. I. Kornis, J. I. Cialdella, M. F. Clothier, V. P. Marshall, D. G. Martin, P. L. McNally, S. A. Mizsak, H. A. Whaley and V. H. Wiley, J. Nat. Prod., 1997, 60, 1139 CrossRef CAS; (c) E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318 CrossRef CAS PubMed; (d) Z. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, J. Am. Chem. Soc., 2014, 136, 14184 CrossRef CAS PubMed; (e) Z. Bian, C. C. Marvin and S. F. Martin, J. Am. Chem. Soc., 2013, 135, 10886 CrossRef CAS PubMed; (f) B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720 CrossRef CAS PubMed.
- S. R. Yong, A. T. Ung, S. G. Pyne, B. M. Skelton and A. H. White, Tetrahedron, 2007, 63, 5579 CrossRef CAS.
- (a) A. Czarna, B. Beck, S. Srivastava, G. M. Popowicz, S. Wolf, Y. Huang, M. Bista, T. A. Holak and A. Domling, Angew. Chem., Int. Ed., 2010, 49, 5352 CrossRef CAS PubMed; (b) V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hubel, D. Rauh and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed; (c) S. Crosignani, C. Jorand-Lebrun, P. Page, G. Campbell, V. Colovray, M. Missotten, Y. Humbert, C. Cleva, J. Arrighi, M. Gaudet, Z. Johnson, P. Ferro and A. Chollet, ACS Med. Chem. Lett., 2011, 2, 644 CrossRef CAS PubMed.
- C. Zhang, P. Dou, Y. You, Z. Wang, M. Zhou, X. Xu and W. Yuan, Tetrahedron, 2019, 75, 130571 CrossRef.
- (a) C. Carson and M. Kerr, Angew. Chem., Int. Ed., 2006, 45, 6560 CrossRef CAS PubMed; (b) L. Moraes, M. Donza, A. Rodrigues, B. Silva, D. Brasil, M. Zoghbi, E. Andrade, G. Guilhon and E. Silva, Molecules, 2015, 20, 22157 CrossRef CAS PubMed; (c) I. Uchida, S. Takase, H. Kayakiri, S. Kiyoto and M. Hashimoto, J. Am. Chem. Soc., 1987, 109, 4108 CrossRef CAS; (d) K. Shimomura, O. Hirai, T. Mizota, S. Matsumoto, J. Mori, F. Shibayama and H. Kikuchi, J. Antibiot., 1987, 40, 600 CrossRef CAS PubMed.
- (a) W. Li, J. Gan and D. Ma, Angew. Chem., Int. Ed., 2009, 48, 8891 CrossRef CAS PubMed; (b) E. Miller, C. Kauffman, P. Jensen and W. Fenical, J. Org. Chem., 2007, 72, 323 CrossRef CAS PubMed; (c) E. Moreno-Clavijo, A. T. Carmona, A. J. Moreno-Vargas, M. A. Rodríguez-Carvajal and I. Robina, Bioorg. Med. Chem., 2010, 18, 4648 CrossRef CAS PubMed; (d) M. Bols, Acc. Chem. Res., 1998, 31, 391 CrossRef; (e) M. Bols, R. Hazell and I. Thomsen, Chem. –Eur. J., 1997, 3, 941 CrossRef; (f) M. J. Alves, F. T. Costa, V. C. Duarte, A. G. Fortes, J. A. Martins and N. M. Micaelo, J. Org. Chem., 2011, 76, 9584 CrossRef CAS PubMed.
- (a) C. Pei, Y. Jiang and M. Shi, Eur. J. Org. Chem., 2012, 4206 CrossRef CAS; (b) H. Yang and M. Shi, Org. Biomol. Chem., 2012, 10, 8236 RSC; (c) G. Ran, P. Wang, W. Du and Y. Chen, Org. Chem. Front., 2016, 3, 861 RSC; (d) P. Xu, J. Liu, L. Shen, Z. Cao, X. Zhao, J. Yan and J. Zhou, Nat. Commun., 2017, 8, 1619 CrossRef PubMed; (e) P. Xu, C. Chen, J. Liu, Y. Song, F. Zhou, J. Yan and J. Zhou, J. Org. Chem., 2018, 83, 12763 CrossRef CAS PubMed.
- B. Liu, K. Li, S. Luo, J. Huang, H. Pang and L. Gong, J. Am. Chem. Soc., 2013, 135, 3323 CrossRef CAS PubMed.
- (a) M. Tong, X. Chen, J. Li, R. Huang, H. Tao and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4680 CrossRef CAS PubMed; (b) J. Li, R. Huang, Y. Xing, G. Qiu, H. Tao and C. Wang, J. Am. Chem. Soc., 2015, 137, 10124 CrossRef CAS PubMed; (c) R. Huang, X. Chang, J. Li and C. Wang, J. Am. Chem. Soc., 2016, 138, 3998 CrossRef CAS PubMed.
- (a) Z. Wang, T. Yang, R. Chen, X. Ma, H. Liu and K. Wang, RSC Adv., 2020, 10, 24288 RSC; (b) B. Gu, S. Wu, H. Xu, W. Yang, Z. Liu and W. Deng, Chin. Chem. Lett., 2021, 32, 672 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2153627, 2153695. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra02759c |
| This journal is © The Royal Society of Chemistry 2022 |