DOI:
10.1039/D2RA02448A
(Paper)
RSC Adv., 2022,
12, 25364-25376
Sulfur vacancies on MoS2 enhanced the activation of peroxymonosulfate through the co-existence of radical and non-radical pathways to degrade organic pollutants in wastewater†
Received
16th April 2022
, Accepted 26th July 2022
First published on 20th September 2022
Abstract
The enhancement of vacancies in catalysts involving Fenton-like reactions is a promising way to remove organic pollutants in wastewater, but sulfur vacancies are rarely involved. In this work, MoS2 containing defect sites were synthesized by a simple high-temperature treatment and then applied for activating peroxymonosulfate to eliminate organic pollutants in wastewater. The structure was characterized by several techniques such as XRD, BET, and XPS. Important influencing factors were systemically investigated. The results indicated that MoS2 with sulfur vacancies possessed a higher catalytic activity than that of the parent MoS2. The annealing temperature of the catalyst had a great effect on the removal of organic pollutants. Besides, the catalytic system had a wide pH range. Quenching and electron paramagnetic resonance (EPR) experiments indicated that the reaction system contained radical and non-radical species. The characterization results revealed that the defect sites in catalysts mainly strengthened the activity of catalysts. This study offers a new heterogeneous catalyst for the removal of organic pollutants via the peroxymonosulfate-based Fenton-like reactions.
1. Introduction
With the development of industries, a large number of organic pollutants are discharged into the water environment, causing a serious problem of water pollution. Many useful technologies including adsorption,1 membranes,2 and advanced oxidation reactions (AORs)3–5 have been developed to dispel organic pollutants. In these ways, the AOR involving peroxymonosulfate (denoted as PMS) as an oxidant is an ideal choice. However, the direct oxidation of organic pollutants by PMS is extremely difficult. Accordingly, it has to introduce external activation factors including heat,6 alkaline conditions,7 and homogeneous8,9 and heterogeneous catalysts.3–5,10–15 Among them, a heterogeneous catalyst for the activation of PMS is a good strategy for repeated use, realization of radical and non-radical routes, etc. The activation of PMS by heterogeneous catalysts containing defect sites has attracted much attention in the past few years.16–34 Among them, the oxygen vacancies are the most studied,16,18–25 which often appear in various catalysts containing oxygen, including pure metal oxides such as Co3O4 16 and ZnO,19 perovskite20 and composited compounds.21–23 For example, Zhao et al.16 reported the role of oxygen vacancies in different Co3O4 crystal planes for the activation of PMS by the formation of 1O2; Gao et al.20 demonstrated the correlation between oxygen-deficient sites in the perovskite and the formation of 1O2 in activated PMS. However, this vacancy was easy to be restored under the aerobic condition. Nitrogen defect sites are the other type of vacancy used for activating PMS.24,25 It often appeared the cases with nitrogen-doped carbon materials. For instance, Wan et al.24 reported the degradation of 4-chlorophenol by N-doped biological carbon-activated PMS. Compared to the nitrogen dopant, the nitrogen vacancy has more affinity with PMS to form a surface polymer. However, the existence of nitrogen vacancies may make the catalyst vulnerable to the attack by reactive oxygen species, resulting in the poor stability of catalyst. Sulfur vacancies used for activating PMS have attracted great attention in recent years.26–31 This case is common in transition metal sulfides. Compared to oxygen and nitrogen vacancies, sulfur vacancies possess more advantages, as it is not only that the valence-changing transition metal ions are usually good activators for PMS but also that the sulfur defect sites themselves can activate both PMS and metal ions. Among them, MoS2 is an ideal catalyst due to its good ability to activate PMS.32–34 Besides, it is responsive to light illumination.35,36 It has been reported that introducing vacancies in MoS2 enhanced the removal ability towards organic pollutants in the presence of PMS.30,31 For example, MoS2 containing defect sites was prepared by a mechanical grinding method,30 and tetracycline was degraded by the above-mentioned catalyst-activated PMS. In this way, the characteristics of MoS2 were optimized, forming sulfur vacancies and exposing more Mo(IV) species. These changes were of great help to degrade tetracycline. However, the promotion was limited. There is no doubt that the development of a novel and effective MoS2-based catalyst with vacancies is highly desirable.
In this work, MoS2 containing sulfur vacancies was synthesized by a high-temperature treatment to activate PMS and then used to degrade RhB in wastewater. Various influencing factors were investigated. The correlation between the structure of the catalyst and the catalytic activity was discussed.
2. Experiments
2.1. Chemicals and materials
Various chemicals and materials such as PMS, peroxydisulfate (PDS), hydrogen peroxide (H2O2, 30 wt%), MoS2 powder, rhodamine B, 5,5-Dimethyl-1-pyrroline N-oxide (DMPO), tert-butanol, tiron, and 4-hydroxy-2,2,6,6-tetramethylpiperidine (TEMP) were purchased from commercial sources. All the above-mentioned chemicals were of analytical grade and employed without further purification.
2.2. Synthesis of catalysts
A series of MoS2 catalysts containing vacancies were synthesized by heat treatment of MoS2 at different temperatures (200, 300, 400 and 500 °C) for different time periods (4, 8 and 12 h) in air atmosphere. They were respectively denoted as MoS2-200, MoS2-300, MoS2-400 and MoS2-500. The original powders were not treated by heat, which was denoted as MoS2. Unless otherwise stated, the treatment time was 4 h. The preparation process of the catalyst is shown in Fig. S1.†
2.3. Characterization of catalysts
The crystal phases of the catalyst were analyzed by X-ray diffraction (XRD) using a D8-Advance X-ray diffractometer (Bruker Corp., Billerica, MA, USA). The specific surface area and pore features of the catalyst were measured by liquid nitrogen physisorption using an Autosorb-iQ/ASAP 2460 apparatus. The morphology of the catalyst was examined by scanning electron microscopy (SEM) using an SU 8100 instrument (Japan). Raman spectra were recorded using a LabRAM HR800. The surface elemental information of catalyst was acquired by X-ray photoelectron spectroscopy (XPS) using an EscaLab 250xi equipment. The concentration of elements in the solution after the reaction was detected by inductively coupled plasma-mass spectrometry (ICP-MS) using an ICP-MS-7700 (Agilent Technologies Co. Ltd., USA). The vacancy of the catalyst and reactive oxygen species were measured by EPR using a Bruker A300.
2.4. Degradation of organic pollutants under different conditions
In brief, a suitable catalyst and PMS were immediately placed into a container containing rhodamine B (denoted as RhB) aqueous solution at room temperature. Afterward, the reaction was carried out. At appropriate time intervals, a suitable volume of the reaction solution was collected and methanol was added into the above-mentioned solution to capture reactive oxygen species. The absorbance of RhB was measured using a photometer. The pH was regulated with dilute H2SO4 or NaOH solution and measured using a pH meter before and after the reaction. According to the experimental requirements, the important reaction parameters and the related chemical reagents were timely adjusted. The quantification of PMS during degradation was measured by a potassium iodide (KI) method, and this is as follows: a 0.5 M KI solution was prepared, 1 mL reaction solution after the centrifugation was added into 1 mL of the KI solution, and finally, the mixture solution was further diluted with a suitable volume of distilled water. After 0.5 h, the absorbance of solution was measured at 351 nm.
3. Results and discussion
3.1. Characterization of different catalysts
The XRD patterns of a series of MoS2 and MoS2-X (X = 200, 300, 400 and 500 °C) catalysts are displayed in Fig. 1 (a). It is observed that the diffraction peaks appeared at 2θ = 14.38°, 32.68°, 39.54°, 49.79° and 58.33°, which were attributed to the characteristic of MoS2 (PDF #37-1492). With increasing temperature below 400 °C, these samples possessed similar diffraction characteristic peaks as MoS2. This indicated that the structure of the treated catalyst remained unchanged relative to the untreated sample. However, the intensity of diffraction characteristic peaks in catalysts continued to reduce below 400 °C, as compared to that of MoS2. This is derived from the loss of the sulfur atoms in MoS2 at high temperatures, thereby resulting in a decrease in the crystallization of catalysts; in other words, sulfur vacancies may be formed. While the treatment temperature arrived at 400 °C, a great change occurred. The intensity of diffraction characteristic peaks for the MoS2-400 catalyst was remarkably reduced. Meanwhile, the diffraction characteristic peaks appeared at 2θ = 12.76°, 23.33°, 25.70°, 27.33°, 38.58° and 38.94°, which were ascribed to the characteristics of MoO3 (PDF #05-0508). The results indicated that the original structure of MoS2 was to some degree damaged after the treatment. This decreased degree became obvious with the further increase in temperature. To further explore the existence of the vacancy, the catalysts were characterized by EPR. The results indicated that the g value of MoS2-300 was slightly changed but the signal intensity of MoS2-300 was greatly altered relative to those of MoS2, as shown in Fig. 1(b). This suggested that the vacancies were indeed formed after the high-temperature treatment of the parent sample. This is in agreement with the results reported in the literature.31
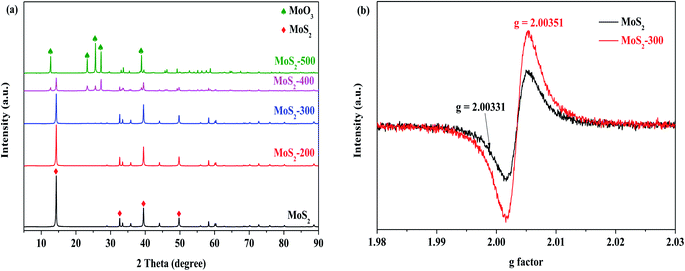 |
| | Fig. 1 (a) XRD patterns of a series of MoS2 catalysts. (b) EPR signal of the sulfur vacancy of MoS2 and MoS2-300. | |
Table 1 presents the results of pore structures of MoS2 and MoS2-300 catalysts. The SBET, Vtotal and DA values of the MoS2 catalyst were 10.54 m2 g−1, 0.0360 cm3 g−1 and 13.64 nm, respectively. After the high-temperature treatment, the above-mentioned values were all reduced. It showed that the pore structure of MoS2 was to some extent destroyed. In combination with the results of RhB degradation, the reduction of SBET did not affect the degradation of RhB. The SEM images of MoS2 and MoS2-300 catalysts are displayed. As observed in Fig. 2(a) and (b), MoS2 exhibited a smooth lamellar structure. After the treatment, the MoS2-300 still kept the lamellar structure but its surface became a little rough. This change is the main reason for the reduction in the SBET value of the catalyst. The above-mentioned results further indicated that the treated catalyst maintained the original structure of MoS2.
Table 1 BET and pore information of different MoS2-based catalystsa
| Catalysts |
SBET (m2 g−1) |
Vtotal (cm3 g−1) |
DA (nm) |
| SBET, Vtotal and DA were respectively denoted as the specific surface area, total pore volume and total pore size of catalysts. |
| MoS2 |
10.54 |
0.0360 |
13.64 |
| MoS2-300 |
8.95 |
0.0185 |
8.29 |
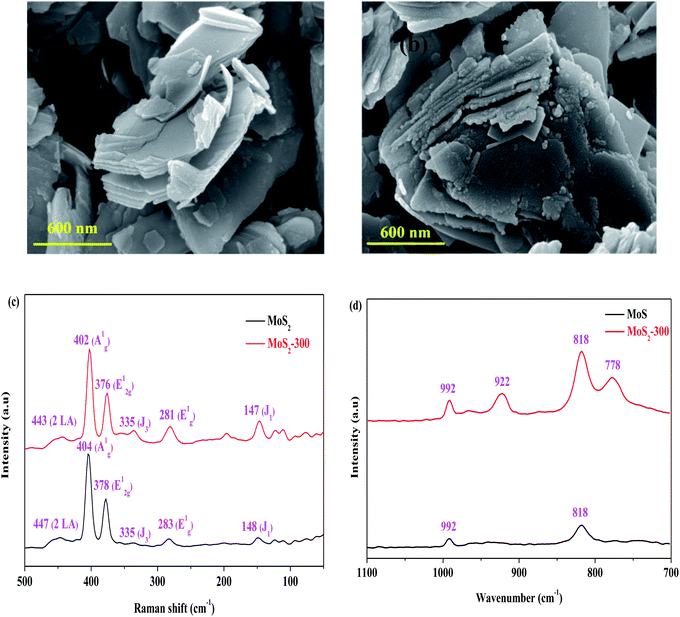 |
| | Fig. 2 (a) SEM image of MoS2. (b) SEM image of MoS2-300. (c) Raman spectra of MoS2. (d) Raman spectra of MoS2-300. | |
The Raman spectra of MoS2 and MoS2-300 catalysts were investigated, and the results are shown in Fig. 2(c) and (d). It is observed from Fig. 2(c) that the two samples exhibited similar spectra in the region of 100–500 cm−1. Generally, the typical spectra regions located at 376–378 cm−1, 402–405 cm−1 and 445–448 cm−1 were respectively assigned to the outer surface of Mo–S (E12g), inner surface of Mo–S (A1g) and substitution acoustic (2 × LA) models.33 These models belonged to the typical characteristic structure of 2H-MoS2. It is the most stable structure of MoS2. In general, the E12g model was selectively excited toward the facial feature, while the A1g model was preferentially excited to the corner feature arising from the polarization.33 In addition to E12g, A1g and 2 × LA, there were three additional models, namely, J1 (148 cm−1), E1g (283 cm−1) and J3 (335 cm−1). J1 and J3 belonged to the super-lattice structure of 1T-MoS2, and E1g belonged to the eight coordinated Mo atoms in 1T-MoS2. The results indicated that the 1T- and 2H-phases co-existed in MoS2, that is, it is one 1T–2H mixture structure. The peak at 992 cm−1 (2 × E12g) belonged to the asymmetric stretching vibration, vas, of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(1) model.33 This may be due to the slight oxidation of the unsaturated Mo and S atoms or the trace incomplete reduction of Mo(VI) during the commercial synthesis of MoS2. Besides, the model (2 × A1g) appeared at 818 cm−1. After treatment at 300 °C, the positions of A1g, E12g and 2 × LA in the sample tended to be smaller wavenumbers, and the strength ratio of A1g and E12g was decreased from 1.87 to 1.72, implying that part of the Mo–S bond was converted from outside to inside. The strength of 2 × E12g and 2 × A1g in the MoS2-300 catalyst became stronger, hinting that more MoO3 species appeared on the surface of the catalyst due to the oxidation during heating treatment. Some new characteristic peaks such as 922 and 778 cm−1, were found in the MoS2-300 catalyst, implying the formation of a new Mo species. In a word, the above-mentioned results showed that the structure of the treated catalyst was partly altered.
O(1) model.33 This may be due to the slight oxidation of the unsaturated Mo and S atoms or the trace incomplete reduction of Mo(VI) during the commercial synthesis of MoS2. Besides, the model (2 × A1g) appeared at 818 cm−1. After treatment at 300 °C, the positions of A1g, E12g and 2 × LA in the sample tended to be smaller wavenumbers, and the strength ratio of A1g and E12g was decreased from 1.87 to 1.72, implying that part of the Mo–S bond was converted from outside to inside. The strength of 2 × E12g and 2 × A1g in the MoS2-300 catalyst became stronger, hinting that more MoO3 species appeared on the surface of the catalyst due to the oxidation during heating treatment. Some new characteristic peaks such as 922 and 778 cm−1, were found in the MoS2-300 catalyst, implying the formation of a new Mo species. In a word, the above-mentioned results showed that the structure of the treated catalyst was partly altered.
Fig. 3 shows the XPS spectra of MoS2 and MoS2-X (X: 300 and 500 °C) catalysts, and some important results are also displayed in Table S1†. In the case of Mo 3d of MoS2, one characteristic peak with 226.80 eV of binding energy appeared, belonging to the sulfur atoms at the end of the Mo–S bond.30–34 The binding energies located at 229.60 and 232.75 eV corresponding to Mo 3d5/2 and Mo 3d3/2 were attributed to the characteristic peaks of Mo(IV).32 These peaks were further divided into four small peaks, corresponding to the characteristic phases of 2H-MoS2 (232.89 and 229.72 eV) and 1T-MoS2 (232.60 and 229.58 eV). Besides, one extremely weak characteristic peak was presented at 235.89 eV, which ascribed to the Mo-based oxides, in other words, it was attributed to the characteristic peaks of Mo(VI).37 As shown in Table S1†, the concentration of Mo(IV) and the ratio of Mo(IV) and Mo(VI) were both decreased after the treatment, especially at 500 °C. However, the ratio of 2H- and 1T-MoS2 first increased and then decreased. It hinted that Mo(IV) did not act as a main catalytic active site. In previous studies,30,32–34 most of the researchers considered that Mo(IV) was in favor of activating PMS. For S 2p of MoS2, there appeared two characteristic peaks located at 163.60 and 162.40 eV of binding energy, corresponding to S2− 2p1/2 and S2− 2p3/2. These peaks were subdivided into four small peaks, which were attributed to the characteristic phases of 2H-MoS2 (163.42 and 162.75 eV) and 1T-MoS2 (163.38 and 162.39 eV). Additionally, one very weak peak with 169.47 eV of binding energy was also observed. It was ascribed to the characteristic peak of SI (–SOn–) species, derived from the oxidation of MoS2.33 With the increase in temperature, the concentration of S2− species decreased, and even disappeared at 500 °C, whereas the concentration of SI species was remarkably increased. This meant that the oxidation of catalysts was extremely serious. For O 1s of MoS2, there appeared two characteristic peaks located at 532.99 and 532.10 eV of binding energy, corresponding to adsorbed molecular water and surface-adsorbed oxygen species,33 respectively. After the treatment at 300 °C, there was an additional characteristic peak located at 531.16 eV, which was attributed to the oxygen coordination like oxygen vacancies.33 It further confirmed that oxygen atoms were introduced by replacing sulfur atoms after the treatment. In a word, the changes in MoS2 appeared after the high-temperature treatment, including the formation of new vacancies and the concentration of different MoS2 phases.
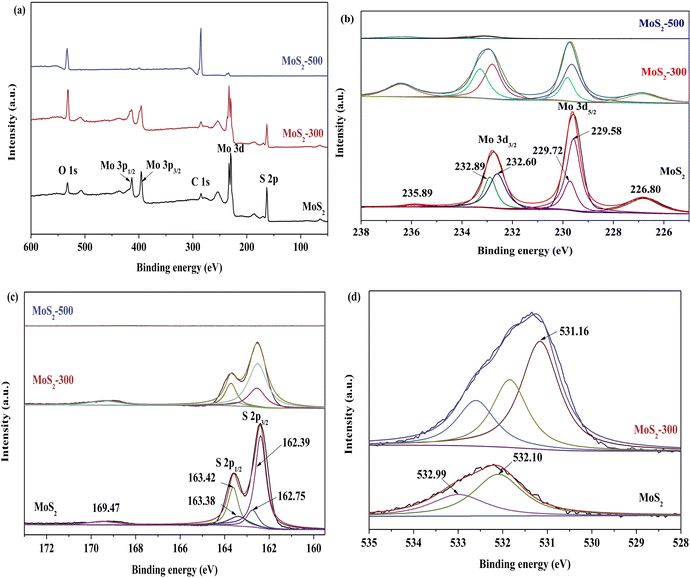 |
| | Fig. 3 XPS spectra of (a) full-range scan of a series of MoS2 catalysts, (b) Mo 3d, (c) S 2p, and (d) O 1s. | |
3.2. Removal of organic pollutants over various catalysts
3.2.1. Removal of RhB in the catalytic systems. Fig. 4 displays the removal of RhB by PMS activated with different MoS2 catalysts. The degradation efficiency of RhB was only about 11% after 30 min of reaction, while MoS2 was employed. After PMS was added, the degradation efficiency of RhB was increased up to 27% at 30 min, hinting that the MoS2-activated PMS could promote the degradation of organic pollutants in solutions. This is consistent with the results reported in the literature.30,32 However, this improvement was limited. In order to further enhance the removal of RhB, MoS2-300 was selected as a heterogeneous catalyst to activate PMS. The degradation efficiency of RhB quickly reached ca. 84% at 30 min. In order to verify the role of MoS2-300, the adsorption experiment was carried out to confirm the contribution of adsorption in this reaction. The result indicated that the degradation efficiency of RhB was about 32% after 30 min of reaction, meaning that MoS2 with defect sites was beneficial for the removal of RhB. Du et al.32 found by using theoretical calculation (DFT) that the surface of MoS2 could strengthen the adsorbed PMS in the presence of sulfur vacancy, and meanwhile, the O–O bonds in PMS could be further extended to form reactive oxygen species. To demonstrate the roles of Mo and sulfur species, several Mo-based catalysts were used to activate PMS to degrade RhB (see Fig. 4(b)). As shown in this figure, while Mo or MoO3 acted as a heterogeneous catalyst, the degradation efficiency of RhB did not exceed 35%. According to the pseudo-first-order kinetics, the reaction rate constant (k) was calculated. As shown in Fig. 4(c), the k value of the MoS2-300/PMS system was remarkably higher than that of other reaction systems. The above-mentioned results indicated that the sulfur defect sites could indeed significantly strengthen the ability of degradation of RhB by activated PMS.
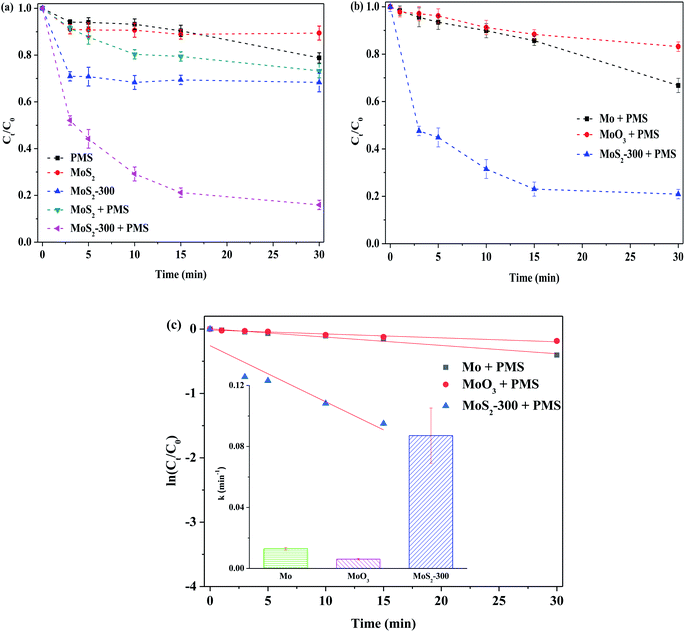 |
| | Fig. 4 Degradation of 10 mg L−1 RhB in different process in the darkness. (a) Adsorption and oxidation of MoS2 and MoS2-300. Reaction conditions: initial pH = 3.0, [PMS] = 1.0 mM, [catal.] = 1.0 g L−1. (b) Oxidation of different Mo-based catalysts. (c) Reaction rate constant of different oxidation systems. Reaction conditions: initial pH = 3.0, [PMS] = 0.5 mM, [catal.] = 1.0 g L−1. | |
Fig. S2† compares the removal of RhB by MoS2-300-activated PMS with or without LED illumination. The degradation efficiency of RhB was about 84% at 30 min in the darkness. After the darkness was switched into LED illumination, the degradation efficiency of RhB was about 86%. It showed that the presence or absence of light illumination had little effect on the removal of RhB. Generally, the defect sites are classified into bulk and surface defect sites. For the photocatalytic reaction, the bulk defect sites often acted as the combination center of photogenerated hole–electron pairs (h+–e−), which had negative influence on the degradation reaction. In this study, MoS2-300 was used as a photocatalyst. Under LED illumination, h+–e− was generated on the MoS2-300 catalyst, in which h+ could directly oxidize RhB, and meanwhile, e− was transferred for PMS and/or dissolved oxygen in the solution to generate reactive oxygen species. The above-mentioned synergy could compensate for the negative effect from the defect sites. Hence, the presence or absence of LED illumination hardly affected the degradation of RhB in this reaction.
Fig. 5(a) shows the removal of RhB by MoS2 and MoS2-X (X: 200, 300, 400 and 500 °C) at different temperatures activating PMS in the darkness. One can see that the degradation of RhB was low, while MoS2 was not treated by heat. With the increase in temperature, the degradation efficiency of RhB was increased. When the temperature arrived at 300 °C, the degradation efficiency of RhB reached the highest value. With the further increase in temperature, the degradation efficiency of RhB started to reduce. As depicted in Fig. 5(b), this change is well reflected on the basis of the relationship between treated temperature and k, showing that the reaction rate is closely related to the treatment temperature of the catalyst. This is because MoS2 is treated at high temperatures, resulting in the formation of defect sites, thus exposing more active sites. In this case, more reactive oxygen species are formed from PMS, which, in turn, enhance the degradation of RhB. The temperature is too high, in this case, the structure of MoS2 is destructed, as confirmed by XRD characterization (see Fig. 1(a)), and hence, it is disadvantageous for the activation of PMS. Normally, MoS2 began to be decomposed into MoO3 at 315 °C. However, the latter was less capable of activating PMS than the former, as evidenced from Fig. 4(b). Accordingly, the degradation efficiency of RhB was decreased, particularly the higher temperature. The degradation of RhB by PMS activated with MoS2-300 for different time periods in the darkness was studied, and the results are displayed in Fig. S3.† It was found that the time was in the range of 4–12 h, and the degradation efficiency of RhB over all these catalysts was maintained at 79–88% after the reaction. This meant that the time had little effect on the degradation of RhB.
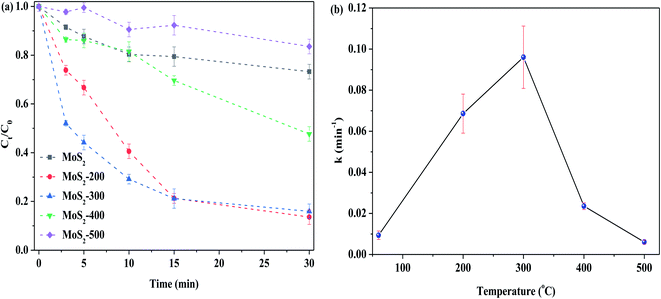 |
| | Fig. 5 (a) Effect of the calcined temperature on the degradation of RhB. (b) Relationship between calcined temperature and reaction rate constant. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [PMS] = 1.0 mM, [catal.] = 1.0 g L−1, in the darkness. | |
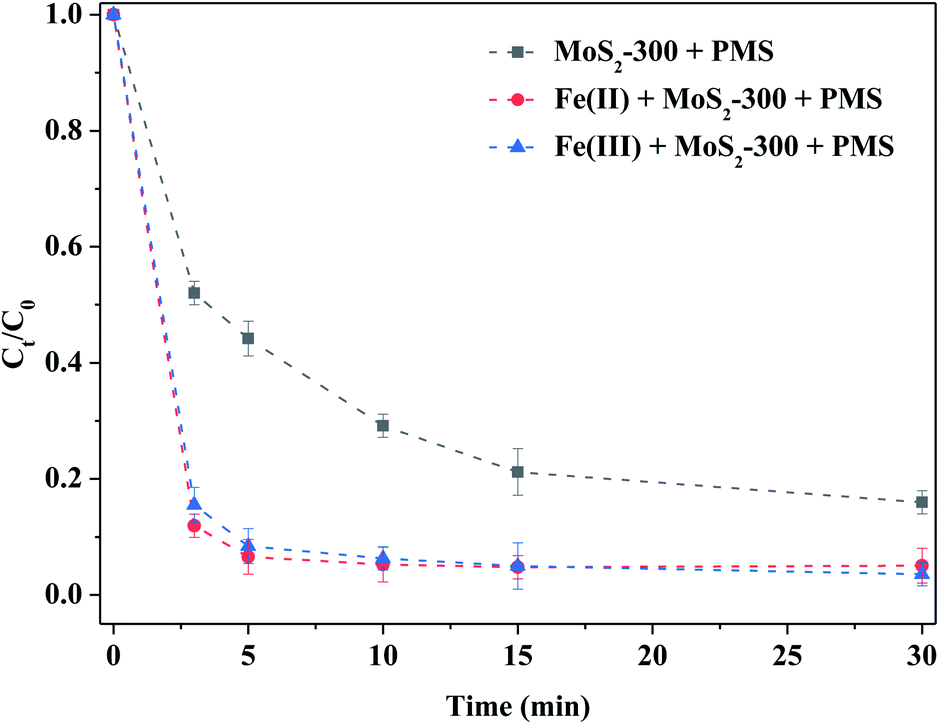
Fig. 6 shows the degradation of RhB by MoS2-300-activated PMS with homogeneous Fe(II) or Fe(III) ions. Compared to the control experiment, Fe(II) or Fe(III) could strengthen the degradation ability of RhB by the MoS2-300/PMS system. For example, the degradation efficiency of RhB reached 90% in both the reaction systems after 5.0 min of reaction, which was higher than that of the control experiment. Kuang et al.31 pointed out that the Fe(III) adsorption energy on MoS2 was −1.83 eV and −3.62 eV in the absence and presence of sulfur vacancies, respectively. That is to say, MoS2 with sulfur vacancies in thermodynamics was more favorable to adsorb Fe(III), and thus, promoted the reaction between Fe(III) and MoS2. As a result, the sulfur defect sites near MoS2-300 could accelerate the cycles of Fe(II)/Fe(III) to produce more reactive oxygen species, as shown in formulas (3-1)–(3-3).31 Consequently, Fe(II) or Fe(III) could boost the ability of removal of RhB by MoS2-300-activated PMS.
| | |
Mo(IV) + 2Fe(III) → Mo(VI) + 2Fe(II)
| (3-1) |
| | |
Mo(VI) + 2HSO5− → Mo(IV) + 2SO5˙− + 2H+
| (3-2) |
| | |
Fe(II) + HSO5− → Fe(III) + OH− + SO4˙−
| (3-3) |
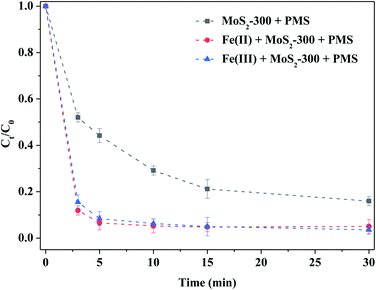 |
| | Fig. 6 Comparison of homogeneous Fe ions activating PMS in the degradation of RhB. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [PMS] = 1.0 mM, [MoS2-300] = 1.0 g L−1, [Fe(II)] = [Fe(III)] = 0.20 mM, in the darkness. | |
Fig. S4† demonstrates the removal of RhB by different oxidants activated with MoS2-300. It is found that the degradation efficiency of RhB was about 80%, while PMS was used as an oxidant, and about 60% of RhB was obtained when PDS or H2O2 was employed as an oxidant. The above-mentioned results indicated that MoS2-300 has a strong activation ability toward typical oxidants. However, their differences were mainly due to the different properties of oxidants. The structure of PDS or H2O2 was symmetrical, whereas that of PMS was asymmetrical. In this case, PMS was easier to be activated than PDS and H2O2. In addition, the types of reactive oxygen species formed by PMS were different from those by PDS and H2O2 under the same conditions.33,34 For instances, while PMS was activated, the reactive oxygen species mainly included SO4˙−, ˙OH and 1O2. On the contrary, while PDS was activated, the main reactive oxygen species were ˙OH, O2˙− and 1O2. In general, SO4˙− had a higher catalytic oxidation capacity than that of ˙OH, O2˙− and 1O2. Besides, the repulsion between MoS2 and the oxidant played an important role. The surface of MoS2 exhibited negative charges,38 and thus, the repulsive force between PDS (S2O82−) and MoS2 became larger than that of PMS (HSO5−) with MoS2. As a result, the MoS2-300 catalyst-activated PMS possessed a higher degradation efficiency of RhB. Fig. S5† displays the removal of different organic dyes (including RhB, methyl blue (MB), methyl orange (MO) and orange II (AOII)) by MoS2-300 activating PMS in the darkness. Most of them were quickly degraded under the same reaction condition. It indicated that this catalytic system had a good removal ability for organic dyes in wastewater.
3.2.2. Optimized reaction conditions. Fig. 7(a) displays the removal of RhB by MoS2-300 activated at different initial pH values. Herein, no buffer solution was chosen, which avoided its interference. While the initial solution pH was 2.0, the degradation efficiency of RhB was about 88% at 30 min. The removal of RhB tended to decrease with the increase in the initial solution pH. In general, the degradation efficiency of RhB was maintained at 69–88% while the initial solution pH was in the range of 2.0–9.0. Obviously, the system possessed a wide pH range. The pKa value of PMS was 9.3;39 in other words, while the solution pH was above 9.3, PMS started to convert into SO52−. Clearly, the conversion of PMS was not one primary factor in the reduction degradation of RhB. While the acidic solution was used, the sulfur atoms on the corner of the MoS2 catalyst reacted with H+ in the solution, and therefore, the sulfur atoms were consumed.40 Concurrently, more sulfur vacancies were exposed, thereby accelerating the electron transfer between PMS and sulfur vacancies. Under alkaline conditions, the above-mentioned trapping reaction did not occur, thus preventing the exposure of sulfur vacancies. Therefore, the removal of RhB was decreased. Nevertheless, whether the initial reaction solution was acidic or alkaline, the final solution pH reached 2.0–3.5 after the reaction, as displayed in Fig. S6.† In this case, it was in favor of adsorbing RhB on the surface of the catalyst. It has been reported that the surface of MoS2 showed negative charges at pH = 2–9,41 and RhB was a cationic dye. Consequently, the MoS2-300 was favorable for the adsorption of RhB in a wide pH range. The roles of sulfur vacancies in MoS2-based catalysts after the high-temperature treatment and during the reaction were discussed. On the basis of the removal of RhB, the order of the catalytic activity is as follows: MoS2-300 > MoS2. The fact told that it was easier to activate PMS in the presence of vacancy in the catalyst, while the solution was in an acidic environment.
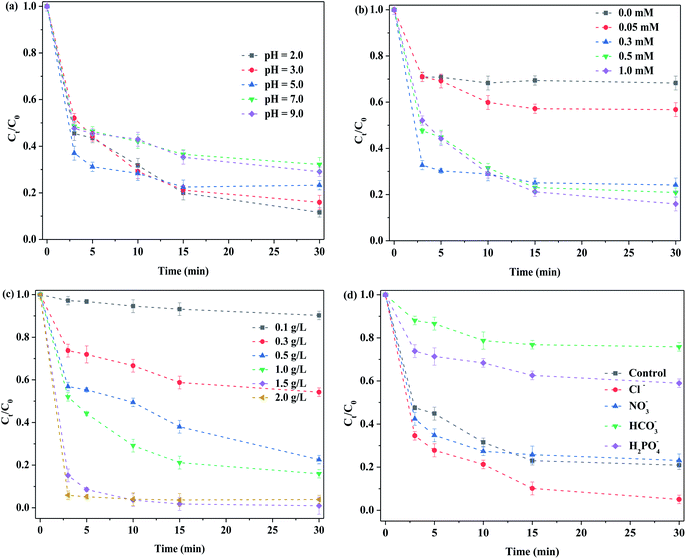 |
| | Fig. 7 Influence of reaction conditions on the degradation of RhB: (a) initial pH; (b) PMS concentration; (c) catalyst concentration; and (d) anions. Reaction conditions: [RhB] = 10 mg L−1, [anions] = 100 mM, in the darkness. | |
Fig. 7(b) demonstrates the effect of concentrations of PMS on the removal of RhB. While PMS was not added into the reaction solution, the degradation efficiency of RhB was about 32% in 30 min, which was mainly attributed to the adsorption of the catalyst. When the concentration of PMS was 0.05 mM, the degradation efficiency of RhB increased from 32% to 43%. It showed that MoS2-300 could activate PMS to form reactive oxygen species, which was beneficial for degrading RhB. The results meant that PMS itself had a limited ability to remove RhB. With the increase in the concentration of PMS, the removal of RhB was further increased. When 0.5 mM of PMS was employed, the degradation efficiency of RhB reached about 79%. By further increasing the concentration of PMS, the degradation efficiency of RhB was slightly increased up to 84%. More reactive oxygen species were generated with the increase in the concentration of PMS, and thus, the degradation efficiency of RhB was increased. The repulsive force between MoS2-300 and PMS was further enhanced, while the higher concentration of PMS was employed.31 Accordingly, the removal of RhB tended to be stable.
Fig. 7(c) displays the effect of dosages of the MoS2-300 catalyst on the removal of RhB. It was found that when the concentration of the catalyst was 0.1 g L−1, the degradation efficiency of RhB was only 9.8% at 30 min. With the increase in the dosage of the catalyst, the removal of RhB was increased. When the dosage of the catalyst arrived at 1.5 g L−1, the degradation efficiency of RhB was over 90% only for 5 min. With the further increase in the dosage of the catalyst, the removal of RhB became stable. The dosage of the catalyst was insufficient, and there were no enough active sites for activating PMS, and thus the removal of RhB was low. With the increase in the dosage of the catalyst, PMS activated by the catalyst produced more reactive oxygen species. Accordingly, the removal of RhB was increased. While the dosage of the catalyst was too high, the concentration of active sites was reduced due to the collision between catalyst particles. Moreover, a limited reactive oxygen species were formed because the PMS concentration was constant. Accordingly, the removal of RhB retained a little change.
Fig. 7(d) displays the influence of various anions on the removal of RhB. The degradation efficiency of RhB was about 80% without the addition of extra anions, while the reaction time was 30 min. While different anions were respectively added, they had different effects on the removal of RhB. For examples, after the addition of Cl−, the removal of RhB was further increased, probably due to the formation of more active species such as Cl˙ in the reaction system,42 as observed in formula (3-4). After the addition of NO3−, the removal of RhB was slightly decreased, which may be attributed to the formation of less reactive oxygen species, as shown in formula (3-5). Similar results were found.30 From this point of view, it is concluded that the main reactive oxygen species in this reaction system were not typical strong oxidizing species such as SO4˙− and ˙OH. However, when HCO3− or H2PO4− was added into the initial solution, the removal of RhB was inhibited in both cases, especially for HCO3−. For instance, for HCO3−, the degradation efficiency of RhB was only about 20% after 60 min of reaction. When the pH of the solution was increased to 8–9, HCO3− could neutralize most of H+ in the solution. As a result, the sulfur atoms in the corner of MoS2 were not captured. Therefore, the interaction between sulfur vacancies and PMS was significantly inhibited. For H2PO4−, the degradation efficiency of RhB was about 40% after 60 min of reaction. On the one hand, when H2PO4− was added, the pH of the solution was increased and it affected the binding of sulfur atoms on the edge of MoS2 by H+. On the other hand, the phosphate in H2PO4− could coordinate closely with Mo species on the MoS2 to form phosphomolybdate.39 In this case, it induced the steric hindrance effect and thus affected the activation of PMS. Accordingly, the removal of RhB was reduced, as compared to the control experiment.
| | |
Cl− + SO4˙− → Cl˙− + SO42−
| (3-4) |
| | |
NO3− + SO4˙− → NO3˙− + SO42−
| (3-5) |
3.2.3. Stability of the catalyst. Fig. 8 shows the stability of the catalyst. One can see that the degradation efficiency of RhB was about 80% after the first reaction. After the reaction, the catalyst was regenerated, that is, the reaction solution was filtered to achieve solid–liquid separation and then dried. Finally, it was calcined at 300 °C. The regenerated catalyst was added according to the reaction condition of the first reaction and then the second reaction was conducted. Under the same reaction condition, the degradation efficiency of RhB was about 80% after 30 min of the reaction. The above-mentioned steps were followed by the multiple reaction and regeneration. After the fifth reaction, the degradation efficiency of RhB still reached about 70% of the initial degradation efficiency. This indicated that the life of catalyst was long. It is noteworthy that the catalyst used in this experiment had to be treated at 300 °C. Under identical reaction conditions, the degradation efficiency of RhB was about 10% while the used catalyst after the first reaction was not calcined, as shown in Fig. S7(a).† Obviously, the reason of the deactivation of catalyst was to a great extent attributed to the covered and/or blocked by the intermediates or products. Besides, some sulfur vacancies were newly formed after the regeneration. Du et al.32 also found that the used MoS2 catalyst needed to be regenerated by H2O2 and the activity of the catalyst was almost recovered. It should be noted that the oxidation of MoS2 became increasingly serious during the reaction and regeneration. In this case, its structure and catalytic activity are difficult to fully be recovered, as evidenced in Fig. S2.† In this study, the initial catalytic activity could be restored by simple calcination without additional chemical treatment. Therefore, the regeneration used in this work had many advantages such as simplicity, safety and easy operation. After the reaction, the used catalyst was characterized by XRD and SEM techniques. As displayed in Fig. S7(b) and (c),† the phase and morphology of catalyst are basically the same before and after the reaction, suggesting that the structure of catalyst remained unchanged. After the reaction, the loss components of the catalyst were detected by the ICP-MS. It is observed from Table S2† that the concentrations of Mo and S elements in the solution were 36.2 and 91.6 mg L−1 after the reaction, respectively. Combined with the catalyst dosage, the loss efficiency of catalyst was 12.8%. This further confirmed that the catalyst possessed a good stability.
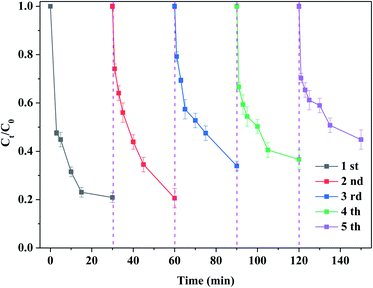 |
| | Fig. 8 Stability of the catalyst. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [PMS] = 0.5 mM, [MoS2-300] = 1.0 g L−1, in the darkness. | |
3.2.4. Identified reactive oxygen species on the removal of RhB. Fig. 9(a) displays the influence of methanol on the removal of RhB. Methanol (MeOH) could capture the SO4˙− and ˙OH species, as there is no significant difference in reaction rate constants.43 In addition, tert-butanol (TBA) was able to identify ˙OH species.31 While methanol with different concentrations (250 and 500 mM) was respectively employed in this system, the degradation efficiency of RhB was decreased from 79% to 48% and 43%. This implied the presence of SO4˙− and ˙OH in this reaction, and they contributed toward the removal of RhB. As shown in Fig. 9(b), while tert-butanol with different concentrations (250 and 500 mM) was used in the reaction solution, the degradation efficiency of RhB was decreased from 79% to 64% and 50%, respectively. It showed the existence of ˙OH in this reaction. Compared to SO4˙−, ˙OH played a greater role in the RhB degradation. The formation of SO4˙− species was derived from the interaction of Mo(IV) with PMS, whereas the ˙OH species originated from the reaction of SO4˙− with H2O, as shown in formulas (3-6) and (3-7):| | |
Mo(IV) + HSO5− → Mo(VI) + SO4˙− + OH−
| (3-6) |
| | |
SO4˙− + H2O → ˙OH + SO42− + H+
| (3-7) |
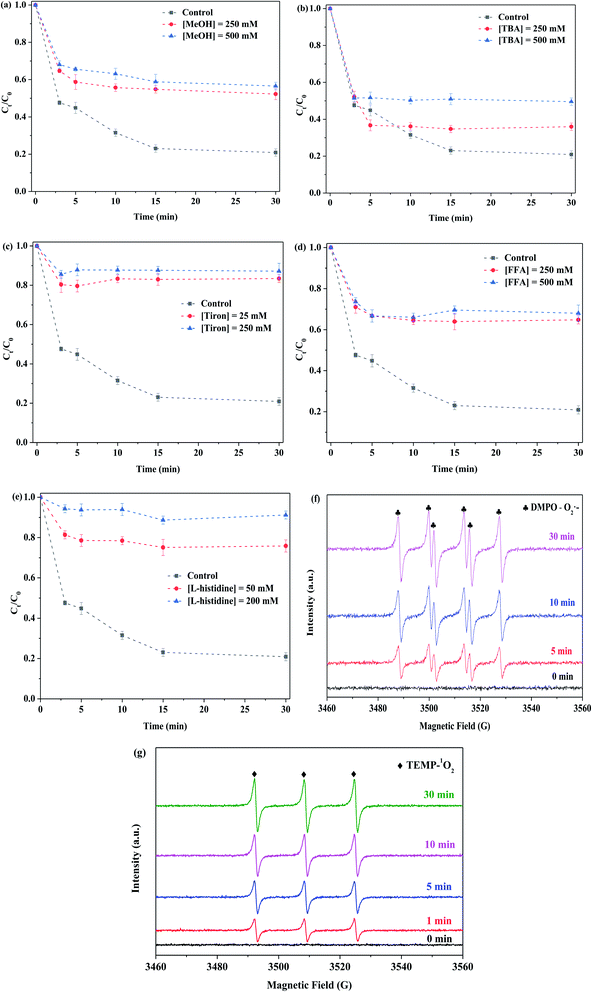 |
| | Fig. 9 Effect of quenchers (a)–(e). (f) and (g) EPR signal of the PMS activation system for the detection of O2˙− and 1O2 in the presence of DMPO and TEMP, respectively. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [PMS] = 0.5 mM, [MoS2-300] = 1.0 g L−1, in the darkness. | |
The influence of tiron on the RhB degradation was studied, and the results are shown in Fig. 9(c). The degradation efficiency of RhB quickly decreased from 79% to 17% and 13%, while tiron (25 and 250 mM) with different concentrations was respectively employed in the reaction solution. It showed that the reaction system contained a large number of O2˙− species, and meanwhile, it played an important role in the removal of RhB. The MoS2-300 catalyst was rich in sulfur defect sites. These vacancies had a good affinity for PMS in water and then could react with each other to generate reactive oxygen species.33 At the liquid–solid interface, the intermediates were finally evolved into O2˙−, as shown in formulas (3-8) and (3-9):
| |
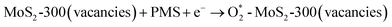 | (3-8) |
| |
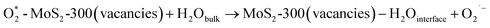 | (3-9) |
The effect of furfuryl alcohol on the RhB degradation was investigated. As shown in Fig. 9(d), furfuryl alcohol (FFA) with different concentrations (250 and 500 mM) was respectively used, and the degradation efficiency of RhB was reduced from 79% to 35% and 32%. Additionally, while L-histidine with different concentrations (50 and 200 mM) was employed in the reaction system, the degradation of RhB was also obviously decreased, as evidenced in Fig. 9(e). This indicated that the 1O2 species existed in this reaction system and played an important role in the degradation of organic pollutants. There were several formed sources of 1O2, and they are as follows: (i) O2˙− as an intermediate,33,43 as shown in formulas (3-10) and (3-11); (ii) the vacancies interacted with PMS;43 and (iii) O2˙− reduced Mo(VI),44 as shown in formulas (3-12):
| | |
2O2˙− + H2O → H2O2 + OH− + 1O2
| (3-10) |
| | |
O2˙− +˙OH + 2H+ → OH− + 1O2
| (3-11) |
| | |
Mo(VI) + O2˙− + 2H+ → Mo(IV) + 1O2
| (3-12) |
In order to confirm the presence of O2˙− and 1O2 in the reaction system, EPR was used to probe the generation of reactive oxygen species. DMPO (methanol as a solvent) was employed to detect the existence of O2˙−, and TEMP was applied for capturing 1O2. The EPR spectrum shown in Fig. 9(f) displays four big peaks and two small peaks for DMPO–O2˙− spin adducts, and the intensity of the relative signals became stronger with the increase in reaction time. Similarly, the remarkable signal for the TEMPO was also found and the corresponding intensity was increased with the prolonged reaction time, as shown in Fig. 9(g). The results indicated that the reaction system contained O2˙− and 1O2.
In a word, the reaction system included free radical species such as SO4˙−, ˙OH and O2˙−, and non-radical species like 1O2. Among them, O2˙− and 1O2 were the main reactive oxygen species, as demonstrated in formula (3-13). In order to verify the source of these species, the variation in the PMS concentration before and after the reaction was investigated, and the results are shown in Fig. S8.† It was found that the decomposition efficiency of PMS reached 100% after the reaction. This indicated that the catalyst had a good activation ability toward PMS, and the source of reactive oxygen species was mainly the decomposition of PMS in this reaction.
| | |
RhB + SO4˙−/˙OH/O2˙−/1O2 → intermediates + CO2 + H2O
| (3-13) |
4. Conclusion
In summary, MoS2 with defect sites has been synthesized by high-temperature treatment. The temperature affected the change in MoS2 phases. It could promote the removal of RhB by activating PMS due to the existence of sulfur vacancies. In a wide range of initial pH, the catalytic system maintained a good degradation capacity. In addition, the foreign anions had different influences on the removal of RhB. Among them, Cl− promoted the degradation reaction, whereas HCO3− suppressed the removal of RhB. After running several reaction and regeneration cycles, the catalyst still retained good stability. It is confirmed that O2˙− and 1O2 were the main active oxygen species in this reaction. It offered a new reaction pathway for the removal of organic pollutants in wastewater.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Hunan Provincial Natural Science Foundation of China (No. 2021JJ50012; 2022JJ50156), Open Project of State Key Laboratory of Safety and Health for Metal Mines (No. 2019-JSKSSYS-04) and Open Project of Fujian Provincial Key Lab of Coastal Basin Environment (Fujian Polytechnic Normal University) (No. S1-KF2010).
References
- S. Zhou, P. Gu, H. Wan, Y. Zhu, N. Li, D. Chen, A. Marcomini, Q. Xu and J. Lu, Preparation of new triptycene- and pentiptycene-based crosslinked polymers and their adsorption behavior towards aqueous dyes and phenolic organic pollutants, Sep. Purif. Technol., 2021, 278, 119495 CrossRef.
- N. Li, W. Wang, C. Ma, L. Zhu, X. Chen, B. Zhang and C. Zhong, A novel conductive rGO/ZnO/PSF membrane with superior water flux for electrocatalytic degradation of organic pollutants, J. Membr. Sci., 2022, 641, 119901 CrossRef CAS.
- T. Zhang, H. Zhu and J.-P. Croué, Production of sulfate radical from peroxymonosulfate induced by a magnetically separable CuFe2O4 spinel in water: efficiency, stability, and mechanism, Environ. Sci. Technol., 2013, 47(6), 2784–2791 CrossRef CAS PubMed.
- T. Zhang, Y. Chen and T. O. Leiknes, Oxidation of refractory benzothiazoles with PMS/CuFe2O4: kinetics and transformation intermediates, Environ. Sci. Technol., 2016, 50(11), 5864–5873 CrossRef CAS.
- X. Ruan, S. Zhou, C. Yin, J. Bai, X. Zhang, A. Khan, A. Xu and X. Li, Insight into the catalytic performance of silver oxides towards peroxymonosulfate activation for pollutants degradation: efficiency, mechanism and stability, Colloids Surf., A, 2022, 642, 128674 CrossRef CAS.
- H. Milh, D. Cabooter and R. Dewil, Role of process parameters in the degradation of sulfamethoxazole by heat-activated peroxymonosulfate oxidation: radical identification and elucidation of the degradation mechanism, Chem. Eng. J., 2021, 422, 130457 CrossRef CAS.
- F. Yang, Y. Huang, C. Fang, Y. Xue, L. Ai, J. Liu and Z. Wang, Peroxymonosulfate/base process in saline wastewater treatment: the fight between alkalinity and chloride ions, Chemosphere, 2018, 199, 84–88 CrossRef CAS PubMed.
- M. Chen, L. Zhu, S. Liu, R. Li, N. Wang and H. Tang, Efficient degradation of organic pollutants by low-level Co2+ catalyzed homogeneous activation of peroxymonosulfate, J. Hazard. Mater., 2019, 371, 456–462 CrossRef CAS PubMed.
- L. Wang, H. Xu, N. Jiang, Z. Wang, J. Jiang and T. Zhang, Trace cupric species triggered decomposition of peroxymonosulfate and degradation of organic pollutants: Cu(III) being the primary and selective intermediate oxidant, Environ. Sci. Technol., 2020, 54(7), 4686–4694 CrossRef CAS PubMed.
- J. Li, M. Li, H. Sun, Z. Ao, S. Wang and S. Liu, Understanding of the oxidation behavior of benzyl alcohol by peroxymonosulfate via carbon nanotubes activation, ACS Catal., 2020, 10(6), 3516–3525 CrossRef CAS.
- A. E. ElMetwally, F. Goodarzi, K. K. Meier, E. M. Zahran, S. Rayat, S. Kegnæs, M. R. Knecht and L. G. Bachas, Cu2S @Bi2S3 double-shelled hollow cages as a nanocatalyst with substantial activity in peroxymonosulfate activation for atrazine degradation, ACS Appl. Nano Mater., 2021, 4(11), 12222–12234 CrossRef CAS.
- Y. Bao, W. Yan, P.-P. Sun, J. Z. Y. Seow, S. K. Lua, W. J. Lee, Y. N. Liang, T.-T. Lim, Z. J. Xu, K. Zhou and X. Hu, Unexpected intrinsic catalytic function of porous boron nitride nanorods for highly efficient peroxymonosulfate activation in water treatment, ACS Appl. Mater. Interfaces, 2022, 14(16), 18409–18419 CrossRef CAS PubMed.
- L. Dong, Y. Li, D. Chen, X. Chen and D. Zhang, Facilitated activation of peroxymonosulfate by loading ZIF-8 on Fe3O4-MnO2 for deep mineralization of bisphenol A, ACS ES&T Water, 2021, 1(2), 417–429 Search PubMed.
- J. Qi, X. Yang, P.-Y. Pan, T. Huang, X. Yang, C.-C. Wang and W. Liu, Interface engineering of Co(OH)2 nanosheets growing on the KNbO3 perovskite based on electronic structure modulation for enhanced peroxymonosulfate activation, Environ. Sci. Technol., 2022, 56(8), 5200–5212 CrossRef CAS PubMed.
- X. Mi, H. Zhong, H. Zhang, S. Xu, Y. Li, H. Wang, S. Zhan and J. C. Crittenden, Facilitating redox cycles of copper species by pollutants in peroxymonosulfate activation, Environ. Sci. Technol., 2022, 56(4), 2637–2646 CrossRef CAS PubMed.
- L. Zhao, J. Zhang, Z. Zhang, T. Wei, J. Wang, J. Ma, Y. Ren and H. Zhang, Co3O4 crystal plane regulation to efficiently activate peroxymonosulfate in water: the role of oxygen vacancies, J. Colloid Interface Sci., 2022, 623, 520–531 CrossRef CAS PubMed.
- W. Wang, Y. Liu, Y. Yue, H. Wang, G. Cheng, C. Gao, C. Chen, Y. Ai, Z. Chen and X. Wang, The confined interlayer growth of ultrathin two-dimensional Fe3O4 nanosheets with enriched oxygen vacancies for peroxymonosulfate activation, ACS Catal., 2021, 11(17), 11256–11265 CrossRef CAS.
- L. Liu, Q. Liu, Y. Wang, J. Huang, W. Wang, L. Duan, X. Yang, X. Yu, X. Han and N. Liu, Nonradical activation of peroxydisulfate promoted by oxygen vacancy-laden NiO for catalytic phenol oxidative polymerization, Appl. Catal., B, 2019, 254, 166–173 CrossRef CAS.
- L. Kong, G. Fang, Z. Fang, Y. Zou, F. Zhu, D. Zhou and J. Zhan, Peroxymonosulfate activation by localized electrons of ZnO oxygen vacancies for contaminant degradation, Chem. Eng. J., 2021, 416, 128996 CrossRef CAS.
- P. Gao, X. Tian, Y. Nie, C. Yang, Z. Zhou and Y. Wang, Promoted peroxymonosulfate activation into singlet oxygen over perovskite for ofloxacin degradation by controlling the oxygen defect concentration, Chem. Eng. J., 2019, 359, 828–839 CrossRef CAS.
- J. You, W. Sun, S. Su, Z. Ao, C. Liu, G. Yao and B. Lai, Degradation of bisphenol A by peroxymonosulfate activated with oxygen vacancy modified nano-NiO-ZnO composite oxides: a typical surface-bound radical system, Chem. Eng. J., 2020, 400, 125915 CrossRef CAS.
- L. Wu, Z. Sun, Y. Zhen, S. Zhu, C. Yang, J. Lu, Y. Tian, D. Zhong and J. Ma, Oxygen vacancy-induced nonradical degradation of organics: critical trigger of oxygen (O2) in the Fe-Co LDH/peroxymonosulfate system, Environ. Sci. Technol., 2021, 55(22), 15400–15411 CrossRef CAS PubMed.
- X. Luo, Y. You, M. Zhong, L. Zhao, Y. Liu, R. Qiu and Z. Huang, Green synthesis of manganese-cobalt-tungsten composite oxides for degradation of doxycycline via efficient activation of peroxymonosulfate, J. Hazard. Mater., 2022, 426, 127803 CrossRef CAS PubMed.
- Z. Wan, Z. Xu, Y. Sun, M. He, D. Hou, X. Cao and D. C. W. Tsang, Critical impact of nitrogen vacancies in nonradical carbocatalysis on nitrogen-doped graphitic biochar, Environ. Sci. Technol., 2021, 55(10), 7004–7014 CrossRef CAS.
- G. Wang, Y. Zhao, H. Ma, C. Zhang, X. Dong and X. Zhang, Enhanced peroxymonosulfate activation on dual active sites of N vacancy modified g-C3N4 under visible-light assistance and its selective removal of organic pollutants, Sci. Total Environ., 2021, 756, 144139 CrossRef CAS PubMed.
- Y. Huang, L. Lai, W. Huang, H. Zhou, J. Li, C. Liu, B. Lai and N. Li, Effective peroxymonosulfate activation by natural molybdenite for enhanced atrazine degradation: role of sulfur vacancy, degradation pathways and mechanism, J. Hazard. Mater., 2022, 435, 128899 CrossRef CAS PubMed.
- L. Wu, P. Guo, X. Wang, H. Li, X. Zhang, K. Chen and P. Zhou, The synergy of sulfur vacancies and heterostructure on CoS@FeS nanosheets for boosting the peroxymonosulfate activation, Chem. Eng. J., 2022, 446, 136759 CrossRef CAS.
- C. Zhou, L. Zhu, L. Deng, H. Zhang, H. Zeng and Z. Shi, Efficient activation of peroxymonosulfate on CuS@MIL-101(Fe) spheres featured with abundant sulfur vacancies for coumarin degradation: performance and mechanisms, Sep. Purif. Technol., 2021, 276, 119404 CrossRef CAS.
- L. Wu, P. Guo, X. Wang, H. Li, A. Li and K. Chen, Mechanism study of CoS2/Fe(III)/peroxymonosulfate catalysis system: the vital role of sulfur vacancies, Chemosphere, 2022, 288, 132646 CrossRef CAS PubMed.
- X. Li, Y. Guo, L. Yan, T. Yan, W. Song, R. Feng and Y. Zhao, Enhanced activation of peroxymonosulfate by ball-milled MoS2 for degradation of tetracycline: boosting molybdenum activity by sulfur vacancies, Chem. Eng. J., 2022, 429, 132234 CrossRef CAS.
- H. Kuang, Z. He, M. Li, R. Huang, Y. Zhang, X. Xu, L. Wang, Y. Chen and S. Zhao, Enhancing co-catalysis of MoS2 for persulfate activation in Fe3+-based advanced oxidation processes via defect engineering, Chem. Eng. J., 2021, 417, 127987 CrossRef CAS.
- M. Du, Q. Yi, J. Ji, Q. Zhu, H. Duan, M. Xing and J. Zhang, Sustainable activation of peroxymonosulfate by the Mo(IV) in MoS2 for the remediation of aromatic organic pollutants, Chin. Chem. Lett., 2020, 31(10), 2803–2808 CrossRef CAS.
- J.-C. E. Yang, M.-P. Zhu, X. Duan, S. Wang, B. Yuan and M.-L. Fu, The mechanistic difference of 1T-2H MoS2 homojunctions in persulfates activation: structure-dependent oxidation pathways, Appl. Catal., B, 2021, 297, 120460 CrossRef CAS.
- H. Zhou, L. Lai, Y. Wan, Y. He, G. Yao and B. Lai, Molybdenum disulfide (MoS2): a versatile activator of both peroxymonosulfate and persulfate for the degradation of carbamazepine, Chem. Eng. J., 2020, 384, 123264 CrossRef CAS.
- S. Karmakar, S. Biswas and P. Kumbhakar, A comparison of temperature dependent photoluminescence and photo-catalytic properties of different MoS2 nanostructures, Appl. Surf. Sci., 2018, 455, 379–391 CrossRef CAS.
- W. Liu, Q. Hu, F. Mo, J. Hu, Y. Feng, H. Tang, H. Ye and S. Miao, Photo-catalytic degradation of methyl orange under visible light by MoS2 nanosheets produced by H2SiO3 exfoliation, J. Mol. Catal. A: Chem., 2014, 395, 322–328 CrossRef CAS.
- B. M. Garland, N. Fairley, N. C. Strandwitz, R. Thorpe, P. Bargiela and J. Baltrusaitis, A study of in situ reduction of MoO3 to MoO2 by X-ray Photoelectron Spectroscopy, Appl. Surf. Sci., 2022, 598, 153827 CrossRef CAS.
- W. M. Divigalpitiya, R. F. Frindt and S. R. Morrison, Inclusion systems of organic molecules in restacked single-layer molybdenum disulfide, Science, 1989, 246, 369–371 CrossRef CAS PubMed.
- J. F. Goodman and P. Robson, Decomposition of inorganic peroxyacids in aqueous alkali, J. Chem. Soc., 1963, 534, 2871–2875 RSC.
- M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng, Y. Zhou, J. Zhang and Y. Yin, Metal sulfides as excellent co-catalysts for H2O2 decomposition in advanced oxidation processes, Chem, 2018, 4, 1359–1372 CAS.
- H. Luo, Y. Cheng, Y. Zeng, K. Luo, D. He and X. Pan, Rapid removal of organic micropollutants by heterogeneous peroxymonosulfate catalysis over a wide pH range: performance, mechanism and economic analysis, Sep. Purif. Technol., 2020, 248, 117023 CrossRef CAS.
- Y. Hong, J. Peng, X. Zhao, Y. Yan, B. Lai and G. Yao, Efficient degradation of atrazine by CoMgAl layered double oxides catalyzed peroxymonosulfate: optimization, degradation pathways and mechanism, Chem. Eng. J., 2019, 370, 354–363 CrossRef CAS.
- J. Ye, J. Dai, D. Yang, C. Li, Y. Yan and Y. Wang, Interfacial engineering of vacancy-rich nitrogen-doped FexOy@MoS2 co-catalytic carbonaceous beads medicated non-radicals for fast catalytic oxidation, J. Hazard. Mater., 2022, 421, 126715 CrossRef CAS PubMed.
- X. Zhou, H. Luo, B. Sheng, X. Chen, Y. Wang, Q. Chen and J. Zhou, Cu2+/Cu+ cycle promoted PMS decomposition with the assistance of Mo for the degradation of organic pollutant, J. Hazard. Mater., 2021, 411, 125050 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Lei Caic,
Chao Xiec,
Gang Lia and
Tian-Jiao Jiangc
*abc,
Lei Caic,
Chao Xiec,
Gang Lia and
Tian-Jiao Jiangc