
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–antitumor activity relationship of hybrid acetogenins focusing on connecting groups between heterocycles and the linker moiety†
Kaito Ohtaa,
Tetsuya Fushimib,
Mutsumi Okamurac,
Akinobu Akatsukac,
Shingo Danc,
Hiroki Iwasakia,
Masayuki Yamashitaa and
Naoto Kojima *a
*a
aKyoto Pharmaceutical University, 1 Misasagi-Shichono-cho, Yamashina-ku, Kyoto 607-8412, Japan. E-mail: kojima@mb.kyoto-phu.ac.jp
bGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
cCancer Chemotherapy Center, Japanese Foundation for Cancer Research, 3-8-1 Ariake-ku, Tokyo 135-8550, Japan
First published on 25th May 2022
Abstract
We studied hybrid molecules of annonaceous acetogenins and mitochondrial complex I-inhibiting insecticides to develop a novel anticancer agent. A structure–antitumor activity relationship study focusing on the connecting groups between the heterocycles and the linker moiety bearing the tetrahydrofuran moiety was conducted. Eleven hybrid acetogenins with 1-methylpyrazole instead of γ-lactone were synthesized and their growth inhibitory activities against 39 human cancer cell lines were evaluated. The nitrogen atom at the 2′-position of the linker moiety was essential for inhibiting cancer growth. The 1-methylpyrazole-5-sulfonamide analog showed potent growth inhibition of NCI-H23, a human lung cancer cell line, in a xenograft mouse assay without critical toxicity. Hence, the results of this study may pave the way for the development of novel anticancer agents, with both selective and broad anticancer activities.
Introduction
Cancer is the leading cause of mortality worldwide, accounting for approximately 10 million deaths in 2020.1 Cancer treatment typically includes chemotherapy, radiation therapy, and/or surgery. More recently, immunotherapy has also been employed for the treatment of cancer. Unfortunately, several other types of cancers remain difficult to treat with known anticancer drugs. Therefore, the development of drugs with novel mechanisms is crucial. Recently, examination of the inhibitors of mitochondrial complex I (reduced nicotinamide-adenine dinucleotide-ubiquinone oxidoreductase) has garnered significant interest.2 For example, Kawada and co-workers reported that mitochondrial complex I inhibitors selectively suppress tumor growth through concomitant acidification of the intra- and extra-cellular environment since tumor tissues comprise more stromal cells, whose energy production depends on oxidative phosphorylation in the mitochondria, rather than normal tissues.3 One such inhibitor, darinaparsin, has been submitted to regulatory authorities designated as a treatment for relapsed or refractory peripheral T-cell lymphoma in June 2021.4Owing to our interest in complex I inhibitors, we have studied the structure–activity relationship (SAR) of annonaceous acetogenins and their analogs.5 Annonaceous acetogenins are polyketides isolated from Annona plants, which show antitumor activity against human cancer cells by inhibiting mitochondrial complex I.6,7 Most of them have one to three tetrahydrofuran (THF) rings in the center of the long hydrocarbon chain and an α,β-unsaturated-γ-lactone ring at the end of the molecule. In contrast, mitochondrial complex I-inhibiting insecticides (e.g., tebufenpyrad) typically comprise N-containing heterocycles with hydrophobic side chains (Fig. 1).8
 | ||
| Fig. 1 Synthesis of hybrid acetogenin 1-methylpyrazole analogs. | ||
We hypothesized that an acetogenin analog with an N-containing heterocycle in place of the γ-lactone ring9 can act as a novel antitumor agent because the structural features of acetogenins and insecticides are similar; in addition, both are known to bind to the ND1 subunit in the mitochondria.10 Solamin was selected as a mother compound because it is a mono-THF acetogenin with a simple structure; however, it comprises all characteristic components of acetogenins.11 We have previously reported that a hybrid12 analog 1 with 1-methylpyrazole showed 80-fold higher growth inhibitory activity against NCI-H23, a human lung cancer cell line, than solamin.13 Subsequently, the effect of the connecting group between heterocycles and hydrophobic parts bearing the THF moiety on their biological activities was examined because 1-methylpyrazole groups of most insecticides are linked to the hydrophobic sections with an amide group. Preliminary data have shown that the connecting group significantly influences the biological activity; for instance, an analog linked via an amide group has a potent growth inhibitory activity against some human cancer cell lines.14 This study reports (1) the synthesis of hybrid acetogenins with sulfonamide and ketomethylene bond as an amido bond's bioisosteres, (2) analysis of the effects of connecting groups on growth inhibitory activities against human cancer cell lines, (3) the effects of substituents on 1-methylpyrazole, and (4) in vivo mice xenograft studies.
Results and discussion
Synthesis of hybrid acetogenins
The synthesis of all analogs 7a–7k was initiated using a common α-tetrahydrofuranyl aldehyde 2 prepared by a method previously reported by us.15 Scheme 1 illustrates the synthesis of analogs 7a–7h. Under Carreira's conditions,16 the asymmetric alkynylation of 2 with 10-azido-1-decyne 3 resulted in a high yield of propargyl alcohol 4, which shows a high diastereoselectivity; this threo/trans/threo stereochemical configuration on 4 is the same as that of the stereoisomer, which is reported to show the most potent anti-tumor activity.17 The reduction of azide and the alkyne moiety of 4 was simultaneously carried out to obtain primary amine 5. Then, amine 5 was condensed to 1-methylpyrazole-5-carboxylic acid with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), resulting in a good yield of amide 6a. Substituted pyrazole-5-carboxamides 6b–6c and pyrazole-4-carboxamide 6d were also prepared using a similar procedure, resulting in moderate yields. The sulfonamides 6e–6f were synthesized from 5 using the corresponding sulfonyl chlorides in the presence of Et3N. Deprotection of tert-butyldimethylsilyl (TBS) ether in 6a–6f under acidic conditions resulted in moderate to good yields of the desired analogs 7a–7f. N-Methyl amide 7g was synthesized by N-methylation of 7a using methyl iodide under basic conditions. The reduction of 7a using BH3·SMe2 in THF under reflux conditions produced amine 7h, albeit with a low yield.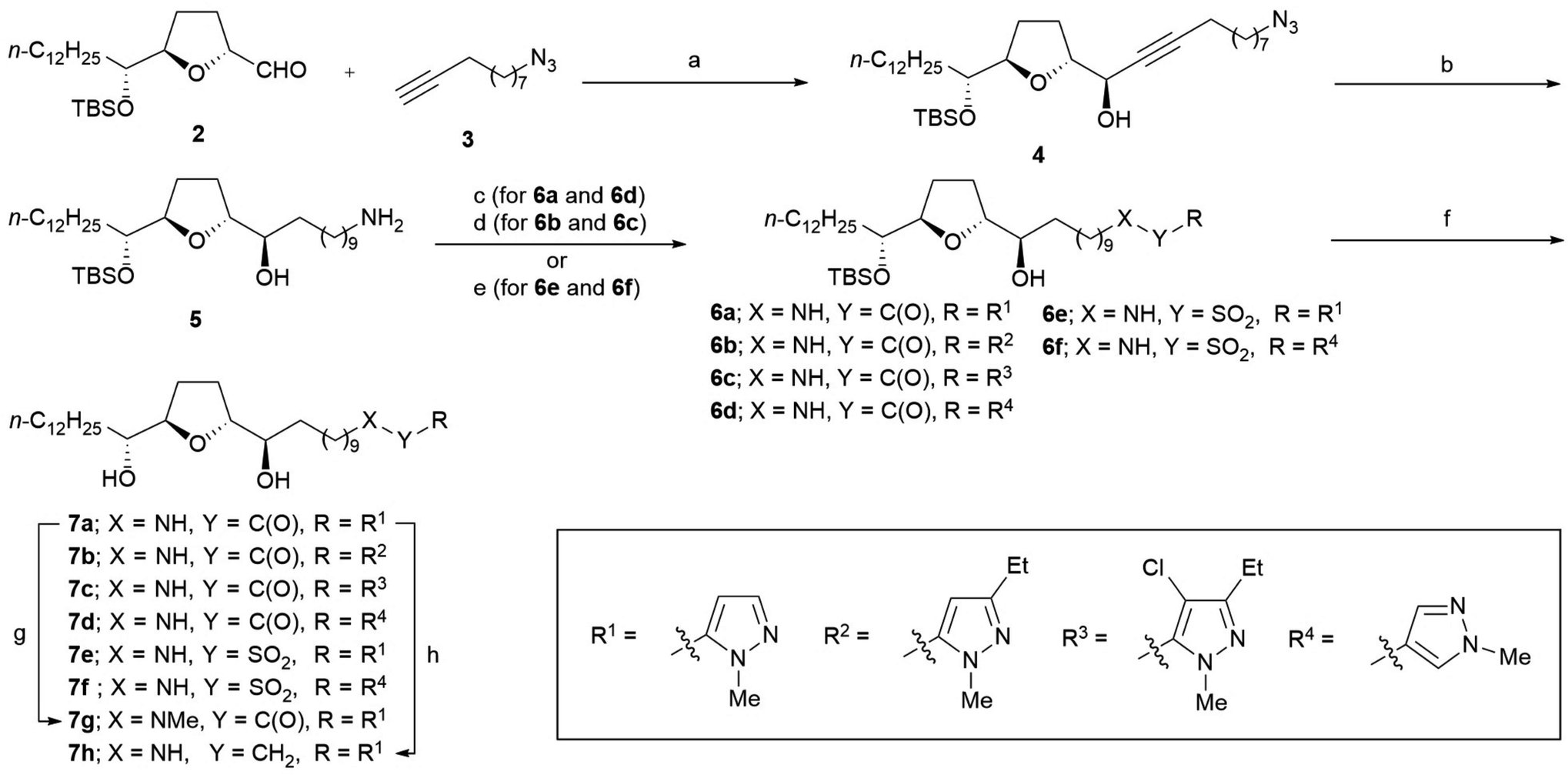 | ||
Scheme 1 Synthesis of analogs 7a–7h. Reagents and conditions: (a) Zn(OTf)2, (1R,2S)-(−)-N-methylephedrine, Et3N, toluene, rt, 84%, dr = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4; (b) H2 (3 atm), Pd(OH)2–C, EtOAc, rt, 79%; (c) R-COOH, EDC, DMAP, THF, rt, 6a (85%); 6d (51%); (d) R-COOH, DCC, DMAP, THF, rt; (e) R-SO2Cl, Et3N, CH2Cl2, rt, 6e (73%); 6f (73%); (f) 48% HF aq., THF/MeCN, rt, 7a (75%); 7b (35% from 4); 7c (50% from 4); 7d (quant.); 7e (93%); 7f (96%); (g) MeI, NaH, DMF, 0 °C, 60%; (h) BH3·SMe2, THF, reflux, 27%. DCC = N,N′-dicyclohexylcarbodiimide, DMAP = N,N-dimethylaminopyridine, EDC = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide. :4; (b) H2 (3 atm), Pd(OH)2–C, EtOAc, rt, 79%; (c) R-COOH, EDC, DMAP, THF, rt, 6a (85%); 6d (51%); (d) R-COOH, DCC, DMAP, THF, rt; (e) R-SO2Cl, Et3N, CH2Cl2, rt, 6e (73%); 6f (73%); (f) 48% HF aq., THF/MeCN, rt, 7a (75%); 7b (35% from 4); 7c (50% from 4); 7d (quant.); 7e (93%); 7f (96%); (g) MeI, NaH, DMF, 0 °C, 60%; (h) BH3·SMe2, THF, reflux, 27%. DCC = N,N′-dicyclohexylcarbodiimide, DMAP = N,N-dimethylaminopyridine, EDC = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide. | ||
The synthesis of inverse amide 7i was initiated via the same reaction as that used for the synthesis of 4 using methyl undec-10-ynoate 8 instead of 3 to obtain propargyl alcohol 9 with high diastereoselectivity (Scheme 2). Good yields of carboxylic acid 11 were obtained via hydrogenation of propargyl alcohol 9, followed by hydrolysis under basic conditions via methyl ester 10. Condensation of 11 with 5-amino-1-methylpyrazole produced amide 12 with a 74% yield. Inverse amide 7i was synthesized via deprotection of TBS ether of 12.
 | ||
| Scheme 2 Synthesis of inverse amide 7i; reagents and conditions: (a) Zn(OTf)2, (1R,2S)-(−)-N-methylephedrine, Et3N, toluene, rt, 77%, dr = 96:4; (b) H2 (1 atm), Pd(OH)2–C, EtOAc, rt, 96%; (c) 1N NaOH, MeOH, reflux, 82%; (d) 5-amino-1-methylpyrazole, EDC, DMAP, THF, 0 °C to rt, 74%; (e) 48% HF aq., THF/MeCN, rt, 88%. | ||
Scheme 3 illustrates the synthesis of the ester 7j. Ester 7j was obtained from 2 using the following steps: (1) asymmetric alkynylation with alkyne 13, (2) hydrogenation of the triple bond, (3) deprotection of TBS ethers, and (4) primary alcohol-selected condensation of 1-methylpyrazole-5-carboxylic acid.
 | ||
| Scheme 3 Synthesis of ester 7j; reagents and conditions: (a) Zn(OTf)2, (1R,2S)-(−)-N-methylephedrine, Et3N, toluene, rt, 68%, dr = 95:5; (b) H2 (3 atm), Pd(OH)2–C, EtOAc, rt, 82%; (c) 48% HF aq., THF/MeCN, rt, 84%; (d) 1-methylpyrazole-5-carboxylic acid, EDC, DMAP, THF, 0 °C to rt, 43% (67% brsm). | ||
Ketomethylene 7k was synthesized from 2 via the following steps: (1) asymmetric alkynylation with alkyne 17, (2) protection of secondary alcohol, (3) reductive deprotection of Bn ester and simultaneous reduction of the triple bond, (4) condensation of 5-iodo-1-methylpyrazole via Weinreb amide, and (5) deprotection of bis-TBS ethers (Scheme 4).
 | ||
| Scheme 4 Synthesis of ketomethylene 7k; reagents and conditions: (a) Zn(OTf)2, (1R,2S)-(−)-N-methylephedrine, Et3N, toluene, rt, 73%, dr = 92:8; (b) TBSOTf, 2,6-lutidine, CH2Cl2, rt, quant.; (c) H2 (1 atm), 10% Pd–C, EtOAc, rt, 96%; (d) N,O-dimethylhydroxylamine, EDC, Et3N, DMAP, CH2Cl2, rt, 94%; (e) 5-iodo-1-methylpyrazole, n-BuLi, THF, −30 °C to rt, 44% (66% brsm); (f) 48% HF aq., THF/MeCN, rt, quant. | ||
Evaluation of growth inhibitory activity against human cancer cells
The growth inhibitory activities of analogs 7a–7k against JFCR39,18 a panel of 39 human cancer cell lines were evaluated. The results are presented in Fig. 2. The activities of analogs 1, 7l,13 23, and solamin19 are shown for comparison. The x-axis represents the logarithm of 1/GI50 (50% growth inhibitory concentration) relative to the control values for the aforementioned 39 human cancer cell lines. | ||
| Fig. 2 Cell growth inhibition was assessed using the sulforhodamine B colorimetric assay by measuring changes in total cellular protein levels following 48 h of treatment with a given test compound. Fingerprints were obtained by calculating 50% growth inhibitory concentration (GI50) values for JFCR39 cell lines. The x-axis represents the logarithm of the 1/GI50 values for 39 human cancer cell lines. Absence of a bar signifies that the GI50 value of the corresponding cell line is higher than 10−4 M. CNS = central nervous system. | ||
First, our comparison of the activities of 1-methylpyrazole-5-yl analogs (1, 7a, 7e, and 7g–7k) revealed that the patterns could be classified into two categories: selective inhibition and broad inhibition. For instance, amide 7a showed selective and very potent activities against some cancer cells, including BSY-1, HCT-116, NCI-H23, DMS114, LOX-IMVI, MKN-B, and MKN-A, but did not inhibit the growth of 12 types of cells in this concentration range. The selective activity was also observed for alkyl 1, ester 7j, and ketomethylene 7k, although their potencies were different. Sulfonamide 7e, 1-methylamide 7g, and methylene amine 7h also showed potent inhibition of DMS114, MKN-B, and MKN-A; however, their selectivities were low because they exhibited moderate activities against all other cancer cells tested. The planarity and/or hydrogen-donating properties of the amide proton attributed to the introduction of the amide bond did not affect their growth inhibitory activity; however, these properties affected their selectivities for specific cancer cell lines. Interestingly, the activity of inverse amide 7i was deficient in contrast to that of amide 7a.
In terms of structural features, analogs with a nitrogen atom at the 2′-position of the side chain (group A: 7a, 7e, 7g, and 7h) tended to be significantly more active than the others (group B: 1 and 7i–7k) (Fig. 3A). These tendencies were observed against approximately half of the cell lines (for example, MKN-B in Fig. 3B; see also Fig. S1†), while activities were not observed against the remaining cell lines (e.g., MKN45 as shown in Fig. 3C; see also Fig. S1†). When the values of lipophilicity (XLOGP3)20 of the partial structure of analogs were calculated (Fig. 3F),21 the activities of the analogs in group A were correlated with their XLOGP3 values for most cells (Fig. S2†). For example, the relationship between the logarithm of 1/GI50 against NCI-H522 and XLOGP3 values can be expressed using linear equation (eqn (1)) (Fig. 3D). In eqn (1) and the following correlation equations, n refers to the number of compounds, r implies correlation coefficient, s refers to standard deviation, and the figures in parentheses indicate 95% confidence intervals. This equation allows for the design of novel analogs; analogs linked via boron amide might be promising since the XLOGP3 value is −0.57. In contrast, interestingly, compounds of group B showed an inverse correlation with those of group A. For example, the activities against NCI-H23 are represented by eqn (2) (Fig. 3E).
| Log(1/GI50) = −1.016(±1.326)XLOGP3 + 5.659(±0.407), n = 4, r2 = 0.845, s = 0.118 | (1) |
| Log(1/GI50) = 0.896(±0.438)XLOGP3 + 4.732(±0.365), n = 4, r2 = 0.975, s = 0.123 | (2) |
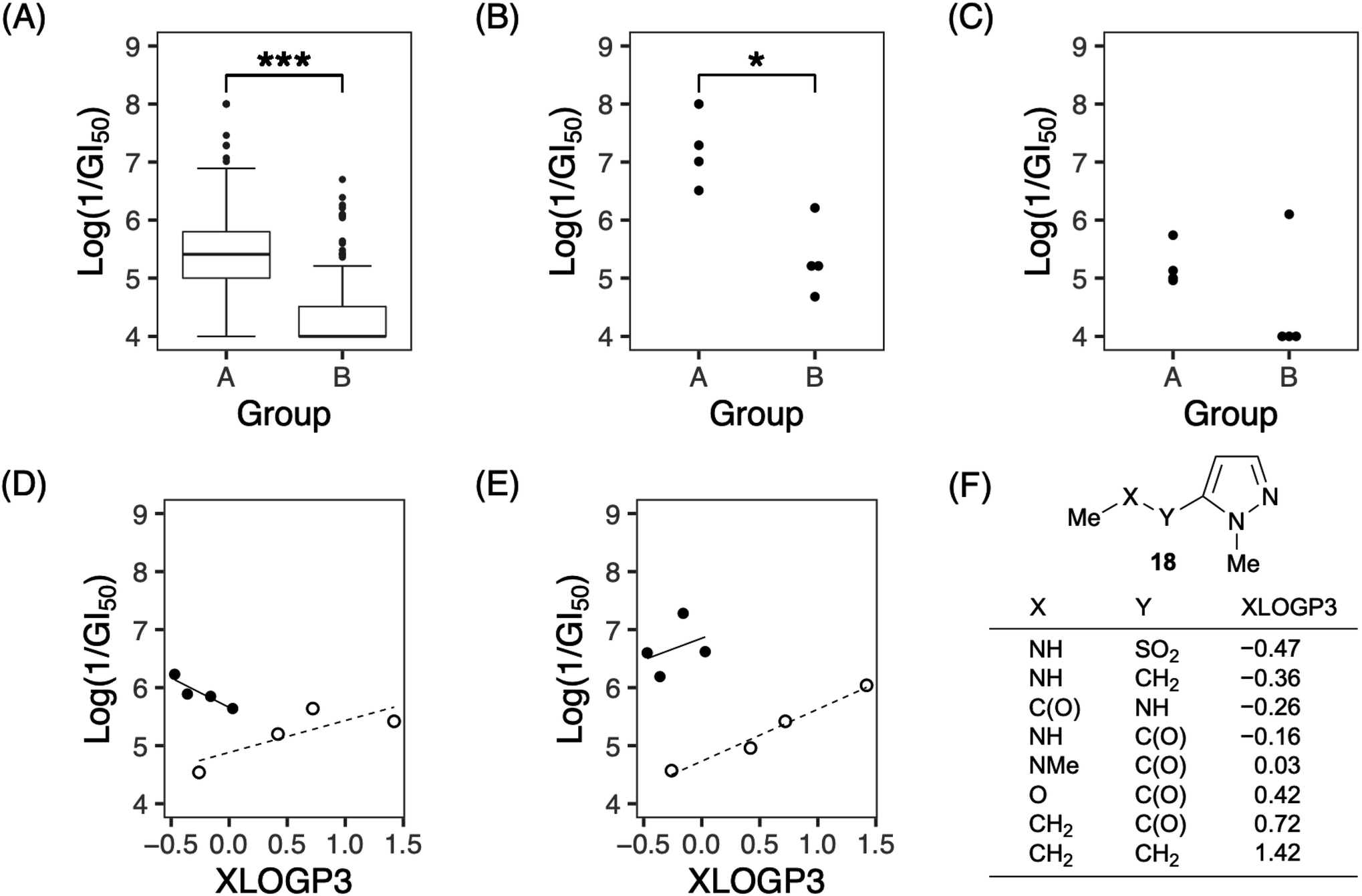 | ||
| Fig. 3 Comparison of growth-inhibitory activities of analogs 1, 7a, 7e, and 7g–7k. (A) Box plot distribution of logarithm of 1/GI50 against all tested cell lines in group A (7a, 7e, 7g and 7h) and B (1 and 7i–7k). Horizontal bars in the middle of each box indicate the median percentages of growth inhibition. The top and bottom of each box represent the upper and lower quartiles, respectively. The whiskers show the 5th and 95th percentiles. Data points outside the 5th and 95th percentiles are shown as dots above and below each plot. (B and C) Each plot shows distribution of logarithm of 1/GI50 against MKN-B (B) or MKN45 (C) in groups A and B. (D and E) Correlation between lipophilicity (XLOGP3) of the partial structure of analogs and logarithm of 1/GI50 against NCI-H522 (D) or NCI-H23 (E). Solid circles represent compounds of group A, and open circles represent compounds of group B. Trend lines represent the expected values predicted by the given QSAR model. (F) Partial structure of our analogs and their XLOGP3 values. P values were determined using the two-sided Mann–Whitney U test (***P < 0.001 and *P < 0.05). | ||
Next, the effect of substituents in 1-methylpyrazole was evaluated (Fig. 2). Both 3-ethylpyrazole 7b and 4-chloro-3-ethylpyrazole 7c, with substituents identical to those of tebufenpyrad, showed selective and potent growth inhibitory activity against some cells. The selective and potent growth inhibitory activity of 7b and 7c was observed against the same cell lines as those observed with 7a, except for LOX-IMVI. Therefore, the connecting groups could be more significant than the substituents of 1-methylpyrazole observed in our analogs. It is possible that carboxylic acid 23 produced by the hydrolysis of 7c was not in its active form because of the lack of the activity, suggesting that the presence of pyrazole moiety at a certain distance from the THF moiety is vital to the activity.
Both 4-pyrazole-carboxamide 7d and 4-pyrazole-sulfonamide 7f showed significantly more potent activities than 4-alkyl-pyrazole 7l, suggesting that the connecting groups observed in our analogs might also play an essential role when present in other analogs.
The aforementioned results revealed that the growth inhibitory activities of our analogs significantly depended on the connecting group between the 1-methylpyrazole moiety and the hydrophobic alkyl chain bearing the THF ring. The importance of the nitrogen atom at the 2′-position of the side chain is unclear; however, the presence of a polar atom in this position may be essential, as 2′-hydroxy solamin, also known as murisolin, showed more potent activity than solamin.22 The positive correlation between the reduction in lipophilicity and activity suggested the presence of a water-soluble pocket at the binding site of our analogs, suggesting that more potent analogs might be discovered using synthetic analogs with less lipophilicity.
In vivo studies using mice xenografts
To clarify the efficacy of our analogs as novel anticancer agents, we conducted in vivo studies using mouse xenografts. Amide 7a and sulfonamide 7e were tested since they showed distinctive fingerprints in in vitro studies: while the former demonstrated selective activity, the latter showed a broad activity. The sulfonamide 7e was selected from the broad inhibitory analogs with similar potencies, 7e, 7g, and 7h, because the total yield of 7e (45% from 2) was higher than those of 7g and 7h (which were 25% and 11%, respectively). NCI-H23 was selected for inoculation into mice because the two analogs 7a and 7e showed almost the same inhibition potency (log(1/GI50 of 7a) = 7.28; log(1/GI50 of 7e) = 6.60). The results for 7a are shown in Fig. 4A and B. The maximum tolerated dose (MTD) of 7a was 5 mg kg−1; this finding can be attributed to its high toxicity. Three administrations of 5 mg kg−1 7a on days 0, 7, and 14 did not show significant antitumor activity. In contrast, no death was observed after administering 100 mg kg−1 of sulfonamide 7e, which was 20-fold higher than that of 7a (Fig. 4D). Furthermore, as shown Fig. 4C, the administration of 7e on day 0 at a dose of 100 mg kg−1 significantly inhibited tumor growth as determined on day 16 (1.43 ± 0.71 compared to tumor size at day 0), whereas the control tumor grew more than six times during this period (5.58 ± 1.56). Hence, the average tumor size in drug-treated mice compared to those in control mice was 27%. Weight loss was observed after the first administration; however, this loss was recovered later, suggesting that 7e was well tolerated. When 100 mg kg−1 of 7e was administered twice on days 0 and 6, weight loss was observed after each administration. However, the bodyweight was eventually recovered, and a more potent tumor growth inhibition effect was observed on day 16 (12% of control). The administration of 7e at a dose less than 100 mg kg−1 can also be expected to show significant tumor growth inhibition, although this was not tested in the present study due to the lack of sufficient amounts of 7e. This result suggests that sulfonamide 7e is a promising novel anticancer lead, although further investigation of molecular properties, such as solubility, membrane permeability, and bioavailability, would be necessary before the analog can be used as an anticancer agent.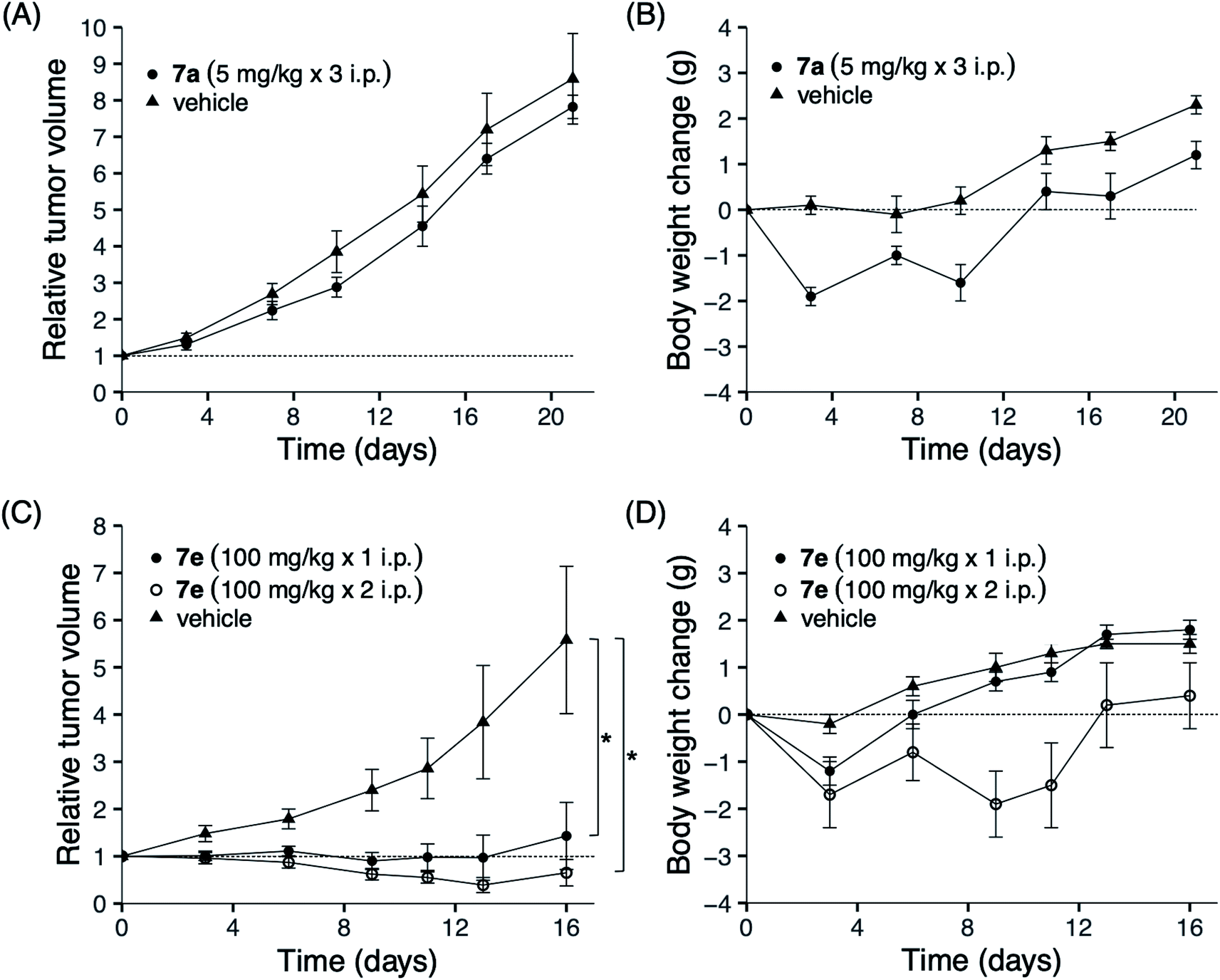 | ||
| Fig. 4 Tumor growth and body weight changes in nude mice bearing a human lung cancer NCI-H23 xenograft. Nude mice were subcutaneously inoculated with a tumor fragment measuring 3 × 3 × 3 mm from a subcutaneous tumor developed. When the tumor reached a volume of 100–300 mm3, the mice were divided randomly into test groups (day 0). (A and B) Amide 7a (5 mg kg−1) was intraperitoneally administered at days 0, 7 and 14. (C and D) Sulfonamide 7e (100 mg kg−1) was intraperitoneally administered on day 0, or on both days 0 and 6. Body weight and relative tumor volume are expressed as means ± SD. *P < 0.05, two-sided Mann–Whitney U test compared with the vehicle-administered control group. Triangle: control group; open and closed circles: administered group. | ||
These results indicate that the connecting group of our analogs contributes not only to the potency of growth inhibitory activity against cancer cells but also to their safety. Sulfonamide groups are known as one of the bioisosteres of the amide bond.23 It is not clear why the conversion of the amide bond into the sulfonamide bond in our analogs avoided the loss of bodyweight of mice; however, the metabolites, the primary amine and/or the carboxylic acid produced by hydrolysis of 7a, might have induced the loss of bodyweight because the sulfonamide bonds are often used in place of amide to improve stability against amide bond hydrolysis by proteolytic enzymes. We previously reported that an acetogenin analog with thiophene in place of γ-lactone in solamin showed potent antitumor activity without acute toxicity.24 Using in vitro data to predict in vivo toxicity may not be accurate; however, we might obtain an analog via searching with amide's bioisostere, which shows a broad spectrum activity against JFCR39 since both the sulfonamide and the thiophene carboxamide analogs show wide spectrum effects. The present SAR study is valuable because it provides another approach to demonstrate that acetogenins might lead to the development of novel anticancer agents.
Conclusions
We reported the synthesis of 11 hybrid acetogenins with various connecting groups between their pyrazole and the linker bearing the THF moiety and their growth inhibitory activities against 39 human cancer cell lines. The results demonstrated that the nitrogen atom at the 2′-position of the linker could be crucial for the inhibition of cancer growth. However, substituents in the pyrazole ring did not affect their activities. Moreover, the connecting groups divided our analogs into two categories in terms of activity: one showed selective activity and the other showed broad activity. Amide 7a, one of the former compounds, caused substantial loss of body weight in mice and did not show significant growth inhibition against NCI-H23 in a mouse xenograft assay, although exhibited potent activity in vitro. In contrast, sulfonamide 7e, one of the latter compounds, showed potent antitumor activity without acute toxicity. These data indicate the immense potential of our hybrid acetogenins as novel antitumor lead compounds. Investigation of their physical properties, bioavailability, mode of action, and structure–activity association are underway.Experimental section
Chemistry
Melting points were measured using a Yanaco MP micro-melting point apparatus and were uncorrected. Optical rotations were measured using a JASCO DIP-360 digital polarimeter or JASCO P-1020 digital polarimeter. NMR spectra were recorded in the specified solvents using Bruker Ascend™ 500 (1H: 500 MHz; 13C: 125 Hz), JEOL JNM-GX-500 (1H: 500 MHz; 13C: 125 Hz), JEOL ECS-400 (1H: 400 MHz; 13C: 100 Hz), JEOL JNM-AL300 (1H: 300 MHz; 13C: 75 Hz), or Bruker Ultrashield™ 300 (1H: 300 MHz; 13C: 75 Hz) spectrometers. Chemical shifts were recorded in ppm relative to that of the internal solvent signal [CDCl3: 7.26 ppm (1H NMR), 77.0 ppm (13C NMR)] or tetramethylsilane [0 ppm] used as the internal standard. The following abbreviations have been used: broad singlet = br s, singlet = s, doublet = d, triplet = t, quartet = q, quintet = qn, sextet = sext, septet = sep, and multiplet = m. IR absorption spectra (FT = diffuse reflectance spectroscopy) were recorded with KBr powder using a Horiba FT-210 IR spectrophotometer or as neat films on NaCl plates using a Shimadzu FTIR-8400S, and only noteworthy absorptions (in cm−1) were listed. Mass spectra were obtained using a JEOL JMS-600H, JEOL JMS-700, JEOL GC-mate II, JEOL SX-102A, or JEOL LCMS-IT-TOF mass spectrometer. Column chromatography was performed using a Kanto Chemical Silica Gel 60 N (spherical, neutral, 63–210 μm) column, and flash column chromatography was performed using a Merck Silica Gel 60 (40–63 μm) column. All air- and moisture-sensitive reactions were carried out in flame-dried glassware in an atmosphere consisting of Ar or N2. All solvents were dried and distilled according to standard procedures if necessary, while organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure using a rotary evaporator.The synthetic procedure and characterization data for analogs 1 and 7l are presented in the ESI.† Analogs 7a and 7g–7j were synthesized using procedures described in our previous report.14
:4) as eluent yielded 6d (9.7 mg, 51%) as a colorless oil. [α]D21 + 9.0 (c 0.63 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.06 (s, 3H), 0.08 (s, 3H), 0.87–0.90 (m, 12H), 1.26–1.66 (m, 42H), 1.91–1.94 (m, 2H), 2.45 (br s, 1H), 3.35–3.39 (m, 1H), 3.37 (dt, 2H, J = 7.3, 6.1 Hz), 3.53–3.57 (m, 1H), 3.77 (dt, 1H, J = 7.3, 6.1 Hz), 3.86 (dt, 1H, J = 8.5, 6.1 Hz), 3.91 (s, 3H), 5.97 (br s, 1H), 7.72 (s, 1H), 7.82 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: −4.6, −4.2, 14.1, 18.2, 22.6, 25.4, 25.6, 25.9 (3C), 26.9, 28.4, 28.5, 29.25, 29.30, 29.4 (2C), 29.49, 29.53, 29.55, 29.59 (2C), 29.63, 29.65, 29.73, 29.8, 31.9, 33.2, 33.4, 39.2, 39.5, 74.1, 75.2, 82.2, 82.4, 119.0, 131.6, 137.6, 162.4; IR (KBr) cm−1: 3316, 1632; MS (FAB) m/z: 678 [M + H]+; HRMS (FAB) m/z: calcd for C39H76N3O4Si: 678.5605; found: 678.5554 [M + H]+.:1) as eluent yielded 6e (350 mg, 73%) as a colorless oil. [α]D25 + 9.0 (c 0.42 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.06 (s, 3H), 0.07 (s, 3H), 0.88 (t, 3H, J = 7.3 Hz), 0.89 (s, 9H), 1.21–1.52 (m, 40H), 1.60–1.68 (m, 2H), 1.91–1.95 (m, 2H), 3.04 (dt, 2H, J = 7.3, 6.1 Hz), 3.35–3.39 (m, 1H), 3.53–3.57 (m, 1H), 3.77 (dt, 1H, J = 7.3, 6.4 Hz), 3.86 (dt, 1H, J = 7.9, 6.1 Hz), 4.09 (s, 3H), 4.81 (t, 1H, J = 6.1 Hz), 6.75 (d, 1H, J = 2.4 Hz), 7.47 (d, 1H, J = 2.4 Hz); 13C NMR (125 MHz, CDCl3) δ: −4.6, −4.1, 14.1, 18.3, 22.7, 25.4, 25.6, 25.9 (3C), 26.4, 28.5, 28.6, 29.0, 29.3 (2C), 29.5, 29.56, 29.59, 29.62 (4C), 29.7 (2C), 29.8, 31.9, 33.2, 33.4, 38.5, 43.3, 74.1, 75.2, 82.3, 82.4, 111.0, 137.6, 138.9; IR (KBr) cm−1: 3292; MS (FAB) m/z: 714 [M + H]+; HRMS (FAB) m/z: calcd for C38H76N3O5SSi: 714.5275; found: 714.5262 [M + H]+.:2, 1.5 mL) with stirring at rt. After stirring for 3 h at same temperature, water was added to the reaction mixture and the mixture was extracted with CH2Cl2. The combined organic layer was washed with brine prior to drying and solvent evaporation. Purification by flash column chromatography over silica gel with CHCl3/MeOH (25:1) as eluent yielded 7b (15.6 mg, 35% over 3 steps) as a white waxy solid. [α]D25 + 8.5 (c 0.55 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.88 (t, 3H, J = 6.7 Hz), 1.22–1.72 (m, 42H), 1.24 (t, 3H, J = 7.9 Hz), 1.95–2.01 (m, 2H), 2.63 (q, 2H, J = 7.9 Hz), 3.35–3.42 (m, 4H), 3.80 (q, 2H, J = 6.7 Hz), 4.11 (s, 3H), 6.00 (br s, 1H), 6.27 (s, 1H); 13C NMR (75 MHz, CDCl3) δ: 13.9, 14.1, 21.2, 22.7, 25.5 (2C), 26.9, 28.7 (2C), 29.2, 29.3, 29.4 (3C), 29.5, 29.57 (2C), 29.62 (4C), 29.7, 31.9, 33.4 (2C), 38.8, 39.5, 74.00, 74.02, 82.61, 82.64, 103.8, 135.9, 152.8, 160.1; IR (KBr) cm−1: 3423, 3286, 1639; MS (FAB) m/z: 592 [M + H]+; HRMS (FAB) m/z: calcd for C35H66N3O4: 592.5053; found: 592.5053 [M + H]+.:1, 0.75 mL) with stirring at rt. After stirring for 2 h at the same temperature, water was added to the reaction mixture and the mixture was extracted with CH2Cl2. The combined organic layer was dried prior to solvent evaporation. Purification using flash column chromatography over silica gel with EtOAc to CHCl3/MeOH (85:15) as eluent yielded 7d (7.5 mg, quant.) as a white waxy solid. Mp. 92.1–93.1 °C; [α]D25 + 11.4 (c 0.57 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.88 (t, 3H, J = 7.0 Hz), 1.26–1.71 (m, 42H), 1.97–2.02 (m, 2H), 2.36 (br s, 2H), 3.36–3.41 (m, 4H), 3.80 (dt, 2H, J = 7.9, 6.1 Hz), 3.92 (s, 3H), 5.73 (br s, 1H), 7.69 (s, 1H), 7.82 (s, 1H); 13C NMR (75 MHz, CDCl3) δ: 14.0, 22.6, 25.5, 25.6, 26.9, 28.7 (2C), 29.2, 29.3, 29.38 (2C), 29.44, 29.56, 29.59 (3C), 29.62 (2C), 29.63, 29.69, 29.72, 31.9, 33.5, 39.2, 39.5, 74.02, 74.03, 82.6, 82.7, 119.0, 131.7, 137.6, 162.5; IR (KBr) cm−1: 3319, 1630; MS (FAB) m/z: 564 [M + H]+; HRMS (FAB) m/z: calcd for C33H62N3O4: 564.4740; found: 564.4743 [M + H]+.![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 2.96 (t, 2H, J = 7.3 Hz), 3.41 (q, 2H, J = 6.1 Hz), 3.80 (td, 2H, J = 7.3, 6.1 Hz), 3.95 (s, 3H), 4.62 (br s, 1H), 7.76 (s, 1H), 7.81 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 14.1, 22.6, 25.5, 25.6, 26.5, 28.7 (2C), 29.0, 29.3 (3C), 29.4 (2C), 29.56 (2C), 29.59, 29.60, 29.62 (2C), 29.7 (2C), 31.9, 33.4, 39.5, 43.2, 74.00, 74.03, 82.61, 82.64, 122.2, 131.7, 138.5; IR (KBr) cm−1: 3447, 3360, 3275; MS (FAB) m/z: 600 [M + H]+; HRMS (FAB) m/z: calcd for C32H62N3O5S: 600.4410; found: 600.4410 [M + H]+:1 to 20:1) as eluent yielded 18 (412 mg, 73%, dr = 92:8) as a colorless oil. [α]D25 + 10.0 (c 0.61 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.06 (s, 3H), 0.08 (s, 3H), 0.87–0.89 (m, 12H), 1.26–1.51 (m, 34H), 1.61–1.79 (m, 3H), 1.88–2.07 (m, 3H), 2.18 (td, 2H, J = 7.3, 2.4 Hz), 2.34 (t, 2H, J = 7.3 Hz), 2.35 (br s, 1H), 3.56–3.59 (m, 1H), 3.89–3.93 (m, 1H), 4.08 (td, 1H, J = 7.3, 3.7 Hz), 4.15–4.17 (m, 1H), 5.11 (s, 2H), 7.26–7.38 (m, 5H); 13C NMR (125 MHz, CDCl3) δ: −4.6, −4.2, 14.1, 18.2, 18.7, 22.7, 24.9, 25.5, 25.9 (3C), 27.6, 28.3, 28.5, 28.8, 29.0, 29.1, 29.2, 29.25, 29.32, 29.57 (2C), 29.60 (2C), 29.65, 29.8, 31.9, 32.9, 34.3, 65.6, 66.0, 74.9, 78.0, 82.5, 82.6, 86.3, 128.1 (3C), 128.5 (2C), 136.1, 173.6; IR (NaCl) cm−1: 3503, 1740; MS (FAB) m/z: 721 [M + Na]+; HRMS (FAB) m/z: calcd for C43H74NaO5Si: 721.5203; found: 721.5222 [M + Na]+.:1) as eluent yielded 19 (478 mg, quant.) as a colorless oil. [α]D25 + 7.3 (c 1.22 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.05 (s, 3H), 0.06 (s, 3H), 0.09 (s, 3H), 0.12 (s, 3H), 0.86–0.90 (m, 21H), 1.25–1.69 (m, 37H), 1.89–1.95 (m, 3H), 2.17 (td, 2H, J = 7.3, 1.8 Hz), 2.35 (t, 2H, J = 7.6 Hz), 3.59 (dt, 1H, J = 7.3, 4.5 Hz), 3.97–4.02 (m, 2H), 4.37 (dt, 1H, J = 5.5, 1.8 Hz), 5.11 (s, 2H), 7.34–7.36 (m, 5H); 13C NMR (125 MHz, CDCl3) δ: −4.9, −4.62, −4.60, −4.2, 14.1, 18.2, 18.3, 18.7, 24.9, 25.8 (3C), 26.0 (3C), 26.9, 27.7, 28.6, 28.8, 29.06, 29.10, 29.2, 29.3 (3C), 29.6 (4C), 29.7 (2C), 29.8, 31.9, 32.5, 34.3, 66.0, 66.3, 74.5, 79.5, 82.1, 82.3, 85.5, 128.1 (3C), 128.5 (2C), 136.1, 173.7; IR (NaCl) cm−1: 1740; MS (FAB) m/z: 835 [M + Na]+; HRMS (FAB) m/z: calcd for C49H88NaO5Si2: 835.6068; found: 835.6078 [M + Na]+.:1) as eluent yielded 20 (190 mg, 96%) as a colorless oil. [α]D25 + 17.3 (c 0.51 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.878 (t, 3H, J = 7.3 Hz), 0.881 (s, 18H), 1.26–1.52 (m, 40H), 1.61–1.72 (m, 4H), 1.81–1.86 (m, 2H), 2.34 (t, 2H, J = 7.3 Hz), 3.56–3.60 (m, 2H), 3.91 (q, 2H, J = 6.1 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.5 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.7, 25.9 (2C), 26.0 (6C), 27.3, 29.1, 29.2, 29.37, 29.43, 29.55, 29.63 (3C), 29.65 (3C), 29.69 (2C), 29.8 (2C), 31.9 (2C), 32.6, 34.0, 74.8 (2C), 81.8 (2C), 179.9; IR (NaCl) cm−1: 1712; MS (FAB) m/z: 749 [M + Na]+; HRMS (FAB) m/z: calcd for C42H86NaO5Si2: 749.5911; found: 749.5895 [M + Na]+.:1 to 10:1) as eluent yielded 21 (12.2 mg, 94%) as a colorless oil. [α]D25 + 10.9 (c 0.57 in CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.88 (t, 3H, J = 6.9 Hz), 0.88 (s, 18H), 1.25–1.72 (m, 44H), 1.78–1.86 (m, 2H), 2.41 (t, 2H, J = 7.3 Hz), 3.18 (s, 3H), 3.55–3.59 (m, 2H), 3.68 (s, 3H), 3.90 (q, 2H, J = 5.9 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.6 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.6 (2C), 25.86, 25.93 (6C), 27.2, 29.3, 29.42, 29.44, 29.5, 29.60 (6C), 29.62 (4C), 29.7 (2C), 29.8, 31.9 (2C), 32.6, 61.1, 74.7 (2C), 81.7 (2C), 174.8; IR (NaCl) cm−1: 1684; MS (FAB) m/z: 792 [M + Na]+; HRMS (FAB) m/z: calcd for C44H91NNaO5Si2: 792.6333; found: 792.6331 [M + Na]+.:1 to EtOAc) as eluent yielded 22 (19.3 mg, 44%, brsm 66%) as a colorless oil. [α]D25 + 24.6 (c 0.32 in CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.86–0.89 (m, 21H), 1.22–1.56 (m, 40H), 1.65–1.72 (m, 4H), 1.80–1.84 (m, 2H), 2.83 (t, 2H, J = 7.3 Hz), 3.58 (dt, 2H, J = 6.8, 4.6 Hz), 3.85–3.94 (m, 2H), 4.17 (s, 3H), 6.82 (d, 1H, J = 2.3 Hz), 7.46 (d, 1H, J = 2.3 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.5 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.3, 25.9, 26.0 (6C), 27.3, 29.3, 29.36, 29.44, 29.5, 29.60 (2C), 29.62 (3C), 29.63 (3C), 29.65 (2C), 29.68, 29.8 (2C), 31.9, 32.6, 40.2, 40.6, 74.8 (2C), 81.8 (2C), 111.2, 137.5, 138.5, 192.0; IR (NaCl) cm−1: 1676; MS (FAB) m/z: 813 [M + Na]+; HRMS (FAB) m/z: calcd for C46H90N2NaO4Si2: 813.6337; found: 813.6335 [M + Na]+.
), 2.96 (t, 2H, J = 7.3 Hz), 3.41 (q, 2H, J = 6.1 Hz), 3.80 (td, 2H, J = 7.3, 6.1 Hz), 3.95 (s, 3H), 4.62 (br s, 1H), 7.76 (s, 1H), 7.81 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 14.1, 22.6, 25.5, 25.6, 26.5, 28.7 (2C), 29.0, 29.3 (3C), 29.4 (2C), 29.56 (2C), 29.59, 29.60, 29.62 (2C), 29.7 (2C), 31.9, 33.4, 39.5, 43.2, 74.00, 74.03, 82.61, 82.64, 122.2, 131.7, 138.5; IR (KBr) cm−1: 3447, 3360, 3275; MS (FAB) m/z: 600 [M + H]+; HRMS (FAB) m/z: calcd for C32H62N3O5S: 600.4410; found: 600.4410 [M + H]+:1 to 20:1) as eluent yielded 18 (412 mg, 73%, dr = 92:8) as a colorless oil. [α]D25 + 10.0 (c 0.61 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.06 (s, 3H), 0.08 (s, 3H), 0.87–0.89 (m, 12H), 1.26–1.51 (m, 34H), 1.61–1.79 (m, 3H), 1.88–2.07 (m, 3H), 2.18 (td, 2H, J = 7.3, 2.4 Hz), 2.34 (t, 2H, J = 7.3 Hz), 2.35 (br s, 1H), 3.56–3.59 (m, 1H), 3.89–3.93 (m, 1H), 4.08 (td, 1H, J = 7.3, 3.7 Hz), 4.15–4.17 (m, 1H), 5.11 (s, 2H), 7.26–7.38 (m, 5H); 13C NMR (125 MHz, CDCl3) δ: −4.6, −4.2, 14.1, 18.2, 18.7, 22.7, 24.9, 25.5, 25.9 (3C), 27.6, 28.3, 28.5, 28.8, 29.0, 29.1, 29.2, 29.25, 29.32, 29.57 (2C), 29.60 (2C), 29.65, 29.8, 31.9, 32.9, 34.3, 65.6, 66.0, 74.9, 78.0, 82.5, 82.6, 86.3, 128.1 (3C), 128.5 (2C), 136.1, 173.6; IR (NaCl) cm−1: 3503, 1740; MS (FAB) m/z: 721 [M + Na]+; HRMS (FAB) m/z: calcd for C43H74NaO5Si: 721.5203; found: 721.5222 [M + Na]+.:1) as eluent yielded 19 (478 mg, quant.) as a colorless oil. [α]D25 + 7.3 (c 1.22 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.05 (s, 3H), 0.06 (s, 3H), 0.09 (s, 3H), 0.12 (s, 3H), 0.86–0.90 (m, 21H), 1.25–1.69 (m, 37H), 1.89–1.95 (m, 3H), 2.17 (td, 2H, J = 7.3, 1.8 Hz), 2.35 (t, 2H, J = 7.6 Hz), 3.59 (dt, 1H, J = 7.3, 4.5 Hz), 3.97–4.02 (m, 2H), 4.37 (dt, 1H, J = 5.5, 1.8 Hz), 5.11 (s, 2H), 7.34–7.36 (m, 5H); 13C NMR (125 MHz, CDCl3) δ: −4.9, −4.62, −4.60, −4.2, 14.1, 18.2, 18.3, 18.7, 24.9, 25.8 (3C), 26.0 (3C), 26.9, 27.7, 28.6, 28.8, 29.06, 29.10, 29.2, 29.3 (3C), 29.6 (4C), 29.7 (2C), 29.8, 31.9, 32.5, 34.3, 66.0, 66.3, 74.5, 79.5, 82.1, 82.3, 85.5, 128.1 (3C), 128.5 (2C), 136.1, 173.7; IR (NaCl) cm−1: 1740; MS (FAB) m/z: 835 [M + Na]+; HRMS (FAB) m/z: calcd for C49H88NaO5Si2: 835.6068; found: 835.6078 [M + Na]+.:1) as eluent yielded 20 (190 mg, 96%) as a colorless oil. [α]D25 + 17.3 (c 0.51 in CHCl3); 1H NMR (500 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.878 (t, 3H, J = 7.3 Hz), 0.881 (s, 18H), 1.26–1.52 (m, 40H), 1.61–1.72 (m, 4H), 1.81–1.86 (m, 2H), 2.34 (t, 2H, J = 7.3 Hz), 3.56–3.60 (m, 2H), 3.91 (q, 2H, J = 6.1 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.5 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.7, 25.9 (2C), 26.0 (6C), 27.3, 29.1, 29.2, 29.37, 29.43, 29.55, 29.63 (3C), 29.65 (3C), 29.69 (2C), 29.8 (2C), 31.9 (2C), 32.6, 34.0, 74.8 (2C), 81.8 (2C), 179.9; IR (NaCl) cm−1: 1712; MS (FAB) m/z: 749 [M + Na]+; HRMS (FAB) m/z: calcd for C42H86NaO5Si2: 749.5911; found: 749.5895 [M + Na]+.:1 to 10:1) as eluent yielded 21 (12.2 mg, 94%) as a colorless oil. [α]D25 + 10.9 (c 0.57 in CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.88 (t, 3H, J = 6.9 Hz), 0.88 (s, 18H), 1.25–1.72 (m, 44H), 1.78–1.86 (m, 2H), 2.41 (t, 2H, J = 7.3 Hz), 3.18 (s, 3H), 3.55–3.59 (m, 2H), 3.68 (s, 3H), 3.90 (q, 2H, J = 5.9 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.6 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.6 (2C), 25.86, 25.93 (6C), 27.2, 29.3, 29.42, 29.44, 29.5, 29.60 (6C), 29.62 (4C), 29.7 (2C), 29.8, 31.9 (2C), 32.6, 61.1, 74.7 (2C), 81.7 (2C), 174.8; IR (NaCl) cm−1: 1684; MS (FAB) m/z: 792 [M + Na]+; HRMS (FAB) m/z: calcd for C44H91NNaO5Si2: 792.6333; found: 792.6331 [M + Na]+.:1 to EtOAc) as eluent yielded 22 (19.3 mg, 44%, brsm 66%) as a colorless oil. [α]D25 + 24.6 (c 0.32 in CHCl3); 1H NMR (400 MHz, CDCl3) δ: 0.05 (s, 6H), 0.06 (s, 6H), 0.86–0.89 (m, 21H), 1.22–1.56 (m, 40H), 1.65–1.72 (m, 4H), 1.80–1.84 (m, 2H), 2.83 (t, 2H, J = 7.3 Hz), 3.58 (dt, 2H, J = 6.8, 4.6 Hz), 3.85–3.94 (m, 2H), 4.17 (s, 3H), 6.82 (d, 1H, J = 2.3 Hz), 7.46 (d, 1H, J = 2.3 Hz); 13C NMR (75 MHz, CDCl3) δ: −4.5 (2C), −4.3 (2C), 14.1, 18.2 (2C), 22.7, 24.3, 25.9, 26.0 (6C), 27.3, 29.3, 29.36, 29.44, 29.5, 29.60 (2C), 29.62 (3C), 29.63 (3C), 29.65 (2C), 29.68, 29.8 (2C), 31.9, 32.6, 40.2, 40.6, 74.8 (2C), 81.8 (2C), 111.2, 137.5, 138.5, 192.0; IR (NaCl) cm−1: 1676; MS (FAB) m/z: 813 [M + Na]+; HRMS (FAB) m/z: calcd for C46H90N2NaO4Si2: 813.6337; found: 813.6335 [M + Na]+.Biology
Statistical analysis
All statistical analyses were performed using Rstudio 2021.09.1 + 372 for Mac,25 which is a graphical user interface for R version 4.1.2 (2021-11-01).26 The differences were considered significant when *P < 0.05, **P < 0.01, or ***P < 0.001.Author contributions
S. D., and N. K. participated in the research design. K. O., T. F., M. O., S. D., and N. K. conducted the experiments. K. O., M. O., S. D., and N. K. performed the data analysis. K. O., M. O., A. A., S. D., H. I., M. Y., and N. K. wrote or contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The in vitro antiproliferative activities of acetogenin analogs against human cancer cell lines were examined by the Screening Committee of Anticancer Drugs supported by a Grant-in-Aid for Scientific Research on Innovative Areas, Scientific Support Programs for Cancer Research, from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). We thank Mr Takahiro Tatsukawa for his technical assistance in the preliminary part of this study and Prof. Tetsuaki Tanaka and Prof. Takehiko Yoshimitsu for their helpful discussions. This study was supported in part by JSPS KAKENHI [grant numbers 25460159, 16K08330 (N. K.), 16H05105 (S. D. and A. A.)], the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2015–2019 [Grant Number S1511024L] (N. K.), a Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research (N. K.), and a research grant from the Nippon Foundation (S. D.).Notes and references
- Cancer Today, https://gco.iarc.fr/today, (accessed january 2022).
- For recent review of mitochondrial complex inhibitors for cancer therapy, see: M. Bedi, M. Ray and A. Ghosh, Mol. Cell. Biochem., 2022, 477, 345–361 CrossRef CAS PubMed.
- J. Yoshida, T. Ohishi, H. Abe, S. Ohba, H. Inoue, I. Usami, M. Amemiya, R. Oriez, C. Sakashita, S. Dan, M. Sugawara, T. Kawaguchi, J. Ueno, Y. Asano, A. Ikeda, M. Takamatsu, G. Amori, Y. Kondoh, K. Honda, H. Osada, T. Noda, T. Watanabe, T. Shimizu, M. Shibasaki and M. Kawada, iScience, 2022, 24, 103497 CrossRef PubMed.
- Solasia Pharma K. K., https://solasia.co.jp/en/pipeline/sp02.html, (accessed January 2022).
- (a) N. Kojima, H. Hayashi, H. Iwasaki and M. Yamashita, Chem. Pharm. Bull., 2020, 68, 675–678 CrossRef CAS PubMed; (b) N. Kojima, M. Abe, Y. Suga, K. Ohtsuki, T. Tanaka, H. Iwasaki, M. Yamashita and H. Miyoshi, Bioorg. Med. Chem. Lett., 2013, 23, 1217–1219 CrossRef CAS PubMed; (c) N. Kojima, Y. Suga, H. Hayashi, T. Yamori, T. Yoshimitsu and T. Tanaka, Bioorg. Med. Chem. Lett., 2011, 21, 5745–5749 CrossRef CAS PubMed; (d) N. Kojima, H. Hayashi, S. Suzuki, H. Tominaga, N. Maezaki, T. Tanaka and T. Yamori, Bioorg. Med. Chem. Lett., 2008, 18, 6451–6453 CrossRef CAS PubMed; (e) N. Maezaki, N. Kojima and T. Tanaka, Synlett, 2006, 7, 993–1003 Search PubMed; (f) N. Kojima, Yakugaku Zasshi, 2004, 124, 673–681 CrossRef CAS.
- For recent review of isolation and biological activity of annonaceous aceotgenins, see: A. Neske, J. R. Hidalgo, N. Cabedo and D. Cortes, Phytochemistry, 2020, 174, 112332 CrossRef CAS.
- For recent review of medicinal chemistry of annonaceous aceotgenins, see: N. Kojima and T. Tanaka, Molecules, 2009, 14, 3621–3661 CrossRef CAS PubMed.
- M. Murai and H. Miyoshi, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 884–891 CrossRef CAS.
- Heterocyclic acetogenin analogs in place of γ-lactone by other groups, see: (a) E. R. Gould, E. F. B. King, S. K. Menzies, A. L. Fraser, L. B. Tulloch, M. K. Zacharova, T. K. Smith and G. J. Florence, Bioorg. Med. Chem., 2017, 25, 6126–6136 CrossRef CAS PubMed; (b) Y. Liu, Q. Xiao, Y. Liu, Z. Li, Y. Qiu, G.-B. Zhou, Z.-J. Yao and S. Jiang, Eur. J. Med. Chem., 2014, 78, 248–258 CrossRef CAS; (c) Y.-J. Chen, S. Jin, J. Xi and Z.-J. Yao, Tetrahedron, 2014, 70, 4921–4928 CrossRef CAS; (d) R. A. Duval, G. Lewin, E. Peris, N. Chahboune, A. Garofano, S. Dröse, D. Cortes, U. Brandt and R. Hocquemiller, Biochemistry, 2006, 45, 2721–2728 CrossRef CAS; (e) R. A. Duval, E. Poupon, V. Romero, E. Peris, G. Lewin, D. Cortes, U. Brandt and R. Hocquemiller, Tetrahedron, 2006, 62, 6248–6257 CrossRef CAS; (f) R. Duval, G. Lewin and R. Hocquemiller, Bioorg. Med. Chem., 2003, 11, 3439–3446 CrossRef CAS.
- I. Y. Shiraishi, M. Murai, N. Sakiyama, K. Ifuku and H. Miyoshi, Biochemistry, 2012, 51, 1953–1963 CrossRef PubMed.
- (a) Y. Nakanishi, F.-R. Chang, C.-C. Liaw, Y.-C. Wu, K. F. Bastow and K.-H. Lee, J. Med. Chem., 2003, 46, 3185–3188 CrossRef CAS PubMed; (b) S. C. Sinha and E. Keinan, J. Am. Chem. Soc., 1993, 115, 4891–4892 CrossRef CAS; (c) S. H. Myint, D. Cortes, A. Laurens, R. Hocquemiller, M. Lebȩuf, A. Cavé, J. Cotte and A.-M. Quéro, Phytochemistry, 1991, 30, 3335–3338 CrossRef CAS.
- For reviews of hybridazation as an approach to new drug, see: (a) H. M. S. Kumar, L. Herrmann and S. B. Tsogoeva, Bioorg. Med. Chem. Lett., 2020, 30, 127514 CrossRef PubMed; (b) G. Bérubé, Expert Opin. Drug Discovery, 2016, 11, 281–305 CrossRef PubMed; (c) K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS.
- N. Kojima, T. Fushimi, N. Maezaki, T. Tanaka and T. Yamori, Bioorg. Med. Chem. Lett., 2008, 18, 1637–1641 CrossRef CAS PubMed.
- N. Kojima, T. Fushimi, T. Tatsukawa, T. Yoshimitsu, T. Tanaka, T. Yamori, S. Dan, H. Iwasaki and M. Yamashita, Eur. J. Med. Chem., 2013, 63, 833–839 CrossRef CAS PubMed.
- (a) N. Kojima, N. Maezaki, H. Tominaga, M. Yanai, D. Urabe and T. Tanaka, Chem.–Eur. J., 2004, 10, 672–680 CrossRef CAS; (b) N. Kojima, N. Maezaki, H. Tominaga, M. Asai, M. Yanai and T. Tanaka, Chem.–Eur. J., 2003, 9, 5428 CrossRef; (c) N. Maezaki, N. Kojima, H. Tominaga, M. Yanai and T. Tanaka, Org. Lett., 2003, 5, 1411–1414 CrossRef CAS PubMed; (d) N. Maezaki, N. Kojima, M. Asai, H. Tominaga and T. Tanaka, Org. Lett., 2002, 4, 2977–2980 CrossRef CAS PubMed.
- D. E. Frantz, R. Fässler and E. M. Carreira, J. Am. Chem. Soc., 2000, 122, 1806–1807 CrossRef CAS.
- T. Matsumoto, N. Kojima, A. Akatsuka, T. Yamori, S. Dan, H. Iwasaki and M. Yamashita, Tetrahedron, 2017, 73, 2359–2366 CrossRef CAS.
- (a) S. Dan, M. Okamura, M. Seki, K. Yamazaki, H. Sugita, M. Okui, Y. Mukai, H. Nishimura, R. Asaka, K. Nomura, Y. Ishikawa and T. Yamori, Cancer Res., 2010, 70, 4982–4994 CrossRef CAS; (b) S. Yaguchi, Y. Fukui, I. Koshimizu, H. Yoshimi, T. Matsuno, H. Gouda, S. Hirono, K. Yamazaki and T. Yamori, J. Natl. Cancer Inst., 2006, 98, 545–556 CrossRef CAS PubMed; (c) T. Yamori, A. Matsunaga, S. Sato, K. Yamazaki, A. Komi, K. Ishizu, I. Mita, H. Edatsugi, Y. Matsuba, K. Takezawa, O. Nakanishi, H. Kohno, Y. Nakajima, H. Komatsu, T. Andoh and T. Tsuruo, Cancer Res., 1999, 59, 4042–4049 CAS.
- N. Kojima, T. Morioka, D. Urabe, M. Yano, Y. Suga, N. Maezaki, A. Ohashi-Kobayashi, Y. Fujimoto, M. Maeda, T. Yamori, T. Yoshimitsu and T. Tanaka, Bioorg. Med. Chem., 2010, 18, 8630–8641 CrossRef CAS.
- T. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. Zhang, Y. Li, R. Wang and L. Lai, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, XLOGP3 values of compounds were calculated using the SwissADME online tool, Sci. Rep., 2017, 7, 42717 CrossRef , (https://www.swissadme.ch).
- N. Maezaki, H. Tominaga, N. Kojima, M. Yanai, D. Urabe, R. Ueki, T. Tanaka and T. Yamori, Chem.–Eur. J., 2005, 11, 6237–6245 CrossRef CAS.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS.
- (a) S. Ando, C. Moyama, N. Kojima, M. Fujita, K. Ohta, Y. Kohno, H. Ii and S. Nakata, Biochem. Biophys. Res. Commun., 2022, 591, 62–67 CrossRef CAS; (b) K. Ohta, A. Akatsuka, S. Dan, H. Iwasaki, M. Yamashita and N. Kojima, Chem. Pharm. Bull., 2021, 69, 1029–1033 CrossRef CAS; (c) T. Matsumoto, A. Akatsuka, S. Dan, H. Iwasaki, M. Yamashita and N. Kojima, Tetrahedron, 2020, 76, 131058 CrossRef CAS; (d) A. Akatsuka, N. Kojima, M. Okamura, S. Dan and T. Yamori, Pharmacol. Res. Perspect., 2016, 4, e00246 Search PubMed; (e) N. Kojima, Y. Suga, T. Matsumoto, T. Tanaka, A. Akatsuka, T. Yamori, S. Dan, H. Iwasaki and M. Yamashita, Bioorg. Med. Chem., 2015, 23, 1276–1283 CrossRef CAS; (f) N. Kojima, T. Fushimi, T. Tatsukawa, T. Tanaka, M. Okamura, A. Akatsuka, T. Yamori, S. Dan, H. Iwasaki and M. Yamashita, Eur. J. Med. Chem., 2014, 86, 684–689 CrossRef CAS PubMed.
- R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2021 Search PubMed.
- RStudio Team, RStudio: Integrated Development Environment for R, RStudio, PBC, Boston, MA, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02399g |
| This journal is © The Royal Society of Chemistry 2022 |