
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved performance of self-reactivated Pt–ThO2/C catalysts in a direct ethanol fuel cell
Yubing Xue a,
Dashu Pana,
Feng Zuob,
Songtao Xiaoa,
Xiang Lia,
Fuyan Loua,
Mingming Lia and
Yinggen Ouyang*a
a,
Dashu Pana,
Feng Zuob,
Songtao Xiaoa,
Xiang Lia,
Fuyan Loua,
Mingming Lia and
Yinggen Ouyang*a
aChina Institute of Atomic Energy, Beijing, 102413, China. E-mail: yubing_xue@163.com
bSchool of Nuclear Science and Technology, Xi'an Jiaotong University, Xi'an, 710049, China
First published on 7th June 2022
Abstract
A novel self-reactivated catalyst Pt–ThO2/C was prepared for the first time by selecting radioactive material ThO2 as the catalytic additive to address the low activity and toxicity of the anode Pt/C catalyst in a direct ethanol fuel cell. The catalytic activity and resistance to CO poisoning of Pt-6.67 wt%ThO2/C were found to be superior to those of Pt/C–NaBH4 in electrochemical workstation and single-cell tests. It is speculated that the exist of ThO2 not only improves the catalytic activity via the synergistic effect of Pt and Th, but also produces a large amount of radiolysis products, OH radicals, due to 232Th which oxidatively desorbs CO from Pt–COads and solves the CO poisoning problem.
Introduction
According to the 2016–2021 China New Energy Industry Development Prospects and Investment Strategic Planning Analysis Report, it is well established that of the proven fossil fuels all over the world, coal could be exploited for 110 years, oil for 53 years and natural gas for 54 years, while burning fossil fuels would cause the emission of 32 billion tonnes of carbon dioxide, 120 million tonnes of sulphur oxides and 100 million tonnes of nitrogen oxides in the next single year. The shortage of fossil fuels and the environmental pollution caused by combustion for power generation are becoming salient. Consequently, there is an urgent need to explore renewable and clean alternative energy sources as well as to develop new power generation technologies to avoid combustion for power generation.1,2 The direct alcohol fuel cell (DAFC), an energy conversion device with compact configuration and convenient operation, can convert the chemical energy of alcohols such as ethanol,3 methanol,4 ethylene glycol,5 propanol6 and butanol7 directly into electricity in an environmentally friendly way, i.e., the combustion products do not contain sulphur oxides or nitrogen oxides which are noxious to the environment and human health.8,9 The DAFC systems have been utilized in vehicles and especially portable electronic devices like laptops, mobile phones and power packs due to the high energy density of alcohol, convenience, durability, and stable power supply.10–13 Direct ethanol fuel cells (DEFCs) are DAFCs using ethanol as the fuel with higher energy density and less toxic intermediate products compared to methanol.14Numerous fundamental studies have been reported that Pt-based catalyst shows the most excellent performance in acidic DEFCs15 and Nafion membranes, the proton exchange membrane (PEM) are commonly applied.16–19 What's more, the design of multimodal porous carbon for the interfacial microporous layer has also been studied to improve the performance of electrocatalyst.20,21 The development of electrocatalysts plays a crucial role in improving the performance of fuel cells. In addition to exploring high-efficient approaches to synthesize catalysts,22 the factors responsible for superior catalytic activity and stability of nanocatalysts have also been inspected to further conduct the synthesis of electrocatalysts.23 And the impacts of ligands on the catalytic activities of like PtRu, PtPd and Pt/C have deepen the understanding of the application of heterogeneous catalysis.24
However, fabrication or manufacture of Pt/C catalyst is the foremost obstacle to the application and commercialization of DEFC because of its high cost (accounts for 36% of the fuel cell's total expenditure) and the performance drop attributed by the poor ability to resist CO poisoning.15,25,26 Ethanol in the anode catalytic layer generates CO-intermediates as a result of incomplete oxidation.27 It has been confirmed by in situ infrared spectroscopy that the intermediate product CO is adsorbed on the Pt surface by linear adsorption, forming stable Pt carbonyl groups (Pt + CO → Pt–COads). As CO covers the active site of Pt, it affects the Pt/C catalytic performance and prevents further dissociation of ethanol, ultimately leading to a dramatic decrease in the overall cell performance.28–31
In addition to scan the poisoned Pt/C electrode in 0.5 mol L−1 H2SO4 solution, there are three other ways to settle the catalyst performance drop caused by the CO poisoning: (1) based on the bifunctional mechanism, the addition of some non-Pt elements to the Pt/C catalyst can contribute to the removal of CO. M. Watanabe et al. explained the detoxification mechanism of Pt–Ru/C catalysts, where Pt adsorbs the intermediate product CO to form Pt carbonyl groups (Pt + CO → Pt–COads) and Ru decomposes and adsorbs OH radicals from water at low potentials (Ru + H2O → Ru–OHads + H+ + e−), which rapidly oxidizes CO on the Pt surface to CO2 for discharge (Pt–COads + Ru–OHads → CO2 + H+ + e−), exposing the Pt active site and restoring the catalytic activity.32 Yunteng Qu et al. prepared a Pt–SnO2/graphene catalyst and tested the i–t curve, finding out that the resistance to CO poisoning was significantly better than Pt/graphene because the formation of adsorbed oxygen-containing groups OHads on the SnO2 surface at low potentials could improve the efficiency of CO oxidation.33 Xiuyu Wang et al. investigated that applying Pt–Au/CNT@TiO2 catalyst prepared by adding TiO2 to Pt–Au/CNT to the system could improve the resistance to poisoning. One reason is that TiO2 increases the electron density of the alloy by interacting with Pt–Au alloy, and some electrons are transferred to the anti-bonding orbital of CO, weakening the bonding strength of the C–O bond while reducing the oxidation overpotential. For another, according to the bifunctional mechanism, the Ti ions in the high valence state decompose water molecules to form OHads species in the adsorbed state, which oxidize CO on the Pt surface to produce CO2.34 There are also Pt–RuO2,35,36 Pt–ZrO2 (ref. 37 and 38) and Pt–WO3 (ref. 39 and 40) that improve the catalyst's resistance to CO poisoning through a bifunctional mechanism. (2) By changing the electronic structure of Pt through the ligand effect, the bond strength between Pt carbonyl groups (Pt–COads) could be weakened, which is conducive to promoting the oxidative desorption of CO and improving the resistance of the catalyst to poisoning. J. R. Kitchin et al. found through density function theory that Pt–Ti, Pt–V, Pt–Cr, Pt–Mn, Pt–Fe, Pt–Co Pt–Ni bimetals can change the electronic structure of the Pt surface through electronic interactions, also known as the ligand effect, thus changing chemical properties and weakening ability of platinum to adsorb CO.41 (3) The introduction of oxygen-containing groups in the catalyst carrier improves the resistance to CO poisoning of the catalyst. Xiaomei Chen et al. prepared the catalyst PtPdNPs/GNs by loading Pt–Pd alloys on graphene nanosheets and prepared catalysts Pt/GNs, Pd/GNs, PtPdNPs/C for comparison. The 1000 second i–t curves were tested to evaluate the resistance of the catalysts to CO poisoning. At the end of the 1000 second test, the ethanol oxidation current density of PtPdNPs/GNs was still much higher than the other catalysts, so the resistance to CO poisoning was better. One of the reasons for the improved resistance to poisoning is that the authors used the Hummers' method to prepare graphene oxide, where the carrier introduces a large number of oxygen-containing groups, which are later reduced to graphene as the catalyst carrier, and the remaining oxygen-containing groups on the carrier help to remove CO from the Pt–Pd alloy. The other reason is that, relative to Pt, Pd is proved to be more oxytropic with lower cost, which can act as an oxidation promoter by forming oxygen-containing species on the Pd metal particles, oxidizing and removing CO from the Pt surface.42
The natural abundance of 232Th is almost 100%, with a half-life of 1.4 × 1010 years, a thorium valence of +4 corresponding to the oxide ThO2, insoluble in water and alkali while soluble in H2SO4 solution.43 Lijin Xu et al.44 mixed SrCO3, BaSO4 and ThO2 powders in a mortar and crushed them into small particles under high pressure, and calcined at 900 °C for 0.5 h to obtain the catalyst. The catalyst was then utilized to catalyze the methane oxidation coupling reaction. During the action of the catalyst, the hydrocarbon bond of methane is broken and the hydrogen is released to interact with oxygen to form water, likewise the C2 double bond (alkene bond) is formed to produce olefins. The addition of ThO2 significantly improves the C2 yield and C2 selectivity relative to SrCO3/BaSO4, since introducing ThO2 substitutes Th4+ for Sr2+ as well as Ba2+, increasing the number of lattice defects and improving the catalytic activity. Ramya Arumugam et al.45 synthesized ThO2/Fe3O4 nanocomposites by hydrothermal methods with larger specific surface area, more active sites and higher stability for the catalytic and removal of triphenylmethane dyes (MG) from the aqueous phase with a degradation rate about 99.53% at pH = 5. Nanostructured N-doped ThO2 was synthesized by Wei, HT et al.46 and the material exhibited significant visible light absorption and high photocatalytic activity for the reduction of Cr(VI) under visible light (wavelength >420 nm) irradiation. Co–ThO2–ZSM-5 bifunctional zeolite catalysts were prepared for catalyzing the conversion of carbon monoxide and hydrogen to hydrocarbons by Murty A. N. et al.47 The catalytic performance (i.e. activity and selectivity) of the composites depended on the content of cobalt on the mesoporous zeolite ZSM-5 and the co-catalytic effect of ThO2. Considering the co-catalytic effect of ThO2 as shown above and the radioactivity property of it, we selected ThO2 to inspect its effect on the Pt/C catalyst.
In this work, we report the catalytic activity, durability, and resistance to CO poisoning of the novel catalyst Pt–ThO2/C in which the catalytic activity of ThO2 was firstly taken into consideration to address the problem of CO poisoning in the anode Pt/C catalyst of DEFC. The catalyst Pt–ThO2/C was synthesized by loading Pt particles onto the ThO2-loaded carbon carrier via impregnation–reduction method and characterized by TEM, EDS, XRD, ICP-MS and XPS. The catalytic performance of the catalyst was tested by an electrochemical workstation and three electrodes. Likewise, Pt–xThO2/C was assembled into a fuel cell to test the maximum cell power density.
Results and discussion
Physical characterization results
 | ||
| Fig. 1 (a) TEM micrograph and particle size distribution of ThO2; TEM micrographs and the Pt particle size distribution of the synthesized catalysts (b) Pt-6.67%ThO2/C, (c) Pt-40%ThO2/C and (d) Pt/C–NaBH4, respectively. | ||
 | ||
| Fig. 2 Pt–ThO2/C catalyst surface distribution of Th, Pt, C and O. | ||
 | ||
| Fig. 3 X-ray diffraction patterns of (a) ThO2 and (b) Pt–xThO2/C. | ||
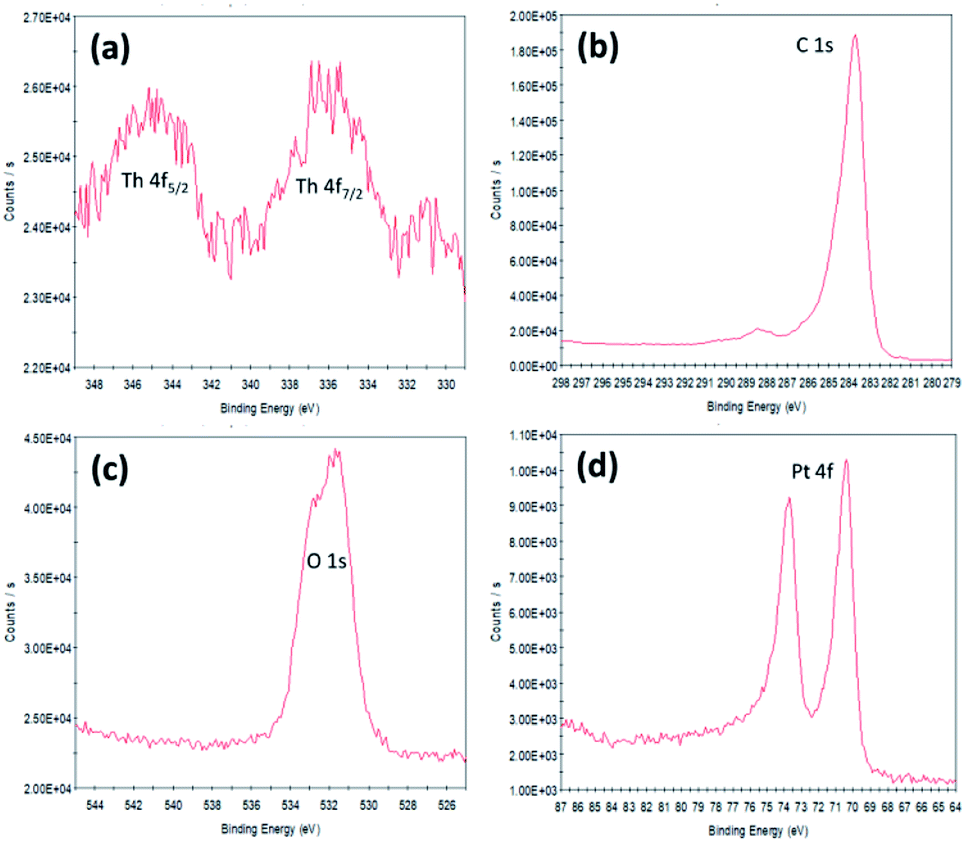 | ||
| Fig. 4 XPS regions (a) Th 4f5/2 and 4f7/2, (b) C 1s, (c) O 1s and (d) Pt 4f lines of Pt-10 wt%ThO2/C. | ||
| Catalysts | Pt | ThO2 |
|---|---|---|
| 20%Pt-40%ThO2/C | 18.9% | 36.3% |
| 20%Pt-20%ThO2/C | 19.0% | 19.4% |
| 20%Pt-10%ThO2/C | 19.3% | 9.9% |
| 20%Pt-6.67%ThO2/C | 18.2% | 6.0% |
| 20%Pt-3.33%ThO2/C | 20.9% | 3.3% |
| 20%Pt-1.67%ThO2/C | 19.5% | 1.5% |
| 20%Pt-0.83%ThO2/C | 19.7% | 0.9% |
| 20%Pt-0.42%ThO2/C | 19.4% | 0.4% |
| 20%Pt-0.21%ThO2/C | 18.9% | 0.1% |
| 20%Pt-0.1%ThO2/C | 19.2% | 0.08% |
| 20%Pt/C–NaBH4 | 19.3% | 0 |
Catalytic performance for ethanol oxidation reaction (EOR)
The cyclic voltametric (CV) curves of the 8 catalysts in a mixture solution of 0.5 mol L−1 H2SO4 and 0.5 mol L−1 C2H5OH are presented in Fig. 5. By comparing the magnitudes of the ethanol oxidation peak current densities of different catalysts, it was found that the addition of ThO2 as a catalytic additive to Pt/C could improve the catalytic activity, and the ethanol oxidation peak current density of Pt-6.67%ThO2/C (3.38 mA cm−2) was significantly higher than Pt/C–NaBH4 (2.34 mA cm−2), which might be due to the synergy between Pt and Th. It can also be observed that the catalytic activity of the material gradually increases as the mass fraction of ThO2 in the catalyst increases from 0.1%. The maximum activity is achieved at a ThO2 mass fraction of 6.67% and subsequent increases in ThO2 content result in a significant decrease in the catalytic activity of the material. This is due to the fact that the mass fraction of Pt in the catalyst is fixed and increasing the ThO2 content leads to a relative reduction in the carbon content and the fact that ThO2 is non-conductive, which seriously affects the conductivity of the catalyst. On the other hand, an increase in ThO2 content leads to an increase in the density of the particles loaded on the carbon, causing agglomeration, which can also affect the catalytic activity. | ||
| Fig. 5 Cyclic voltamograms of ethanol oxidation on catalyst systems in a mixture of 0.5 mol L−1 H2SO4 + 0.5 mol L−1 C2H5OH solution at 25 °C at a scan rate of 50 mV s−1. | ||
The retention rates of catalytic activity of prepared 11 catalysts after scanning 200 circles CV in a mixture solution of 0.5 mol L−1 H2SO4 and 0.5 mol L−1 C2H5OH are listed in Table 2, which are used to evaluate the durability performance of the catalysts. By comparing the activity retention rate of the different catalysts on the 200th circle, it can be seen that the activity of the catalysts mostly remained above 90%, with only a few percentage points of decay. This may be due to the fact that the Pt particles prepared by the reduction of H2PtCl6 by NaBH4 have a larger particle size and are lost less in acidic media. The catalytic activity of Pt-6.67%ThO2/C was maintained at 95.5%, an increase compared to Pt/C–NaBH4 (92.7%), indicating that the addition of ThO2 can improve catalyst durability.
| Catalysts | 1st ethanol oxidation peak current density mA cm−2 | 200th ethanol oxidation peak current density mA cm−2 |
|---|---|---|
| 20%Pt-40%ThO2/C | 2.16 (100%) | 1.88 (87.0%) |
| 20%Pt-20%ThO2/C | 2.36 (100%) | 2.12 (89.8%) |
| 20%Pt-10%ThO2/C | 2.57 (100%) | 2.34 (91.0%) |
| 20%Pt-6.67%ThO2/C | 3.38 (100%) | 3.23 (95.5%) |
| 20%Pt-3.33%ThO2/C | 3.25 (100%) | 3.09 (95.0%) |
| 20%Pt-1.67%ThO2/C | 3.19 (100%) | 3.11 (97.4%) |
| 20%Pt-0.83%ThO2/C | 2.82 (100%) | 2.74 (97.1%) |
| 20%Pt-0.42%ThO2/C | 3.01 (100%) | 2.94 (97.6%) |
| 20%Pt-0.21%ThO2/C | 2.64 (100%) | 2.58 (97.7%) |
| 20%Pt-0.1%ThO2/C | 2.40 (100%) | 2.29 (95.4%) |
| 20%Pt/C–NaBH4 | 2.34 (100%) | 2.17 (92.7%) |
Fig. 6 presents the i–t curves of 11 catalysts in a mixture solution of 0.5 mol L−1 H2SO4 and 0.5 mol L−1 C2H5OH at a constant potential of 0.6 V (vs. SCE). Initially for a very short period of time, the intermediate product CO species from the oxidation of ethanol adsorb onto the active sites of Pt, forming a stable platinum carbonyl group that covers the active site of Pt, preventing further dissociation of ethanol and causing catalyst poisoning, which severely affects the catalytic activity and manifests itself as a sharp drop in the current curve. Afterwards, the adsorption and oxidative desorption of CO on the Pt surface gradually tends to a dynamic equilibrium due to the detoxification effect of ThO2, and the current curve decreases to a flattening. It is obvious that the current curve decreases more slowly with time for Pt-6.67%ThO2/C compared to the other catalysts. After 3000 s of constant voltage testing, the current density of Pt-6.67%ThO2/C was 0.89 mA cm−2, which was still significantly higher than Pt/C–NaBH4 (0.41 mA cm−2), indicating that the addition of ThO2 enable the catalyst to reactivate the catalyst by itself. The reason for this is that α particles released by 232Th irradiates water to produce OH radicals (H2O + *α → ˙OH + H+ + e−), which rapidly removes COads (Pt–COads + ˙OH → Pt + CO2 + H+ + e−) and releases the active site of Pt. Thus, we call the whole process self-reactivation and the Pt–xThO2/C the self-reactivated catalyst.
 | ||
| Fig. 6 Amperometric i–t curves of ethanol oxidation on catalyst systems in a mixture of 0.5 mol L−1 H2SO4 + 0.5 mol L−1 C2H5OH solution at 25 °C at 0.6 V (vs. SCE) for 3000 s. | ||
Fig. 7 shows the chronopotentiometry curves of Pt-6.67%ThO2/C and Pt/C–NaBH4 in a mixture solution of 0.5 mol L−1 H2SO4 and 0.5 mol L−1 C2H5OH at a constant current of 0.1 mA. Within a very short initial period of time, the electrode potential rises sharply to the ethanol oxidation onset potential. As the test time increases, the electrode potential first rises slowly. The intermediate product CO produced during the oxidation of ethanol gradually accumulates on the catalyst surface, making the electrocatalytic activity of the catalyst decrease and the overpotential of ethanol oxidation increase, and in order to meet the set current conditions, the electrode potential must rise accordingly. As the catalyst is further poisoned, the catalyst surface is almost completely covered with toxic species and the electrocatalytic oxidation of ethanol is almost impossible, at which point the electrode potential rises sharply until it reaches the oxygen precipitation potential as a means of meeting the set current demand. The resistance of the catalyst to CO poisoning is evaluated on the basis of the time taken for the electrode potential to rise sharply to a sudden jump in potential. Pt-6.67%ThO2/C shows a potential jump time of 337 s and Pt/C–NaBH4 shows a potential jump time of 60 s, so Pt-6.67%ThO2/C is more resistant to CO poisoning, most likely due to the self-reactivation effect mentioned above.
 | ||
| Fig. 7 Chronopotentiometry curves of ethanol oxidation on catalyst systems in a mixture of 0.5 mol L−1 H2SO4 and 0.5 mol L−1 C2H5OH solution at 25 °C at 0.1 mA. | ||
Single cell performance
To further investigate the modification effect of ThO2 on Pt/C catalyst, Pt-6.67%ThO2/C and Pt/C–NaBH4 were used as anode catalyst to assemble single cell and respectively test the cell V–i polarization curves. Fig. 8 shows the V–i polarization curves and power density curves for the two catalysts. The V–i curve is polarized for three main reasons, one is activation polarization, when a chemical reaction occurs on the electrode surface to output current, the reaction resistance must be overcome, the reaction resistance leads to polarization. Secondly, ohmic polarization, when the battery is operated, the flow of ions and electrons will be subject to resistance to produce a voltage drop, and resistance leads to polarization. Third is the concentration difference polarization, if the rate of transport of ethanol to the electrode is less than the rate of consumption, it will cause the concentration of ethanol on the surface of the electrode to be lower than its own concentration, this concentration difference leads to polarization. | ||
| Fig. 8 Cell voltage and power density as a function of current density for the single cell with Pt-6.67%ThO2/C catalyst (anode) and Pt/C–NaBH4 catalyst (anode) operating at 60 °C. | ||
Fig. 8 V–i curves also show that the polarization of Pt/C–NaBH4 is more obviously than Pt-6.67%ThO2/C. Moreover, according to the power density curves, it can be found that the maximum power density of single cell is 20.52 mW cm−2 for Pt-6.67%ThO2/C and 18.96 mW cm−2 for Pt/C–NaBH4. So it is further clear from the fuel cell test results that the addition of ThO2 can improve Pt/C catalytic performance.
Experimental
Materials
Cabot carbon (XC-72) was purchased from Suzhou Sinero. Th(NO3)4·4H2O (AR) was purchased from Changsha Jingkang. H2PtCl6·6H2O (AR), Na3C6H5O7·2H2O (AR), C2H5OH (GR), NaBH4 (AR) were purchased from Aladdin reagent. NaOH (AR) and H2SO4 (AR) were purchased from sinopharm chemical reagent. C3H8O (AR) was purchased from Macklin reagent. Nafion (5 wt%) was purchased from Shanghai Hesen.Synthesis of Pt–ThO2/C catalysts
Physical characterizations
The microstructure of Pt–xThO2/C Pt–xThO2/C and Pt/C–NaBH4 was observed by the transmission electron microscopy (TEM) measurement with JEM-F200 manufactured by Japan Electronics Corporation. The sample was prepared by sonicating the material into anhydrous ethanol and then adding a small amount of the suspension drop by drop onto a copper grid to dry for measurement.Energy dispersive spectroscopy (EDS) tests were carried up on a SU8020 manufactured in Hitachi of Japan to roughly observe the existence and distribution of the elements on the surface of the catalyst Pt–xThO2/C.
The physical phases present in the material was characterized by the X-ray diffraction technique (XRD) on a Bruker D8 ADVANCE, manufactured in Germany, equipped with a Cu-Kα X-ray source and operated at a voltage of 40 kV, a current of 40 mA and a test angle of 5–90°.
The X-ray photoelectron spectroscopy (XPS) was obtained on a VG Multilab 2000 using Al Kα (1486.7 eV) as the exciting radiation to further confirm the presence and the chemical states of the elements at the materials' surface.
Inductively coupled plasma mass spectrometer (ICP-MS) measurements were conducted on an Agilent 7850 ICP-MS from Agilen of Japan to analyze the elemental content of the solution.
Electrochemical measurements
An electrochemical workstation with a three-electrode system was used to test the catalytic performance of the catalysts. The electrochemical workstation used for the experiments was manufactured by Shanghai Chenhua, model CHI760E. The three electrodes are, in order, a saturated calomel electrode as reference electrode, model CHI150, with a saturated KCl solution inside the saturated calomel electrode; a platinum wire electrode as counter electrode, model CHI115; and a glassy carbon electrode as working electrode, model CHI104, with a 3 mm diameter glassy carbon in the middle part of the glassy carbon electrode. All three electrodes were purchased from Shanghai Chenhua. When the electrochemical workstation is in operation, the three electrodes form two circuits, where the working electrode loaded with catalyst and the reference electrode form a circuit to test the working electrode potential, as the reference electrode potential is known to be constant. For example, the standard electrode potential of a saturated calomel electrode is 0.2412 V. The potential output from the workstation is the difference in potential between the working and reference electrodes. The working electrode and counter electrode form a circuit to test the working electrode current, this is known as the “three electrodes and two circuits”. The working electrode is prepared as follows: using in turn 0.3 μm and 0.05 μm Al2O3 powder, the electrode is polished on a polishing flannel with the aim of polishing the glassy carbon to a mirror-like surface without scratches and the PTFE material wrapped around the glassy carbon to a very hydrophobic surface. Weigh 10 mg of the catalyst to be tested, add to 2.4 mL of ultra-pure water, 0.1 mL of Nafion, 2.5 mL of C3H8O and sonicate for 30 min to obtain a 2 mg mL−1 slurry of catalyst. Pipette 5 μL of the slurry onto the glassy carbon electrode and allow drying naturally as a working electrode.Fuel cell measurements
The catalyst to be tested was ultrasonically dispersed with the Nafion solution in C3H8O under ice bath conditions. The catalyst suspension was then sprayed onto a PTFE film as the catalytic layer and subsequently hot-pressed onto both sides of a Nafion 115 film at 120 °C and 10 MPa maintained for 120 s. The outermost side used carbon cloth (H2315T10AC1 NOK, Japan) as the diffusion layer to form a membrane electrode (MEA) with an effective area of 2 × 2.5 cm2. A homemade catalyst with a Pt loading of 1.2 mg cm−2 was used for the cell anode and a commercial Pt/C with a Pt loading of 1.2 mg cm−2 was used for the cathode, with a Nafion solution (5 wt%) content of 15 wt% for the anode and 10 wt% for the cathode. The cell operating temperature was set at 60 °C with 2 mol L−1 C2H5OH passing through the anode at a flow rate of 1.5 sccm and O2 passing through the cathode at a flow rate of 400 sccm. Prior to testing, a 0.5 mol L−1 H2SO4 solution was passed from the anode for 30 min at 55 °C for reducing the internal resistance of the proton exchange membrane. The test was then started after passing 0.5 sccm of 1.5 mol L−1 C2H5OH solution on the anode side and 800 sccm of O2 on the cathode and discharging at 100 mA cm−2 for 10 hours for stabilizing the internal mass transfer channels of the catalytic layer.Conclusions
To settle the low activity and susceptibility to CO poisoning of Pt/C anode catalysts for DEFC, a radioactive ThO2 catalytic additive prepared by hydrothermal method was introduced into the Pt/C catalyst at first time. TEM, EDS, XRD, XPS and ICP-MS characterization revealed that Pt and ThO2 were successfully loaded on the carbon carrier and evenly distributed, while the Pt particles were poorly dispersed on the carbon and showed significant agglomeration due to the relatively rapid and violent reaction of NaBH4 in reducing H2PtCl6.The catalyst performance was determined by electrochemical workstation equipped with three electrodes. It was illustrated by the results that the catalysts prepared with 20% mass fraction of Pt and 6.67% mass fraction of ThO2 showed the best catalytic activity and resistance to CO poisoning which was significantly improved the performance compared to Pt/C–NaBH4. The increases mentioned above are partly due to the synergistic effect of Pt and Th. Moreover, Pt–xThO2/C is a self-reactivated catalyst, i.e., α particles released by the radioactive 232Th irradiate water to produce a large amount of strongly oxidizing hydroxyl radicals, which oxidize the CO adsorbed on Pt surface, expose the active site of Pt and promote further dissociation of ethanol. The results of the single cell operating performance of Pt-6.67%ThO2/C and Pt/C–NaBH4 used as anode catalysts in DEFC respectively illustrated that the cell polarization was much more severe for Pt/C–NaBH4, and the maximum power density of the cell was higher for Pt-6.67%ThO2/C than that of Pt/C–NaBH4, further confirming the impressive self-reactivation effect of ThO2 on Pt/C catalyst.
Author contributions
Yubing Xue: formal analysis, investigation, visualization, writing – original draft. Dashu Pan: formal analysis, validation. Feng Zuo: data curation, investigation. Songtao Xiao: methodology, project administration. Xiang Li: formal analysis. Fuyan Lou: resources. Mingming Li: writing – review&editing. Yinggen Ouyang: funding acquisition, conceptualization, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support for this project provided by the National Natural Science Foundation of China.Notes and references
- H. Zeng and H. Li, IOP Conf. Ser.: Earth Environ. Sci, 2020, 440, 042010 CrossRef.
- X. Xu, Z. Wei, Q. Ji, C. Wang and G. Gao, Resour. Policy, 2019, 63, 101470 CrossRef.
- J. Seweryn and A. Lewera, Appl. Catal., B, 2014, 144, 129–134 CrossRef CAS.
- J. R. C. Salgado, V. A. Paganin, E. R. Gonzalez, M. F. Montemor, I. Tacchini, A. Anson, M. A. Salvador, P. Ferreira, F. M. L. Figueiredo and M. G. S. Ferreira, Int. J. Hydrogen Energy, 2013, 38, 910–920 CrossRef CAS.
- V. Livshits, M. Philosoph and E. Peled, J. Power Sources, 2008, 178, 687–691 CrossRef CAS.
- A. Anis, S. Al-Zahrani and F. A. El Aleem, Int. J. Electrochem. Sci., 2012, 7, 6221–6233 CAS.
- V. K. Puthiyapura, D. J. L. Brett, A. E. Russell, W. Lin and C. Hardacre, ACS Appl. Mater. Interfaces, 2016, 8, 12859–12870 CrossRef CAS PubMed.
- U. Lucia, Renewable Sustainable Energy Rev., 2014, 30, 164–169 CrossRef CAS.
- O. Z. Sharaf and M. F. Orhan, Renewable Sustainable Energy Rev., 2014, 32, 810–853 CrossRef CAS.
- T. Maiyalagan and S. Pasupathi, Mater. Sci. Forum, 2010, 657, 143–189 CAS.
- D. M. Fadzillah, S. K. Kamarudin, M. A. Zainoodin and M. S. Masdar, Int. J. Hydrogen Energy, 2019, 44, 3031–3054 CrossRef CAS.
- S. P. S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 97, 1–12 CrossRef CAS.
- S. Song, W. Zhou, Z. Liang, R. Cai, G. Sun, Q. Xin, V. Stergiopoulos and P. Tsiakaras, Appl. Catal., B, 2005, 55, 65–72 CrossRef CAS.
- S. Samad, K. S. Loh, W. Y. Wong, T. K. Lee, J. Sunarso, S. T. Chong and W. R. W. Daud, Int. J. Hydrogen Energy, 2018, 43, 7823–7854 CrossRef CAS.
- M. Ercelik, A. Ozden, Y. Devrim and C. O. Colpan, Int. J. Hydrogen Energy, 2017, 42, 2658–2668 CrossRef CAS.
- Y. Devrim, S. Erkan, N. Bac and I. Eroglu, Int. J. Energy Res., 2012, 37, 435–442 CrossRef.
- B. Fang, L. Daniel, A. Bonakdarpour and D. P. Wilkinson, Adv. Mater. Interfaces, 2022, 2200349 CrossRef.
- G. Liu, Z. Yang, X. Wang and B. Fang, Nanomaterials, 2021, 11, 3462 CrossRef CAS PubMed.
- A. Nouri-Khorasani, A. Bonakdarpour, B. Fang and D. P. Wilkinson, ACS Appl. Mater. Interfaces, 2022, 14, 9084–9096 CrossRef PubMed.
- B. Fang, L. Daniel, A. Bonakdarpour, R. Govindarajan, J. Sharman and D. P. Wilkinson, Small, 2021, 17, 2102288 CrossRef CAS PubMed.
- M. S. Kim, B. Fang, N. K. Chaudhari, M. Song, T. S. Bae and J. S. Yu, Electrochim. Acta, 2010, 55, 4543–4550 CrossRef CAS.
- L. Lu, B. Wang, D. Wu, S. Zou and B. Fang, Nanoscale, 2021, 13, 3709–3722 RSC.
- L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- V. B. Oliveira, J. P. Pereira and A. M. F. R. Pinto, Energy, 2017, 133, 652–665 CrossRef CAS.
- M. A. F. Akhairi and S. K. Kamarudin, Int. J. Hydrogen Energy, 2016, 41, 4214–4228 CrossRef CAS.
- K. Miyazaki, T. Matsumiya, T. Abe, H. Kurata, T. Fukutsuka, K. Kojima and Z. Ogumi, Electrochim. Acta, 2011, 56, 7610–7614 CrossRef CAS.
- Z. Zakaria, S. K. Kamarudin and S. N. Timmiati, Appl. Energy, 2016, 163, 334–342 CrossRef CAS.
- A. Arshad, H. Ali, A. Habib and M. A. Bashir, Therm. Sci. Eng. Prog., 2019, 9, 308–321 CrossRef.
- R. Ali and A. Pasha, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 376, 012103 Search PubMed.
- K. A. Page and B. W. Rowe, Polymers for Energy Storage and Delivery: Polyelectrolytes for Batteries and Fuel Cells, 2012, 1096, pp. 147–164 Search PubMed.
- M. Watanabe and S. Motoo, J. Electroanal. Chem. Interfacial Electrochem., 1975, 60, 275–283 CrossRef CAS.
- Y. Qu, Y. Gao, L. Wang, J. Rao and G. Yin, Chemistry, 2016, 22, 193–198 CrossRef CAS PubMed.
- X. Wang, J. Zhang and H. Zhu, Chin. J. Catal., 2011, 32, 74–79 CrossRef CAS.
- Y. Gu and W. Wong, J. Electrochem. Soc., 2006, 153, A1714–A1718 CrossRef CAS.
- Z. Chen, X. Qiu, B. Lu, S. Zhang, W. Zhu and L. Chen, Electrochem. Commun., 2005, 7, 593–596 CrossRef CAS.
- Y. Bai, J. Wu, J. Xi, J. Wang, W. Zhu, L. Chen and X. Qiu, Electrochem. Commun., 2005, 7, 1087–1090 CrossRef CAS.
- H. Song, X. Qiu and F. Li, Appl. Catal., A, 2009, 364, 1–7 CrossRef CAS.
- V. Raghuveer and B. Viswanathan, J. Power Sources, 2005, 144, 1–10 CrossRef CAS.
- D. Zhang, Z. Ma, G. Wang, K. Kongstantinov, X. Yuan and H. Liu, Electrochem. Solid-State Lett., 2006, 9, A423–A426 CrossRef CAS.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS PubMed.
- X. Chen, Z. Cai, X. Chen X and M. Oyama, J. Mater. Chem. A, 2014, 2, 315–320 RSC.
- A. H. Alomari, M. A. Saleh, S. Hashim, A. Alsayaheen, I. Abdeldin and A. Abukashabeh, Radiat. Phys. Chem., 2020, 170, 108660 CrossRef CAS.
- X. Lijin, Q. Fali and L. Shaoje, J. Nat. Gas Chem., 1996, 5, 136 Search PubMed.
- R. Arumugam, P. Periakaruppan and R. Selvanathan, Int. J. Pharm. Sci. Res., 2019, 10, 2902–2910 CAS.
- H. T. Wei, Y. T. Wen and Y. C. Zhang, Catal. Commun., 2017, 99, 66–70 CrossRef CAS.
- A. N. Murty, A. A. Williams, R. T. Obermyer and V. U. S. Rao, J. Appl. Phys., 1987, 61, 4361–4363 CrossRef CAS.
- A. C. Ferreira, A. P. Goncalves, T. A. Gasche, A. M. Ferraria, A. M. Botelho do Rego, M. R. Correia, A. M. Bola and J. B. Branco, J. Alloys Compd., 2010, 497, 249–258 CrossRef CAS.
- F. J. Pereira, M. A. Castro, M. D. Vázquez, L. Debán and A. J. Aller, J. Lumin., 2017, 184, 169–178 CrossRef CAS.
- Y. Huentupil, G. Cabello-Guzmán, B. Chornich, R. Arancibia and G. E. Buono-Core, Polyhedron, 2019, 157, 225–231 CrossRef CAS.
- H. Iida and A. Igarashi, Appl. Catal., A, 2006, 298, 152–160 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |