
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D printed interdigitated supercapacitor using reduced graphene oxide-MnOx/Mn3O4 based electrodes†
Mahshid Mokhtarnejad‡
abc,
Erick L. Ribeiro‡abc,
Dibyendu Mukherjee *abc and
Bamin Khomami*ab
*abc and
Bamin Khomami*ab
aDepartment of Chemical & Biomolecular Engineering, University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: bkhomami@utk.edu; dmukherj@utk.edu
bMaterial Research and Innovation Laboratory (MRAIL), University of Tennessee, Knoxville, Tennessee 37996, USA
cNano-BioMaterials Laboratory for Energy, Energetics & Environment (nbml-E3), University of Tennessee, Knoxville, Tennessee 37996, USA
First published on 13th June 2022
Abstract
In this study hybrid nanocomposites (HNCs) based on manganese oxides (MnOx/Mn3O4) and reduced graphene oxide (rGO) are synthesized as active electrodes for energy storage devices. Comprehensive structural characterizations demonstrate that the active material is composed of MnOx/Mn3O4 nanorods and nanoparticles embedded in rGO nanosheets. The development of such novel structures is facilitated by the extreme synthesis conditions (high temperatures and pressures) of the liquid-confined plasma plume present in the Laser Ablation Synthesis in Solution (LASiS) technique. Specifically, functional characterizations demonstrate that the performance of the active layer is highly correlated with the MnOx/Mn3O4 to rGO ratio and the morphology of MnOx/Mn3O4 nanostructures in HNCs. To that end, active layer inks comprising HNC samples prepared under optimal laser ablation time windows, when interfaced with a percolated conductive network of electronic grade graphene and carbon nanofibers (CNFs) mixture, indicate superior supercapacitance for functional electrodes fabricated via sequential inkjet printing of the substrate, current collector layer, active material layer, and gel polymer electrolyte layer. Electrochemical characterizations unequivocally reveal that the electrode with the LASiS synthesized MnOx/Mn3O4–rGO composite exhibits significantly higher specific capacitance compared to the ones produced with commercially available Mn3O4–graphene NCs. Moreover, the galvanostatic charge–discharge (GCD) experiments with the LASiS synthesized HNCs show a significantly larger charge storage capacity (325 F g−1) in comparison to NCs synthesized with commercially available Mn3O4–graphene (189 F g−1). Overall, this study has paved the way for use of LASiS-based synthesized functional material in combination with additive manufacturing techniques for all-printed electronics with superior performance.
1 Introduction
The increased global need for renewable energy technologies has inspired much research including development of next generation electrochemical energy storage devices such as supercapacitors (SCs).1,2 However, many challenges including durability, cost, and environmental impact have to be addressed before these technologies are adopted on the global scale. SCs, either double-layer (EDLCs) and/or faradaic (pseudocapacitors), are an attractive alternative to typical batteries due to their higher power density and longer life cycles. Specifically, SCs have a much higher energy density than conventional capacitors and can undergo multiple charge/discharge cycles at ultra-fast rates. They also offer higher power density and extended lifetime compared to conventional batteries. However, their charge storage capacity is typically much lower than batteries,3 partly due to utilization of carbonaceous material-based electrodes with an inherent lower energy density.4 Hence, despite the aforementioned advantages of SCs, SC-based energy storage devices are still not widely utilized as an alternative to traditional energy storage systems.5 To that end, in recent years utilization of green electrochemical active materials with high specific capacitance, namely, transition metal oxides, e.g., MnO, RuO, NiO, and Co3O4 has attracted much attention. However, this class of material lack many desirable properties of carbonaceous materials, including high specific surface area, high electrical and thermal conductivity, chemical stability, and low cost.6 Consequently, more recent studies have been mainly focused on novel synthesis routes to develop nanostructured hybrid materials composed of transition metal oxide and various nanostructures of carbon to exploit their synergistic effects in electrochemical energy storage devices.6In addition to the aforementioned limitations of current SCs, the areal capacitance of SCs is limited due to the low material loading per unit area on the substrate. Increasing the thickness of the electrode is not a viable solution to this problem as the capacitance significantly decreases due to the sluggish ion diffusion in the bulk structure. To that end, three-dimensional (3D) electrodes with a high aspect ratio provide much promise.7 Although the present energy storage manufacturing technologies can be used to produce the main components of SCs, namely, electrode, electrolyte, current collector, and device assembly, many challenges including optimal electrode configuration and economic consideration for large scale adaptation of this technology remain at large.8,9 Amongst existing technologies, 3D printing is an alternative for the manufacturing energy storage devices.10 Accordingly, the fabrication of SCs via sequential printing provides a synergistic advantage that brings the excellent durability and longevity of SCs with the precision and efficiency of 3D printing techniques.11 To that end, additive manufacturing (AM) has been adopted to produce SCs via sequential printing of individual layers.12 Specifically, inkjet printing provides the precision and low cost required for mass production of SCs with improved energy and power densities.13,14
Graphene and reduced graphene oxide (rGO) inks have emerged as carbonaceous material of choice for printing electronics devices via inkjet printing.15 Although GO and rGO have exceptionally large specific surface area, good chemical and thermal stability, and high electrical and thermal conductivity, their low energy density limits their utility as electrodes with high supercapacitance.16 In general, electrodes composed solely of such materials fail to meet the energy density desired for most practical applications. However, combination of transition metal oxides, namely, manganese oxide (Mn-oxide) species known for its high specific capacitance with graphene/rGO has been successfully used in an array of applications, including sensors,17 and supercapacitors.18 It should be noted that much attention has been focused on Mn-oxide as it is well-known for its unique and highly diverse valence and oxidation states (up to VII) of Mn, along with its rich redox chemistry. To that end, diverse classes of Mn-oxides are highly promising candidates for electrode materials in supercapacitors. However, most if not all the state-of-the art chemical synthesis routes currently used to produce high-quality Mn-oxide/rGO-interfaced HNCs are costly and complex.19
Our group has previously developed and patented the Laser Ablation Synthesis in Solution-Galvanic Replacement Reaction (LASiS-GRR) technique20,21 for the synthesis of diverse classes of intermetallic nanoalloys22 and nanocomposites as electrocatalysts,23 and have specifically, adopted LASiS-based techniques for the synthesis of Metal Oxides (MOx), Metal Organic frameworks (MOFs),24 and MOF-derived HNCs.25 The current study aims to introduce a simple, rapid, and inexpensive method to fabricate reduced graphene oxide interfaced with nanostructured manganese-based metal oxides via LASiS and evaluate their application as advanced electrodes for supercapacitors. To that end, MnOx/Mn3O4 has been selected as the metal oxide components for the HNC, owing to its well-known theoretically high specific capacitance and widespread use as electrodes in supercapacitor device. Specifically, herein, we demonstrate that selective tuning of the functionality of MnOx/Mn3O4–rGO HNCs by tailoring their structure–property relations via LASiS-driven routes enable rational design and fabrication of efficient SC electrodes using metal-oxide/carbon-based HNCs. In addition, we also demonstrate the successful integration of optimally synthesized HNCs with suitable solvents, viscosity modifiers, commercial graphene, and binders to prepare active layer inks for the fabrication of sequential inkjet printed supercapacitors with exceptional properties. Overall, this study has paved the way for use of LASiS-based synthesized functional material in combination with standard AM techniques for the fabrication of printed electronics with superior performance.
2 Methodology
2.1 Synthesis of MnOx/Mn3O4–rGO HNCs via LASiS-GRR
All chemicals and supplies used in this study were purchased from Sigma-Aldrich. GO was prepared from graphite flakes through one-pot synthesis by the improved Hummer's method.26 Manganese (Mn) pellets (99.95% pure, 1′′ diameter in 0.125′′ thickness), were purchased from Kurt J. Lesker company. The LASiS-driven synthesis technique employed here is identical to what has been described in detail and reported in our previous studies.6,24,25,27 Specifically, the in-house prepared solution of GO suspensions in deionized water, with a concentration of 20 mg L−1, was bath sonicated for approximately 60 minutes and then, transferred to the LASiS reactor cell. The Mn pallet was mounted on a holder at the bottom of the reactor cell and ablated using a pulsed Nd-YAG Laser (1064 nm) with 168 mJ per pulse energy at 10 Hz repetition rates for specific time windows between 6–10 minutes. The final product was in turn decanted for 24 h using deionized water and collected after the suspension was centrifuged at 4700 rpm for 22 minutes.The entire HNCs synthesis process is schematically depicted in Fig. 1.
 | ||
| Fig. 1 LASiS-based HNCs of nanostructured MnOx/Mn3O4 interfaced with rGO with enhanced supercapacitive properties. | ||
2.2 All-printed supercapacitor: methods and materials
The advantages of the 3D printed SCs in this study include rapid prototyping of 3D electrode architectures and improved device performance. High porosity and large specific surface area are other significant benefits of 3D-printed electrodes, which not only can improve the contact area between electrodes and electrolyte but also can increase the electrochemical performance. Furthermore as reported in the current study, manufacturing the entire device via a 3D printing technique, reduces the cost and complexity of manufacturing facility, while increasing versatility and production capacity.In our design, conductive silver (Ag) nanoparticle pastes with the resistivity of 1.59 μΩ cm at 20 °C serves as the current collector layer of the printed design schematically shown in Fig. 2; the Ag layer was printed and annealed at 120 °C for 1 h. The active layer of the faradaic SC is composed of Mn-oxide/carbon-based electroactive materials interfaced with electronic grade graphene nanoflakes from IG LLC and, carbon nanofibers (CNFs), ethyl-cellulose (E-C) and terpineol from Sigma-Aldrich. The electroactive components comprise either a commercial-grade Mn3O4–graphene nanocomposites (NCs) (99.5% purity, Mn3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :graphene = 1:1) obtained from US Research Nanomaterials Inc., or an in-house LASiS-derived MnOx/Mn3O4–rGO HNCs to allow direct comparison of their respective supercapacitive performance. Specifically, to ensure effective charge transport via a percolated conductive network, graphene nanoflakes (0.9 g) and CNFs (0.1 g) are added to both the commercial Mn3O4–graphene NCs and LASiS-made MnOx/Mn3O4–rGO HNCs (0.2 g). In turn, the respective active layers were dispersed in 45 mL of methanol. The solution was bath-sonicated for 1 h followed by evaporation of methanol at 40 °C under vacuum conditions for approximately 12 h. The resulting powder was dispersed in 3 mL of terpineol by manual stirring. Finally, 0.5 g of E-C was added to the powder and terpinol solution by manual stirring for several minutes. The gel polymer electrolyte (GPE) layer was prepared by dissolving polyvinylidene difluoride (PVDF) and lithium perchlorate (LiClO4, 0.25 M) as the salt (PVDF:LiClO4 = 2:1) in propylene carbonate at 140 °C with continuous stirring for 1 h. After the GPE layer solution turned into a viscous gel upon cooling, it was printed on the active layer.
:graphene = 1:1) obtained from US Research Nanomaterials Inc., or an in-house LASiS-derived MnOx/Mn3O4–rGO HNCs to allow direct comparison of their respective supercapacitive performance. Specifically, to ensure effective charge transport via a percolated conductive network, graphene nanoflakes (0.9 g) and CNFs (0.1 g) are added to both the commercial Mn3O4–graphene NCs and LASiS-made MnOx/Mn3O4–rGO HNCs (0.2 g). In turn, the respective active layers were dispersed in 45 mL of methanol. The solution was bath-sonicated for 1 h followed by evaporation of methanol at 40 °C under vacuum conditions for approximately 12 h. The resulting powder was dispersed in 3 mL of terpineol by manual stirring. Finally, 0.5 g of E-C was added to the powder and terpinol solution by manual stirring for several minutes. The gel polymer electrolyte (GPE) layer was prepared by dissolving polyvinylidene difluoride (PVDF) and lithium perchlorate (LiClO4, 0.25 M) as the salt (PVDF:LiClO4 = 2:1) in propylene carbonate at 140 °C with continuous stirring for 1 h. After the GPE layer solution turned into a viscous gel upon cooling, it was printed on the active layer.
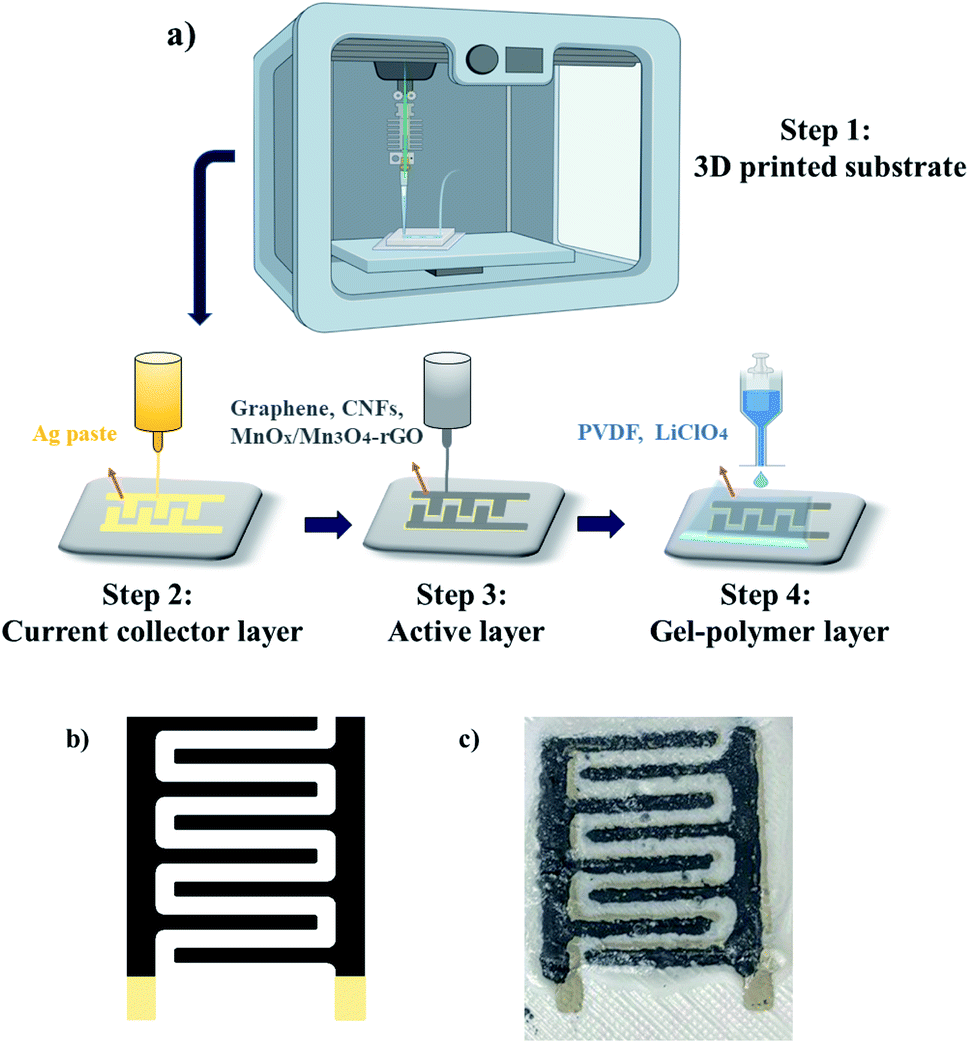 | ||
| Fig. 2 (a) Schematic illustration of the sequential layer-on-layer procedure for the 3D printing of SC electrodes; (b) software generated interdigitated patterns for the current collector and active layers, (c) final all-printed SC electrode design composed of printed substrate, current collector, active, and the GPE layers. | ||
The device patterns were assembled by sequential 3D printing procedure (one layer at a time) shown schematically in Fig. 2(a). Printing patterns for the active and current collector layers were initially developed in Solidworks software (2018, Dassault Systémes SolidWorks Corp., MA, USA). Interdigitated patterns of the current collector and active layer comprise eight lines in total (size and dimensions are discussed in ESI Fig. S1 and S4†), four lines for the positive and four for the negative electrode (Fig. 2(b)). Evidently, the size and shape of the pattern can be customized depending on the system requirements. After trying various printed designs on different substrates, the fillet pattern in Fig. 2(b) was selected for a combination of reasons, namely, simplicity, printer compatibility, and reproducibility. Upon investigation of the printing results on various substrates, acrylonitrile butadiene styrene (ABS) was selected as the substrate. The substrate was printed using filament printing methods with a large format fused deposition (FD) printer (Model: Raise3D Pro2 3D Printer (Fig. S2†)) by heating the printing nozzle to the specific temperature required for printing of ABS filament, namely, 240 °C. It should be noted that printing of ABS filament requires nozzle temperature of 210–250 °C, as well as a heated bed with a temperature range of 80 to 110 °C. Furthermore, ABS needed to be printed in an enclosed print chamber and cooled down slowly to avoid any possible shrinkage and/or wrapping.28 The reason to utilize our custom 3D printed substrates is to maximize the benefits of an all-printed device fabrication process and to avoid the challenges in implementation of prefabricated substrates as illustrated in ESI Fig. S3.† After printing the ABS substrate, the substrate was kept at room temperature for 10 minutes to allow solidification. Later, the substrate was rinsed with methanol and propylene to remove all possible contamination. The surface roughness of the printed substrate was maintained to facilitate ink adhesion (Fig. S2†).
In turn, sequential layer-on-layer printing was used to assemble the final all-printed SC design (Fig. 2(c)). The current-collector, active layer, and the GPE layer were printed by the Engine HR printer (Model: Hydra 16A printer of Hyrel 3D EHR system). The silver (Ag) paste was printed on the substrate as a current collector layer using an inkjet printer based on the pattern designed on Solidworks software and was annealed at 120 °C for 1 h. Due to its high conductivity, the Ag current-collector facilitates a rapid charge transfer from the active layer. The active layer of the SC is intended to provide a fast surface redox reaction in the printed system. It should be noted that the viscosity, cure temperatures, software parameters, and MnOx/Mn3O4 particle sizes were tailored to allow optimal printed designs and prevent printing failures such as shortcuts between electrodes due to “oozing” of the ink. The active layer is printed on the Ag layer and then subjected to a thermal treatment at 80 °C for 1 h. Specifically, the active layer was prepared by dispersing the desired HNCs in the previously prepared paste composed of graphene, CNFs, and E-C. Finally, the GPE layer was printed on the top of the active layer and dried at room temperature overnight. The aforesaid process was identically repeated for fabricating the control electrodes comprised of commercial Mn3O4–graphene NCs for a comparison of SC performances.
2.3 Structural, compositional, and morphological characterization
The HNCs were first characterized by: (1) X-ray diffraction (XRD) analysis with a Phillips X'Pert-Pro diffractometer with a Cu Kα monochromatized radiation source (λ = 0.1541 nm) operating at 45 kV, and 40 mA, (2) Philips XL-30ESEM scanning electron microscope equipped with energy-dispersive X-ray (EDX) spectroscopy, (3) Nicolet iS50 FT-IR Spectrometer, and (4) Helios 5UX dual beam FIB/SEM.2.4 Electrochemical characterization
A conventional, three-compartment electrochemical (EC) cell from Pine Instruments, LLC containing a saturated double junction Ag/AgCl reference electrode and a Pt counter electrode was utilized for all EC characterizations of the HNCs. All EC characterizations were carried out with a three electrode configuration for the ink prepared from the laser-based HNCs and with a two-electrode configuration directly on the electrodes of the printed device. Specifically, cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) experiments were performed using a research-grade potentiostat (SP-150, Biologic instrument).
 | (1) |
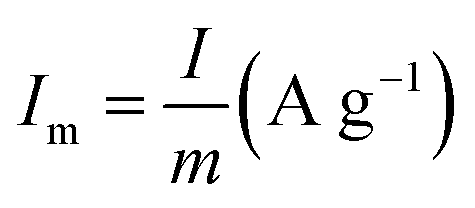 is the current density (current per unit mass) during discharge, m (g) is the weight loading of the electroactive material, and ΔV (V) is the potential drop.
is the current density (current per unit mass) during discharge, m (g) is the weight loading of the electroactive material, and ΔV (V) is the potential drop.2.5 Characterization of all-printed SCs
Thermogravimetric analysis (TGA) was performed on a standard TA Instrument (Q50 15 GA) by heating the samples at a rate of 10 °C min−1 from ambient temperature to 600 °C and 800 °C in an inert N2 environment to ascertain the thermal properties and stability of the Laser-derived (MnOx/Mn3O4–rGO)/commercial (Mn3O4–graphene) HNCs, ink, binder, and solvent. In addition, the data is used to estimate the wt% of the electroactive component.3 Results and discussions
3.1 LASiS synthesized MnOx/Mn3O4–rGO HNCs
:MnO2:Mn3O4 is calculated to be 28:8.7:63 in the sample prepared under 8 minutes of ablation time. In the case of the commercial HNC, only clear diffraction patterns of crystalline Mn3O4 are observed. On the other hand, XRD patterns of samples prepared under 9 and 10 minutes of ablation times indicate negligible presence of MnO2 crystal structures in the HNCs (see Fig. S7†). The wt% of the different Mn-oxide crystalline structures present in the composite MnOx/Mn3O4 samples prepared with different ablation time is summarized in Table 1. In addition, Fig. 3(b) depicts the FT-IR spectra of pure Mn3O4 and MnOx/Mn3O4–rGO samples prepared with 8 minutes ablation time. Hereon, the vibration bands around 595, 445 and 481 cm−1 are attributed to the stretching of the Mn–O bonds in Mn3O4 NPs and MnOx/Mn3O4–rGO HNCs.29,30 The presence of the Mn–O stretching vibration on the HNCs suggests the successful integration of the MnOx/Mn3O4 with the rGO. These FT-IR results also advocate that the MnOx/Mn3O4 nanostructured materials are embedded within the rGo nanosheets. In addition, results are also in full agreement with the SEM (Fig. 4) and XRD data (Fig. 3(a)) further highlighting the formation of MnOx/Mn3O4–rGO HNCs.
 | ||
| Fig. 3 (a) XRD patterns for commercial Mn3O4–graphene NCs and MnOx/Mn3O4–rGO HNCs prepared under 8 minute ablation time along with the standard peak patterns for crystalline MnO, MnO2 and Mn3O4 structures. (b) FT-IR spectrum of Mn3O4 (red) and synthesized HNCs (MnOx/Mn3O4–rGO) under 8 min ablation time (black). | ||
 | ||
| Fig. 4 SEM micrographs of MnOx/Mn3O4–rGO HNCs prepared via LASiS under (a) 6 minute (b) 7 minute (c) 8 minute (d) 9 min, and (e) 10 minute ablation time. | ||
Correspondingly, SEM micrographs of HNCs synthesized under different ablation times (6–10 minutes) are shown in Fig. 4. The figure depicts uniformly grafted MnOx nanorods (NRs) and trace amounts of spherical nanoparticles (NPs) on the rGO nanosheets. It also shows that the sample synthesized under 8 minutes ablation time has higher number densities of more well-dispersed NRs in the ensuing products. The NRs observed in SEM images can be attributed to MnO2 and/or Mn3O4 structures based on XRD patterns, whereas the spherical particles are primarily MnO NPs.
Formation of the observed MnOx/Mn3O4 NRs can originate from the nucleation of the “tunnel-like” α-MnO2 polymorphic structures from the LASiS-induced plasma dynamics. Such structures comprised of the characteristic corner-shared [MnO6] octahedral arrangements are known to be stabilized at nanoscale dimensions and have been reported to emerge in typical hydrothermal synthesis routes.31–34 Hence, it can be posited that these α-MnO2 crystallites can in turn undergo edge-to-edge growth resulting in tunnel-like structures that form the 1D NRs through solution-phase reactions. To that end, SEM images (Fig. 4) together with XRD patterns (Fig. 3(a) and S5†), support the observation of the α-MnO2 crystal structures (referenced to the standard XRD patterns for α-MnO2 in Fig. 3(a)) and the formation of nanorods in the 8 minute ablation during the LASiS process. Specifically, the incipient α-MnO2 rod-shaped seeding sites can subsequently serve as the template for the growth of the Mn-rich phases. These Mn-rich species in turn experience faradaic reactions, resulting in a mixed oxide composition, namely, MnO, MnO2 and Mn3O4 stabilized at the nanoscale dimension. Herein, it is noted that the extent of conversion of Mn-rich phases in the incipient α-MnO2 structures into Mn3O4 phases via faradaic reactions depend on the available initial concentrations of the α-MnO2 phase from the LASiS process. Hence the resultant MnO2 concentrations (wt%) are different in the samples prepared under different ablation times (see Table 1). To that end, the aforesaid structural analyses from XRD measurements (Fig. 3(a) and S5†) coupled with observations from the SEM micrographs (Fig. 4) strongly suggest that the interplay between the specific wt% balance of the 3 manganese oxide phases (MnO, Mn3O4 and MnO2) formed during the high-energy laser ablation processes plays a critical role in the capacitance behavior of the samples prepared under different ablation times. Hence, significantly higher amount of MnO2 (∼8.7 wt%) in the NRs typically formed under 8 minutes of ablation time (see Table 1) can be ascribed to the aforementioned structure-morphology evolution promoted by the specific laser-induced plasma dynamics at the solution interface. The formation of this significant amount of MnO2 in the 8 minute ablation samples will be shown to be critical in explaining the enhanced supercapacitance of the HNCs when inked onto the electrodes.
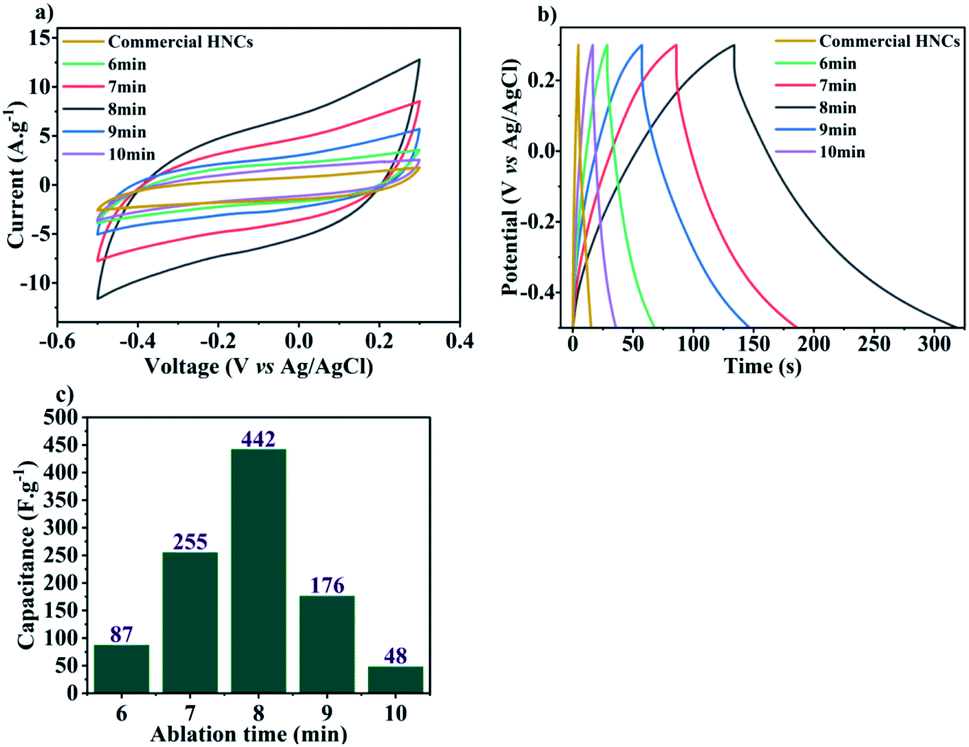 | ||
| Fig. 5 (a) CV curves in 0.5 M Na2SO4, and (b) discharge curves at 2 A g−1 for LASiS-synthesized MnOx/Mn3O4–rGO and commercially-made HNCs; (c) calculated capacitance values (F g−1) for MnOx/Mn3O4–rGO HNCs synthesized under varying ablation times (6–10 minutes). | ||
A significant insight from the aforesaid results is the critical role of ablation times during LASiS in tailoring the relative volume loading, composition (wt% distribution of MnO, MnO2, Mn3O4) and structure of the as-synthesized MnOx/Mn3O4 NRs in the rGO matrix which, in turn, tunes the capacitive properties of synthesized HNCs by altering their ion diffusion pathways, conductivity and ultimately, charge transport properties. To that end, a higher density of MnOx/Mn3O4 NRs allows more charges to be stored due to the increased rate of faradaic reactions with the electrolyte as a result of the increased interfacial area for electrolyte interactions. These observations need to be considered in conjunction with the fact that, at a critical concentration, redox reaction pathways can be stymied due to compromised charge transfer processes through the NCs due to the relatively low conductive nature of the oxide species. Therefore, the conductive nature stemming from the existence of rGO is crucial for enhancing the supercapacitive properties of MnOx–rGO HNCs. The rapid charge transfer in the conductive sp2 carbon network delivered by the rGO nanosheets suppresses charge accumulation on the MnOx/Mn3O4 nanostructured surfaces, thereby enhancing the electrode/electrolyte interfacial charge transport and higher power outputs. Furthermore, the distinctly enhanced supercapacitive and discharge curve behaviors shown above (see Fig. 5(a) and (b)) for the HNCs synthesized under 7–9 minute ablation times (with the best SC performance from the 8 minute ablation sample) can be directly correlated to the emergence of a significantly higher number of well-dispersed MnOx/Mn3O4 NRs (seen from Fig. 4) and the significantly higher amount (wt%) of MnO2 present in the 8 minute ablation sample (see Table 1). Hence, it can be posited that the excess interfacial areas and tubular channel-like pathways for the NRs comprised of higher α-MnO2 contents, that emerges from the high-energy LASiS-induced processes, play a significant role in promoting superior charge transport and fast ion diffusion properties. It would be worth highlighting that previous studies have conclusively established that the tubular structures of the α-MnO2 phase make them promising candidates as SC electrode materials. This can be attributed to their optimal tunnel spaces that can essentially control the electrolyte ion adsorption rates on the crystalline MnO2 structures on inner tunnel surfaces which, in turn, has an immediate impact on the charge storage process during the redox reactions.36–39
As mentioned before, charge transfer mechanisms and faradaic reaction rates are both essential elements in optimizing the supercapacitive properties of Mn oxide–rGO HNCs. Hence, we have investigated the optimal ratio of MnOx/Mn3O4 NRs to the carbonaceous rGO matrices via TGA experiments on the as-synthesized HNC samples. Fig. 7(a) depicts the thermal properties of LASiS-derived HNCs and commercial NCs at a scan rate of 10 °C up to 800 °C under N2 atmosphere. The Mn3O4–graphene and MnOx/Mn3O4–rGO plots show a two-step thermal degradation process with about 26 and 24.8 wt% initial weight loss up to around 550 °C, respectively. The removal of adsorbed water/moisture and elimination of some residual oxygen containing elements from GO could be attributed to the initial weight loss of commercial and laser derived HNCs up to 220 °C. The sharp weight loss at higher temperatures (between 350–500 °C) may be ascribed to the decomposition of the oxygen containing functional groups present in the GO sheets and successful reduction process of GO. Finally above 550 °C, the residue weight percentage of manganese oxide in the Mn3O4–graphene NCs and MnOx/Mn3O4–rGO HNCs is 74 and 75.2 wt%, respectively which is mainly ascribed to the higher residual metal oxides mass present in the LASiS synthesized HNCs at 800 °C. Thus, the aforesaid TGA analyses for Mn-oxide: rGO compositional ratios can be directly correlated to the effective supercapacitive properties seen in Fig. 5 and the SEM images in Fig. 3(a). Specifically, a clear symbiotic faradaic and conductive relation is required for superior capacitive properties. Hence, faradaic properties of MnOx/Mn3O4–rGO HNCs that arise from Mn oxide and rGO is crucial to the conductivity of the electrode material.
3.2 Printed supercapacitor
 | ||
| Fig. 6 Structure of the SC ink material shown by the SEM images of: (a) percolated networks of CNFs and MnOx/Mn3O4 NRs on graphene nanosheets before heat treatment, and (b) printed active layer containing E-C binder after heat treatment at 80 °C. | ||
 | ||
| Fig. 7 Physicochemical properties of the printed ink and HNCs analyzed via TGA of: (a) commercial (red) and LASiS-synthesized (blue) HNCs, and (b) printed ink (after vacuum drying of solvents), solvent, and binder. | ||
For ease of visualization, the enlarged inset in Fig. 6(a) indicates the color-coded MnOx/Mn3O4 NRs and the rGO nanoflakes. Furthermore, the composition of the active SC components quantified via the wt% of CNFs, electronic grade graphene nanoflakes and the electroactive MnOx/Mn3O4–rGO HNCs, are shown in Table 2.
| SC ink Material | CNF | Graphene | Laser derived HNCs |
|---|---|---|---|
| Weight percent (%) | 9 | 75 | 16 |
In order to investigate the thermal stability of the all-printed SC electrode layers after the 80 °C annealing treatments (as described in the Methodology section 2.5), subsequent TGA was also performed on the printed ink with the active materials in an N2 environment. The TGA test results shown in Fig. 7(b) are intended to establish the thermal stability of the MnOx/Mn3O4–rGO content in the ink material and the HNCs structures. The initial large mass change of ∼60 wt% in the SC ink denotes the evaporation of the organic solvent (terpineol) at an onset temperature of ∼200 °C. The second significant mass change of ∼20 wt% occurs between 200–350 °C that indicates the complete degradation of the binder. From 350 °C forward, the solvents are completely evaporated, and the mass loss shows the thermal decomposition of the E-C in the binder. These results confirm that the processed active layer used in the printed SC ink after the annealing process is composed of ∼20 wt% of the as-synthesized HNCs. Since there is no significant mass change in samples annealed at 100 °C (Fig. 7(b)), it can be concluded that the inks did not undergo any thermal decomposition during the annealing process.
 | ||
| Fig. 8 Electrochemical performances of all-printed SCs as recorded from: (a) CV curves at 20 mV s−1 scanning rate for the printed SCs containing commercial (red) and LASiS synthesized (blue) MnOxMn3O4–rGO HNCs and (b) discharge curves at 1 A g−1. | ||
4 Conclusions
In this study, the supercapacitive properties of HNCs comprised of rGO interfaced with nanostructured MnOx/Mn3O4 synthesized via Laser Ablation Synthesis in Solution (LASiS) using various ablation times (6–10 minutes) in tandem with a post-processing thermal annealing process is investigated. Detailed structural characterizations revealed that the electroactive nonstructures embedded in the rGO 2D matrices are primarily composed of MnOx/Mn3O4 species with varying mass concentrations (wt%) of MnO, MnO2 and Mn3O4 oxide phases. Functional characterizations demonstrated the influence of varying ablation times during the LASiS process on the structure–composition properties of the ensuing HNCs – namely, in the form of the optimal concentration ratios for: (1) rGO:MnOx/Mn3O4 NRs, and (2) MnO:MnO2:Mn3O4 structures (wt%) which, in turn, govern the characteristic capacitive behaviors of the HNC-based electrode inks. Specifically, electrochemical characterizations indicated that HNCs samples carrying significantly higher MnO2 contents (∼9 wt%) in the MnOx/Mn3O4 NRs, and prepared under 8 minute ablation time, exhibited the highest supercapacitive properties with capacitance of ∼442 F g−1. We also implemented direct inkjet printing techniques for the successful fabrication of advanced SC electrodes comprising the active layer ink composed of HNC-based electroactive materials interfaced with a percolated conductive network of electronic grade graphene nanosheets and CNFs. In conclusion, the electrochemical characterization of the all-printed SC electrode material fabricated from LASiS-derived MnOx/Mn3O4–rGO HNCs showed a higher specific capacitance of Csp ∼325 F g−1 owing to higher surface area and redox reactions, as compared to standard commercially-made Mn3O4–graphene NCs ∼189 F g−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express special thanks to Dr Rigoberto Advincula for providing access to his 3D printers located in his laboratory housed in the Chemical and Biomolecular Engineering department at University of Tennessee, Knoxville. We are also grateful to IG LLC for providing the electronic grade graphene used in this study. SEM and XRD was performed at the Institute for Advanced Materials & Manufacturing (IAMM) Diffraction Facility and Electron Microscopy, and Advanced Microscopy and Imaging Center (AMIC) located at the University of Tennessee, Knoxville.Notes and references
- B. K. Kim, S. Sy, A. Yu and J. Zhang, Handbook of clean energy systems, 2015, pp. 1–25 Search PubMed.
- D. K. Niakolas, M. Daletou, S. G. Neophytides and C. G. Vayenas, Ambio, 2016, 45, 32–37 CrossRef PubMed.
- M.-L. Seol, I. Nam, E. L. Ribeiro, B. Segel, D. Lee, T. Palma, H. Wu, D. Mukherjee, B. Khomami and C. Hill, et al., ACS Appl. Energy Mater., 2020, 3, 4965–4973 CrossRef CAS.
- W. Zhou, X. Liu, K. Zhou and J. Jia, Nanomaterials in Advanced Batteries and Supercapacitors, Springer, 2016, pp. 271–315 Search PubMed.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- S. Hu, E. L. Ribeiro, S. A. Davari, M. Tian, D. Mukherjee and B. Khomami, RSC Adv., 2017, 7, 33166–33176 RSC.
- S. H. Park, G. Goodall and W. S. Kim, Mater. Des., 2020, 193, 108797 CrossRef CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 1–10 Search PubMed.
- X. Tian, J. Jin, S. Yuan, C. K. Chua, S. B. Tor and K. Zhou, Adv. Energy Mater., 2017, 7, 1700127 CrossRef.
- A. Ambrosi and M. Pumera, Chem. Soc. Rev., 2016, 45, 2740–2755 RSC.
- V. Egorov, U. Gulzar, Y. Zhang, S. Breen and C. O'Dwyer, Adv. Mater., 2020, 32, 2000556 CrossRef CAS PubMed.
- A. H. Espera, J. R. C. Dizon, Q. Chen and R. C. Advincula, Prog. Addit. Manuf., 2019, 4, 245–267 CrossRef.
- P. Chang, H. Mei, S. Zhou, K. G. Dassios and L. Cheng, J. Mater. Chem. A, 2019, 7, 4230–4258 RSC.
- P. Calvert, Chem. Mater., 2001, 13, 3299–3305 CrossRef CAS.
- L. T. Le, M. H. Ervin, H. Qiu, B. E. Fuchs and W. Y. Lee, Electrochem. Commun., 2011, 13, 355–358 CrossRef CAS.
- R. Liang, Y. Du, P. Xiao, J. Cheng, S. Yuan, Y. Chen, J. Yuan and J. Chen, Nanomaterials, 2021, 11, 1248 CrossRef CAS PubMed.
- M. Mabrook, C. Pearson, A. Jombert, D. Zeze and M. Petty, Carbon, 2009, 47, 752–757 CrossRef CAS.
- O.-S. Kwon, H. Kim, H. Ko, J. Lee, B. Lee, C.-H. Jung, J.-H. Choi and K. Shin, Carbon, 2013, 58, 116–127 CrossRef CAS.
- R. Dong, Q. Ye, L. Kuang, X. Lu, Y. Zhang, X. Zhang, G. Tan, Y. Wen and F. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 9508–9516 CrossRef CAS PubMed.
- D. Mukherjee and S. Hu, Compositions, systems and methods for producing nanoalloys and/or nanocomposites using tandem laser ablation synthesis in solution-galvanic replacement reaction, US Pat.,10326146, 2019 Search PubMed.
- D. Mukherjee and S. Hu, Compositions, systems and methods for producing nanoalloys and/or nanocomposites using tandem laser ablation synthesis in solution-galvanic replacement reaction, US Pat., 11127956, 2021 Search PubMed.
- S. Hu, K. Cheng, E. L. Ribeiro, K. Park, B. Khomami and D. Mukherjee, Catal. Sci. Technol., 2017, 7, 2074–2086 RSC.
- S. Hu, G. Goenaga, C. Melton, T. A. Zawodzinski and D. Mukherjee, Appl. Catal. B Environ., 2016, 182, 286–296 CrossRef CAS.
- E. L. Ribeiro, S. A. Davari, S. Hu, D. Mukherjee and B. Khomami, Mater. Chem. Front., 2019, 3, 1302–1309 RSC.
- E. L. Ribeiro, E. M. Davis, M. Mokhtarnejad, S. Hu, D. Mukherjee and B. Khomami, Catal. Sci. Technol., 2021, 11, 3002–3013 RSC.
- D. C. Marcano, D. V. Kosynkin, Z. S. Sun, A. Alemany, L. B. Lu and J. M. Tour, Improved synthesis of graphene oxide, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed.
- S. Hu, C. Melton and D. Mukherjee, Phys. Chem. Chem. Phys., 2014, 16, 24034–24044 RSC.
- J. Ramian, J. Ramian and D. Dziob, Materials, 2021, 14, 7070 CrossRef CAS PubMed.
- Y. Kong, R. Jiao, S. Zeng, C. Cui, H. Li, S. Xu and L. Wang, Nanomaterials, 2020, 10, 367 CrossRef CAS PubMed.
- M. Jarvin, S. A. Kumar, G. Vinodhkumar, E. Manikandan and S. Inbanathan, Mater. Lett., 2021, 305, 130750 CrossRef CAS.
- H. Wang, J. Zhang, X. Hang, X. Zhang, J. Xie, B. Pan and Y. Xie, Angew. Chem., 2015, 127, 1211–1215 CrossRef.
- Q. Feng, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 319–333 RSC.
- X. Wang and Y. Li, Chem. -Eur J., 2003, 9, 300–306 CrossRef CAS PubMed.
- N. Tang, X. Tian, C. Yang, Z. Pi and Q. Han, J. Phys. Chem. Solids, 2010, 71, 258–262 CrossRef CAS.
- Q. Cheng and A. Brajter-Toth, Anal. Chem., 1995, 67, 2767–2775 CrossRef CAS.
- C. Xu, F. Kang, B. Li and H. Du, J. Mater. Res., 2010, 25, 1421–1432 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CrossRef CAS.
- Z. Chen, Z. Jiao, D. Pan, Z. Li, M. Wu, C.-H. Shek, C. L. Wu and J. K. Lai, Chem. Rev., 2012, 112, 3833–3855 CrossRef CAS PubMed.
- Y. Shen, R. Zerger, R. DeGuzman, S. Suib, L. McCurdy, D. Potter and C. O’young, Science, 1993, 260, 511–515 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02009b |
| ‡ Mahshid Mokhtarnejad and Erick L. Ribeiro contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2022 |