
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the functional mechanism of zinc ions of PARP1 binding with single strand breaks and double strand breaks†
Shuya Suna,
Xin Wangb,
Rongfeng Lina and
Kai Wang *bc
*bc
aSchool of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, No. 232, Waihuan East Road, Guangzhou 510006, China
bSchool of Agriculture and Biology, Zhongkai University of Agriculture and Engineering, Guangzhou 510000, P. R. China. E-mail: wangkai@zhku.edu.cn
cAbinitio Technology Company, Ltd, Guangzhou 510640, P. R. China
First published on 29th June 2022
Abstract
Poly(ADP-ribose)polymerase 1 (PARP1) is a key target for the treatment of cancer-related diseases, and plays an important role in biological processes such as DNA repair, regulating a variety of metabolic and signal transduction processes. Understanding the dynamic binding mechanisms between each domain of PARP1 and DNA is of great significance to deepen the understanding on the function of PARP1 and to facilitate the design of inhibitors. Herein, strategies such as classical molecular dynamics simulation, conformational analysis, binding free energy calculation and energy decomposition were used to shed light on the binding mechanisms of different DNA binding domains (DBDs, including ZnF1, ZnF2 and ZnF3) in PARP1 with DNA and on the influences of zinc ions on the binding process. On one hand, during binding with DNA, ZnF2 tends to expand its space to identify the DNA damage sites and ZnF1/ZnF2 recognizes the interfaces on both sides of DNA damage rather than one side during the process of DNA repair. More importantly, the stable secondary structure of L2 of ZnF2 (PRO146 to MET153) is the key conformational change for ZnF1 and ZnF2 to recognize DNA damage. Meanwhile, ZnF3 has little effect on the binding mechanisms of PARP1. On the other hand, for the structural differences of DBD domains, zinc ions in ZnF1 and ZnF2 (Zn1 and Zn2) have an impact not only on the conformational changes of PARP1, but also on the conformational changes brought by the interaction of double strand breaks (DSB) and single strand breaks (SSB). And meanwhile, Zn3 also has little effect on ZnF3 for the system of ZnF3/DSB. The findings presented in this work deepen the understanding on the functional mechanism of PARP1 and provide a theoretical basis for further study on the interaction between different inhibitors and DBD domains to design more potential inhibitors.
Introduction
PARP1 (poly(ADP ribose)polymerase 1) is a popular target with great potential to study anticancer drugs in current scientific research, which plays an important role in DNA repair, maintaining genomic integrity, regulating a variety of metabolic and signal transduction processes.1–8 In the past decades, the research on PARP1 has achieved great milestones, including the discovery that PARP1 catalyzes the production of PAR (poly(ADP-ribose)) and the elucidation of its three-dimensional structures and functions of different domains.7,9–14 In addition, subsequent studies described the purification of PARP1 and revealed the possible relationship between PARP1 and DNA repair.15–18 In the development of inhibitors, four PARP inhibitors have been approved by the regulatory authorities by the FDA (such as nivolumab and atezolizumab) which can effectively treat patients with ovarian cancer and breast cancer with mutation of breast cancer susceptibility gene (BRCA) of PARP.19–22 What's more, a variety of inhibitors have been used in (pre-)clinical research showing potential therapeutic for other cancers or non-neoplastic diseases, which can not only block DNA damage repair, but also play a role through new mechanisms such as immune activation.22–24 According to NextPharma, there are dozens of PARP projects under research in the world, which is one of the key directions in the field of drug research and development.15,20,23,25–27 Nevertheless, the speed and efficiency of research and development of PARP inhibitors cannot meet the needs of contemporary society, which is limited by the knowledge on PARP1.So far, the structure and function of PARP1 domains have been widely studied. PARP1 is composed of a single peptide chain with a length of 1014 amino acids, which can be divided into six domains. Three zinc-finger motifs (ZnF1, ZnF2, ZnF3, 2–372) consist of N-terminal DNA binding domain (DBD) which is responsible for the connection of whole protein and for the activation of PARP1;10,11,28 the BRCA1 C-terminal (BRCT) domain (385–476) has been found to combine with DNA structure to mediate DNA transfer which connects DBD and subsequent tryptophan–glycine–arginine-rich (WGR) domain (542–638) with main sites of self-modification; C-terminal catalytic domain (CAT, 662–1014) contains NAD+ binding sites and catalytic sites for the synthesis of PAR, including a regulatory helical subdomain (HD, 662–779) and ADP-ribosyl transferase (ART, 788–1014).29,30 At present, all domains of PARP1 are crystallized which helpful to reveal the recognition mechanism of DBD binding with DNA.11,28 At the same time, the database contains many active crystal structures of inhibitors binding to PARP1, which can be used to develop more scoring function or to well predict the binding affinities of new inhibitors for PARP1.
Although some works reveal the recognition mechanism of DBD binding with DNA or CAT binding with inhibitors, the key detailed conformational changes of DBD during binding with DNA which is helpful to describe the interactions between inhibitors and each domain of PARP1 still remain in mystery.11,28,29 What's more, the influence of zinc ions in PARP1 on the binding process of DBD with DNA has not been revealed. Clarifying this issue may assist in elucidating the function and mechanism of PARP1, and in providing new ideas and theoretical support for the development of inhibitors. To reveal the binding mechanism of these molecules, many methods which can effectively describe intermolecular interactions and binding modes have been developed, including experimental methods of fluorescence and circular dichroism spectroscopy, and theoretical calculation methods such as molecular docking and molecular dynamics (MD) simulation.31–34 To enrich the scientific research mentioned above, in this paper, we studied the dynamic binding mechanism between each domain of DBD with DSB or SSB by classical MD simulations and studied the conformational change and key residues difference between 3 zinc-finger (ZnF1, ZnF2, ZnF3) binding with DSB or SSB in the presence or absence of zinc ion.
Materials and methods
Construction of initial models
Crystal structures of ZnF1 and ZnF2 bound to a multidomain PARP1 structure were downloaded from Protein Data Bank. The complexes of DBD with DSB and DBD with SSB (2n8a, 4av1, 4opx, Fig. 1a–c) were used to study the conformational changes and binding free energy changes caused by zinc ions. 15 systems were constructed to examine the conformational changes and the recognition mechanism of DBD, as is shown in Table 1. 11 modified systems were constructed to reveal the conformational change and key residues difference for ZnF1, ZnF2, ZnF3 binding with DSB or SSB with the absence of Zn1, Zn2 or Zn3 (Zn1, Zn2 and Zn3 belong to ZnF1, ZnF2 and ZnF3 respectively, Fig. 1d–f), as is described in Table 2. The structural optimization and the protonation process of potential protonated residues of each model were performed and missing hydrogen atoms were added for each model by the leap module of Amber20.37,38 After that, TIP3P water environment with boundary extension of 10 Å cubic water box was constructed for each model. In order to neutralize the charge of all systems, appropriate amounts of chlorine and sodium ions were added by Amber20. The zinc parameters39 and distance restraints with 1000 kcal (mol−1·Å−1) were applied for Zn1, Zn2 and Zn3 to keep their 4-fold coordination features. In addition, OL15 and FF19SB force fields were parameterized for all the systems.40,41 | ||
| Fig. 1 The crystal structure of 3 domains of PARP1 bound with DNA and zinc coordination of ZnF1, ZnF2 and ZnF3. (a) Crystal structure of ZnF1 and ZnF2 binding with DSB (4av1.pdb28). (b) Crystal structure of ZnF1 and ZnF2 binding with SSB (2n8a.pdb35). (c) Crystal structure of ZnF1 and ZnF2 binding with DSB (4opx.pdb36). (d) Structure of ZnF1 and the atom of Zn1 coordination region of ZnF1 (2l30.pdb10). (e) Structure of ZnF2 and Zn2 coordination of ZnF2 (2l31.pdb10). (f) Structure of ZnF3 and Zn3 coordination of ZnF3 (2riq.pdb11). | ||
| Name | Protein | PDB code |
|---|---|---|
| M01 | ZnF1 monomer | 2l30 |
| M02 | ZnF2 monomer | 2l31 |
| M03 | ZnF3 monomer | 2riq |
| M04 | ZnF1 monomer | 4av1 |
| M05 | ZnF2 monomer | |
| M06 | DSB monomer | |
| M07 | ZnF1/ZnF2/DSB complex | |
| M08 | ZnF1–ZnF2 monomer | 2n8a |
| M09 | SSB monomer | |
| M10 | ZnF1–ZnF2/SSB complex | |
| M11 | ZnF1 monomer | 4opx |
| M12 | ZnF3 monomer | |
| M13 | CAT monomer | |
| M14 | DSB monomer | |
| M15 | ZNF1/ZNF3/CAT complex |
| Number | Name | Protein | Removed atoms |
|---|---|---|---|
| 1 | M01-1 | ZnF1 monomer | Zn1 |
| 2 | M02-1 | ZnF2 monomer | Zn2 |
| 3 | M03-1 | ZnF3 monomer | Zn3 |
| 4 | M04-1 | ZnF1 monomer | Zn1 |
| 5 | M05-1 | ZnF2 monomer | Zn2 |
| 6 | M07-1 | ZnF1/ZnF2/DSB complex | Zn1, Zn2 |
| 7 | M08-1 | ZnF1–ZnF2 monomer | Zn1, Zn2 |
| 8 | M10-1 | ZnF1–ZnF2/SSB complex | Zn1, Zn2 |
| 9 | M11-1 | ZnF1 monomer | Zn1 |
| 10 | M12-1 | ZnF3 monomer | Zn3 |
| 11 | M15-1 | ZNF1/ZNF3/CAT complex | Zn1, Zn3 |
MD simulation
All MD simulations were performed by Amber20 software with GPU acceleration. Cut-off distance for van der Waals (VDW) and electrostatic non-bonded interactions was set to be 12 Å. In addition, long-range electrostatic interactions were calculated by particle mesh Ewald (PME) method.42 SHAKE algorithm was restricted for all covalent bonds of hydrogen atoms. Periodic boundary condition (PBC) was employed for all MD simulations. Multi-step structural optimization was carried out to obtain more stable and reasonable initial systems. The first step was to optimize water molecules by restricting all protein, DNA, ligand molecules and zinc ions. The second step was releasing the side chain of protein and DNA molecules for optimizing the protein side chains and solvent molecules. The third step is all released for optimization sampling results except the restraints of zinc coordination. After energy minimization, the system temperature was raised from 0 K to 310 K in 50 ps by the regular ensemble (NVT). Subsequently, the solution density of the system was brought to closer to normal by isothermal isobaric ensemble (NPT) for 100 ps. Finally, the system was equilibrated by NPT at 310 K under 1.0 atm pressure for 100 ns. The temperature and pressure were controlled by the Langevin dynamics method and the Berendsen pressure scaling algorithm,43 respectively. The Beeman method were used to integrate Newtonian equations of motion. All systems reach equilibrium at the end of the equilibration, as shown by Fig. S1 and S2.†Structure analysis and definition of key loops
The root-mean-square deviation (RMSD) parameters were calculated by Cpptraj44 to describe the dynamic stability and conformational change of protein and DNA. The root-mean-square fluctuation (RMSF) parameters were evaluated by VMD45 to estimate key residue fluctuations of each protein during the simulations. The binding energy of each complex were calculated using the MM-GBSA method.46In order to clarify the conformational changes of ZnF1 and ZnF2 domains in the process of recognizing DNA, two key loop structures are defined as L1 and L2 (as shown in Fig. 2) based on the structure analysis of ZnF1 and ZnF2 binding with DNA (as shown in Fig. 1a and b). Among them, the function of L2 of ZnF1 is to identify the gap of DNA damage or interaction with L2 of ZnF2, and the L1 of ZnF1 overlaps with the zinc coordination region to stabilize the binding interaction between ZnF1 and DNA. Similarly, for ZnF2, L2 recognizes the gap of DNA damage and L1 stabilizes the binding interaction between ZnF2 and DNA. In addition, the distance between Cα atom in Cys21 of L1 and Cα atom in Met43 of L2 (denoted by d1 in Fig. 2a) was used to characterize the conformational changes of ZnF1 structure. Similarly, d2 which was defined as the distance between Cα atom in Cys125 and the Cα atom in Gln150 was used to describe the conformational changes of ZnF2 (Fig. 2b).
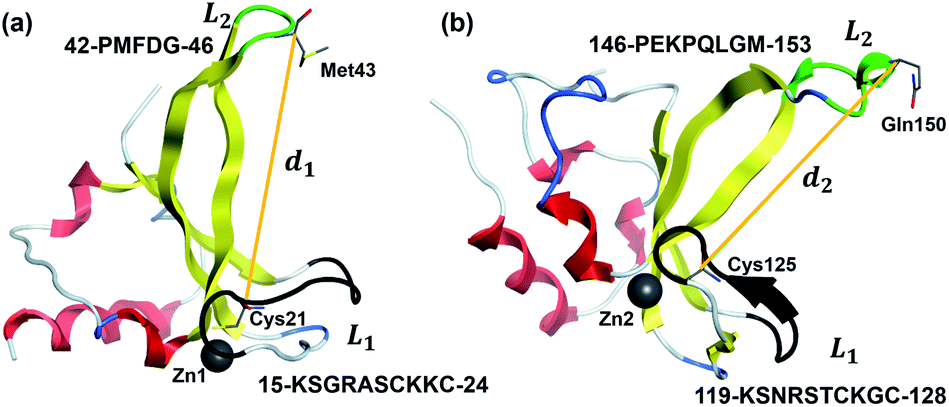 | ||
| Fig. 2 The definition of key loop structures in ZnF1 and ZnF2. The region that recognizes DNA damage was defined as L1, which is depicted as green cartoon and the region that stabilizes the DNA binding process was defined as L2, which is depicted as black cartoon. The α-helixes, β-sheets and other loops of ZnF1 and ZnF2 are marked by red, yellow, and white, respectively. Zinc atoms are depicted as gray ball. (a) d1 describes the distance of Cα atom in Cys21 and Cα atom in Met43. The sequences of L1 and L2 of ZnF1 are PMFDG and KSGRASCKKC, respectively. (b) d2 describes the distance of Cα atom in Cys125 and Cα atom in Gln150. The sequences of L1 and L2 of ZnF2 are PEKPQLGM and KSNRSTCKGC, respectively. | ||
Results and discussion
Exploring the role of zinc ion of ZnF1/ZnF2/ZnF3 monomer
The functions of three zinc ions in ZnF1, ZnF2 and ZnF3 were explored through MD simulations of the systems of M01, M02, M03, M01-1, M02-1 and M03-1 (Table 1). As shown in Fig. 3a–c, RMSF values indicate structure difference between monomer structure and its structure without zinc atom. The larger the RMSF the higher the flexibility is. In addition, Fig. 3d–f show the 3D structures of these systems. The functions of zinc ions in ZnF1, ZnF2 and ZnF3 can be elucidated by the comparison of RMSFs and 3D structures for systems with and without zinc ions, which will be discussed separately as follows. | ||
| Fig. 3 RMSFs and structure comparation of ZnFn and ZnFn(-) (n = 1, 2, 3). ‘(-)’ stands for removing zinc atom. (a)–(c) The RMSFs of per-residue for all systems. (d)–(e) The Comparison of 3D structures of ZnFn and ZnFn(-) after 100 ns MD simulation. The green cartoon model is ZnFn, and the yellow one is ZnFn(-). Blue and red cartoon correspond to the loop area in ZnFn and ZnFn(-), respectively. | ||
Firstly, for ZnF1, Zn1 can stabilize the structure of L2. The absence of Zn1 directly enhances the flexibility of L1 and L2 (Fig. 3a). Obviously, Zn1 can stabilize the atoms within their coordination region and indirectly stabilize the structure of L1 and L2. In order to describe the influence of zinc ion on conformational change and dynamic mechanisms of zinc ion more clearly, the distance between L1 and L2 are calculated to exhibit the conformational change (Fig. 4a). It can be noted that the loss of Zn1 has a greater impact on d1, making d1 of ZnF1(-) larger than ZnF1.
 | ||
| Fig. 4 (a) The comparison of d1 of ZnF1 and ZnF1(-) during 100 ns MD simulation. (b) The comparison of d2 of ZnF2 and ZnF2(-) during 100 ns MD simulation. | ||
Secondly, the role played by zinc ion in ZnF2 is different from that in ZnF1. Specifically, unlike Zn1 in ZnF1 which makes L1 and L2 more flexible, Zn2 in ZnF2 reduces the flexibility of L1 but increases the flexibility of L2 (Fig. 3b). In addition, L1 of ZnF2 and ZnF2(-) points to two different directions, indicating a more obvious conformational change compared with ZnF1 (Fig. 3e). The loss of Zn2 in ZnF2 results in the 90° rotation of L1 while the stability of the surrounding amino acid structure is maintained. Meanwhile, d2 is calculated to describe the conformational change between L1 and L2. In the early stage of MD simulation, d2 of ZnF2(-) fluctuates significantly. However, after the MD simulation is stable in the later stage, the 90° flip of L1 has little effect on d2 of ZnF2(-) between L1 and L2.
Finally, for ZnF3, the comparison of RMSF values show that the loss of Zn3 directly leads to the increase of the flexibility of the loop region connected to zinc coordination, and its sequence is in the range of 311 to 322 (denoted by L1 in Fig. 3c and f), while other protein structures are relatively stable. However, the study on monomer structure is not enough to explain the relationship between its structure and function. Therefore, the binding effect of ZnF1/ZnF2/ZnF3 binding with DNA is further studied.
Conformational change and key interaction analysis of ZnF1/ZnF2/ZnF3 binding with SSB/DSB
In order to describe the interaction between DBD of PARP1 and DNA more accurately, crystal structure rather than homologous modeling structure was used in MD simulations to study the interaction between ZnF1/ZnF2/ZnF3 and DNA. Three systems were used to describe the interaction between DBD and DNA. The first is the system of ZnF1 and ZnF2 interacting with DSB, in which ZnF1 and ZnF2 exist separately (4av1); the second is the interaction system of ZnF1–ZnF2 structure bound with SSB, in which ZnF1 and ZnF2 are connected by a loop structure (2n8a); the third one is the system of ZnF1/ZnF3/CAT bound with DSB (4opx). As shown in Table 1, for the model of M04 to M18, the monomers in all protein-DNA interaction systems are individually simulated by MD simulations and compared with their bound systems. Then the detailed conformational changes of ZnF1, ZnF2 and ZnF3 were analyzed between the state of monomer and the state of binding with DNA to describe the dynamic mechanism of DNA recognition process for DBD of PARP1.For the comparation of RMSF values of ZnF1 and ZnF1/DSB, L1 loop of ZnF1 shows a certain degree of reduction in flexibility after binding to DSB (Fig. 5a). However, L1 loop of ZnF2 shows a notable decrease after binding to DSB (Fig. 5b). Furthermore, the distance between L1 and L2 was examined to describe the structure difference caused by the interaction with DSB. Fig. 5c and d show the variation curves of d1 and d2 during the process of MD simulation. Among them, d1 and d2 of monomer change more greatly and maintain a slightly higher values than their counterparts of complexes. The changes of d1 and d2 can be noted by the comparison of 3D structures. Fig. 5e shows the superposition structure of ZnF1 between monomer and the complex of ZnF1/DSB. The relative position of L2 of ZnF1 changes slightly while its flexibility changes greatly. In contrast, for ZnF2, both the relative position and structural flexibility of L2 loop structure exhibit obvious changes (Fig. 5f). However, whether in ZnF1 or ZnF2, the relative position and structural flexibility of L1 structure keep stable. Therefore, the dynamic changes of L2 in position and structural flexibility are helpful for ZnF1 and ZnF2 to recognize and obtain DNA damage sites and the dynamic stability of L1 loop is helpful to grasp and stabilize DNA structure.
 | ||
| Fig. 5 The binding interaction analysis between ZnF1/ZnF2 and DSB. (a) Comparison of RMSF values of ZnF1 between ZnF1 monomer and ZnF1/DSB complex. (b) Comparison of RMSF values of ZnF2 between ZnF2 monomer and ZnF2/DSB complex. (c) The comparison of d1 of ZnF1 during 100 ns MD simulation. (d) The comparison of d2 of ZnF2 during 100 ns MD simulation. (e) and (f) The comparison of 3D structures of ZnF1/ZnF2 binding with DSB after 100 ns MD simulation. The green cartoon model is ZnF1/ZnF2 of the systems of ZnF1/ZnF2/DSB, and the yellow one is ZnF1 or ZnF2 monomer. Blue and red cartoon correspond to L1 and L2 between ZnF1/ZnF2/DSB and ZnF1 or ZnF2, respectively. (g) and (h) The per-residue energy decomposition of ZnF1 in the systems of ZnF1/DSB and ZnF2 in ZnF2/DSB. | ||
The binding energies of ZnF1 and ZnF2 with DSB are −38.74 kcal mol−1 and −63.40 kcal mol−1, respectively, as calculated by the method of MMGBSA.42 ZnF2 has stronger binding ability to DSB than ZnF1. This result is consistent with the previous experimental hypothesis that ZnF2 plays the major role in gap recognition.47 Similarly, L1 and L2 loop structures are of great significance for the combination of DSB based on the data of decomposition of energy contribution. According to Fig. 5g and h, the regions that mainly interact with DSB are near L1 and L2 loops. In addition, the side chain of Arg34 in ZnF1 and Arg138 in ZnF2 form stable hydrogen bonds with the backbone atoms of DSB. The simulated interactions between key amino acid residues and nucleic residues of DSB are consistent with previous experimental studies.28 Our simulations reveal the dynamic changes of their binding process and subtle conformational differences. In order to better describe the relationship between ZnF1 and ZnF2, the binding energy between ZnF1 and ZnF2 is calculated based on the MD trajectories of ZnF1/ZnF2/DSB system. The binding energy is −6.32 kcal mol−1 which means that the binding ability between ZnF1 and ZnF2 is relatively weak in the system of ZnF1/ZnF2/DSB. However, the effect of ZnF1 and ZnF2 binding to DSB cannot be fully explained due to the lack of connection structure between ZnF1 and ZnF2. In order to deal with this problem, the system of ZnF1–ZnF2 binding with SSB was used to study the dynamic changes of ZnF1 and ZnF2 conformations (the model of M08-M10 in Table 1).
Although, ZnF1 and ZnF2 are connected through a long loop structure in the system of ZnF1–ZnF2/SSB, the RMSF of that long loop is excluded to eliminate the influence caused by its high structural flexibility. Fig. 6a shows that the structural flexibility of L1 and L2 of ZnF1 decreases after combining with SSB. Meanwhile, d1 fluctuates in a smaller range after ZnF1 binding with SSB (Fig. 6b). According to the observation of 3D structure in Fig. 6c, there are certain position differences in the superposition of L2 between the states of ZnF1 and ZnF1/SSB. What's more, the per-residue energy decomposition diagram (Fig. 6d) shows that the binding ability of L1 with SSB is stronger than that of L2, which can also explain why L2 has a larger conformational flexibility of is than L1. For ZnF2, there is almost no difference in the change of structural flexibility (Fig. 6e), but the distance between L1 and L2 shows that d2 changes smoothly for the systems of ZnF2 and ZnF2/SSB. In addition, d2 of ZnF2 are than d2 of ZnF2/SSB. It can be inferred that there is a certain conformational change after ZnF2 binding SSB. As shown in Fig. 6f and g, the main conformational change occurs mainly in L2 region of ZnF2. The secondary structure of L2 forms a relatively stable structure of beta sheet after binding with SSB, while it remains a loop structure in the monomer state. According to Fig. 6d and h, Arg34 and Arg138 can also form hydrogen bonds with the main chain atoms of SSB structure,28 same as that in the system of ZnF1/ZnF2/DSB (Fig. 5g and h).
 | ||
| Fig. 6 The binding interaction analysis between ZnF1–ZnF2 and SSB. (a) and (e) Comparison of RMSF values of ZnF1 and ZnF2 between ZnF1–ZnF2 monomer and ZnF1–ZnF2/SSB complex. (b) The comparison of d1 of ZnF1 during 100 ns MD simulation. (f) The comparison of d2 of ZnF2 during 100 ns MD simulation. (c) and (g) The comparison of 3D structures of ZnF1–ZnF2 binding with SSB after 100 ns MD simulation. The green cartoon model is ZnF1/ZnF2 of the system of ZnF1–ZnF2/SSB, and the yellow one is ZnF1 or ZnF2 monomer. Blue and red cartoon correspond to L1 and L2 between ZnF1–ZnF2/SSB and ZnF1–ZnF2, respectively. (d) and (h) The per-residue energy decomposition of ZnF1 and ZnF2 in the system of ZnF1–ZnF2/DSB. | ||
The total binding energy of ZnF1–ZnF2/SSB is −119.09 kcal mol−1. Since ZnF1 and ZnF2 are connected, the energy decomposition contribution values are calculated for ZnF1 and ZnF2 based on the energy decomposition of ZnF1–ZnF2/SSB. Lys97 is used to distinguish ZnF1 and ZnF2 and their energy decomposition contribution values are −35.20 kcal mol−1 and −56.41 kcal mol−1, respectively. That is, ZnF2 has stronger binding ability to SSB than ZnF1, which is consistent with the conclusion that ZnF2 has stronger binding ability to DSB than ZnF1. This result also proves that ZnF2 has a stronger binding ability to DNA than ZnF1. A stable interaction between the two key loops in the binding process of ZnF2 with DSB and SSB exists, which is consistent with the experimental hypothesis that ZnF2 plays the major role in gap recognition.28,29,47 In addition, the binding ability of L2 in ZnF1/SSB is stronger than that in ZnF1/DSB (Fig. 5g and 6d). This indicates that ZnF1 and ZnF2 recognize the broken gap on both sides of DNA damage rather than one side on the process of DNA recognition. ZnF2 can realize its recognition and stable function on the process of binding to broken DNA damage, while ZnF1 plays its function on stabilizing DSB structure and plays its function of stabilizing and identifying SSB damage gap. In conclusion, it reveals that L2 of ZnF1 and ZnF2 play an important role in identifying DNA damage gaps and the synergistic existence of L2 in ZnF1 and in ZnF2 is conducive to the repair of DNA damage. What's more, it explains why DSB can exert stronger force on the DBD domains of PARP1 than SSB, resulting in the stability of DSB structure and DBD domains which makes DSB unable to be repaired.
The main function of ZnF3 is to stabilize DBD domains and subsequent CAT domain, as demonstrated previously. Therefore, the interaction mechanism among ZnF1, ZnF3, CAT and DSB are studied to explore the influence mechanism of ZnF1/ZnF3 binding to DSB. RMSF of ZnF1 and ZnF1 in ZnF1/ZnF3/DSB in Fig. 7a are consistent with the conclusion of ZnF1 combined with DSB in Fig. 5a, which means the mainly change of structural flexibility after binding with DNA is reflected in the loop region of L2 of ZnF1. However, according to the energy decomposition contribution diagram in Fig. 7c, L2 of ZnF1 shows a strong ability to combine DSB and its contribution is consistent with Fig. 6d but different with Fig. 5g. It means that ZnF1 can stabilize and identify DSB for the lack of ZnF2, which proved that ZnF1 and ZnF2 recognize the interfaces on both sides of DNA damage rather than one side on the process of DNA recognition. For ZnF3, it can be seen from Fig. 7b and d that only two amino acids (Ser274 and Gly275) of ZnF3 interact with DSB and their structure also becomes more stable when ZnF3 combined with DSB.11 The total binding energy of ZnF3/DSB is 2.68 kcal mol−1, which indicates the weak interaction of ZnF3 with DSB in the system of ZnF3/DSB. In this part, we revealed the structural differences and functional mechanisms of the interaction of ZnF1/ZnF2/ZnF3 in DBD binding with DSB and SSB, and next, we will explore the structural changes brought by zinc ions in each domain during these binding processes.
 | ||
| Fig. 7 The binding interaction analysis of the system of ZnF1/ZnF3/CAT/DSB. (a) and (b) Comparison of RMSF values of ZnF1 and ZnF3 between ZnF1 or ZnF3 monomer and ZnF1/ZnF3/CAT/DSB. (c) and (d) The comparison of 3D structures of ZnF1/ZnF3 binding with DSB after 100 ns MD simulation. The green cartoon model is ZnF1/ZnF3 of the systems of ZnF1/ZnF3/DSB, and the yellow one is ZnF1 or ZnF3 monomer. Blue and red cartoon correspond to the loop area in L1 and L2, respectively. (e) and (f) The per-residue energy contribution spectrums of ZnF1 and ZnF3 in the system of ZnF1/ZnF3/CAT/DSB. | ||
The dynamics analysis of ZnF1(-)/ZnF2(-)/ZnF3(-) binding with SSB/DSB
The effect of zinc ions on the structure of the three monomers of DBD have been revealed in Subsection 3.1. However, the impact of zinc ions on the binding process of DBD with DNA is not clear, which will be unveiled in this subsection. For this purpose, eight models (no. 4–11 shown in Table 2) are designed.Firstly, as shown in Fig. 8a, the loss of Zn1 just slightly affects the structural flexibility near L1 of ZnF1(-). Therefore, in terms of structural flexibility, the absence of Zn1 does not have a significant impact on the process of binding DSB. However, d1 of ZnF1(-) maintains higher values than that in ZnF1 during the MD simulation from 40 ns to 100 ns (Fig. 8b). In addition, ZnF1 and ZnF1(-) differ significantly in structure between L1 and L2 according to structural superposition of ZnF1 and ZnF1(-) shown in Fig. 8c, to be specific, the horizontal positions of both L1 and L2 are obviously different between cases of ZnF1 and ZnF1(-). Moreover, as to the interaction with DSB, the difference between cases of ZnF1 and ZnF1(-) lies in the interaction between amino acid residues near L1 and DSB according to the contribution diagram of energy decomposition (Fig. 8d). This is due to the conformational change caused by the absence of Zn1. Interestingly, the lack of Zn2 promotes the structural flexibility of L2, but does not affect the that of L1, which directly related to the region of Zn2 coordination (Fig. 8e). Moreover, the difference in d2 between ZnF2 and ZnF2(-) is slight as shown in Fig. 8f. The reason for this phenomenon is that L2 forms a stable helical structure in ZnF2(-), and the combination with DSB keeps the structure of L1 unchanged (Fig. 8g). In addition, the energy decomposition in L2 increases significantly (as shown in Fig. 8h), which indicates that the helical structure formed by L2 in ZnF2(-) strengthens the binding ability between L2 and DSB. What's more, there is a slight difference near L2 region between ZnF2(-) and ZnF2 in Fig. 8h, including of the residues of Ile154, Arg156 and Trp157. This observation is supported by the binding free energies shown in Table 3. The binding free energy increased significantly between ZnF1(-)/ZnF2(-)/DSB and ZnF1/ZnF2/DSB, in which ZnF1 changes from −38.74 to −51.34 kcal mol−1 and ZnF2 changes from −63.40 to −100.42 kcal mol−1. According to each energy term, the influence of Zn1 on the binding energy of ZnF1 to DSB is mostly caused by electrostatic energy and polar interaction. In contrast, the effect of Zn2 on the binding energy of ZnF2/DSB originates mainly from VDW energy and non-polar interaction. In conclusion, the absence of Zn1 and Zn2 leads not only to the enhancement of the binding ability between ZnF1/ZnF2 and DSB, but also to that L2 of ZnF2 form more stable secondary structure to interact with DSB. In addition, more residues around L2 are involved in the interaction of DSB.
 | ||
| Fig. 8 The binding interaction analysis between ZnF1(-)/ZnF2(-) and DSB. (a) Comparison of RMSF values of ZnF1 between ZnF1/DSB and ZnF1(-)/DSB complex. (b) The comparison of d1 of ZnF1 during 100 ns MD simulation. (c) The comparison of 3D structures of ZnF1 or ZnF1(-) binding with DSB after 100 ns MD simulation. The green cartoon model is ZnF1 in the system of ZnF1/ZnF2/DSB, and the yellow one is ZnF1(-) in ZnF1(-)/ZnF2(-)/DSB. Blue and red cartoon correspond to L1 and L2 between ZnF1/ZnF2/DSB and ZnF1(-)/ZnF2(-)/DSB, respectively. (d) The per-residue energy decomposition of ZnF1 in ZnF1/DSB and ZnF1(-) in ZnF1(-)/DSB. (e) Comparison of RMSF values of ZnF2 between ZnF2/DSB and ZnF2(-)/DSB complex. (f) The comparison of d2 of ZnF2 during 100 ns MD simulation. (g) The comparison of 3D structures of ZnF1 or ZnF1(-) binding with DSB after 100 ns MD simulation. (h) The per-residue energy decomposition of ZnF2 in ZnF2/DSB and ZnF2(-) in ZnF2(-)/DSB. | ||
| Terms | ZnF1/DSB | ZnF2/DSB | ZnF1(-)/DSB | ZnF2(-)/DSB |
|---|---|---|---|---|
| a van der Waals energy.b Electrostatic energy.c Polar solvation free energy.d Non-polar solvation free energy.e Calculated Gibbs free energy. All the energy terms are in kcal mol−1. | ||||
| ΔEvdwa | −39.87 | −62.86 | −43.72 | −74.78 |
| ΔEeleb | −654.67 | −955.34 | −819.00 | −1000.11 |
| ΔEpolarc | 661.57 | 962.58 | 817.29 | 984.45 |
| ΔEnonpolard | −5.77 | −7.78 | −5.91 | −9.98 |
| ΔGe | −38.74 | −63.40 | −51.34 | −100.42 |
Secondly, the influence on the conformational configuration and binding energy induced by the loss of Zn1 and Zn2 is studied. The difference between RMSF of ZnF1 in ZnF1/SSB and that of ZnF1(-) in ZnF1(-)/SSB is slight (Fig. 9a), similar with the impact caused by the loss of Zn2 (Fig. 9e). However, the influence triggered by the loss of zinc ions on d1 and d2 is significantly different. Specifically, d1 of ZnF1(-)/SSB is much larger than that of ZnF1/SSB (Fig. 9b), whereas d2 of ZnF2(-)/SSB is almost identical to that of ZnF2/SSB (Fig. 9f). It can be noted from the structural superposition diagram of ZnF1 and ZnF1(-) shown in (Fig. 9c) that the deletion of Zn1 has a great impact on the structural position of L1 and L2. In particular, compared with L2 in ZnF1/SSB, L2 in ZnF1(-)/SSB seems to rotate for 90°. Moreover, the energy decomposition shown in Fig. 9d demonstrates that the amino acid residues in L1 are enhanced and L2 are weakened on the combination of ZnF1(-) and SSB. In contrast, the loss of Zn2 does not induce obvious conformational changes as evidenced by the overlapping structure shown in Fig. 9g. A slight difference between the energy decomposition of ZnF2/SSB and ZnF2(-)/SSB is observed near L2, including the increase of the residues of Lys148 and Gln150 and the decrease of Trp157 (Fig. 9h). In addition, as shown in Table 4, the lack of Zn1 and Zn2 weakens the VDW interaction and nonpolar energy, but significantly increases electrostatic interaction and polar energy, leading to a significant increase in the binding free energy. To be specific, the binding free energy increases from −119.09 kcal mol−1 (for ZnF1–ZnF2/SSB) to 145.08 kcal mol−1 (for ZnF1–ZnF2(-:-)/SSB).
 | ||
| Fig. 9 The binding interaction analysis between ZnF1–ZnF2(-:-) and SSB. (a) Comparison of RMSF values of ZnF1 between ZnF1/SSB and ZnF1(-)/SSB complex. (b) The comparison of d1 of ZnF1 during 100 ns MD simulation. (c) The comparison of 3D structures of ZnF1 or ZnF1(-) binding with SSB after 100 ns MD simulation. The green cartoon model is ZnF1 in the system of ZnF1–ZnF2/SSB, and the yellow one is ZnF1(-) in ZnF1–ZnF2(-:-)/SSB. Blue and red cartoon correspond to L1 and L2 between ZnF1–ZnF2/SSB and ZnF1–ZnF2(-:-)/SSB, respectively. (d) The per-residue energy decomposition of ZnF1 in ZnF1/SSB and ZnF1(-) in ZnF1(-)/SSB. (e) Comparison of RMSF values of ZnF2 between ZnF2/SSB and ZnF2(-)/SSB complex. (f) The comparison of d2 of ZnF2 during 100 ns MD simulation. (g) The comparison of 3D structures of ZnF1 or ZnF1(-) binding with SSB after 100 ns MD simulation. (h) The per-residue energy decomposition of ZnF2 in ZnF2/SSB and ZnF2(-) in ZnF2(-)/SSB. | ||
| Terms | ZnF1–ZnF2/SSB | ZnF1–ZnF2(-:-)/SSB |
|---|---|---|
| ΔEvdw | −139.21 | −112.84 |
| ΔEele | −2511.84 | −2647.44 |
| ΔEpolar | 2549.79 | 2630.35 |
| ΔEnonpolar | −17.83 | −15.15 |
| ΔG | −119.09 | −145.08 |
The influences exerted by the loss Zn1 and Zn2 on the process of ZnF1 and ZnF2 binding to DSB or SSB can be summarized as follows. On one hand, the increase in binding free energy after the deletion of Zn1 and Zn2 for ZnF1–ZnF2/DSB is 49.62 kcal mol−1 while that for ZnF1/ZnF2 with SSB is only 25.99 kcal mol−1, implying a major difference in the process of ZnF1/ZnF2 binding to DSB/SSB. On the other hand, the absence of zinc ions has different effects on the conformational changes of ZnF1 and ZnF2. In the binding process with DSB, the lack of Zn1 and Zn2 directly affects the horizontal movement of L2 of ZnF1 and the secondary structure of L2 of ZnF2. However, for the binding process with SSB, the lack of Zn1 and Zn2 induces a 90° rotation of L2, but has little effect on the structure of L2 in ZnF2. In conclusion, zinc ions have an impact not only on the conformational changes of ZnF1 and ZnF2 (Fig. 3), but also on the conformational changes brought by the interaction of DSB and SSB.
Finally, in the same manner, the effect of Zn1 and Zn3 on the binding process of ZnF1/ZnF3 to DSB in explored. For ZnF1 in ZnF1/ZnF3/DSB, the behavior of ZnF1 is similar to that of ZnF2 in ZnF1/ZnF2/DSB (Fig. 8). When ZnF1 exists alone, the functions of ZnF1 and ZnF2 are similar, which means that in the structural system of 4av1, L1 and L2 of ZnF2 completely interact with the fracture surface of DSB, while ZnF1 only stabilizes DSB. However, in the system of 4opx, ZnF1 has enough space to realize the interaction for L1 and L2 binding to DSB (Fig. S3†). It can be concluded that Zn1 has little effect on RMSF, structural changes and energy decomposition diagram between ZnF1/DSB and ZnF1(-)/DSB. Similarly, due to the sequence differences between ZnF1 and ZnF2, the stability of L2 of ZnF2 plays an important role in the process of binding DNA (Fig. S3c†). On the other hand, for ZnF3, the lack of Zn3 directly leads to the increase of structural flexibility of ZnF3 (Fig. S3b†), especially for L1 connected by Zn3 which has obvious changes in secondary structure (Fig. S3d†). In addition, due to the weak interaction between ZnF3 and DSB (Table S1†), the lack of Zn3 has no effect on the interaction of any amino acid residues of ZnF3. What's more, Zn1 has most impact on electrostatic interaction and polar energy in the binding process of ZnF1 with DSB, which is consistent with the previous conclusion. By analyzing the conformational changes of Zn1, Zn2 and Zn3 ions on the process of DBD binding with DSB or SSB, the recognition function and mechanism of ZnF1 and ZnF2 can be clarified under different conditions.
Fig. 10 is a schematic diagram describing the binding mechanism and per-residue energy analysis of ZnF1/ZnF2 combined with DSB and SSB. In Fig. 10a, L2 of ZnF1 is far away from the DSB which shows a weak ability to bind with DSB. The lack of Zn1 induces a significant increase in the space between L1 and L2 of ZnF1(-) (Fig. 10b). What's more, the binding ability of L1 with DSB increases to a certain extent, which is consistent with the contribution value of energy decomposition in Fig. 10e. It describes that the energy value of ZnF1 in L1 increases significantly after the loss of Zn1, while the L2 region maintains little contribution. For the binding process of ZnF2 and DSB, the most obvious change is the formation of stable secondary structure of L1 and L2 of ZnF2. The per residue energy contribution spectrum in Fig. 10e proves that the lack of Zn2 leads to the formation of a stable secondary structure in L2 region, which can greatly increase the binding ability between L2 of ZnF2 and DSB. On the other hand, for the binding mode of SSB, in Fig. 10c, L1 of ZnF1 and ZnF2 are responsible for stabilizing the entire DNA structure, while L2 of ZnF1 and ZnF2 are located at the broken gap of SSB, which is conducive to repairing the damaged DNA structure. The loss of zinc ions in ZnF1 results in the movement of L1 towards SSB and the deviation of L2 from the SSB gap towards the interior of the SSB damaged pocket. Consequently, SSB gap is increased (Fig. 10d).
 | ||
| Fig. 10 Schematic diagram of different conformational changes and binding mechanisms of ZnF1/ZnF2 with DSB or SSB. Gray protein model stands for ZnF1 and cyan one is ZnF2. Green model stands for DNA. The L1 and L2 of ZnF1 and ZnF2 are marked red. (a) The binding mode of ZnF1/ZnF2/DSB after 100 ns MD simulation. (b) The binding mode of ZnF1(-)/ZnF2(-)/DSB after 100 ns MD simulation. (c) The binding mode of ZnF1–ZnF2/SSB after 100 ns MD simulation. (d) The binding mode of ZnF1–ZnF2(-:-)/SSB after 100 ns MD simulation. (e) The comparison of per residue energy contribution spectrum of ZnF1/ZnF2 and ZnF1(-)/ZnF2(-) binding with DSB. (f) The comparison of per residue energy contribution spectrum of ZnF1–ZnF2 and ZnF1–ZnF2(-:-) binding with SSB. | ||
Correspondingly, for the lack of Zn2 in ZnF2, the formation of stable secondary structure of L2 leads to the increase of binding ability between ZnF2 and SSB, consistent with the per residue energy contribution spectrum in Fig. 10f. Meanwhile, the change for L1 of ZnF2 is not obvious compared with L2 of ZnF2. In addition, the per-nucleotide-residue energy contribution spectrum of DSB and SSB were described at Fig. S4,† showing key-residue interaction difference for DNA. For DSB, the difference of binding energy contribution is mainly reflected in the end of DSB binding with ZnF2 and ZnF2(-), while for SSB, it is mainly reflected in the interaction region at the broken gap of SSB. In brief, differences in conformational changes and binding mechanisms bring dynamics details on the recognition of DNA for PARP1. L2 of ZnF1 and ZnF2 change greatly during the binding of ZnF1/ZnF2 to DSB, and d2 of ZnF2 increases to enlarge the space to interact with DSB. What's more, L2 of ZnF1 forms a stable secondary structure to identify damaged DNA gaps. Meanwhile, the larger the structural flexibility of L2 in ZnF2, the greater the distance changes of d2 for ZnF2 which obtains a larger space to interact with SSB. On the other hand, the absence of Zn1 and Zn2 leads to the increase of structural flexibility of most key loop regions in the state of monomer. For the complex structure of ZnF1(-)/ZnF2(-)/DSB, the main changes are the increase of d1 of ZnF1 and the formation of stable secondary structure in L2 of ZnF2. In addition, the increase of d1 of ZnF1 is the main difference between ZnF1–ZnF2/SSB and ZnF1–ZnF2(-:-)/SSB.
Conclusions
In the current study, the detailed structural changes DBD in PARP1 during the binding process with DNA were investigated and the dynamic conformational differences of DBD caused by zinc ions were revealed. Firstly, during the binding of DBD with DSB, the positional and structural changes of L1 are helpful for ZnF1 and ZnF2 to recognize and obtain DNA damage sites and the stable of L2 loop is helpful to grasp and stabilize DNA structure. Secondly, during the binding process of DBD with SSB, L2 of ZnF1 and ZnF2 play an important role in identifying DNA damage gaps. ZnF1 and ZnF2 recognize the interfaces on both sides of DNA damage rather than one side on the process of DNA repair. Thirdly, Zn1 and Zn2 have an impact not only on the conformational changes of ZnF1 and ZnF2, but also on the conformational changes brought by the interaction of DSB and SSB. What's more, ZnF3 has little effect on the binding mechanisms of PARP1, and Zn3 has little effect on ZnF3. Further MM/GBSA and energy decomposition explored the differences in key residues among these interacting systems.In conclusion, for ZnF1, whether it binds to DNA or not, the lack of Zn1 can directly expand the space between L1 and L2. And meanwhile, the increase of d2 in ZnF2 is of great significance for ZnF1 and ZnF2 to recognize DNA. Specifically, enough space is the prerequisite for ZnF2 to effectively bind with DNA. More importantly, the stable secondary structure of L2 is the key conformational change for ZnF1 and ZnF2 to recognize DNA damage, which is of great significance for PARP1 to realize its repair function.
These findings can not only deepen the understanding on the biological functional mechanism of PARP1 but also provides theoretical basis to further study the interaction mechanisms between inhibitors and PARP1.
Author contributions
S. Sun performed the MD simulations and analysed the data. X. Wang performed the calculation of MMGBSA. R. Lin discussed the project. K. Wang supervised the whole project and wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (21803079).Notes and references
- M. Xiao, J. Guo, L. Xie, C. Yang, L. Gong, Z. Wang and J. Cai, Mol. Cancer Res., 2020, 18, 436–447 CrossRef CAS PubMed.
- L. K. Saha, Y. Murai, S. Saha, U. Jo, M. Tsuda, S. Takeda and Y. Pommier, Nucleic Acids Res., 2021, 49, 10493–10506 CrossRef CAS PubMed.
- Y. Hu, J. Lin, H. Fang, J. Fang, C. Li, W. Chen, S. Liu, S. Ondrejka, Z. Gong, F. Reu, J. Maciejewski, Q. Yi and J. J. Zhao, Leukemia, 2018, 32, 2250–2262 CrossRef CAS PubMed.
- J. Murai, S. Y. Huang, B. B. Das, A. Renaud, Y. Zhang, J. H. Doroshow, J. Ji, S. Takeda and Y. Pommier, Cancer Res., 2012, 72, 5588–5599 CrossRef CAS PubMed.
- F. A. Weber, G. Bartolomei, M. O. Hottiger and P. Cinelli, Stem Cells, 2013, 31, 2364–2373 CrossRef CAS PubMed.
- J. M. Rodriguez-Vargas, F. J. Oliver-Pozo and F. Dantzer, Oxid. Med. Cell. Longevity, 2019, 2019, 2641712 Search PubMed.
- P. A. Loeffler, M. J. Cuneo, G. A. Mueller, E. F. DeRose, S. A. Gabel and R. E. London, BMC Struct. Biol., 2011, 11, 37 CrossRef CAS PubMed.
- M. Engbrecht and A. Mangerich, Cancers, 2020, 12(7), 1813 CrossRef CAS PubMed.
- Y. Liu, X. Xu, H. Yang, E. Xu, S. Wu, W. Wei and J. Chen, Analyst, 2018, 143, 2501–2507 RSC.
- S. Eustermann, H. Videler, J. C. Yang, P. T. Cole, D. Gruszka, D. Veprintsev and D. Neuhaus, J. Mol. Biol., 2011, 407, 149–170 CrossRef CAS PubMed.
- M. F. Langelier, K. M. Servent, E. E. Rogers and J. M. Pascal, J. Biol. Chem., 2008, 283, 4105–4114 CrossRef CAS PubMed.
- M. F. Langelier, J. L. Planck, S. Roy and J. M. Pascal, J. Biol. Chem., 2011, 286, 10690–10701 CrossRef CAS PubMed.
- D. Slade, M. S. Dunstan, E. Barkauskaite, R. Weston, P. Lafite, N. Dixon, M. Ahel, D. Leys and I. Ahel, Nature, 2011, 477, 616–620 CrossRef CAS PubMed.
- I. Kameshita, Z. Matsuda, T. Taniguchi and Y. Shizuta, J. Biol. Chem., 1984, 259, 4770–4776 CrossRef CAS PubMed.
- H. D. Chen, C. H. Chen, Y. T. Wang, N. Guo, Y. N. Tian, X. J. Huan, S. S. Song, J. X. He and Z. H. Miao, Int. J. Cancer, 2019, 145, 714–727 CrossRef CAS PubMed.
- C. Liu, A. Vyas, M. A. Kassab, A. K. Singh and X. Yu, Nucleic Acids Res., 2017, 45, 8129–8141 CrossRef CAS PubMed.
- A. Ray Chaudhuri and A. Nussenzweig, Nat. Rev. Mol. Cell Biol., 2017, 18, 610–621 CrossRef CAS PubMed.
- E. A. Maltseva, N. I. Rechkunova, M. V. Sukhanova and O. I. Lavrik, J. Biol. Chem., 2015, 290, 21811–21820 CrossRef CAS PubMed.
- D. Stępiński, Histochem. Cell Biol., 2018, 150, 607–629 CrossRef PubMed.
- H. Zhu, M. Wei, J. Xu, J. Hua, C. Liang, Q. Meng, Y. Zhang, J. Liu, B. Zhang, X. Yu and S. Shi, Mol. Cancer, 2020, 19, 49 CrossRef CAS PubMed.
- P. C. Fong, D. S. Boss, T. A. Yap, A. Tutt, P. Wu, M. Mergui-Roelvink, P. Mortimer, H. Swaisland, A. Lau, M. J. O'Connor, A. Ashworth, J. Carmichael, S. B. Kaye, J. H. Schellens and J. S. de Bono, N. Engl. J. Med., 2009, 361, 123–134 CrossRef CAS PubMed.
- A. Aili, J. Wen, L. Xue and J. Wang, Front. Oncol., 2021, 11, 712765 CrossRef PubMed.
- J. Mateo, C. J. Lord, V. Serra, A. Tutt, J. Balmana, M. Castroviejo-Bermejo, C. Cruz, A. Oaknin, S. B. Kaye and J. S. de Bono, Ann. Oncol., 2019, 30, 1437–1447 CrossRef CAS PubMed.
- U. K. Velagapudi, M. F. Langelier, C. Delgado-Martin, M. E. Diolaiti, S. Bakker, A. Ashworth, B. A. Patel, X. Shao, J. M. Pascal and T. T. Talele, J. Med. Chem., 2019, 62, 5330–5357 CrossRef CAS PubMed.
- R. E. Salmas, A. Unlu, M. Yurtsever, S. Y. Noskov and S. Durdagi, J. Enzyme Inhib. Med. Chem., 2016, 31, 112–120 CrossRef CAS PubMed.
- K. C. Chen, M. F. Sun and C. Y. Chen, Evidence-Based Complementary Altern. Med., 2014, 2014, 917605 Search PubMed.
- N. J. Curtin and C. Szabo, Nat. Rev. Drug Discovery, 2020, 19, 711–736 CrossRef CAS PubMed.
- A. A. E. Ali, G. Timinszky, R. Arribas-Bosacoma, M. Kozlowski, P. O. Hassa, M. Hassler, A. G. Ladurner, L. H. Pearl and A. W. Oliver, Nat. Struct. Mol. Biol., 2012, 19, 685–692 CrossRef CAS PubMed.
- J. Rudolph, J. Mahadevan and K. Luger, Biochemistry, 2020, 59, 2003–2011 CrossRef CAS PubMed.
- S. Hurtado-Bages, G. Knobloch, A. G. Ladurner and M. Buschbeck, Mol. Metab., 2020, 38, 100950 CrossRef CAS PubMed.
- J. K. Maurya, M. U. Mir, U. K. Singh, N. Maurya, N. Dohare, S. Patel, A. Ali and R. Patel, Biopolymers, 2015, 103, 406–415 CrossRef PubMed.
- J. K. Maurya, M. U. Mir, N. Maurya, N. Dohare, A. Ali and R. Patel, J. Biomol. Struct. Dyn., 2016, 34, 2130–2145 CrossRef CAS PubMed.
- M. Kumari, N. Dohare, N. Maurya, R. Dohare and R. Patel, J. Biomol. Struct. Dyn., 2017, 35, 2016–2030 CrossRef CAS PubMed.
- R. Patel, N. Maurya, M. U. D. Parray, N. Farooq, A. Siddique, K. L. Verma and N. Dohare, J. Mol. Recognit., 2018, 31(11), e2734 CrossRef PubMed.
- S. Eustermann, W. F. Wu, M. F. Langelier, J. C. Yang, L. E. Easton, A. A. Riccio, J. M. Pascal and D. Neuhaus, Mol. Cell, 2015, 60, 742–754 CrossRef CAS PubMed.
- M. R. Patel, A. Bhatt, J. D. Steffen, A. Chergui, J. Murai, Y. Pommier, J. M. Pascal, L. D. Trombetta, F. R. Fronczek and T. T. Talele, J. Med. Chem., 2014, 57, 5579–5601 CrossRef CAS PubMed.
- H. M. A. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, C. Jin, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xue, D. M. York, S. Zhao and P. A. Kollman, Amber 2022, University of California, San Francisco, 2021 Search PubMed.
- D. A. Case, T. E. Cheatham 3rd, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- M. B. Peters, Y. Yang, B. Wang, L. Fusti-Molnar, M. N. Weaver and K. M. Merz Jr, J. Chem. Theory Comput., 2010, 6, 2935–2947 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- R. Galindo-Murillo, J. C. Robertson, M. Zgarbova, J. Sponer, M. Otyepka, P. Jurecka and T. E. Cheatham 3rd, J. Chem. Theory Comput., 2016, 12, 4114–4127 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Gotz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- D. R. Roe and T. E. Cheatham 3rd, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- L. Xu, H. Sun, Y. Li, J. Wang and T. Hou, J. Phys. Chem. B, 2013, 117, 8408–8421 CrossRef CAS PubMed.
- G. Gradwohl, J. M. Menissier de Murcia, M. Molinete, F. Simonin, M. Koken, J. H. Hoeijmakers and G. de Murcia, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2990–2994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02683j |
| This journal is © The Royal Society of Chemistry 2022 |