
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mild and concise synthesis of aryloxy phosphoramidate prodrug of alcohols via transesterification reaction†
Hanglu Yingab,
Jie Yaoab,
Fan Wu *ab,
Yufen Zhaoab and
Feng Ni*ab
*ab,
Yufen Zhaoab and
Feng Ni*ab
aInstitute of Drug Discovery Technology, Ningbo University, Ningbo, Zhejiang 315211, P. R. China. E-mail: wufan@nbu.edu.cn; nifeng@nbu.edu.cn
bQian Xuesen Collaborative Research Center of Astrochemistry and Space Life Sciences, Ningbo University, Ningbo, Zhejiang 315211, P. R. China
First published on 29th April 2022
Abstract
A synthesis of aryloxy phosphoramidate prodrug of alcohols enabled by a transesterification strategy is described here. This reaction operates under mild conditions and thus has excellent functional group tolerance. This method provides an efficient and practical solution to the rapid construction of the aryloxy phosphoramidate prodrugs library for potential SAR studies.
The phosphate and phosphonate prodrug strategy is widely recognized as an effective approach to improve the physicochemical and pharmacological properties of therapeutic nucleosides and sugars.1 Over the last few decades, the ProTide prodrug strategy pioneered by Prof. Chris McGuigan has emerged as a powerful platform for developing nucleotide therapeutics.2 This prodrug approach has been extensively studied in the antiviral and anticancer fields, enabling the discovery and development of three FDA-approved antiviral drugs and several clinical candidates (Fig. 1a).3 Whereas early efforts focused mainly on nucleoside-based medicines, many recent discoveries suggested that this technology can be applied to non-nucleoside substrates as well.4 The nature of different components of the phosphoramidate core is essential for the prodrug's potency, especially for the case of non-nucleoside drug candidates in which other amino acid motifs are more effective than the commonly used L-alanine.5 As a result, SAR studies of amino acid ester and aryl moieties would be necessary to identify the optimal combination of these masking groups when applying ProTide technology to a new therapeutic chemical entity. Consequently, an efficient method capable of rapidly assembling aryloxy phosphoramidate prodrug library from parent drug would be very attractive to the discovery of new ProTide prodrugs.
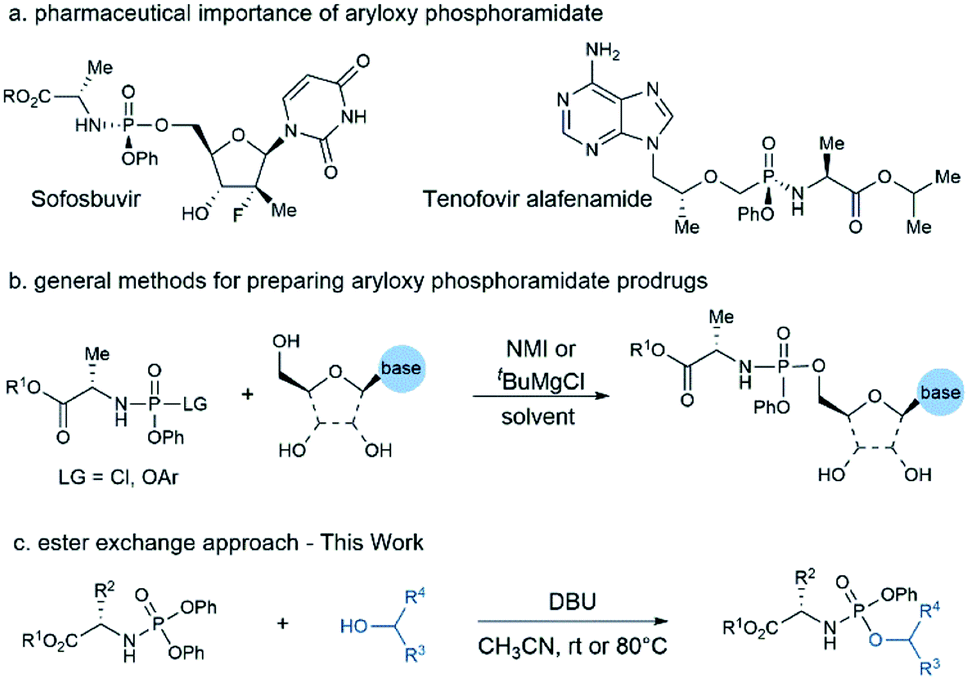 | ||
| Fig. 1 Pharmaceutical importance and the synthetic methods of aryloxy phosphoramidate prodrugs. | ||
Current methods for the preparation of aryloxy phosphoramidate structures include: (a) phosphorylating the nucleoside with phosphorochloridate reagent,6 (b) ester exchange between nucleoside and a diarylphosphite followed by oxidative amination,7 (c) coupling of the amino acid ester with a nucleoside aryl phosphate or its derivatives.8 Among these methods, coupling nucleosides with phosphorochloridate reagents in the presence of either N-methylimidazole (NMI) or tert-butyl magnesium chloride (tBuMgCl) is the most popular strategy for ProTide prodrug construction (Fig. 1b). Although this method has enabled the synthesis of numerous nucleoside prodrugs, regioselectivity and diastereoselectivity issues associated with this approach remained challenging.9 As a result, numerous efforts have been devoted to develop methods with high regioselectivity10 and diastereoselectivity.11 While these advances offer a variety of choices on batch synthesis of designated compounds, other issues remain to be addressed. First, the current method mainly focused on accessing phosphoramidate prodrugs containing L-alanine and nucleoside. The compatibility of these methods towards other amino acid motif and non-nucleosides substrates are less investigated. Second, compatible substrate nucleoside bases are limited due to the high reactivity of phosphorylating reagents such as phosphorchloridate or diarylphosphite, which have to involve protection of the nucleobase moiety sometimes.
Given the growing interest in expanding ProTide technology to other therapeutic areas and the unmet need for rapid access to the prodrug library for hit screenings or SAR studies, we wonder if we can develop a practical method capable of preparing aryloxy phosphoramidates prodrugs from both nucleoside and non-nucleoside substrates. We reasoned that N-diphenyl phosphoryl amino acid ester could serve as a mild phosphorylating reagent and afford aryloxy phosphoramidate prodrugs via a simple transesterification process. Herein, we report our discovery on a DBU promoted synthesis of aryloxy phosphoramidates prodrugs from stable and readily available N-diphenylphosphoryl amino acid esters and alcoholic substrates (Fig. 1c).
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (eq.) | 2 (eq.) | Base (eq.) | Solvent | Yieldb (%) |
a Reaction conditions: phosphorylating reagent 1 (0.3–0.6 mmol), stavudine 2 (0.2 mmol), base (1.0–3.0 equiv.) and solvent (1 mL) at room temperature, t = 36 h.b NMR yield using triethyl phosphate as the internal standard. The yield shown in parentheses is isolated yield, and the product is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of RP and SP diastereoisomers. :1 mixture of RP and SP diastereoisomers. |
|||||
| 1 | 1.5 | 1.0 | DIPEA (2.0) | CH3CN | — |
| 2 | 1.5 | 1.0 | K2CO3 (2.0) | CH3CN | — |
| 3 | 1.5 | 1.0 | NaOH (2.0) | CH3CN | 11 |
| 4 | 1.5 | 1.0 | DBU (2.0) | CH3CN | 78 |
| 5 | 2.0 | 1.0 | DBU (2.0) | CH3CN | 91(88) |
| 6 | 3.0 | 1.0 | DBU (2.0) | CH3CN | 92 |
| 7 | 2.0 | 1.0 | DBU (1.0) | CH3CN | 65 |
| 8 | 2.0 | 1.0 | DBU (3.0) | CH3CN | 83 |
| 9 | 2.0 | 1.0 | DBU (2.0) | DCM | 71 |
| 10 | 2.0 | 1.0 | DBU (2.0) | DMF | 27 |
| 11 | 2.0 | 1.0 | DBU (2.0) | THF | 36 |
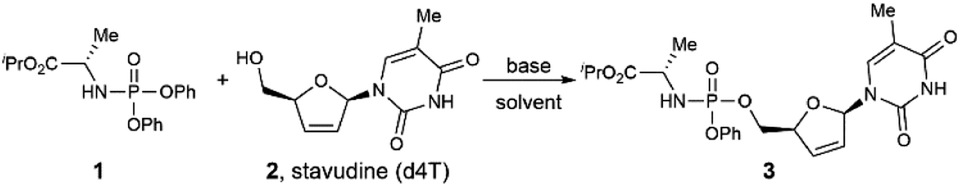
We began our study by examining the reaction of phosphoramidate 1 and stavudine (d4T, anti-HIV drug) in the presence of diisopropylethylamine (DIPEA) as the base in acetonitrile (CH3CN) at 25 °C (Table 1, entry 1). Unfortunately, no product was detected, and all starting materials were recovered. After screening several other bases (entries 2–4), we found that inorganic bases such as K2CO3 and NaOH promoted the reaction but in low yield. In contrast, strong organic base 1,8-diazabicyclo 5.4.0 undec-7-ene (DBU) afforded desired product in a much higher yield. This result suggests that efficient deprotonation of the hydroxyl group is essential to drive this reaction. Screening of other similar bases such as 1,5-diazabicyclo [4.3.0]non-5-ene (DBN), 1,1,3,3-tetramethyl-guanidine (TMG), and 1,5,7-triazabicyclo [4.4.0]dec-5-ene (TBD) did not improve the reaction efficiency.12 Increasing the equivalents of phosphorylating reagent 1 to 2.0 equivalent or 3.0 equivalent afforded the target product in 91% and 92% yield, respectively (entries 5–6). Stoichiometry optimization on base revealed that 2.0 equivalent of DBU was optimal, with 1.0 or 3.0 equivalent of DBU gave impaired yield (entries 7–8). In addition, solvent is proved to be vital for a productive reaction, as replacing CH3CN with DCM, DMF, and THF only afford product 3 in 27% to 71% yield (entries 9–11). To test the scalability of this strategy, we conducted a 1.5 mmol scale synthesis of product 3 under the standard conditions. This reaction afforded the desired product in 85% isolated yield, demonstrating the scalability and potential synthetic application of this protocol.13
While previous studies suggested that L-alanine was optimal for antiviral and anticancer ProTide prodrugs, some new SAR data obtained in the studies of non-nucleoside drug candidates indicated that other amino acids, in some cases, might be more effective than L-alanine.5 Therefore, we decided to explore the scope of N-diphenylphosphoryl amino acid esters (Table 2). Under standard conditions, a variety of L-alanine esters were tolerated, affording desired aryloxy phosphoramidates prodrugs in good yields (products 4–6). Moreover, substrates containing nonpolar amino acid residue such as valine, isoleucine, phenylalanine proceeded smoothly to afford the target products with good efficiency (products 7–9). In addition, N-diphenylphosphoryl tryptophan or O-protected serine were well tolerated in the standard conditions, affording desired product in high yield (products 10–11).
| a Standard reaction conditions, 36 h. The yield shown here is isolated yield, and the product is a 1:1 mixture of RP and SP diastereoisomers. |
|---|
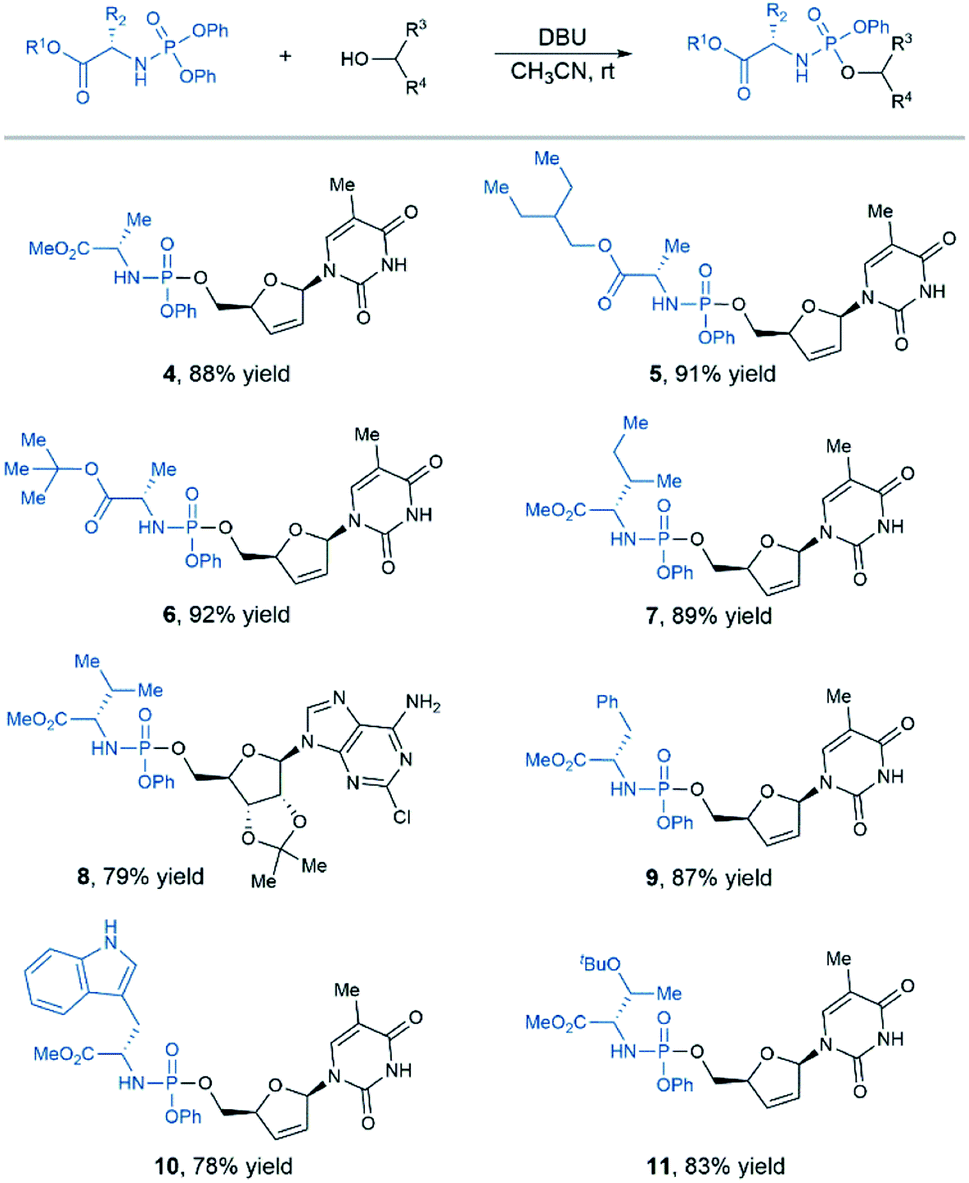 |
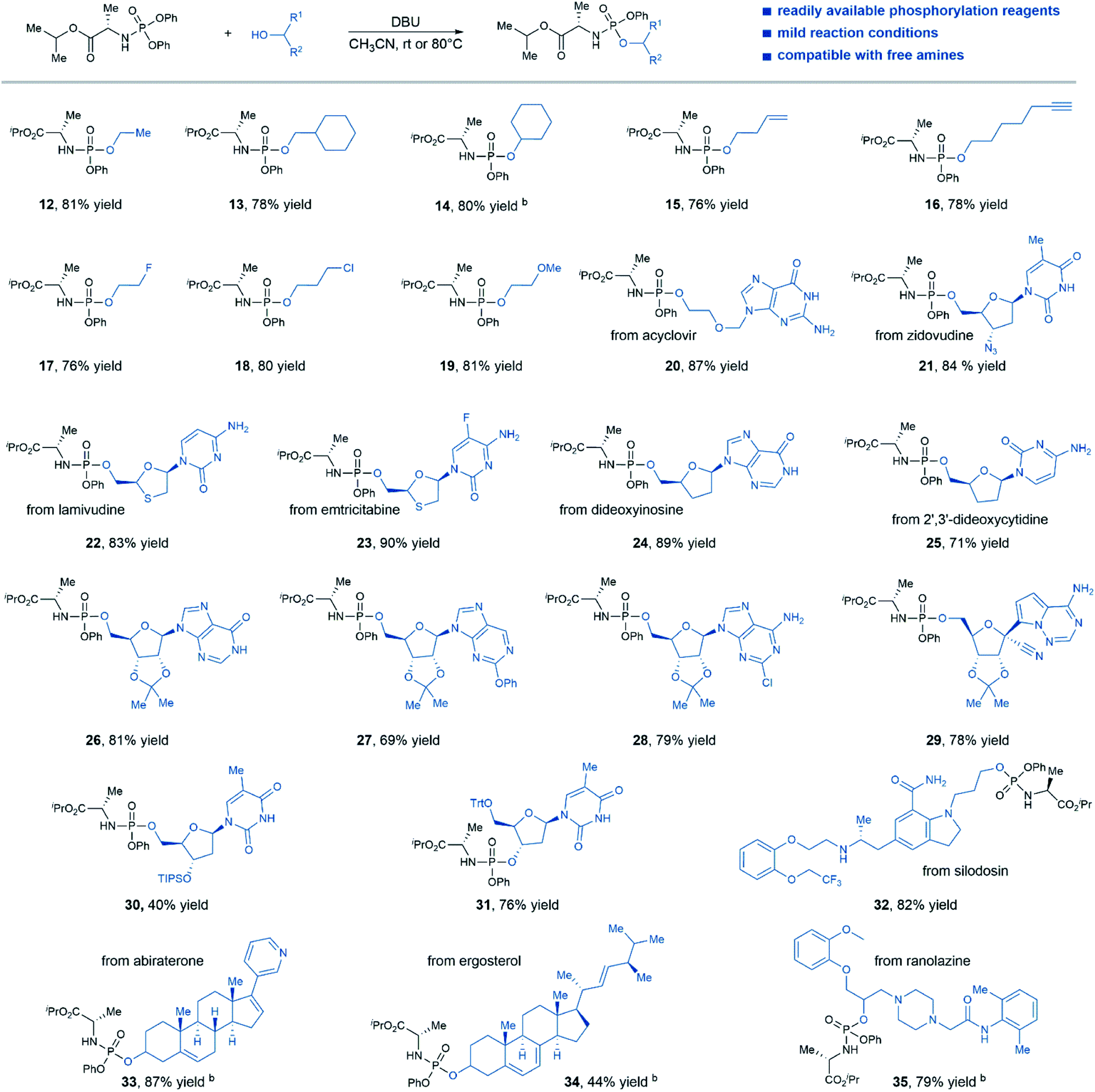
Satisfied with the scope of N-diphenylphosphoryl amino acid esters, we then investigated the generality of this protocol regarding the coupling of various alcohol substrates with phosphorylation reagent 1 (Table 3). A range of alcohols with different steric environments were smoothly converted to the desired products in consistently good yields (products 12–14). In the case of the secondary alcohol, an elevated temperature (80 °C) was required for efficient transformation (product 14). Substrates bearing functional groups such as alkene (product 15), alkyne (product 16), halides (product 17–18), and ether (product 19) were well tolerated in this protocol and delivered the desired product with good efficiency (76–81% yield). Nucleoside substrates that contain only 5′-hydroxyl group, such as acyclovir (product 20), zidovudine (product 21), lamivudine (product 22), emtricitabine (product 23), dideoxyinosine (product 24), and 2′,3′-dideoxycytidine (product 25) were all viable substrates that afforded desired aryloxy triester phosphoramidates prodrugs in good yields. Notably, the nucleophilic nitrogens on those nucleosides were well tolerated under our reaction condition, no N-phosphorylation by-products were observed. Moreover, nucleoside substrates with protected 2′,3′-hydroxyl groups were also effective for product formation (products 26–29, 69–81% yield). Low conversion was observed when substrates bearing 2′,3′-hydroxyl groups were directly subjected to the reaction conditions, likely due to poor solubility of this type of substrates in CH3CN. Although treating nucleoside bearing unprotected 3′,5′-hydroxyl groups with the standard reaction conditions gave a mixture of regioisomers, mono-phosphorylation of 5′ or 3′-hydroxyl group can be achieved by protecting the other hydroxyl group (products 30 and 31). In addition, non-nucleoside medicines such as silodosin, abiraterone, ergosterol, and ranolazine were also viable substrates for this reaction, afforded desired prodrug products in good yields (products 32–35), except for ergosterol. The amine and amide functional groups in these complex drugs were well tolerated in this protocol without side reactions, thus demonstrating the potential application of this method to the synthesis of aryloxy phosphoramidates prodrug of non-nucleoside drug candidates.
In conclusion, we have developed a mild and concise protocol for synthesizing aryloxy phosphoramidate prodrugs of alcohols and nucleosides. This method utilizes diphenyl phosphoramidates as the phosphoryl donor to realize the phosphorylation of a range of primary or secondary alcohols. This method operates under mild conditions and has good functional group tolerance, thus enabling the synthesis of a range of prodrug analogues of nucleosides containing nucleophilic functional groups. We believe this work will provide a valuable tool for the rapid construction of the aryloxy phosphoramidate prodrug library for hit screenings or SAR studies, which is essential for discovery of potent ProTide drug candidates.
Author contributions
F. W. and F. N. directed the project and wrote the manuscript. F. W. and F. N. conceived the idea and designed the experiments. H. Y. and J. Y. performed the experiments. All the authors participated in the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank supports from National Natural Science Foundation of China (91856126, 21778042), Scientific Research Grant of Ningbo University (215-432000282) and Ningbo Top Talent Project (215-432094250).Notes and references
- For selected reviews, see: (a) A. J. Wiemer and D. F. Wiemer, Top. Curr. Chem., 2015, 360, 115–160 CrossRef CAS PubMed; (b) K. M. Heidel and C. S. Dowd, Future Med. Chem., 2019, 11, 1625–1643 CrossRef CAS PubMed.
- For selected reviews (a) D. Cahard, C. McGuigan and J. Balzarini, Mini-Rev. Med. Chem., 2004, 4, 371–381 CrossRef CAS PubMed; (b) A. S. Alanazi, E. James and Y. Mehellou, ACS Med. Chem. Lett., 2019, 10, 2–5 CrossRef CAS PubMed; (c) M. Serpi and F. Pertusati, Expert Opin. Drug Discovery, 2021, 16, 1149–1161 CrossRef CAS PubMed.
- (a) P. J. Thornton, H. Kadri, A. Miccoli and Y. Mehellou, J. Med. Chem., 2016, 59, 10400–10410 CrossRef CAS PubMed; (b) Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS PubMed.
- (a) C. McGuigan, M. Serpi, R. Bibbo, H. Roberts, C. Hughes, B. Caterson, A. T. Gibert and C. R. A. Verson, J. Med. Chem., 2008, 51, 5807–5812 CrossRef CAS PubMed; (b) M. Serpi, R. Bibbo, S. Rat, H. Roberts, C. Hughes, B. Caterson, M. J. Alcaraz, A. T. Gibert, C. R. Verson and C. McGuigan, J. Med. Chem., 2012, 55, 4629–4639 CrossRef CAS PubMed; (c) N. Hamon, M. Quintiliani, J. Balzarini and C. McGuigan, Bioorg. Med. Chem. Lett., 2013, 23, 2555–2559 CrossRef CAS PubMed; (d) N. Hamon, M. Slusarczyk, M. Serpi, J. Balzarini and C. McGuigan, Bioorg. Med. Chem., 2015, 23, 829–838 CrossRef CAS PubMed; (e) E. James, F. Pertusati, A. Brancale and C. McGuigan, Bioorg. Med. Chem. Lett., 2017, 27, 1371–1378 CrossRef CAS PubMed; (f) L. Osgerby, Y.-C. Lai, P. J. Thornton, J. Amalfitano, C. S. Le Duff, I. Jabeen, H. Kadri, A. Miccoli, J. H. R. Tucker, M. M. K. Muqit and Y. Mehellou, J. Med. Chem., 2017, 60, 3518–3524 CrossRef CAS PubMed; (g) M. S. Davey, R. Malde, R. C. Mykura, A. T. Baker, T. E. Taher, C. S. Le Duff, B. E. Willcox and Y. Mehellou, J. Med. Chem., 2018, 61, 2111–2117 CrossRef CAS PubMed; (h) N. A. Lentini, B. J. Foust, C.-H. C. Hsiao, A. J. Wiemer and D. F. Wiemer, J. Med. Chem., 2018, 61, 8658–8669 CrossRef CAS PubMed.
- (a) L.-J. Gao, S. De Jonghe, D. Daelemans and P. Herdewijn, Bioorg. Med. Chem. Lett., 2016, 26, 2142–2146 CrossRef CAS PubMed; (b) C. Morozzi, J. Sedláková, M. Serpi, M. Avigliano, R. Carbajo, L. Sandoval, Y. Valles-Ayoub, P. Crutcher, S. Thomas and F. Pertusati, J. Med. Chem., 2019, 62, 8178–8193 CrossRef CAS PubMed.
- For selected examples of NMI-mediated coupling of nucleoside with phosphorochloridate reagents, see: (a) C. McGuigan, R. N. Pathirana, N. Mahmood, K. G. Devine and A. J. Hay, Antiviral Res., 1992, 17, 311–321 CrossRef CAS PubMed; (b) P. Franchetti, L. Cappellacci, M. Grifantini, L. Messini, G. A. Sheikha, A. G. Loi, E. Tramontano, A. De Mantis, M. G. Spiga and P. La Colla, J. Med. Chem., 1994, 37, 3534–3541 CrossRef CAS PubMed; (c) C. McGuigan, P. W. Sutton, D. Cahard, K. Turner, G. O'Leary, Y. Wang, M. Gumbleton, E. De Clercq and J. Balzarini, Antiviral Chem. Chemother., 1998, 9, 473–479 CrossRef CAS PubMed. for selected examples of t-BuMgCl-mediated synthesis of aryloxyphosphoramidate prodrugs, see: (d) C. McGuigan, O. M. Wedgwood, E. De Clercq and J. Balzarini, Bioorg. Med. Chem. Lett., 1996, 6, 2359–2362 CrossRef; (e) Y. Mehellou, C. McGuigan, A. Brancale and J. Balzarini, Bioorg. Med. Chem. Lett., 2007, 17, 3666–3669 CrossRef CAS PubMed.
- For selected examples, see: (a) X. Li, H. Fu, Y. Jiang, Y. Zhao and J. Liu, Synlett, 2004, 14, 2600–2602 Search PubMed; (b) M. Ora, J. Ojanpera and H. Lonnberg, Chem.–Eur. J., 2007, 13, 8591–8599 CrossRef CAS PubMed.
- For selected examples, see: (a) H. Chapman, M. Kernan, E. Prisbe, J. Rohloff, M. Sparacino, T. Terhorst and R. Yu, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 621–628 CrossRef CAS PubMed; (b) R. L. Mackman, A. S. Ray, H. C. Hui, L. Zhang, G. Birkus, C. G. Boojamra, M. C. Desai, J. L. Douglas, Y. Gao, D. Grant, G. Laflamme, K.-Y. Lin, D. Y. Markevitch, R. Mishra, M. McDermott, R. Pakdaman, O. V. Petrakovsky, J. E. Vela and T. Cihlar, Bioorg. Med. Chem., 2010, 18, 3606–3617 CrossRef CAS PubMed.
- For a comprehensive review, see: (a) U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed. For the challenges and recent progress of controlling P-stereogenic centers, see: (b) X. Ye, L. Peng, X. Bao, C.-H. Tan and H. Wang, Green Synth., 2021, 2, 6–18 CrossRef.
- For selected examples see: (a) B. Simmons, Z. Liu, A. Klapars, A. Bellomo and S. M. Silverman, Org. Lett., 2017, 19, 2218–2221 CrossRef CAS PubMed; (b) D. A. Glazier, J. M. Schroeder, S. A. Blaszczyk and W. Tang, Adv. Synth. Catal., 2019, 361, 3729–3732 CrossRef CAS.
- For selected examples see: (a) F. Pertusati and C. McGuigan, Chem. Commun., 2015, 51, 8070–8073 RSC; (b) K. Tran, G. L. Beutner, M. Schmidt, J. Janey, K. Chen, V. Rosso and M. D. Eastgate, J. Org. Chem., 2015, 80, 4994–5003 CrossRef CAS PubMed; (c) D. A. DiRocco, Y. Ji, E. C. Sherer, A. Klapars, M. Reibarkh, J. Dropinski, R. Mathew, P. Maligres, A. M. Hyde, J. Limanto, A. Brunskill, R. T. Ruck, L.-C. Campeau and I. W. Davies, Science, 2017, 356, 426–430 CrossRef CAS PubMed; (d) E. Cini, G. Barreca, L. Carcone, F. Manetti, M. Rasparini and M. Taddei, Eur. J. Org. Chem., 2018, 2622–2628 CrossRef CAS; (e) M. Wang, L. Zhang, X. Huo, Z. Zhang, Q. Yuan, P. Li, J. Chen, Y. Zou, Z. Wu and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20814–20819 CrossRef CAS PubMed; (f) V. Gannedi, B. Villuri, S. N. Reddy, C.-C. Ku, C.-H. Wong and S.-C. Hung, J. Org. Chem., 2021, 86, 4977–4985 CrossRef CAS PubMed; (g) A. Klapars, J. Y. L. Chung, J. Limanto, R. Calabria, L.-C. Campeau, K. R. Campos, W. Chen, S. M. Dalby, T. A. Davis, D. A. DiRocco, A. M. Hyde, A. M. Kassim, M. U. Larsen, G. Liu, P. E. Maligres, A. Moment, F. Peng, R. T. Ruck, M. Shevlin, B. L. Simmons, Z. J. Song, L. Tan, T. J. Wright and S. L. Zultanski, Chem. Sci., 2021, 12, 9031–9036 RSC.
- See ESI (S3) for details.†.
- Details about reaction can be found in the ESI (S34).†.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01995g |
| This journal is © The Royal Society of Chemistry 2022 |