
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antioxidant properties of butylated phenol with oxadiazole and hydrazone moiety at ortho position supported by DFT study†
Raied M. Shakiraab,
Muhammad Kumayl Abd Wahaba,
Nurdiana Nordin *a and
Azhar Ariffin*a
*a and
Azhar Ariffin*a
aDepartment of Chemistry, Faculty of Science, Universiti Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: ndiana13@um.edu.my; azhar70@um.edu.my; Fax: +60 7967 4193; Tel: +60 7967 7022 Tel: +60 7967 4080
bDepartment of Chemistry, Ibn Al-Haitham University of Baghdad, Baghdad, Iraq
First published on 9th June 2022
Abstract
Two series of 1,3,4-oxadiazole derivatives at the sixth position of the 2,4-di-tert-butylphenol group were synthesized. The antioxidant properties were evaluated by DPPH and FRAP assays. Compound 3 showed significant antioxidant activity, while its alkyl derivatives exhibited decreased antioxidant activity in both assays. The preferential antioxidant mechanism of the reactive antioxidant molecules prepared from the further reaction of compound 3 to produce compounds 4 and 6 was investigated using density functional theory. Calculating their comprehensive reactivity descriptors was used to assess their antioxidant reactivity. According to the calculated descriptors, compounds 4c and 6d are the most reactive antioxidants within their own group compared to the other derivative moieties. The results are identical to ascorbic acid's, indicating that they have similar activity. The experimental data and the calculated descriptors are in good agreement. The nature of the substituents and their positions have a significant impact on the derivatives' antioxidant capabilities.
Introduction
Excessive production of reactive free radicals in the body, which are produced by a variety of enzymatic and nonenzymatic activities, causes substantial macromolecule damage in protein, DNA, membrane lipids, and carbohydrates, resulting in cellular damage. The oxidative stress that results is thought to play a role in the ageing process and the development of degenerative diseases such as type 2 diabetes, inflammation, neurological diseases, and malignancies.1–3 As a result, there is a growing interest in using medicinal substances with antioxidant activity to block these unwanted oxidative events by trapping free-radical intermediates produced during oxidative reactions.4–7Recently, many reports have emphasised that phenol derivatives play a very important role in human health and in therapeutics,8–10 as well as in industry,11 where they are employed as stabilisers,12–14 via their antioxidant activity. Hindered phenols are notably interesting antioxidant compounds,15,16 in which the di-tert-butylphenol can be considered a crucial starting material in preparing antioxidant compounds.17,18 Many derivatives of the di-tert-butylphenol group (containing a heterocyclic group) have been observed to show good biological activity (in addition to antioxidant activity), such as anti-inflammatory activity,19 the growth inhibition of lung cancer20 and the development of cyclooxygenase-2 inhibitors.21 This antioxidant activity is not only confined to di-tert-butyl groups flanking the hydroxyl group (such as 2,6-DTBP) but also the semi-hindered phenol with one tert-butyl group bonded to the hydroxyl in position 2 and the other at position 4.22,23
Conversely, 2,4-di-tert-butylphenol is known as an antioxidant, and it has been reported that its antioxidant activity is lower than that of BHT (butylated hydroxytoluene) and that the tert-butyl group in the para position can reduce the compound's antioxidant ability.24 Furthermore, compounds that can be classified as strong antioxidants usually share common structural features. For example, such compounds often have a hindered phenol25 or multiple phenolic hydroxyl groups, e.g., flavonoids,26,27 or have a fully conjugated π system, e.g., carotenoids.28 It is interesting to note that substituted groups may also influence the scavenging ability of such compounds, as mentioned previously, which indicates the existence of a close relationship between chemical structure and the ability to scavenge free radicals. The 1,3,4-oxadiazoles and their 2,5-disubstituted derivatives function well as development materials due to their large spectrum of various biological activities, such as anti-inflammatory,29 anticancer,30 antibacterial,31 antifungal,18 antihypertensive,32 antitubercular,33 anti-HIV34 and antioxidant activities.35 Moreover, some oxadiazoles derivatives have exhibited antioxidant activities even though they do not contain any phenol group.36
In this study, we synthesised a 1,3,4-oxadiazole ring at position six of 2,4-di-tert-butyl phenol. We aim to accomplish the phenol's antioxidant activity (to be approaching that of BHT or slightly less) by increasing the steric hindrances surrounding its hydroxyl while increasing free radical stability (which is formed after the phenol donates its hydrogen atom). Hydrazones of 2,4-DTBP (as an intermediate in the formation the oxadiazole ring) have also been investigated for their antioxidant ability. The synthesised compounds are illustrated in Fig. 1 and 2. In addition, we performed a systematic analysis of seven synthesised compounds of 1,3,4-oxadiazole ring at position six of 2,4-di-tert-butyl phenol derivatives by means of structural conformational and electron analyses of protonated (neutral) and deprotonated forms. In the context of conceptual DFT, the global reactivity descriptors of neutral compounds have been derived. These calculations may result in the implementation of biological antioxidants by illustrating the SAR and radical scavenging mechanisms of these naturally occurring chemicals.
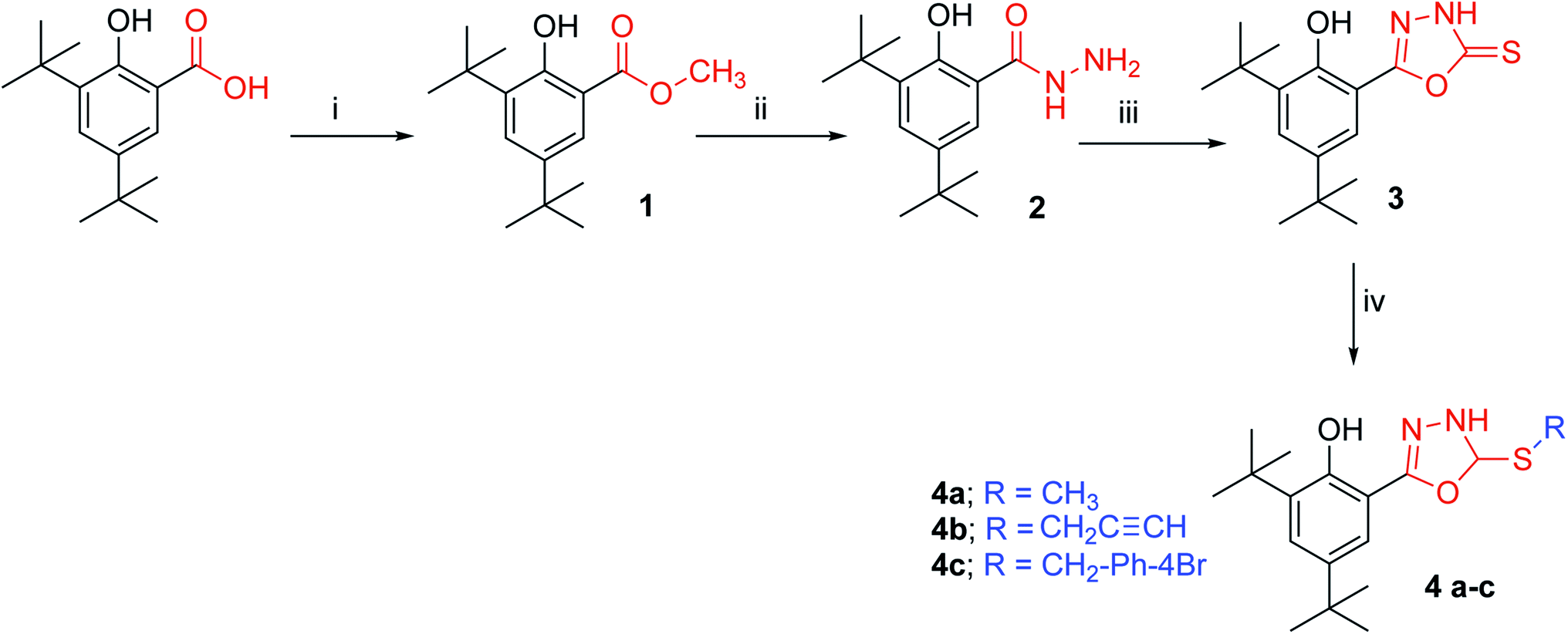 | ||
| Fig. 1 Reaction pathway for the synthesis of compounds 3 and 4a–c (i) MeI, NaHCO3, DMF, reflux 9 h. (ii) Heating to melting point, N2H4·H2O. (iii) CS2, KOH, EtOH, reflux 3 h. (vi) RX, K2CO3, acetone, r.t. | ||
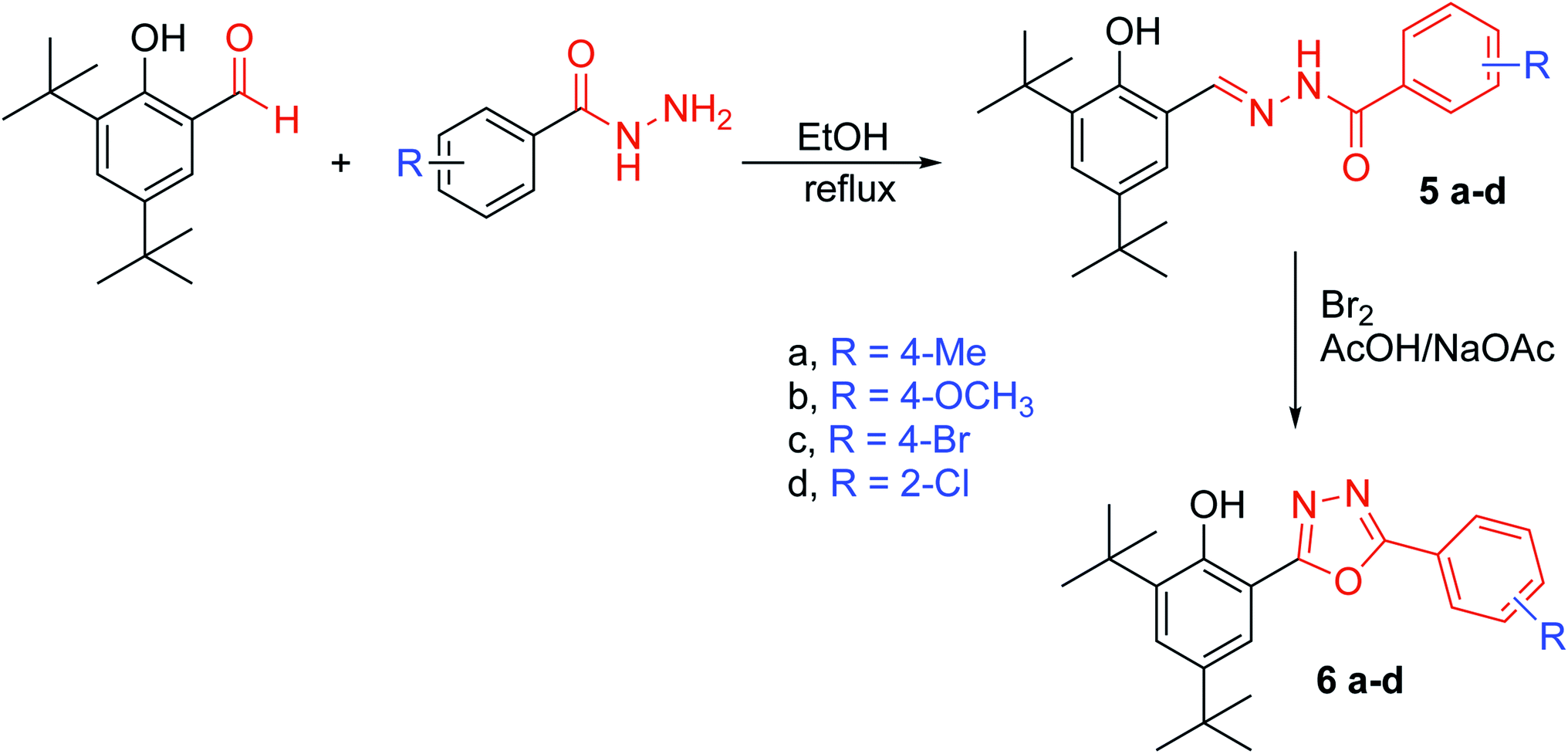 | ||
| Fig. 2 Reaction pathway for the synthesis of compounds 5a–d and 6a–d. | ||
Experimental section
Materials
Unless otherwise noted, all materials were purchased from commercial suppliers and used without purification. The melting points of the materials were determined by the open capillary tube method using MEL-TEMP II apparatus and were uncorrected. The purities of compounds were verified by thin layer chromatography (silica gel TLC) using plates from Merck, and spots were located under iodine vapour and UV radiation. IR spectra were recorded using a PerkinElmer 400 Fourier Transform Infrared (FTIR) Spectrometer. NMR spectra were recorded on a JEOL-ECA 400 MHz, JEOL-Lambda 400 or Bruker 400AVN MHz spectrometer, with CDCl3 and DMSO-d6 used as the solvent and TMS as the internal standard. Mass spectra were recorded using an Agilent 5975 for EI/MS and Finnigan TSQ7000 for HREI/MS (NUS Singapore). UV spectroscopy Power Wave X340 (BIO-TEK Instruments, Inc.) was used to record the FRAP and DPPH assays.Synthesis of methyl 3,5-di-tert-butyl-salicylate (1)
A 100 mL round-bottom flask was charged with 3,5-di-tert-butyl-salicylic acid (3.75 g, 15 mmol) and sodium hydrogen carbonate (1.3 g, 15.5 mmol) in 20 mL dry DMF. Excess methyl iodide was added. The mixture was refluxed for 9 h, and the reaction was monitored by TLC using a hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate solution mixed in a 5:1 ratio. Excess solvent was evaporated under reduced pressure. 25 mL distilled water was added to the solid residue and extracted with 50 mL ethyl acetate. The organic layer was washed with water and dried over anhydrous magnesium sulphate. The solvent was evaporated under reduced pressure. The crude product was recrystallised from ethanol–hexane to afford a pure white solid ester. Yield 3.76 g (94%); mp 72–74 °C; νmax (KBr)/cm−1: 3105 (CHaromatic), 2957, 2908, 2870 (CHaliphatic), 1727 (C
:ethyl acetate solution mixed in a 5:1 ratio. Excess solvent was evaporated under reduced pressure. 25 mL distilled water was added to the solid residue and extracted with 50 mL ethyl acetate. The organic layer was washed with water and dried over anhydrous magnesium sulphate. The solvent was evaporated under reduced pressure. The crude product was recrystallised from ethanol–hexane to afford a pure white solid ester. Yield 3.76 g (94%); mp 72–74 °C; νmax (KBr)/cm−1: 3105 (CHaromatic), 2957, 2908, 2870 (CHaliphatic), 1727 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1600, 1559 (CC), 1215 (C–O). 1H NMR (CDCl3) δ 1.28 (s, 9H, t-Bu), 1.41 (s, 9H, t-Bu), 3.92 (s, 3H, OCH3), 7.57 (d, 1H, J 2.76), 7.78 (d, 1H, J 2.76), 11.32 (s, 1H, OH). 13C NMR (CDCl3) δ 29.47 (3C), 31.50 (3C), 34.38 (C), 35.25 (C), 52.28 (C, OCH3), 110.50 (C), 124.69 (C), 131 (C), 137.51 (C), 140.96 (C), 159.79 (C), 176.05 (C, CO). HREIMS found 264.1725 [M˙+] (calc. for C16H24O3, 264.1725).
O) 1600, 1559 (CC), 1215 (C–O). 1H NMR (CDCl3) δ 1.28 (s, 9H, t-Bu), 1.41 (s, 9H, t-Bu), 3.92 (s, 3H, OCH3), 7.57 (d, 1H, J 2.76), 7.78 (d, 1H, J 2.76), 11.32 (s, 1H, OH). 13C NMR (CDCl3) δ 29.47 (3C), 31.50 (3C), 34.38 (C), 35.25 (C), 52.28 (C, OCH3), 110.50 (C), 124.69 (C), 131 (C), 137.51 (C), 140.96 (C), 159.79 (C), 176.05 (C, CO). HREIMS found 264.1725 [M˙+] (calc. for C16H24O3, 264.1725).
Synthesis of 3,5-di-tert-butyl-salicylic hydrazide (2)
Methyl 3,5-di-tert-butyl-salicylate (2.64 g, 10 mmol) was heated to its melting point in a 50 mL round-bottom flask. When the ester has melted, 5 mL of hydrazine hydrate was added dropwise, and the mixture was heated to 70–75 °C for 2 h. Absolute ethanol was added until a clear solution appeared and then refluxed for another 3 h. Upon cooling, the white precipitate was filtered and washed. Recrystallisation of the crude product from aqueous ethanol afforded a white precipitate. Yield 2.53 g (96%); mp 192–194 °C; νmax (KBr)/cm−1: 3675 (OHphenol), 3316, 3208, 3192 (NHNH2), 2960, 2871 (CHaliphatic), 1626 (CO), 1593 (CC). 1H NMR (DMSO-d6) δ 1.26 (s, 9H, t-Bu), 1.47 (9H, s, t-Bu), 4.93 (brs, 2H, NH2), 7.35 (1H, s), 7.83 (s, 1H), 10.28 (brs, 1H, NH), 13.40 (brs, 1H, OH). 13C NMR (DMSO-d6) δ 29.68 (3C), 31.79 (3C), 34.61 (C), 35.14 (C), 112.82 (C), 121.21 (C), 127.92 (C), 136.83 (C), 140.22 (C), 157.82 (C), 170.11 (C). HREIMS found 264.1838 [M˙+] (calc. for C15H24O2N2, 264.1838).
Synthesis of 2,4-di-tert-butyl-6-(5-thio-4-hydro-1,3,4-oxadiazol-2-yl)phenol (3)
To a solution of hydrazide (1 g, 3.75 mmol) and excess carbon disulphide (0.6 mL) in absolute ethanol, potassium hydroxide (0.21 g, 3.75 mmol) was added in one portion at ambient temperatures. The mixture was stirred and refluxed for 3 h, and the solvent was removed under vacuum. Distilled water (25 mL) was added to the residue and stirred for another 15 minutes. The residue was filtered, and the filtrate was acidified with 5% hydrochloric acid and then filtered once more. The white precipitate was washed with water and recrystallised with ethanol. Yield 0.91 g (79%); mp 222–224 °C; νmax (KBr)/cm−1: 3187 (bs, OH, NH), 3092 (CHaromatic), 2956 (CHaliphatic), 1618 (CN), 1594, 1580 (CC), 1270 (CS), 1218 (C–O), 1095 (C–O–C). 1H NMR (CDCl3) δ 1.27 (s, 9H, t-Bu), 1.38 (s, 9H, t-Bu), 7.41 (d, 1H, J 2.23), 7.45 (d, 1H, J 2.20), 9.1 (brs, 1H, OH). 13C NMR (CDCl3) δ 29.85 (3C), 31.66 (3C), 34.61 (C), 35.48 (C), 109.48 (C), 122.13 (C), 128.34 (C), 137.88 (C), 142.35 (C), 152.96 (C), 161.11 (C, CN), 177.10 (C, CS). HREIMS found 306.1402 [M˙+] (calc. for C16H22N2O2S, 306.1402).
General alkylation 2,4-di-tert-butyl-6-(5-thio-4-hydro-1,3,4-oxadiazol-2-yl) phenol (4a–c)
Alkyl halide (1 mmol) was added in small portions to a stirred suspension of 1,3,4-oxadiazol (0.31 g, 1 mmol) in dry acetone and anhydrous potassium carbonate (0.14 g, 1 mmol). The mixture was left to stir overnight at ambient temperature. The solvent was evaporated and the residue extracted with 25 mL chloroform. The residue was dried over anhydrous magnesium sulphate and recrystallised from a suitable solvent.N), 1595, 1482 (CC), 1178 (C–O), 1095 (C–O–C). 1H NMR (CDCl3) δ 1.31 (s, 9H, t-Bu), 1.44 (s, 9H, t-Bu), 2.78 (s, 3H, SCH3), 7.46 (d, 1H, J 2.44), 7.52 (d, 1H, J 2.44), 10.21 (brs, 1H, OH). 13C NMR (CDCl3) δ 14.77 (C, SCH3), 29.48 (3C), 31.53 (3C), 34.46 (C), 35.39 (C), 107.47 (C), 120.64 (C), 128.46 (C), 137.46 (C), 141.79 (C), 154.30 (C), 164.12 & 166.35 (C7, C8, CN). HREIMS found 320.1562 [M˙+] (calc. for C17H24N2O2S, 320.1558).![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 3153(OHphenol), 2951, 2868 (CHaliphatic), 2164(CC), 1617 (CN), 1995, 1557 (CC), 1178 (C–O), 1095 (C–O–C); 1H NMR (DMSO-d6) δ 1.29 (s, 9H, t-Bu), 1.41 (s, 9H, t-Bu), 3.37 (t, 1H, J 2.67), 4.23 (d, 2H, J 2.68), 7.48 (d, 1H, J 2.44), 7.59 (d, 1H, J 2.44), 10.18 (brs, 1H, OH). 13C NMR (CDCl3) δ 20.95 (SCH2 C), 29.21 (3C), 31.09 (3C), 34.06 (C), 34.88 (C)75.16 ((CH)CH), 78.92 (CCH), 107.69 (C), 121.13 (C), 128.16 (C), 136.90 (C), 141.90 (C4), 153.30 (C), 161.98 & 166.00 (C7 & C8). HREIMS found 344.1558 [M˙+] (calc. for C19H24N2O2S, 344.1558).N), 1995, 1845 (CC), 1265 (C–O), 1081 (C–O–C). 1H NMR (CDCl3) δ 1.32 (s, 9H, t-Bu), 1.45 (s, 9H, t-Bu), 4.46 (s, 2H), 7.35 (d, 2H, J 8.41), 7.36–7.50 (m, 4H). 13C NMR (CDCl3) δ 29.39 (3C), 31.44 (3C), 34.38 (C), 35.32 (C), 36.16 (C), 107.27 (C), 120.57 (C), 122.26 (C), 128.52 (C), 130.76 (2C), 131.99 (2C), 134.64 (C), 137.43 (C), 141.75 (C), 154.26 (C), 162.38 & 166.40 (2C, CN). HREIMS found 474.0960 [M˙+] (calc. for C23H27BrN2O2S, 474.0956).
CH), 3153(OHphenol), 2951, 2868 (CHaliphatic), 2164(CC), 1617 (CN), 1995, 1557 (CC), 1178 (C–O), 1095 (C–O–C); 1H NMR (DMSO-d6) δ 1.29 (s, 9H, t-Bu), 1.41 (s, 9H, t-Bu), 3.37 (t, 1H, J 2.67), 4.23 (d, 2H, J 2.68), 7.48 (d, 1H, J 2.44), 7.59 (d, 1H, J 2.44), 10.18 (brs, 1H, OH). 13C NMR (CDCl3) δ 20.95 (SCH2 C), 29.21 (3C), 31.09 (3C), 34.06 (C), 34.88 (C)75.16 ((CH)CH), 78.92 (CCH), 107.69 (C), 121.13 (C), 128.16 (C), 136.90 (C), 141.90 (C4), 153.30 (C), 161.98 & 166.00 (C7 & C8). HREIMS found 344.1558 [M˙+] (calc. for C19H24N2O2S, 344.1558).N), 1995, 1845 (CC), 1265 (C–O), 1081 (C–O–C). 1H NMR (CDCl3) δ 1.32 (s, 9H, t-Bu), 1.45 (s, 9H, t-Bu), 4.46 (s, 2H), 7.35 (d, 2H, J 8.41), 7.36–7.50 (m, 4H). 13C NMR (CDCl3) δ 29.39 (3C), 31.44 (3C), 34.38 (C), 35.32 (C), 36.16 (C), 107.27 (C), 120.57 (C), 122.26 (C), 128.52 (C), 130.76 (2C), 131.99 (2C), 134.64 (C), 137.43 (C), 141.75 (C), 154.26 (C), 162.38 & 166.40 (2C, CN). HREIMS found 474.0960 [M˙+] (calc. for C23H27BrN2O2S, 474.0956).General synthesis of N′-[(3,5-di-tert-butyl-2-hydroxyphenyl)methylidene]- substituted benzohydrazide (5a–d)
To a warm stirring solution of aryl hydrazide (3 mmol) in 20 mL absolute ethanol, 3,5-di-tert-butyl-salicylaldehyde (0.70 g, 3 mmol) was added in small portions and refluxed for 7 h. Upon cooling, the mixture was stored overnight in a refrigerator at 5 °C. The precipitate was washed with cold ethanol and recrystallised from a suitable solvent.O), 1611 (CN), 1595, 1551 (CC), 1231 (C–O). 1H NMR (DMSO-d6) δ 1.27 (s, 9H, t-Bu), 1.40 (s, 9H, t-Bu), 2.38 (s, 3H, p-CH3), 7.20 (d, 1H, J 2.4), 7.30 (d, 1H, J 2.2), 7.35 (d, 2H, J 8.1), 7.84 (d, 2H, J 8.1), 8.56 (s, 1H, CHN), 12.14 (brs, 1H, NH), 12.29 (brs, 1H, OH). 13C NMR (DMSO-d6) δ 21.61 (C, p-CH3), 29.82 (3C), 31.83 (3C), 34.42 (C), 35.19 (C), 117 (C), 126.04 (C), 126.24 (C), 128.20 (2C), 129.67 (2C), 130.26 (C), 136.17 (C), 140.29 (C), 142.75 (C), 151.54 (C, C7, CN), 155.54 (C), 163.16 (C, CO). HREIMS found 366.2310 [M˙+] (calc. for C23H30N2O2, 366.2307).O), 1619 (CN), 1596, 1572 (CC), 1237 (C–O), 1095 (Ar–O–CH3). 1H NMR (DMSO-d6) δ 1.24 (s, 9H, t-Bu), 1.37 (s, 9H, t-Bu), 3.80 (s, 3H, OCH3), 7.05 (d, 2H, J 9.1), 7.16 (d, 1H, J 2.28), 7.26 (d, 1H, J 2.28), 7.89 (d, 2H, J 8.72), 8.51 (s, 1H, CHN), 12.05 (brs, 1H, NH), 12.28 (brs, 1H, OH). 13C NMR (DMSO-d6) δ 29.30 (3C), 31.31 (3C), 33.88 (C), 34.67 (C), 55.52 (C, OCH3), 114.00 (2C), 117.18 (C), 124.73 (C), 125.56 (C), 125.77 (C), 129.73 (2C), 135.78 (C), 140.45 (C), 150.83 (C), 154.0 (C), 162.41 (C), 162.74 & 162.80 (2C). HREIMS found 382.2256 [M+] (calc. for C23H30N2O3, 382.2256).O), 1621 (CN), 1995, 1558 (CC), 1236 (C–O); 1H NMR (DMSO-d6) δ 1.27 (s, 9H, t-Bu), 1.40 (s, 9H, t-Bu), 7.22 (d, 1H, J 2.2), 7.31 (d, 1H, J 2.2), 7.78 (d, 2H, J 8.5), 7.88 (d, 2H, J 8.5), 8.56 (s 1H, CN), 12.23 (1H, brs, NH), 12.28 (1H, brs, OH). 13C NMR (CDCl3) δ 29.81 (3C), 31.82 (3C), 34.44 (C), 35.18 (C), 117.38 (C), 125.38 (C), 126.25 (C), 126.44 (C), 130.22 (2C), 132.13 (3C), 136.01 (C), 140.91 (C), 152.06 (C), 155.04 (C), 162.30 (C). HREIMS found 430.1084 [M˙+] (calc. for C22H27BrN2O3, 430.1256).O), 1611 (CN), 1231 (C–O); . 1H NMR (DMSO-d6) δ 1.28 (s, 9H, t-Bu), 1.42 (s, 9H, t-Bu), 7.23 (d, 1H, J 2.38), 7.32 (d, 1H, J 2.26), 7.46–7.63 (4H, m), 8.42 (1H, s, CHN), 10.15 (1H, brs, NH), 12.23 (1H, brs, OH). 13C NMR (DMSO-d6) δ 29.26 (3C), 31.26 (3C), 33.87 (C), 34.62 (C), 116.81 (C), 125.70 (C), 125.89 (C), 127.30 (C), 129.42 (1C, C10), 129.79 (1C, C13), 130.47 (1C, C9), 131.58 (C), 134.69 (C), 135.63 (C), 140.41 (C), 151.42 (C, CHN), 154.78 (C), 162.38 (C, CO), HREIMS found 386.1763 [M˙+] (calc. for C22H27ClN2O2, 386.1761).General synthesis of 2,4-di-tert-butyl-6-[5-(substitutephenyl)-1,3,4-oxadiazol-2-yl]phenol (6a–d)
To a solution of N′-[(3,5-di-tert-butyl-2-hydroxyphenyl) methylidene]-substituted benzohydrazide (2 mmol) in 10 mL glacial acetic acid and anhydrous sodium acetate (2 mmol) in a 50 mL round-bottom flask, bromine (1 mmol in 3 mL ACOH) was added dropwise at ambient temperature with vigorous stirring. The mixture was stirred for 1 h and then refluxed further for 2 h. Upon cooling, the mixture was poured into 50 mL ice water. The resulting precipitate was collected and washed with distilled water, dried and either purified by column chorography or recrystallised with a suitable solvent.N), 1590, 1552 (CC), 1219 (C–O); 1H NMR (CDCl3) δ 1.40 (s, 9H, t-Bu), 1.44 (s, 9H, t-Bu), 2.43 (s, 3H, CH3), 7.33 (d, 2H, J 8.0), 7.49 (d, 1H, J 2.44), 7.67 (d, 2H, J 2.44), 8.02 (d, 2H, J 8.0), 10.57 (brs, 1H, OH). 13C NMR (CDCl3) δ 21.68 (C, p-CH3), 29.41 (3C), 31.47 (3C), 34.37 (C), 35.32 (C), 107.63 (C), 120.50 (C), 120.68 (C), 126.99 (2C), 128.37 (C3), 129.82 (2C), 137.45 (C), 141.60 (C) 142.58 (C), 154.68 (C), 163.16, 164.97 (C7 & C8). HREIMS found 364.2162 [M+] (calc. for C23H28N2O2, 364.2151).N), 1585, 1547 (CC), 1222 (C–O), 1170 (O–CH3). 1H NMR (CDCl3) δ 1.37 (s, 9H, t-Bu), 1.48 (s, 9H, t-Bu), 3.87 (s, 3H, OCH3), 7.06 (d, 2H, J 8.8), 7.51 (d, 1H, J 2.2), 7.68 (d, 2H, J 2.4), 8.10 (d, 2H, J 8.8), 10.58 (brs, 1H, OH). 13C NMR (CDCl3) δ 29.42 (3C), 31.48 (3C), 34.38 (C), 35.33 (C), 3.51 (C, OCH3), 107.69 (C), 114.60 (2C), 115.96 (C), 120.46 (C), 128.30 (C), 128.87 (2C), 137.44 (C), 141.58 (C), 154.63 (C), 162.62 (C), 162.56, 164.78 (C7 & C8). HREIMS found 380.2102 [M˙+] (calc. for C23H28N2O3, 380.2100).N), 1587, 1552 (CC), 1214 (C–O), 1H NMR (CDCl3) δ 1.37 (s, 9H, t-Bu), 1.48 (s, 9H, t-Bu), 7.53 (d, 1H, J 2.2), 7.67 (d, 1H, J 2.4), 7.70 (d, 2H, J 8.5), 8.03 (d, 2H, J 8.5), 10.51 (brs, 1H, OH); 13C NMR (CDCl3) δ 29.42 (3C), 31.47 (3C), 34.41 (C), 35.36 (C), 113.50 (C), 120.51 (C), 122.41 (C), 126.73 (C), 128.45 (2C), 128.76 (C), 132.64 (2C), 137.64 (C), 141.77 (C), 154.84 (C), 162.94 & 165.42 (C7 & C8), HREIMS, found 428.1102 [M˙+] (calc. for C22H25BrN2O2, 428.1099).N), 1585, 1547 (CC), 1221 (C–O), 1H NMR (CDCl3) δ 1.34 (s, 9H, t-Bu), 1.48 (s, 9H, t-Bu), 7.70–7.44 (m, 4H), 7.69 (d, 1H, J 2.4), 8.07 (dd, 1H, J 7.56, 1.95), 10.46 (brs, 1H, OH). 13C NMR (CDCl3) δ 29.46 (3C), 31.47 (3C), 34.41 (C), 35.39 (C), 107.42 (C), 120.86 (C), 122.82 (C), 127.24 (C), 128.7 (C), 131.27 (C, C10), 131.46 (C), 132.67 (C), 133.39 (C), 137.60 (C), 141.83 (C), 154.84 (C), 161.43 (C), 165.66 (C). HREIMS found 384.1605 [M˙+] (calc. for C22H25ClN2O2, 384.1605).DPPH radical scavenging activity
The DPPH free radical scavenging assay was carried out using Brand–Williams' method37,38 with slight adjustments. 195 μL of a 100 μM methanolic DPPH solution was mixed with 50 μL of the test compounds at various doses (0–1000 μg mL−1) to make the reaction mixture. Initially, the test chemicals were dissolved in dimethyl sulphoxide (DMSO). Positive controls were BHT and ascorbic acid, which were tested in tandem. The absorbance of the reaction mixture was measured at 570 nm after 60 minutes of incubation in the dark at room temperature. As a result of the lower absorbance, the colour of the reaction mixture changed from purple to yellow. The following equation was used to compute the radical scavenging activity:
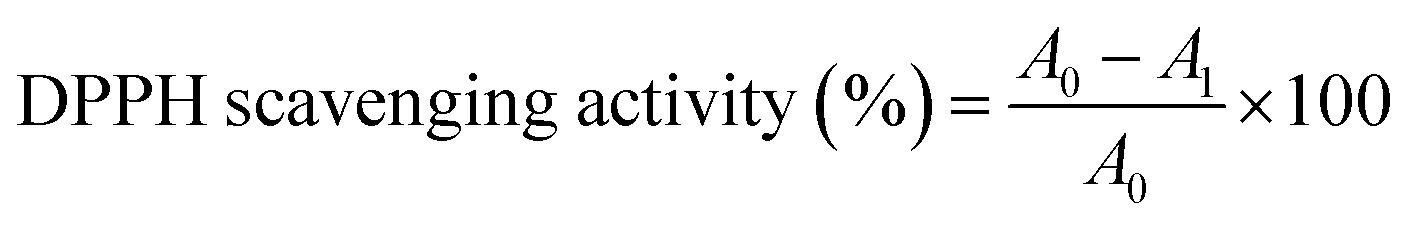 | (1) |
Ferric reducing antioxidant power activity (FRAP)
FRAP was determined according to the method described by Benzie and Strain38,42 with slight modifications. Three reagents were initially prepared as follows: 300 mM acetate buffer (pH = 3.6), 10 mM 2,4,6-tripyridyl-s-triazine (TPTZ) in 40 mM HCl and 20 mM FeCl3.The acetate buffer was mixed with the TPTZ solution in 20 mM FeCl3 at a ratio of 10:1:1 (v/v/v) to prepare a fresh FRAP working solution. Five microliters of the samples or standards were mixed with 300 mL of the FRAP reagent and then incubated at 37 °C for 30 minutes. The absorbance of the stained product was then measured at 595 nm. Based on a calibration curve prepared with ferrous sulfate (FeSO), the results were determined (0–1 mM). The results were expressed in FRAP value.
Statistical analysis
All of the analyses were carried out in triplicate. The data was presented as a mean with a standard deviation. The association between the antioxidant activity of the synthesised compounds in the DPPH and FRAP antioxidant assays was investigated using a correlation coefficient test. At the 0.05 level, the trend is significant. OriginPro software, version 2016, was used to statistically analyse the data.Computational details
The Gaussian 09 programme package was used to accomplish all of the computations provided here.43 All naturally occurring systems, including neutrals, radicals, radical cations, and anions, were subjected to geometry optimisation and frequency analysis in vacuum. At the B3LYP/6-311++g(d,p) level of theory,44,45 they were entirely optimised in their electronic ground states, as evidenced by the absence of imaginary frequencies. Open-shell systems lacking spin impurities, such as radicals and radical cations, were subjected to unconstrained calculations. In all situations, the expectation value of 〈S2〉 operator was around 0.750. At the same level of this structural optimisation, the HOMO and LUMO frontier molecular orbitals were depicted for neutral compounds, and global reactivity descriptors were derived.Results and discussion
Chemistry
All compounds were characterised by IR, 1H NMR, 13C NMR, EIMs and HREIMs. HSQC was used for 4c to distinguish between the SCH2 carbon from the three similar peaks at 34.38, 35.32 and 36.16 ppm. HMBC was used for 5b to distinguish between OH and NH protons in hydrazones. HMBC was also used for 6a to distinguish between the carbons of the oxadiazole ring.N and 1246 cm−1 for CS. From NMR spectroscopic data, all protons were accounted for compounds 4a–c were synthesised by the alkylation of compound 3 which were selectively reacted at the thione group (without affecting the phenol group) at ambient temperature. The HSQC of compound 4c displayed the correlation between the protons of the SCH2 group and their carbons (C–H) at 4.4–36.16 ppm; therefore, the other two peaks must belong to the quaternary carbons at the di-tert-butyl group. The hydrazones 5a–d were successfully synthesised in high yield. The HMBC of compound 5b was used to distinguish between the protons of OH and NH through long-distance coupling for J3 and weak coupling for J2. The most important correlations were those between the hydrogen of phenol hydroxyl at 12.27 ppm with C6, C2 and C1, whereas the NH at 12.04 ppm showed correlation with carbonyl group C8. The proton of the imine group (CHN) showed a correlation with C1 and C6. In addition, the C1 displayed a long-range correlation with C7, C3 and C5. The protons of the 4-bromo phenyl ring also displayed their own correlation, whereas the H10 exhibited a correlation with C12 and C8 even though H11 showed a correlation with H9. The cyclisation of hydrazones was carried out in the presence of bromine as an oxidising agent. The formation of oxadiazoles 6a–d was confirmed by all spectroscopic data available. Furthermore, HMBC was used to distinguish between the two carbons of the oxadiazole ring, C7 and C8. Compound 6a was used as an example to illustrate our results. The correlation between H10 and C12 and C8 as well as the correlation between H5 and C3, C1 and C7 can be clearly observed in the HMBC spectrum. These data help us to distinguish between the two carbons in the oxadiazole ring. Detailed information can be accessed in the ESI† for the respective HSQC (Fig. 9†) and HMBC (Fig. 21†). The structure of compounds 6a–d were further confirmed by the fragmentation pattern shown on the EIM spectra, which revealed the loss of isocyanic acid (see Section 1.2 of the ESI†).Evaluation of antioxidant assay
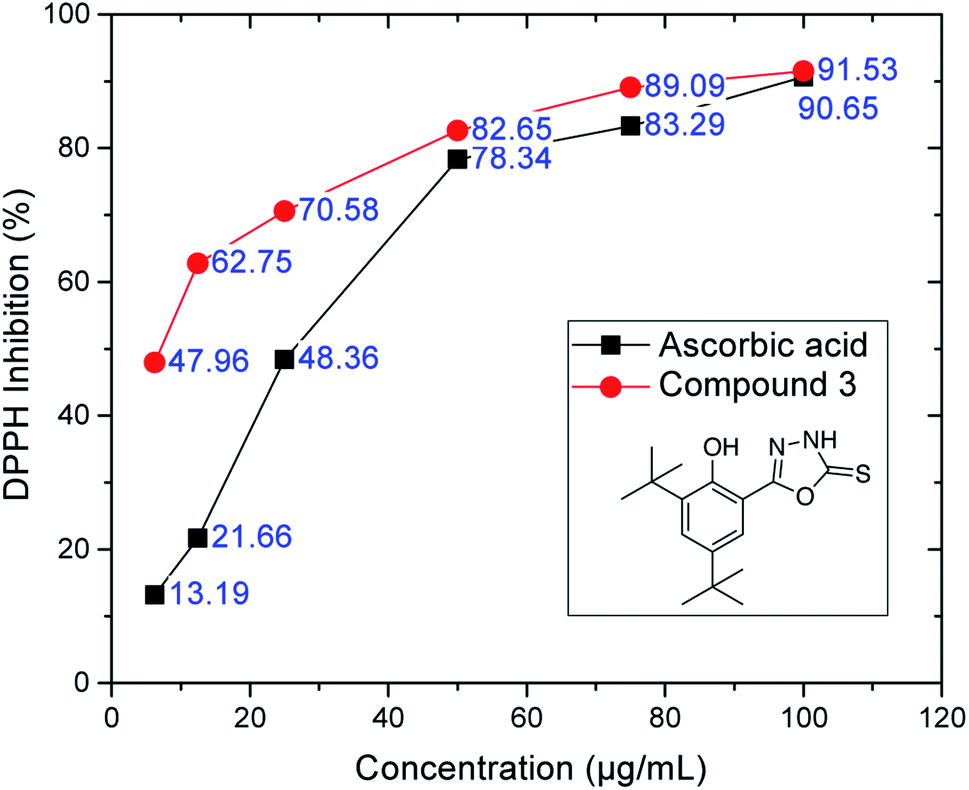 | ||
| Fig. 3 DPPH inhibition of oxadiazole-thione 3 at six different concentrations. Ascorbic acid as a reference compound. | ||
However, the thio-oxadiazole compounds (4a–c) exhibited lower antioxidant activity than compound 3, which led us to conclude that the oxadiazole-thione groups behave as better free radical scavengers.47 The NHCS group can be regarded as a part of the thiourea system, which is known as an effective antioxidant.48 The similarity of this case with that of phenethyl-5-bromo-pyridyl thiourea49 further supports our conclusion. In the series of thio-oxadiazole, compound 4c (IC50: 43.62 ± 0.12 μg mL−1), with 4-bromo benzyl group next to the thio group afforded better antioxidant activity when compared to the activities observed with methyl (4a, IC50: >100 μg mL−1) or propargyl groups (4c, IC50: 43.62 ± 0.12 μg mL−1). In the next series, only hydrazone 5a recorded a good IC50 value (73.77 ± 0.11 μg mL−1), which is comparable to BHT. In the series of phenyloxadiazoles, two compounds (6a, IC50: 72.47 ± 0.28 μg mL; 6d, IC50: 77.42 ± 0.80 μg mL−1), recorded comparable IC50 values to that of BHT (IC50: 79.84 ± 0.15 μg mL−1). Their phenyloxadiazoles (6a–d) exhibited slightly lower antioxidant activity than that of the hydrazones. In this case, different functional groups and heterocyclic rings clearly plays an important role in increasing and decreasing the antioxidant activity, as depicted in Table 1.
| Compound | R | Inhibition ± SDa 100 μg mL−1 | IC50 ± SEMb μg mL−1 | FRAP valuec μM |
|---|---|---|---|---|
| a Standard deviation (SD) value in FRAP was between 0.01–0.16.b SEM Standard error of the mean and IC50: 50% of effective concentration.c Dosage: 100 μg mL−1. | ||||
| 3 | — | 91.53 ± 0.11 | 6.13 ± 0.63 | 4612.78 |
| 4a | CH3 | 40.85 ± 0.17 | >100 | 297.78 |
| 4b | CH2CCH |
37.63 ± 0.64 | >100 | 291.94 |
| 4c | CH2Ph–4Br | 62.59 ± 0.92 | 43.62 ± 0.12 | 1016.16 |
| 5a | 4-CH3 | 61.98 ± 0.61 | 73.77 ± 0.11 | 538.67 |
| 5b | 4-OCH3 | 51.48 ± 0.29 | >100 | 511.17 |
| 5c | 4-Br | 36.11 ± 0.16 | >100 | 278.67 |
| 5d | 2-Cl | 48.36 ± 0.13 | >100 | 433.67 |
| 6a | 4-CH3 | 61.79 ± 0.37 | 72.47 ± 0.28 | 520.33 |
| 6b | 4-OCH3 | 44.76 ± 0.19 | >100 | 359.50 |
| 6c | 4-Br | 26.46 ± 0.44 | >100 | 112.83 |
| 6d | 2-Cl | 54.68 ± 0.50 | 77.42 ± 0.80 | 468.67 |
| BHT | — | 66.03 ± 0.22 | 79.84 ± 0.15 | 488.30 |
| Ascorbic acid | — | 90.65 ± 0.27 | 22.71 ± 0.23 | 848.90 |
| Quercetin | — | — | — | 2090.60 |
| Gallic acid | — | — | — | 2421.10 |
Looking at the percentage of DPPH inhibition, the effect of the substituted group in compounds 5a–d ranked as follows: 4-methyl > 4-methoxy > 2-chloro > 4-bromo; on the other hand, compounds 6a–d showed the following sequence: 4-methyl > 2-chloro > 4-methoxy > 4-bromo. Compounds 6a–d share the same basic structures; the only difference is the position of the substituted group at the phenyl ring. This finding led us to conclude that the nature and position of each substituted group play some role in enhancing or inhibiting the antioxidant ability of the hydrazones and phenyloxadiazoles.
Compound 3 exhibited the best antioxidant activity among other compounds with the FRAP value of 4612.78 μM, which was also significantly higher as compared to the reference compounds gallic acid (2421.10 μM), quercetin (2090.60 μM), BHT (488.3 μM) and ascorbic acid (848.9 μM) (see Fig. 4). The high potency activity of compound 3 can be explained by the stability of the resonance of its radical structures (Scheme 32†). The activation of the hydroxyl group, by electron transfer followed by removal of proton, leads to the activation of the thiol group.52 This reaction allows one mole of oxadiazole-thione 3 to reduce two moles of Fe3+ ion, and consequently, give a high FRAP value and potent antioxidant property for oxadiazole-thione 3 (Scheme 1).
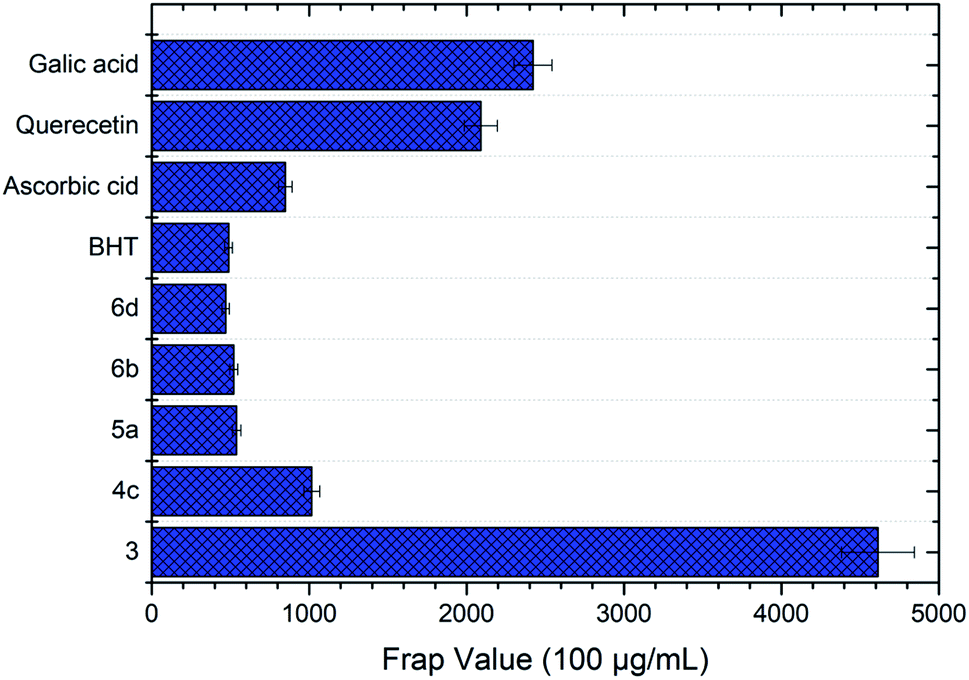 | ||
| Fig. 4 FRAP value of the oxadiazole-thione 3, the thio-oxadiazole series (4c), the hydrazone series (5a) and the phenyloxadiazole series (6b & 6d) at concentrations of 100 μg mL−1. BHT, ascorbic acid, quercetin and gallic acid as reference compounds. | ||
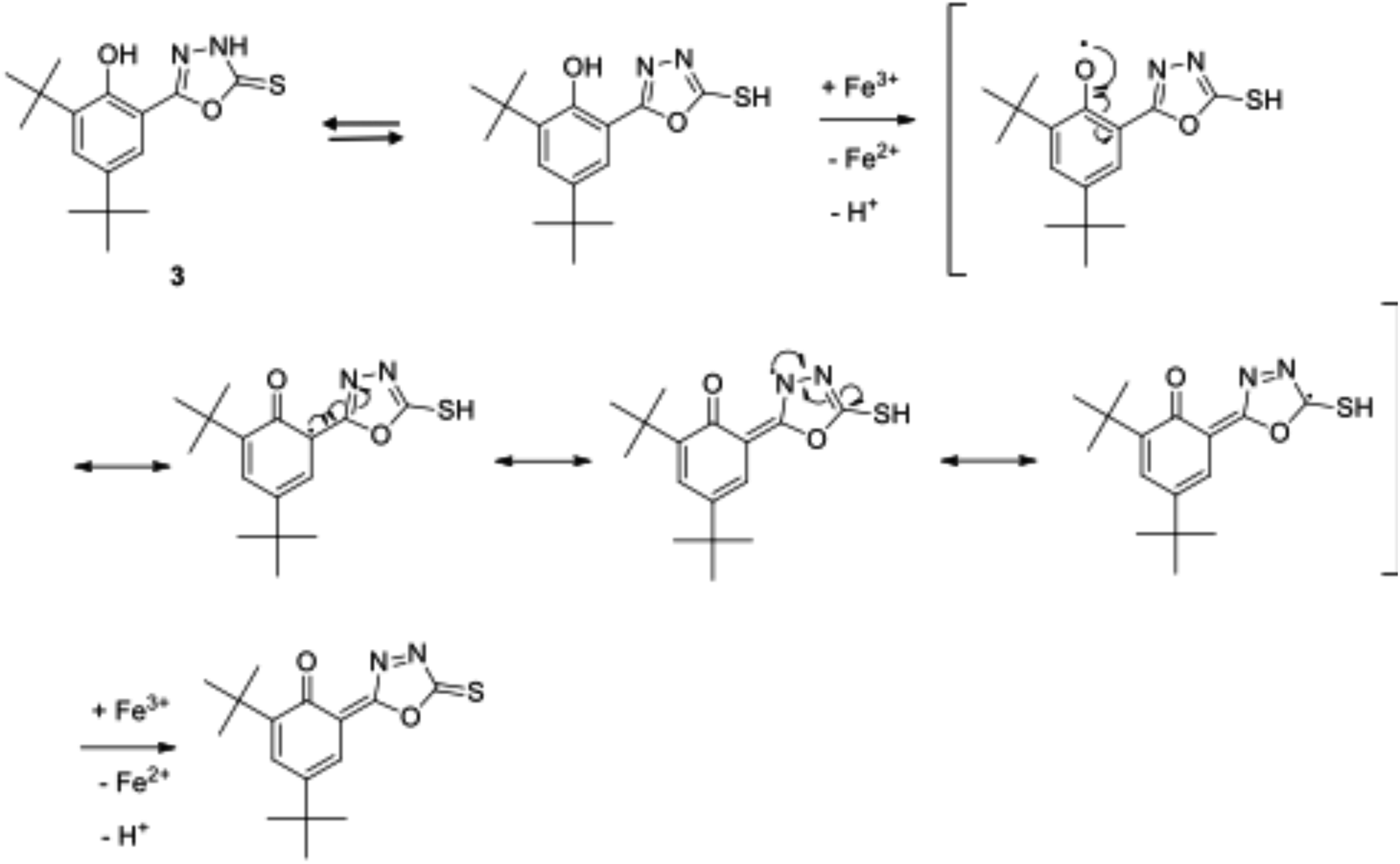 | ||
| Scheme 1 Resonance structures of the radical form of oxadiazole-thione 3. | ||
In the series of thio-oxadiazole compounds, compound 4c, with 4-bromobenzyl substituent, recorded the highest FRAP value at 1016.16 μM, significantly higher than compound 4a (297.78 μM) and 4b (291.94 μM), which were consistent to DPPH results. Similarly, as in DPPH results, compound 5a showed the highest antioxidant property in FRAP assay (538.67 μM) as compared to compound 5b (511.17 μM), 5c (278.67 μM) and 5d (433.67 μM). In the series of phenyloxadiazole compounds, compound 6a, with 4-methyl substituent, displayed the best antioxidant activity with the FRAP value of 520.33 μM, as compared to compound 6b (359.50 μM), 6c (112.82 μM), and 6d (468.67 μM). The results of FRAP assay further reinforced that oxadiazole-thiones possessed higher antioxidant properties as compared to thio-oxadiazoles, hydrazones and phenyloxadiazoles, which were previously exhibited in DPPH assay.
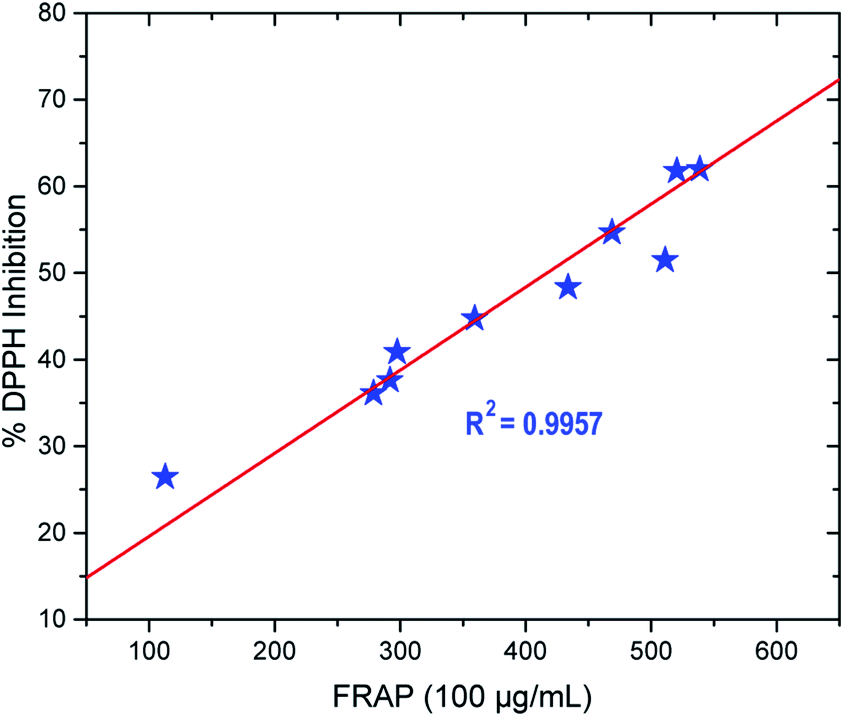 | ||
| Fig. 5 Pearson correlation analysis between DPPH radical scavenging and iron (III) reducing activities of the synthesised compounds for the series of thio-oxadiazole (4a–c), hydrazones (5a–d) and phenyloxadiazole (6a–d). The correlation is significant at the 0.05 level. | ||
Density functional theory
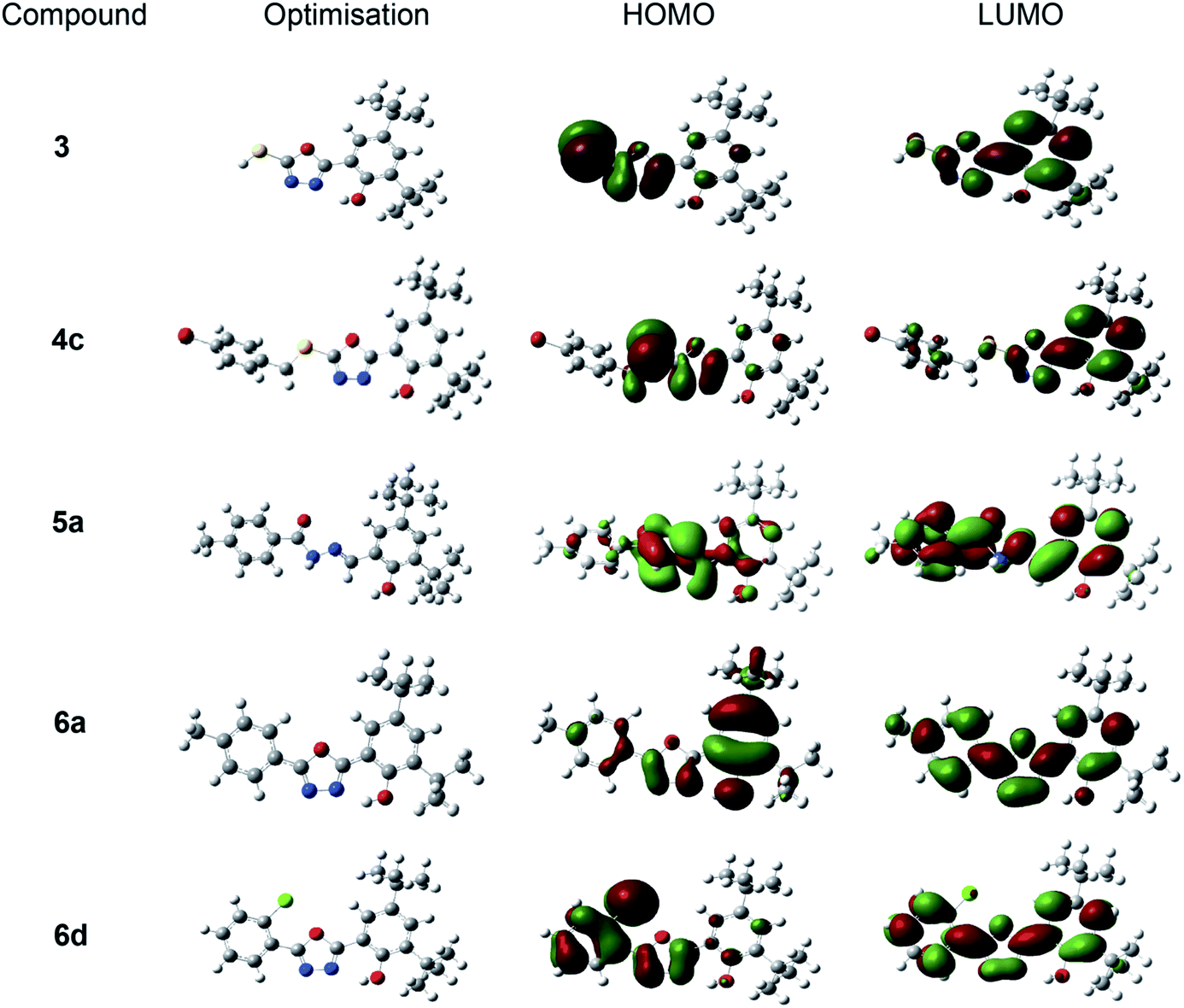 | ||
| Fig. 6 Depictions of the optimised conformations of the most stable structures and frontier molecular orbital plots (HOMO and LUMO) of oxadiazole-thione 3, thio-oxadiazole 4c, hydrazone 5a and phenyloxadiazole 6a & 6d in the gas phase at B3lyp/6-311++g(d,p). | ||
Fig. 6 depicts the gas-phase electron density distribution of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for oxadiazole-thione 3, thio-oxadiazole (4c), hydrazone 5a and the phenyloxadiazole (6a & 6d), which were calculated based on their good antioxidant activity. The HOMO plots demonstrate that HOMOs are localised on the oxadiazole ring and hydrazones bonds in the investigated systems. As a result, the oxadiazole ring and hydrazone would be the most likely reaction site for free radicals to assault and remove an electron. The distribution of LUMOs, on the other hand, demonstrates that the entire molecules are heavily contributed. Compound with smaller molecular orbital gap at the Frontier is more polarised,58 thus, it is expected to have higher intermolecular charge transfer between election donors and acceptors, which increases/decreases its biological activity.59
Table 2 shows that the oxadiazole-thione, 3 has a smaller HOMO–LUMO gap (0.085 a.u) compared to a series of phenyloxadiazolephenyloxadiazole; phenyloxadiazole 6a (0.173 a.u) and 6d (0.170 a.u), followed by hydrazone thio-oxadiazole 4c (0.804 a.u) and 5a (0.182 a.u.), indicating that it has high polarizability. The polarizability of molecules is used to determine the relative tendency of charge distribution. In other words, in the presence of a weak external electric field, the system electronic cloud can be deformed from its typical shape.60
| Molecule | εHOMOa | εLUMOa | ΔEHOMO–LUMOa | Dipole momenta | BDEb | IPb |
|---|---|---|---|---|---|---|
| a All parameters in atomic unit.b All values in kcal mol−1. | ||||||
| 3 | −0.233 | −0.148 | 0.085 | 3.615 | 50.05 | 145.88 |
| 4c | −0.244 | −0.064 | 0.180 | 2.216 | 55.65 | 153.01 |
| 5a | −0.241 | −0.059 | 0.182 | 4.621 | 54.85 | 155.16 |
| 6a | −0.242 | −0.069 | 0.173 | 4.678 | 59.98 | 155.38 |
| 6d | −0.247 | −0.077 | 0.170 | 3.774 | 59.55 | 156.72 |
| BHT55 | — | — | — | — | 70.14 | 171.90 |
| Ascorbic acid55 | — | — | — | — | 74.54 | 196.82 |
| Quercetin56 | — | — | — | — | 65.80 | 150.32 |
| Gallic acid57 | — | — | — | — | 77.10 | 146.70 |
Table 2 shows the calculated BDE values of the OH bond for the compounds in the gas phase. It is shown that oxadiazole-thione 3 has the lowest BDE value (50.05 kcal mol−1), indicating the highest radical scavenging activity as compared to the other compounds, which is consistent with the experimental results of the DPPH assay.
The pattern of BDE values of thio-oxadiazole 4c (55.65 kcal mol−1) and phenyloxadiazoles 6a & 6d (59.98, 59.55 kcal mol−1) is consistent with the result of DPPH assay, in which thio-oxadiazole 4c displayed higher antiradical activity as compared to phenyloxadiazoles 6a and 6b. Meanwhile, the similarity of the BDE values of phenyloxadiazoles 6a and 6d are also reflected in their IC50 values in DPPH assay (72.47 μg mL−1, 77.42 μg mL−1). Only the BDE value of hydrazone 5a (54.85 kcal mol−1) which is slightly lower than thio-oxadiazole 4c, could not explained its activity in DPPH assay (73.77 μg mL−1), which is higher than thio-oxadiazole 4c.
In comparison to the BDE value of BHT as the reference (70.14 kcal mol−1), oxadiazole-thione 3 and thio-oxadiazole 4c recorded lower BDE values, which are in agreement to the results of DPPH assay. From the BDE values, it can be deduced that the oxadiazole-thione group at the ortho position promotes the HAT mechanism of the butylated phenol group, and this is followed by the hydrazones, thio-oxadiazoles, and phenyloxadiazoles groups at lower extent, respectively.
Other than BDE, another parameter that can measure antioxidant property is ionisation potential (IP) or proton dissociation enthalpy (PDE), which describes the energetics of the SET-PT process.62 Compound with lower IP value has higher likelihood to produce superoxide anion radical, by immediately passing the electron to the environment.63 The calculated IP of the compounds are recorded in Table 2.
The obtained results reveal that the IP values of the five selected synthesised compounds are almost equivalent. Oxadiazole-thione 3 has the lowest IP value (145.88 kcal mol−1), as compared to the rest of the synthesised and reference compounds, and this is reflected in the results of FRAP assay. Meanwhile, thio-oxadiazole 4c, which recorded higher IP value (153.01 kcal mol−1) than quercetin (150.32 kcal mol−1) and gallic acid (146.70 kcal mol−1), but lower than compounds 5a (155.16 kcal mol−1), 6a (155.38 kcal mol−1) and 6d (156.72 kcal mol−1), has a consistent pattern in the FRAP assay, in which it recorded lower activity that quercetin and gallic acid, but higher than compounds 5a, 6a and 6d. Similar to BDE parameter, the IP values showed that the oxadiazole-thione group at the ortho position promotes the SET-PT mechanism of the butylated phenol group, followed by the thio-oxadiazoles, hydrazones and phenyloxadiazoles groups at lower extent, respectively.
Conclusions
12 derivatives of butylated phenol with four different groups at the ortho position were synthesised and studied for their antioxidant properties. Oxadiazole-thione 3, which features oxadiazole-thione ring at the ortho position, recorded the best result in both DPPH (IC50: 6.13 μg mL−1) and FRAP (FRAP value: 4612.78) assays, which were significantly better than the reference compounds, including BHT, ascorbic acid, quercetin and gallic acid. The potent antioxidant activity of oxadiazole-thione 3 was explained by the stability of the resonance structure of its radical form, as well as the calculated parameters BDE and IP. Based on the experimental and calculated results, the order of groups at the ortho position of butylated phenol that contribute to higher antioxidant activity can be written as follow: oxadiazole-thione > thio-oxadiazole > hydrazone > phenyloxadiazole. Oxadiazole-thione 3 is a good candidate to be further investigated and developed as an antioxidant.Author contributions
The authors report the following individual contributions to this work: formal analysis: RS, NN, AA; funding acquisition and project administration: NN, AA; investigation: RS, NN; resources: NN, AA; software: NN; supervision: AA, NN; conceptualisation, data curation, methodology, validation, writing – original draft, writing – review and editing: all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A, R. M. S. and M. A. A. would like to thank the University of Malaya for a UMRG grant (RP021B-14AFR) and an IIRG grant (IIRG005B-19FNW). N. N. acknowledge funding from the RU grant (ST018-2021) and FRGS grant (FP118-2020). The authors would like to show their gratitude to Prof. Dr Amir Atto and Prof. Dr Mahmood Ameen Abdulla for their contributions and advices. All authors would like to acknowledge the support of the University of Malaya (UM), providing the infrastructure to realise this work.Notes and references
- D. Krishnaiah, R. Sarbatly and R. Nithyanandam, Food Bioprod. Process., 2011, 89, 217–233 CrossRef CAS.
- T. Wang, J. Gu, P.-F. Wu, F. Wang, Z. Xiong, Y.-J. Yang, W.-N. Wu, L.-D. Dong and J.-G. Chen, Free Radicals Biol. Med., 2009, 47, 229–240 CrossRef CAS PubMed.
- A. C. Maritim, aR. A. Sanders and J. B. Watkins III, J. Biochem. Mol. Toxicol., 2003, 17, 24–38 CrossRef CAS PubMed.
- A. Galano and J. Raúl Alvarez-Idaboy, Int. J. Quantum Chem., 2019, 119, e25665 CrossRef.
- O. Dangles, C. Dufour, C. Tonnelé and P. Trouillas, Recent Advances of Polyphenol Research, 2017, vol. 5, pp. 1–35 Search PubMed.
- M. Hahn, M. Baierle, M. F. Charão, G. B. Bubols, F. S. Gravina, P. Zielinsky, M. D. Arbo and S. Cristina Garcia, Drug Chem. Toxicol., 2017, 40, 368–374 CrossRef CAS PubMed.
- J. A. Ross and C. M. Kasum, Annu. Rev. Nutr., 2002, 22, 19–34 CrossRef CAS PubMed.
- N. Pap, M. Fidelis, L. Azevedo, M. A. V. do Carmo, D. Wang, A. Mocan, E. P. R. Pereira, D. Xavier-Santos, A. S. Sant'Ana and B. Yang, et al., Curr. Opin. Food Sci., 2021, 42, 167–186 CrossRef.
- A. Durazzo, M. Lucarini, E. B. Souto, C. Cicala, E. Caiazzo, A. A. Izzo, E. Novellino and A. Santini, Phytother. Res., 2019, 33, 2221–2243 CrossRef PubMed.
- N. Kumar and N. Goel, Biotechnol. Rep., 2019, 24, e00370 CrossRef PubMed.
- Y. Jiang, Z. Fang, W. Leonard and P. Zhang, J. Funct. Foods, 2021, 77, 104340 CrossRef CAS.
- A. Dargahi, M. R. Samarghandi, A. Shabanloo, M. M. Mahmoudi and H. Z. Nasab, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01601-y.
- H. Park and D. Kim, Prog. Org. Coat., 2021, 157, 106077 CrossRef CAS.
- D. Trache and A. F. Tarchoun, J. Mater. Sci., 2018, 53, 100–123 CrossRef CAS.
- M. A. H. Salahuddin, A. Ismail, N. K. Kassim, M. Hamid and M. S. M. Ali, Food Chem., 2020, 331, 127240 CrossRef CAS PubMed.
- S. Rojas-Lema, S. Torres-Giner, L. Quiles-Carrillo, J. Gomez-Caturla, D. Garcia-Garcia and R. Balart, Antioxidants, 2021, 10, 14 CrossRef CAS PubMed.
- S. Cui, Y. Yu, T. Zhan, C. Zhang and S. Zhuang, Sci. Total Environ., 2021, 778, 146322 CrossRef CAS PubMed.
- O. Prakash, M. Kumar, R. Kumar, C. Sharma and K. R. Aneja, Eur. J. Med. Chem., 2010, 45, 4252–4257 CrossRef CAS PubMed.
- M. Kuchana, D. R. Bethapudi, R. K. Ediga and Y. Sisapuram, J. Appl. Pharm. Sci., 2019, 9, 105–110 CAS.
- Y. W. Song, Y. Lim and S. K. Cho, Biochim. Biophys. Acta, Mol. Cell Res., 2018, 1865, 675–683 CrossRef CAS PubMed.
- R. C. Lino, D. P. B. da Silva, I. F. Florentino, D. M. da Silva, J. L. R. Martins, D. da Costa Batista, K. C. de Siqueira Leite, B. Villavicencio, G. A. Vasconcelos, A. L. P. Silva and Others, Eur. J. Pharm. Sci., 2017, 106, 231–243 CrossRef CAS PubMed.
- S. L. Manju, K. R. Ethiraj, G. Elias and Others, Eur. J. Pharm. Sci., 2018, 121, 356–381 CrossRef PubMed.
- E. Pontiki, D. Hadjipavlou-Litina, K. Litinas, O. Nicolotti and A. Carotti, Eur. J. Med. Chem., 2011, 46, 191–200 CrossRef CAS PubMed.
- M.-A. Yoon, T.-S. Jeong, D.-S. Park, M.-Z. Xu, H.-W. Oh, K.-B. Song, W. S. Lee and H.-Y. Park, Biol. Pharm. Bull., 2006, 29, 735–739 CrossRef CAS PubMed.
- S. A. Saoud, K. F. Ali and R. M. Shakir, Orient. J. Chem., 2017, 33, 1781 CrossRef CAS.
- L. Chen, H. Cao, Q. Huang, J. Xiao and H. Teng, Crit. Rev. Food Sci. Nutr., 2021, 1–13 Search PubMed.
- H. Lou, L. Hu, H. Lu, T. Wei and Q. Chen, Molecules, 2021, 26, 4522 CrossRef CAS PubMed.
- G. Lopez-Gamez, P. Elez-Martinez, O. Martin-Belloso and R. Soliva-Fortuny, Food Chem., 2021, 364, 130377 CrossRef CAS PubMed.
- E. Durgashivaprasad, G. Mathew, S. Sebastian, S. A. M. Reddy, J. Mudgal and G. K. Nampurath, Indian J. Pharmacol., 2014, 46, 521 CrossRef PubMed.
- C. E. Stecoza, G. M. Nitulescu, C. Draghici, M. T. Caproiu, O. T. Olaru, M. Bostan and M. Mihaila, Pharmaceuticals, 2021, 14, 438 CrossRef CAS PubMed.
- R. Das and D. K. Mehta, Drug Res., 2021, 71, 26–35 CrossRef CAS PubMed.
- B. N. P. Kumar, K. N. Mohana, L. Mallesha and B. Veeresh, Med. Chem. Res., 2014, 23, 3363–3373 CrossRef.
- S. A. Khan, P. Ahuja and A. Husain, J. Chin. Chem. Soc., 2017, 64, 918–924 CrossRef CAS.
- F. Hassanzadeh, E. Jafari, M. Zarabi, G. Khodarahmi and G. Vaseghi, Res. Pharm. Sci., 2020, 15, 454 CrossRef PubMed.
- A. M. Rabie, New J. Chem., 2021, 45, 761–771 RSC.
- V. Kontham, B. Ippakayala and D. Madhu, Arabian J. Chem., 2021, 14, 103163 CrossRef CAS.
- W. Brand-Williams, M.-E. Cuvelier and C. Berset, LWT--Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- H. S. Kareem, A. Ariffin, N. Nordin, T. Heidelberg, A. Abdul-Aziz, K. W. Kong and W. A. Yehye, Eur. J. Med. Chem., 2015, 103, 497–505 CrossRef CAS PubMed.
- Y. Ünver, K. Sancak, F. Çelik, E. Birinci, M. Küçük, S. Soylu and N. A. Burnaz, Eur. J. Med. Chem., 2014, 84, 639–650 CrossRef PubMed.
- G. Wang, Y. Xue, L. An, Y. Zheng, Y. Dou, L. Zhang and Y. Liu, Food Chem., 2015, 171, 89–97 CrossRef CAS PubMed.
- S. Shenvi, K. Kumar, K. S. Hatti, K. Rijesh, L. Diwakar and G. C. Reddy, Eur. J. Med. Chem., 2013, 62, 435–442 CrossRef CAS PubMed.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70–76 CrossRef CAS PubMed.
- M. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson and Others, Gaussian 09, Revision d. 01, Gaussian, 2009 Search PubMed.
- R. Manne and T. Åberg, Chem. Phys. Lett., 1970, 7, 282–284 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93–98 CrossRef CAS.
- K. Pyrzynska and A. Pękal, Anal. Methods, 2013, 5, 4288–4295 RSC.
- M. S. Chernovyants, T. S. Kolesnikova and A. O. Karginova, Talanta, 2016, 149, 319–325 CrossRef CAS PubMed.
- H. E. Hashem, A. E.-G. E. Amr, E. S. Nossier, E. A. Elsayed and E. M. Azmy, Molecules, 2020, 25, 2766 CrossRef CAS PubMed.
- K. Harrouche, A. Lahouel, M. Belghobsi, B. Pirotte and S. Khelili, Can. J. Chem., 2019, 97, 824–832 CrossRef CAS.
- N. Liang and D. D. Kitts, Molecules, 2014, 19, 19180–19208 CrossRef PubMed.
- M. Spiegel, K. Kapusta, W. Kołodziejczyk, J. Saloni, B. Żbikowska, G. A. Hill and Z. Sroka, Molecules, 2020, 25, 3088 CrossRef CAS PubMed.
- K. A. Wojtunik-Kulesza, Molecules, 2020, 25, 5267 CrossRef CAS PubMed.
- A. Hocquet and M. Langgård, J. Mol. Model., 1998, 4, 94–112 CrossRef CAS.
- HyperChem(TM) Professional 8.0, Hypercube, Inc., Gainesville, 2001 Search PubMed.
- H. Boulebd, J. Mol. Struct., 2020, 1201, 127210 CrossRef CAS.
- A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- E. Klein, Acta Chim. Slovaca, 2016, 9, 114–123 CrossRef.
- D. H. Ess and K. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- R. El Bahnasawy, E. El Shereafy and T. Kashar, J. Therm. Anal., 1993, 39, 65–74 CrossRef CAS.
- H. A. Kurtz and D. S. Dudis, Rev. Comput. Chem., 1998, 241–279 CAS.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS PubMed.
- A. P. Vafiadis and E. G. Bakalbassis, Chem. Phys., 2005, 316, 195–204 CrossRef CAS.
- D. A. Pratt, G. A. DiLabio, G. Brigati, G. F. Pedulli and L. Valgimigli, J. Am. Chem. Soc., 2001, 123, 4625–4626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02140d |
| This journal is © The Royal Society of Chemistry 2022 |